
Contents
Sigma-1 Receptor-Modulated Neuroinflammation in Neurological Diseases
オンラインで公開2018年9月20日
要旨
シグマ-1受容体(Sig-1R)が多くの神経精神疾患の重要な薬物標的であることを示す多くの証拠がある。シグマ-1Rは中枢神経系(CNS)に富む。神経細胞に加えて、大脳ミクログリアとアストロサイトの両方がSig-1Rを発現している。Sig-1Rs の活性化は、ミトコンドリア機能の促進、酸化ストレスの減少、神経免疫機能の調節など、複数のメカニズムを介して神経細胞の生存を促進し、強力な神経保護効果を引き出すことが知られている。本総説では、神経炎症の調節におけるシグマ-1Rの新たな役割に注目し、神経変性疾患の病態生理や治療におけるシグマ-1R調節性神経炎症の最近の進歩について論じる。
キーワード:シグマ-1受容体、神経炎症、神経疾患、ミクログリア、アストロサイト
序論
シグマ-1受容体(Sig-1R)は小胞体(小胞体)膜貫通型タンパク質である。シグマ-1Rは、シャペロン機能を発揮し、中枢神経系(CNS)における生理・病理学的プロセスを調節する。シャペロン蛋白質であるSig-1Rは、他の蛋白質パートナーとは独立した生物学的機能を発揮しない。Sig-1Rは、ニューロン、アストロサイト、ミクログリアおよびオリゴデンドロサイトを含む脳細胞において広く発現している(Gundlach et al 1986; Alonso et al 2000; Hayashi and Su, 2003a,b, 2004; Gekker et al 2006; Ruscher et al 2011; Zhao et al 2014)。数多くの研究は、Sig-1Rが、カルシウム恒常性およびグルタミン酸活性の調節、反応性種産生の減衰、ERおよびミトコンドリア機能の調節、反応性グリア症の調節、および神経細胞の可塑性を含む複数のメカニズムを介して、神経変性疾患における神経細胞の生存を促進し、神経細胞機能を回復したことを示している(Nguyen et al 2015,2017;RuscherおよびWieloch 2015)。Sig-1Rに加えて、シグマ受容体シグマ-2(Sig-2R)もまた、神経疾患において重要な役割を果たすことが示されている(詳細は、Guo and Zhen, 2015からのレビューを参照のこと)。
神経炎症とは、中枢神経系におけるミクログリア細胞、アストログリア細胞および内皮細胞によって媒介される炎症反応を指す。神経炎症の間、ミクログリアが媒介する炎症性応答は、活性化シグナルに応じて二重の効果を示す(Hanisch and Kettenmann, 2007; Xiong er al)。 一方では、組織の損傷または機能不全に応答するミクログリアの活性化は、ファゴサイトーシスおよびプロ炎症性メディエーターの放出をもたらす。他方では、ミクログリアは、その分極状態に応じて抗炎症性および再生性のミリュウを促進することがある(Patel et al 2013)。例えば、ミクログリアは様々な機能状態とともに顕著な形態学的変化を示し、脳虚血後に親炎症性または抗炎症性の機能を示す。したがって、活性化されたミクログリアは、中枢神経系に対して神経毒性または神経保護作用を示す可能性がある。多くの研究は、神経炎症を標的とすることが、神経変性疾患を治療するための治療的介入を意味することを示唆している。Sig-1Rsがグリア細胞で発現しているという事実は、神経炎症機能障害を伴う中枢神経疾患の治療において、Sig-1Rsが潜在的なターゲットであることを示唆している。本レビュー記事では、主要な神経変性疾患におけるSig-1Rsの神経炎症への影響に関する現在の知見をレビューする。
シグマ-1受容体とアストロサイト
