
Contents
Roles of sigma-1 receptors in Alzheimer’s disease
www.ncbi.nlm.nih.gov/pmc/articles/PMC4484039/
2015年4月15日にオンラインで公開
要旨
アルツハイマー病は進行性の神経変性疾患であり、世界中で老人性認知症の主な原因となっている。アルツハイマー病は進行性の神経変性疾患であり、世界中で老人性認知症の原因となっている。しかし、シグマ-1(シグマ-1)受容体は、長年にわたり、複数の神経疾患や精神疾患に関与してきた。このレビューでは、シグマ-1受容体の機能に関する現在の理解を論じる。
脂質ラフト、セクレターゼ、キナーゼ、神経受容体、イオンチャネルの調節を通して、シグマ-1受容体は細胞内シグナル伝達、TCAサイクル、酸化ストレス、ニューロン可塑性、神経伝達物質放出などに影響を与える。これらの知見をもとに、シグマ-1受容体とアルツハイマー病との関連性を示唆している。
また、認知機能障害の治療薬として期待されるシグマ-1受容体アゴニストが、アルツハイマー病の進行に伴うアミロイドβ神経毒性に対する強力な神経保護効果と抗健忘症効果を示すことを示すエビデンスを詳細に示している。その証拠は、動物モデル、ヒトでの前臨床試験、および完全な臨床試験から得られている。さらに、この受容体に関する解決すべき疑問も提示されている。
NMDARとの関連では、シグマ-1受容体の活性化は、アルツハイマー病に対する2つの全く異なる影響をもたらす可能性がある。アルツハイマー病の初期段階でのシグマ-1剤の活用は、まだ見落とされていた治療の機会である。本論文は、アルツハイマー病におけるシグマ-1受容体についてのさらなる研究への道を開くかもしれない。
キーワード
シグマ-1受容体、アルツハイマー病、病態、アミロイドβ神経毒性、NMDA受容体
序論
アルツハイマー病は、進行性の認知機能障害と行動障害によって特徴づけられる、陰湿な発症を伴う神経変性疾患である。これまでのところ、アルツハイマー病の最も広く受け入れられている病態は、アミロイドβ沈着と高リン酸化タウ蛋白質の神経原線維のもつれである。しかし、既存の薬剤では認知障害を効果的に逆転させることはできない。最近では、シグマ-1受容体が認知機能の改善、特に抗健忘症作用と神経保護作用を示すようになっていた[1]。死後早期の研究では、アルツハイマー病患者の海馬CA1領域でシグマ-1受容体が減少していることが報告されている[2]。その後、三科は、(11C)SA4503を用いたポジトロン断層撮影法(PET)を用いて、アルツハイマー病の初期段階でシグマ-1受容体が消失していることを発見した。前頭葉、側頭葉、後頭葉、小脳、視床で結合電位が44~60%有意に低下していた[3]。このようなシグマ-1受容体密度の変化をもとに、以下の研究では、シグマ-1受容体アゴニストがアルツハイマー病の認知機能障害を有意に低下させることが確認されている。このように、アルツハイマー病の進行におけるシグマ-1受容体の効果と治療の展望を浮き彫りにすることを目指している。
シグマ-1受容体の特徴と生物学的効果
シグマ(シグマ)受容体は、オピオイド受容体のサブタイプとして初めて同定された[4]。オピオイド受容体やフェンシクリジン結合部位との違いを提案したQuirion R以降、独自に受容体ファミリーを確立した[5]。シグマ受容体は体内、特に中枢神経系に豊富に存在し、脊髄に高密度に分布している。脊髄、ポンズ、延髄、赤核、小脳、海馬に高密度に分布し、大脳皮質、視床下部には中密度に、大脳基底核、視床部には低密度に分布している[6]。シグマ1受容体とシグマ2受容体を比較した研究では、大きさ、分布、リガンド親和性に劇的な違いがあることが明らかになった[7]。現在までに、シグマ-1受容体はモルモットとヒトでクローン化されており[8,9]、ラットとマウスでもクローン化されている[10,11]。その遺伝子は223アミノ酸のタンパク質をコードしており、2つの膜貫通ドメインを持ち、短いN末端付近に典型的な小胞体局在シグナルを持っている[12]。しかし、これまでのところ、この受容体に特異的に結合することができる哺乳類のタンパク質は存在しない。最近、Izzolらは、アミロイドβ1-42がシグマ-2/PGRMC1受容体に結合した後、シナプス毒性を示すことを発見した[13]。しかし、一般的には、アルツハイマー病の進行にはシグマ-1受容体がより重要な役割を果たしていると考えられている。
平常時、シグマ-1受容体は主にミトコンドリア関連小胞体膜(MAM)上に局在し、カルシウムイオンに対して高い感度を持つBipシャペロン構造を形成している。コカインや鎮痛剤などのアゴニストによって活性化されると、シグマ-1受容体はBiPから分離し、MAMから細胞の他の部分イノシトール三リン酸(IP3)受容体、N-メチル-D-アスパラギン酸受容体(NMDA)受容体、ドーパミン(DA)受容体、イオンチャネルの調節を通して、シグマ-1受容体はTCAサイクルに影響を与えることができる。酸化ストレス[14]、ミトコンドリア機能、神経細胞の可塑性、5-ヒドロキシトリプタミン、グルタミン酸、ドーパミン、ノルエピネフリン、アセチルコリン、γ-アミノ酪酸などの神経伝達物質の放出[15]。
アルツハイマー病の進行におけるシグマ-1受容体の潜在的なメカニズム
アルツハイマー病の病因や病態に関するエビデンスはここ数十年の間に蓄積されていたが、その正確な原因は完全には解明されなかった。ここでは、シグマ-1受容体とアルツハイマー病を結びつける重要な細胞メカニズムを示唆している(図1)。
図1 アルツハイマー病の職業におけるシグマ-1受容体の可能性のメカニズム
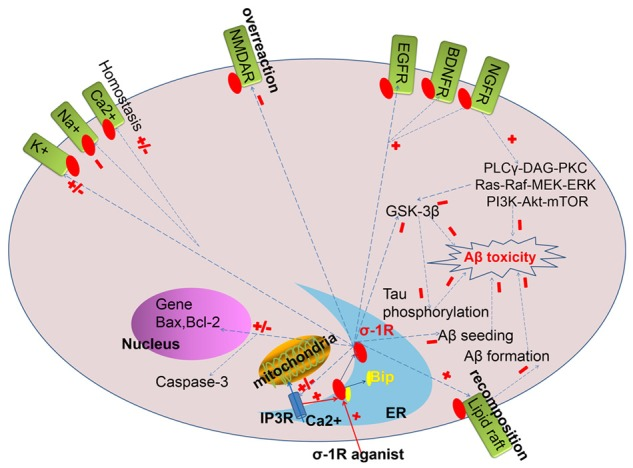
アミロイドβカスケード仮説
アルツハイマー病は遺伝的に不均一性を有する多遺伝子遺伝性疾患と考えられており、一般的には家族性アルツハイマー病と散発性アルツハイマー病に分けられる。現在では、プレセニリン-1(PS-1)プレセニリン-2(PS-2)アミロイド前駆体タンパク質(APP)の3つの常染色体優性遺伝子が若年性アルツハイマー病と関連していることが明らかになっている。しかし、遅発型アルツハイマー病の素因遺伝子は、APOEε4(APOEε4)という1つの遺伝子のみが普遍的に認められている。最近では、大規模なデータセットで全ゲノムを解析する新しい技術により、ABCA7,BIN1,CASS4,CD33,CD2AP、HLA-DRB5-DBR1などを含む、遅発性アルツハイマー病リスクに関連する新しい遺伝子が明らかになっていた[16]。残念ながら、現在までのところ、アルツハイマー病関連遺伝子とシグマ-1受容体を組み合わせたエビデンスはない。アルツハイマー病発症リスクとシグマ-1受容体遺伝子の変異多型との相関関係についての研究は数件しか報告されていない。いくつかの典型的な研究については、本稿の後続のセクションで議論する。発見された4つのアルツハイマー病遺伝子のうち、APP、PS-1,PS-2の変異はアミロイドβの生成を増加させ、APOEε4はアミロイドβの沈着をもたらすことがわかっている。アミロイドβの産生とクリアランスのアンバランスが神経細胞の変性や認知症の初期イベントであることを示唆しており、これはアルツハイマー病につながる病態の他の原因の共通経路でもある。
アミロイドβ神経毒性を緩和するシグマ-1受容体の性質を説明するために、様々なメカニズムが考えられる。