アストロサイトの活性化は神経炎症の重要な特徴と考えられている。障害に応じて、反応性アストロサイトは増殖し、外サイト性グリオトランスミッター、プロ炎症性サイトカイン、およびBDNFなどの神経栄養因子を放出する。Sig-1Rはアストロサイトにおいて高度に発現している(Ruscher et al 2011;Francardo et al 2014)。初代ラットアストロサイトにおいて、Sig-1RアンタゴニストBD1047の前処理は、メタンフェタミン媒介によるSig-1RおよびGFAP発現のアップレギュレーションを阻害する(Zhang et al 2015a)。WTアストロサイトと比較して、Sig-1R KOマウスから分離された一次アストロサイトにおいて、メタンフェタミンはGFAP発現を増加させることができなかった。さらに、アストロサイト由来のラット細胞株であるラットC6グリオーマ細胞において、メタンフェタミンは核内への核内因子κB(NF-κB)p65転座を誘導した。この効果は、BD1047によって逆転した(Zhang et al 2015b)。NF-κB p65シグナル伝達は、神経炎症を含む多様な病理学的プロセスに寄与する。神経障害性マウスでは、BD1047によるSig-1Rの遮断は、おそらく慢性狭窄損傷によって誘導されるアストロサイトの活性化を阻害することを介して、機械的アロディニアを予防する。p-p38の調節がその根底にあるメカニズムである可能性がある(Moon et al 2014)。運動ニューロン変性のマウスモデルでは、Sig-1RアゴニストPRE-084を慢性的に投与すると、反応性アストロサイトーシスも減少した(Peviani et al 2014)。Sig-1R KOマウスから分離された神経細胞-グリア混合培養物において、GFAP発現はWTと比較して増強された(Weng et al 2017)。これらの結果は、Sig-1Rが神経炎症を調節することを示している。一方、炎症性ストレスに応答して、グリア細胞は、傷害を改善するためにBDNFを放出する可能性がある(Béjot et al 2011;Zhang et al 2012;Chen et al 2015)。例えば、新規なSig-1Rアゴニストは、ラット初代皮質アストロサイトからのBDNF放出を刺激した(Malik et al 2015)。一貫して、機能的に選択的なSig-1Rアゴニストとして作用するハロペリドール(還元ハロペリドール)の2つの代謝物は、ヒトアストロサイトを刺激してBDNFを分泌させる。Dalwadi et al 2017)。支援として、最近同定されたSig-1RアロステリックモジュレーターであるSKF83959(Guo et al 2013)が、BDNF産生を刺激することを介して急速に抗うつ効果を誘発することを発見した(Wang et al 2016)。
シグマ-1受容体とミクログリア
脳および脊髄に常駐するマクロファージであるミクログリアは、神経炎症の主要なメディエーターであり、神経変性疾患において有益な役割と有害な役割の両方を果たしている。蓄積された証拠は、神経変性疾患および回復におけるミクログリアの二重の役割を示唆しており、それはミクログリアの機能的表現型に依存する(Hu et al 2015; Xiong et al 2016; Lan et al 2017; Ma et al 2017; Deczkowska et al 2018)。ミクログリアは、最近挑戦されたが、典型的には古典的なM1表現型および代替的なM2表現型に分類される(Ransohoff, 2016)。M1分極したミクログリアは、伝統的に親炎症性で中枢神経系に有害であると考えられているが、M2分極したミクログリアは抗炎症性であり、神経細胞の修復および再生を促進することにより中枢神経系に保護的である。ミクログリア細胞の活性化は、脳卒中、アルツハイマー病 (AD)パーキンソン病(PD)などの多くの神経疾患における病理学的発達に寄与する。M1ミクログリア分極の阻害およびM2分極の促進は、神経疾患および神経変性疾患の創薬および治療のための重要なアプローチと考えられている(Jin et al 2014;Zhang et al 2017b)。末梢免疫系において、Sig-1R活性化は、様々な刺激によって誘導される炎症性応答を強力に抑制する(Bourrié et al 2002)。