- シグマ-1受容体は、脂質ラフトの再構成を調節し、アミロイドβフィブリン形成を阻害することにより、アミロイドβ毒性に対する効果的な保護を有する可能性がある[17]。アスパラチルプロテアーゼβ-セクレターゼ(BACE-1)およびγ-セクレターゼ[18]の相当量は、アミロイドβ産生が優先的に起こる脂質ラフトに局在している。さらに、アミロイド線維形成の開始に関与していると提案されている種であるGM1ガングリオシド結合アミロイドβタンパク質(GM1/アミロイドβ)もまた、細胞膜上の脂質ラフト内に関連している[19,20]。慢性的に活性化されたシグマ-1受容体は、小胞体(小胞体)から細胞膜上の脂質ラフトへと移動し、脂質ラフトの再結合に関与し、タンパク質結合型受容体のシグナル伝達、タンパク質の生合成や内分泌輸送などの細胞機能に影響を与える可能性がある[21]。したがって、持続的なシグマ-1受容体の活性化は、アミロイドβの産生や輸送に積極的に関与する結合部位である脂質ラフトに優先的に移動する可能性がある。
- 海馬ニューロンにおけるアミロイドβ25-35誘導性アミロイドβ1-42の播種を減衰させる[22]。アミロイドβ25-35の投与は、APP発現の有意な増加とβ-セクレターゼ経路の活性化を引き起こし、その結果、海馬の内因性アミロイドβペプチド含有量を増加させ、これをアミロイドβ播種と呼ぶ[23]。 シグマ-1受容体は、Aktとグリコーゲン合成酵素キナーゼ-3β(GSK-3β)活性を調節することにより、このプロセスをブロックすることができる[22]。
- 海馬や前頭前野などの様々な脳領域におけるNMDAR応答を介したアミロイドβに対する認知保護を促進する[24-26]。化合物を直接脳内に全身投与するか、血液脳関門を越えてNMDA受容体の機能を薬理学的に阻害すると、空間学習や受動的回避学習が障害される[27,28]。シグマ受容体は、海馬背側CA3[29]領域の錐体ニューロンをNMDA(興奮性活性化)とNMDA依存性CA1シナプスに増強する[30]ことができ、学習能力や空間記憶の記憶に不可欠である。また、シグマ-1受容体はアミロイドβに関連したNMDARの神経毒性を防ぐことができる[31]。
- シグマ-1受容体は小胞体だけでなく、核膜やプラズマ膜、ミトコンドリアにも局在しており、その結合部位はカルシウム動員の調節に関与している[32-36]。さらに、シグマ-1受容体は、細胞内カルシウムの恒常性を維持することで、ミトコンドリアや神経の生存性を保護している。
- シグマ-1受容体の過剰発現は、PLCγ-DAG-PKC、Ras-Raf-MEK-ERK、PI3K-Akt-mTORシグナル伝達経路を制御することにより、脂質ラフトに関連したNGF、EGF、BDNF効果[1]やNGF誘発神経保護を増強する[37]。脂質ラフトに局在する成長因子受容体[21]が発見されて以来、シグマ-1受容体の持続的な活性化は細胞の生存率に有益であり、したがって、アミロイドβ神経毒性に対する神経保護または神経細胞の回復を促進する可能性がある。さらに、シグマ-1受容体は、海馬における軸索および樹状突起の成長を促進することができる[38]。
- プロアポトーシス遺伝子Baxおよび死滅プロテアーゼcaspase-3の発現を減少させることにより、アミロイドβ25-35誘発アポトーシスを緩和する一方で、抗アポトーシス遺伝子Bcl-2レベルを温存し、結果として細胞生存率を向上させる。
タウタンパク質仮説
広く神経系で発現し、タウタンパク質は、微小管アセンブリを触媒し、微小管構造を安定化するために、微小管関連タンパク質である。アルツハイマー病の定義病理学的特性であること、タウ症は、タンパク質分解酵素と過リン酸化タウ、ペアらせん状フィラメント(PHF-タウ)神経原線維タングル(NFTs)堆積物と、その結果、神経細胞死の後続の形成への抵抗性の増加で構成されている可能性がある。グリコーゲン合成酵素キナーゼ-3(GSK-3)は、タウの高リン酸化を媒介する主要なキナーゼであることが示されている[22]。しかし、GSK-3の過剰活性化の根底にある分子過程と、その潜在的な関連性は明らかにされていない。(i) P(Ser473)-Akt/Akt比の低下を阻止し、(ii) アミロイドβインキュベーションで抑制されたPI3K/AktおよびPLCγ/PKCシグナル伝達経路[39]を促進することにより、GSK-3βのSer9残基をリン酸化する。このようにして、神経細胞の細胞骨格の安定性を維持するために、タウの高リン酸化と神経原線維のもつれを緩和することが可能となった。