Sig-1RリガンドSR31747AおよびSSR125329Aは、抗炎症性サイトカインインターロイキン-10(IL-10)のリポ多糖類(LPS)またはブドウ球菌性腸毒素B(SEB)誘発血清放出を増強し、それと同時に、プロ炎症性サイトカイン腫瘍壊死因子-α(TNF-α;Bourrie et al 1995; Bourrié et al 2017b. 1995; Bourrié et al 1996,2002)。) 対照的に、SR31747Aは、LPS刺激されたRAW 264.7マクロファージにおけるNOおよびIL-10放出の減少を用量依存的に誘導した(Gannon et al 2001)。この矛盾した結果は、末梢マクロファージがSR31747Aによる抗炎症性IL-10産生の増加に応答する細胞ではない可能性を示唆している。Sig-1Rは、ミクログリアにおいて発現され(Gekker et al 2006)中枢神経系において抗炎症作用を媒介する。エビデンスは、Sig-1Rがミクログリアの分極を調節しうることを示している。Chaoは、Sig-1RアンタゴニストBD1047でBV-2細胞を前処理すると、メタンフェタミン誘発のM1マーカー(iNOS)とM2マーカーの比率の増加が有意に減少したことを報告した(Chao et al 2017)。試験管内試験研究では、Sig-1Rs刺激がLPSに対するミクログリアの形態学的、遊走性および炎症性応答を抑制したことが示されている(Hall et al 2009)。Sig-1Rのリガンドと考えられるアフォバゾールは、ATPおよびUTPに応答してミクログリアの活性化および遊走を抑制する。アフォバゾールのこれらの効果は、Sig-1RまたはSig-2Rアンタゴニストのいずれかによって減衰された(Cuevas et al 2011)。Sig-1RノックアウトマウスをPRE-084で処置しても、抗神経炎症効果を誘発しなかったという強い証拠がある(Franchardo et al 2014)。同様に、LPS刺激されたマウスBV2ミクログリア細胞を用いて、強力なアロステリックSig-1RモジュレーターSKF83959が、TNF-α、インターロイキン-1β(IL-1β)および誘導性一酸化窒素合成酵素(iNOS)を含むプロ炎症性メディエーターの発現、NOの放出、および活性酸素種の生成を抑制することを最近の報告で示した(Wu et al 2015)。特に、SKF83959の抗炎症作用は、選択的Sig-1Rアンタゴニスト(BD1047またはBD1063)によってブロックされ、Sig-1R介在事象を示唆した。さらに、SKF83959によるミクログリア活性化の抑制は、MAPK/ERKまたはIKK/IκBシグナル伝達経路のいずれかに依存しないことを示した。この知見は、Sig-1Rアゴニストである(+)-ペンタゾシンが、網膜ミクログリアにおいてLPS誘導MAPK/ERK経路の阻害を介してミクログリア活性化を抑制したという報告とは一致しなかった(Zhao et al 2014)。さらに、特定の用量(1μMおよび10μM)のみの(+)-ペンタゾシンは、低酸素/再酸素化条件下のBV2ミクログリアにおいて、ERK1/2経路を介したアポトーシス細胞死を減少させた(Heiss et al 2016)。このことは、Sig-1Rのアロステリックモジュレーターが、受容体アゴニストとは異なるメカニズムでミクログリア媒介の神経炎症を調節していることを示唆しており、興味深い。
シグ-1受容体と脳卒中
脳卒中は、世界的に高い罹患率と死亡率をもたらす重篤な脳血管疾患である。残念ながら、有効な脳卒中治療法は限られている。臨床では,急性期の虚血性脳卒中に対しては,組織プラスミノーゲンアクチベーター(tPA)を用いた血栓溶解療法が唯一の治療法である.しかし、tPAの治療領域が狭いことから、臨床応用には限界がある。そのため,有効な脳卒中治療薬の開発が急務となっている.虚血性脳卒中は,大脳動脈閉塞による局所的な脳虚血を特徴とする脳卒中の主要なサブタイプである.局所的な脳血流の低下と酸素不足が炎症反応を誘発し、脳に不可逆的な損傷を与える。また、脳の再灌流では、血液と酸素の回復が遅れることで、より重篤な炎症反応が誘発され、脳に二次的な損傷を与えることになる。そのため、脳梗塞後の神経炎症を制御することは、脳虚血の治療介入の可能性があると考えられている。興味深いことに、急性脳卒中後のペンブラニューロンにおけるSig-1R発現の増加が最近報告された(Zhang et al 2017a)。