神経伝達物質仮説
アセチルコリン放出の調節
コリン作動性神経伝達物質は、認知機能の根底にある主要なプロセスであり、特に記憶の保存に重要な役割を果たしている。前脳基底部、基底核、大脳皮質、扁桃体複合体、海馬のコリン作動性ニューロンは、学習や記憶の形成に不可欠であることが観察されている[40,41]。正常な老化では、いくつかの皮質コリン作動性活性はわずかに失われるが、アルツハイマー病や関連する認知症の患者ではアセチルコリンレベルが著しく低下するが、これはコリンアセチルトランスフェラーゼ(chAT)とアセチルコリンエステラーゼ(AchE)の対応する減少に起因する可能性がある[42-44]。
試験管内試験でも生体内試験でも、シグマ-1受容体はアセチルコリン放出の強力な調節因子である。これらのタンパク質は、ラット前頭皮質および海馬からの3H-アセチルコリンのKCl誘発または電気的誘発放出をアップレギュレートすることができる[45-48]。一方、線条体でのアセチルコリン放出はわずかに影響を受けていた[49-51]。したがって、AChE阻害薬で見られる望ましくない副作用が少ないシグマ-1受容体は、アルツハイマー病に関与しているコリン作動性のアミロイドβ誘発性機能障害を緩和すると考えられる[52]。
グルタミン酸放出の調節
コリン作動性のよく知られた欠損に加えて、神経伝達物質であるグルタミン酸もまた、アルツハイマー病において減少することがある。シグマ-1受容体はグルタミン酸レベルを調節するための戦略を表している。(i) 脳由来神経栄養因子(BDNF)に関連したグルタミン酸放出は、薬理学的活性化だけでなく、シグマ-1受容体の過剰発現によっても増強される。これは、PLCγ/IP3経路の関与を介して起こると考えられる[55]。(ii)小胞体から放出される細胞内Ca2+レベルの上昇は、自発的なグルタミン酸放出に重要な役割を果たしている[56,57]。(iii) また、この効果はα-1アドレナリン受容体とドーパミン受容体を介して媒介されているようである[57,58]。
このように、前頭皮質と海馬のシグマ-1受容体によるグルタミン酸レベルの調節は、抗健忘症作用の基礎となる付加的なメカニズムである可能性がある。
炎症仮説
McGeerら[59]は、アルツハイマー病はおそらく免疫応答と炎症応答の不適切な活性化に起因すると提唱しているので、アルツハイマー病における疾患修飾レジメンの導入のための機会の窓が表示される。炎症病理は細胞外アミロイドβ沈着物から生じ、後にタウの凝集体によって増強されることがある。活性化されたミクログリア[60]、アストロサイトおよび炎症性因子によって駆動され、過剰反応は、疾患が進行するにつれて増加し、最終的には、正常な神経組織を攻撃するために「間違った方向」に結果をもたらし、シナプスの損傷およびニューロンの死を引き起こす。
中枢免疫系の免疫球に豊富に発現しているシグマ-1受容体の活性化は、プロ炎症性および抗炎症性の恒常性を維持するだけでなく[61]、神経細胞の生存性を維持することができる[62]。考えられるメカニズムは以下の通りである。
- アミロイドβ25-35誘発アポトーシスからミクログリアの活性化と損傷を減少させる;ミクログリアは器官型海馬スライスに存在する唯一の免疫細胞型である。細胞内カルシウムシグナルを遮断することで、細胞骨格再配列、遊走、サイトカイン産生などのミクログリア活性化の多くの側面が抑制された。また、リポ多糖類(LPS)単球化学吸引性タンパク質1(MCP-1)アデノシン三リン酸(ATP)などの活性化剤は、ミクログリアに対する活性化効果を失う可能性がある[63]。
- 抗炎症性サイトカインIL-10を増強することでT細胞介在性免疫を予防する[64]。
要約すると、シグマ受容体を欠くと、神経細胞はアミロイドβ媒介アミロイド毒性に対してより脆弱になる。細胞内脂質代謝、細胞骨格ネットワーク、免疫応答がより損傷を受けやすくなり、その結果、アルツハイマー病の病因に大きく寄与する神経変性と酸化ストレスによる神経死を加速させることになる。
シグマ-1受容体とアミロイドβ神経毒性に関する研究
アルツハイマー病の特徴的な変化は、タウタンパク質の高リン酸化によるアミロイドβ沈着と神経原線維のもつれからなる。アミロイドβの神経毒性は凝集状態と可溶性オリゴマーβの両方に由来しており、タウタンパク質の過リン酸化を刺激する能力を持つ細胞外アミロイドβ凝集がアルツハイマー病の主要な分子機構と考えられている。しかし、アミロイドβがどのようにしてシグマ受容体の発現に作用しているのかはまだ明らかになっていない。現在のところ、アルツハイマー病を伴うシグマ受容体の研究は、主に内因性リガンドと外因性リガンドを含む受容体リガンドに限定されている。
内因性リガンド
ニューロステロイドは、デヒドロエピアンドロステロンや黄体ホルモンなどのシグマ受容体の内因性リガンドである可能性がある[66]。シグマ受容体に対するレベルが不十分で親和性が低いことから、内因性リガンドの称号は議論の的となっている[67]。プレグネノロン(PREG)とデヒドロエピアンドロステロン(DHEA)は、生理的条件下でシグマ-1受容体リガンドであり、コリン作動性とNMDAR依存性の方法で記憶力と学習能力を向上させることができる[56]。海馬におけるPREGのレベルは、ネズミや高齢ラットの記憶能力と強い相関があることが早くから実証されている[52]。例えば、マウスの海馬と扁桃体にPREGSを訓練後に注射すると、足の衝撃による能動的回避訓練の記憶力が向上することが明らかになった[68]。一方、DHEAの投与は、アミロイドβ25-35によって誘発される記憶障害を改善する可能性もある。特に、DHEAは軸索、樹状突起の増殖を改善し[38]、さらには神経幹細胞の生存率を改善する[69]。
ニューロステロイドによる正の認知効果の正確なメカニズムは完全には解明されていないが、内因性薬物は、γ-アミノ酪酸型A(GABAA)受容体[70]、N-メチル-アスパラギン酸(NMDA)受容体[71]、および電位依存性Ca2+チャネル[72]などの一連のイオンチャネルを調節していることが示唆されている。Meyer DA [56]は、PREGSが選択的にシナプス前端末からのmEPSCsとグルタミン酸放出の頻度の頑健な増加につながることを発見し、これはシナプス前Gi/oタンパク質結合シグマ-1様受容体の活性化によって引き起こされる細胞内Ca2+レベルの上昇に依存している可能性がある。さらに、PREGは最近、アミロイドβ25-35マウスにおけるPI3K/AktおよびRas/ERKシグナルの減少を救済することが示されている[31]一方、DHEAはシグマ-1受容体を活性化し、アミロイドβ25-35障害を受けた新生児ニューロンにおけるPI3K-Akt-mTOR-p70S6kシグナルの変調を介して神経突起の成長を促進する[37]。
外因性リガンド
外因性シグマ-1受容体アゴニストは、薬理学的および病理学的モデルの両方において、潜在的な抗健忘症および神経保護効果を示す。Marrazzo, Aは、最初にシグマ-1受容体アゴニストPRE-084とMR-22(-)が、NMDA受容体遮断の関与なしに、アミロイドβを介した皮質ニューロンの毒性を減少させ、アルツハイマー病の進行を遅らせることができることを証明した[73]。彼らは、シグマ-1タンパク質が興奮死以外の神経変性過程を阻害する可能性があると予測した。これに基づき、軽度から中等度のアルツハイマー病の対症療法として認可されているドネペジルは、コリン作動性とシグマ-1アゴニスティック作用により、アミロイドβおよびグルタミン酸の毒性を減衰させ[74]、軸索増殖を増強させることが明らかになった[1]。