証拠は、Sig-1Rリガンドが脳虚血に対して神経保護効果を発揮することを示唆している。文献は、Sig-Rの活性化によって付与される脳卒中に対する神経保護効果が、炎症性応答の調節に起因し得ることを示している。例えば、脳虚血では、単球化学吸引性プロテイン-1(MCP-1)やIL-1βなどの炎症性サイトカインのmRNAレベルとタンパク質発現が強く増強された。また、Sig-1R アゴニストであるジメモルファンを再灌流開始時に投与すると、NF-κB シグナル伝達経路の阻害に伴い、MCP-1 および IL-1β の mRNA およびタンパク質発現が有意に減少した(Shen er al)。 一貫して、生体内試験でのLPS誘発エンドトキシンショックモデルにおいて、マウスの血漿TNF-α濃度、好中球浸潤、LPSによって誘導される酸化ストレスをジメモルファンで処理することで有意に減少させた。これらの結果は、マウスにおけるLPS誘発エンドトキシンショックに対するジメモルファンの保護効果に、ジメモルファンの抗炎症作用が寄与していることを示唆している(Wang et al 2008)。最近、塞栓性脳梗塞発症後3時間後および24時間後にSig-1RアゴニストPRE-084を投与すると、脳虚血後にプロ炎症性サイトカインを阻害し、IL-10およびIL-4などの抗炎症性サイトカインを増強することにより、梗塞容積が有意に減少し、神経学的欠損が改善されることが示された(AllahtavacoliおよびJarrrott 2011)。この観察と一致して、Sig-1Rの活性化は、実験的脳卒中後の虚血半球のミクログリア/マクロファージにおけるIba1の発現に影響を与える(Ruscher et al 2012)。
興味深いことに、局所脳虚血モデルにおいて、高選択性Sig-1RアンタゴニストであるSIRA(E-52862/MR309)を脳室内または静脈内に投与すると、中大脳動脈閉塞(MCAO)後のマウスの梗塞サイズおよび神経学的障害が有意に減少したことを示す最近の報告でもある。SIRAによって発揮される神経保護効果の根底にあるメカニズムは、メタロプロテアーゼ-9(MMP-9)発現、アストログリア症およびミクログリア増殖の有意な減少と関連している可能性がある(Sánchez-Blázquez et al 2018)。Sig-1Rアゴニストとアンタゴニストが、脳卒中においてどのようにして、そしてなぜ同様の神経保護効果または神経回復効果を産生したのかをさらに研究することは、非常に重要になるであろう。
シグマ-1受容体と外傷性脳損傷(外傷性脳損傷
脳卒中と同様に、外傷後の急性脳損傷も長期的な神経障害を引き起こす可能性がある。最近、制御された皮質衝撃(制御皮質衝撃)モデルを用いた研究が行われ、Sig-1R選択的アゴニストであるPRE-084が、ミクログリアの活性化、タンパク質に対する硝化ストレスおよび酸化ストレスを低減することで、外傷性脳損傷(外傷性脳損傷)において有益な役割を果たすことが示された(Dong et al 2016)。今回の報告では、外傷性脳損傷傷害の15分後にPRE-084をマウスに全身投与し、外傷性脳損傷後28日後の動物の脳浮腫と行動生化学的測定を評価した。その結果、PRE-084は脳浮腫を有意に減少させ、神経学的障害を減衰させた。外傷性脳損傷に対するPRE-084の神経保護効果の根底にあるメカニズムの一つは、Iba1免疫反応の低下に示されるミクログリアの活性化の抑制である可能性がある。しかし、Sig-1 R拮抗薬の併用がPRE-084の神経保護効果を阻害するかどうか、また、ミクログリア/マクロファージの分極状態を区別するかどうかについては、さらなる検討が必要である。
シグマ-1受容体とパーキンソン病
また、Sig-1Rアゴニストの神経保護効果や神経回復効果は、パーキンソン病の実験モデルにおいても報告されている。パーキンソン病におけるSig-1Rの重要な機能的役割はこれまでにも報告されている(Mishina er al)。 最近の研究では、Sig-1Rアゴニストの潜在的な抗パーキンソン効果が報告されている。PRE-084の慢性的な治療は、6-ヒドロキシドパミン(6-OHDA)レシオン化マウスPDモデルにおける運動障害を改善する。CD68陽性ミクログリアおよびマクロファージの数の有意な減少が、PRE-084を投与したマウスにおいて検出された(Francardo et al 2014)。これらの知見は、Sig-1R活性化による神経炎症の減少が、PDモデルにおける機能回復を説明する可能性を示唆している。さらに、パーキンソン病患者および実験動物におけるレボドパ誘発性ジスキネジアにおけるSig-1Rリガンドの潜在的な有益な効果も報告されている(Paquette et al 2009;Fox et al 2017)。このデータは、Sig-1RのモジュレーションがPD治療の新たな薬物標的となりうる可能性を示唆している。
シグマ-1受容体と筋萎縮性側索硬化症
筋萎縮性側索硬化症(ALS)は、脊髄と脳の運動ニューロンが徐々に失われていくことを特徴とする致死的な神経変性疾患である(Boillée er al)。 変異した、誤って折り畳まれたタンパク質の細胞内蓄積は、ALSおよび関連疾患の主要な病理学的特徴である。1993年の画期的な発見は、スーパーオキシドジスムターゼ1(SOD1)の変異がALSの20%を占めることを報告した(Rosen et al 1993)。また、ALSは非細胞自律性疾患として認識されている。変異型SOD1を発現する運動ニューロンは、初期の疾患発症および早期の疾患進行を誘導するが、その後の疾患進行には寄与しない。興味深いことに、変異型SOD1を発現するミクログリアやアストロサイトは、発症時期に影響を与えることなく、発症後の病気の進行を悪化させることが知られている(Ilieva er al)。
中枢神経系では、Sig-1Rは運動ニューロンで高度に発現している(Mavlyutov et al 2010)。若年性ALSではSig-1RのALS原因変異(E102Q)が発見されている(Al-Saif et al 2011)。ニューロ2A細胞における変異型シグ-1R-E102Qの発現は、ミトコンドリアATP産生を低下させ、ミトコンドリア構造を破壊し、ERストレス誘発性神経細胞死を悪化させる(Fukunaga et al 2015)。ミトコンドリア依存性のシグナル伝達は、自然免疫応答および適応免疫応答を制御するので、グリア細胞におけるミトコンドリアの機能不全は、少なくとも部分的には神経炎症を制御する可能性が高い。Sig-1Rの蓄積は、ALS患者のα運動ニューロンのC末端およびER構造の肥大化においても観察され、またSOD1トランスジェニックマウスにおいても観察された(Prause et al 2013)。SOD1*G93AマウスのALSモデルでSig-1Rをノックアウトするとマウスの寿命が短くなり、Sig-1Rの欠乏がALSの病理学的進行を悪化させることが示唆された(Mavlyutov et al 2013)。一方、Sig-1RアゴニストPRE-084の投与は、SOD1*G93AマウスALSモデルにおいてALSの進行を遅らせることが示され、脊髄のミクログリアマーカーIba1-1免疫反応性を有意に低下させたが、アストログリア反応性には影響を与えなかった(Mancuso et al 2012)。さらに、Sig-1RアンタゴニストBD1036との併用投与により、SOD1マウスにおけるPRE-084によって誘導されたミクログリア反応性の低下が逆転した。運動ニューロン疾患モデルのウォブラーマウスを用いた別の研究では、PRE-084を慢性的に投与することで、運動性能に有益な効果が示され、運動ニューロンの生存率が改善された。この観察は他の研究(Ono et al 2014; Tagashira et al 2014)でも支持されており、Sig-1R機能が病態の発生に関与しているだけでなく、ALS治療の有望なドラッグターゲットであることを示している。さらに、PRE-084 SOD1変異マウスを慢性的に治療すると、別のマクロファージ/ミクログリアマーカーCD11bの発現が増加した。特に、M2表現型マーカーCD206陽性細胞数は、ミクログリア/マクロファージマーカーCD68免疫反応性の増加と同時に、PRE-084投与マウスの白質中で有意に増加した(Peviani er al)。 このことから、ミクログリア/マクロファージの分極の調節が、ALSにおけるSig-1R活性化の神経根治効果に関与している可能性が示唆された。ミクログリアの活性化を調節することは、Sig-1RアゴニストのALS治療における有益な効果にも寄与している可能性が示唆された。
シグマ-1受容体とその他の神経変性疾患
また、シグマ-1Rは、アルツハイマー病、多発性硬化症(MS)ハンチントン病にも広く応用されている。最近、新規な選択的アロステリックシグマ-1受容体アゴニストAF710Bは、APPトランスジェニックアルツハイマー病ラットにおいて、おそらく神経炎症抑制を介して認知障害を元に戻すことが報告された(Hall et al 2018)。以前の試験管内試験研究でも、Sig-1R/Sig-2R混合アゴニストであるアフォバゾールが、アミロイドβ25-35に曝露されたラット一次ミクログリアにおいてミクログリア活性化を減少させたことが実証されている(Behensky et al 2013)。MSの実験的自己免疫性脳脊髄炎(EAE)モデルは、中枢神経系における炎症および脱髄を特徴とする。Oxombre et al 2015)は、新規のSig-1RアゴニストがEAEにおける炎症の大きさを減少させることで保護を与えることを示している。
ヒトの脳におけるシグマ-1受容体
北一 et al 2000)は、in situハイブリダイゼーションを用いて、Sig-1R遺伝子がマウス、モルモット、ヒトの脳で発現しており、主に海馬、視床下部、大部分の皮質に分布していることを報告している。1996年以降、Sig-1Rはヒトおよび他のいくつかの種でクローニングされている(Hanner et al 1996;Kekuda et al 1996;Pan et al 1998;Seth et al 1998)。また、疾患リスクの増加とSig-1R遺伝子変異多型との間の相関も報告されている。例えば、Sig-1RのTT-P遺伝子変異がアルツハイマー病に対する危険因子であることが判明している(Fehér er al)。 しかし、この結果を確認するためには、より大きなサンプルサイズの臨床研究が必要である。これまでに、いくつかのSig-1Rアゴニスト(SA4503およびANAVEX2-73)が神経変性疾患の臨床試験に入っている(Lahmy et al 2013;Urfer et al 2014)。これらの取り組みはすべて、神経変性疾患治療のための新しい道筋を提供する可能性がある。
おわりに
Sig-1Rは中枢神経系に広く発現している。Sig-1Rの発現と機能の変化は、多くの神経疾患の病態生理と関連している。Sig-1Rはグリアの機能制御に重要な役割を果たしている。数多くの研究により、Sig-1Rが神経炎症、その結果として神経保護に深い影響を与えることが示されている(図(図1)1.1)。その結果、Sig-1Rは、特にALS、脳卒中、PD、アルツハイマー病などの神経炎症関連疾患の治療薬ターゲットとなる可能性がある。今後の展望としては、特異的かつ高選択性のSig-1RまたはSig-2Rリガンドの開発が必要である。一方、Sig-1RやSig-2Rの機能の複雑さや分布の豊かさを考えると、Sig-1Rのアロステリックモジュレーターは、選択性が高く、かつ不要な影響が少ないことが知られているため、薬剤開発の面でユニークな利点があると考えられる。
図1 シグマ-1受容体(Sig-1R)の神経炎症における潜在的な役割を示す模式モデル
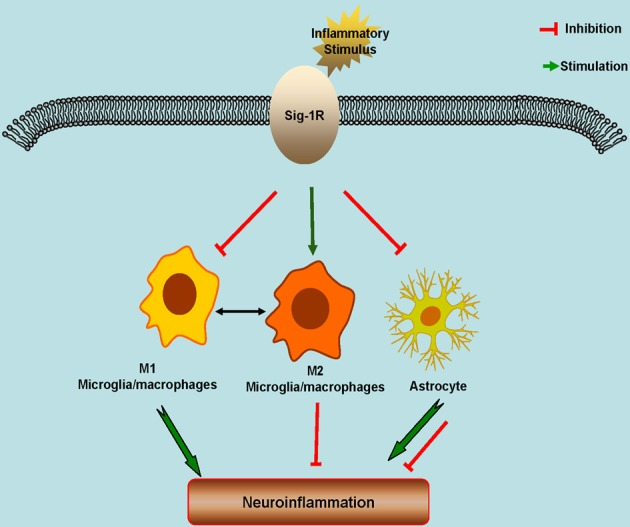
Sig-1Rは、神経細胞、アストロサイト、ミクログリア、オリゴデンドロサイトなどの脳細胞に広く発現している。Sig-1Rの活性化は、M2ミクログリアの修復/再生表現型を促進し、M1ミクログリアの表現型と炎症性刺激に対するアストロサイト応答を抑制する。