さらに、アフォバゾールは、アミロイドβに反応してBax遺伝子、Bcl-2発現、死滅プロテアーゼカスパーゼ-3レベルを調節することで、ニューロンのアポトーシスを緩和することが知られている。さらに、アフォバゾールはアミロイドβ25-35で培養したミクログリアにおいて、ミクログリアの活性化を低下させ、ATPシグナル伝達の破綻を抑制することから、アミロイドβ曝露後のミクログリアの機能を強力に保持することが示唆された。また、アフォバゾールは細胞内の炎症性および抗炎症性のホメオスタシスを維持し[61]、NGFによる神経突起の増殖を促進する[75]。最近では、シグマ-1/ムスカリン受容体の混合リガンドであるANAVEX2-73が、アルツハイマー病におけるGSK-3βの過剰活性化を効率的に減少させ、タウの過剰リン酸化とアミロイドβ1-42シードの両方を抑制することが明らかにされている[22]。まだ(+)ペンタゾシンとSA4503は、二相ベル型用量応答曲線の薬理学的にアミロイド負荷を緩和している。上記のシグマ-1受容体の保護効果は、ハロペリドールやBMY-14802のようなシグマR拮抗薬によって大部分が遮断される。
また、外因性リガンドの認知機能改善への役割については、自由に動くラットの生体内微小透析により、(+)-SKF10,047が前頭皮質と海馬の細胞外アセチルコリンを立体選択的に増加させることが示されており、これはハロペリドールによっても阻害されることが示されている[48,76]。また、シグマ-1アゴニストは、コリン作動性破壊、アミロイドβ投与、正常老化、老化促進マウス(SAM)グルタミン酸/セロトニン作動性、カルシウムチャネル欠損などの薬理学的および病理学的モデルにおいて、抗健忘症効果を示している[1]。興味深いことに、ほとんどの行動テストにおいて、シグマ-1受容体リガンドは対照群の学習能力を促進したり阻害したりしないことから、シグマ-1受容体が活性化されるのは病理学的条件下であることが示唆されている[1]。
全体として、シグマ-1受容体アゴニストは、認知機能障害の治療薬として有望な化合物であり、アミロイドβ神経毒性に対する強力な神経保護効果と抗健忘症作用を示している。
シグマ-1受容体と精神病症状に関する研究
後天的な認知機能障害の特徴に加えて、アルツハイマー病は患者の職業上の社会活動や家族活動に徐々に悪い影響を与える。アルツハイマー病後期には行動障害、攻撃性、幻覚が現れるのに対し、様々なタイプの抑うつ性障害や不安障害のような初期の非認知的表現が発現することがある[77]。これらの精神症状に対して有効な臨床薬はまだ存在しない。シグマ受容体、特にシグマ-1サブタイプの役割は、神経精神疾患における病態生理の標的として長い間同定されてきた。しかし、アルツハイマー病精神病症状に対するシグマ-1受容体の前臨床および臨床的証拠は、現在のところ乏しい。
行動モデルは、シグマ-1受容体に結合するいくつかのリガンドが「抗うつおよび抗不安」のような特性を有することを示唆している[78]。Urani A [79]は、アルツハイマー病ラットに条件付恐怖ストレス試験を実施したところ、イグメシンと(+)-SKF-10,047がストレスによる運動抑制を有意に減少させることを示し、外因性のシグマ-1受容体アゴニストがアルツハイマー病に関連した抑うつ症状を緩和する可能性があることを示唆している。また、抗精神病活性を有するシグマ-1受容体拮抗薬であるBMY-14802は、ラペー核内の体外性5-HT(1A)受容体との直接的な相互作用により、ラペー後面および海馬からの5-HT放出を減少させることで、抗不安作用を示す可能性がある[80]。一方、OPC-14523の急性抗うつ薬様作用では、シグマと5-HT(1A)受容体の相乗的な刺激が要求されている[81]。
いくつかの抗精神病薬の初期臨床試験では、シグマ-1タンパク質に対して一定の親和性が示されている[82]。例えば、シグマ-1受容体拮抗薬であるハロペリドール[83]は、焦燥感、敵意、攻撃性を抑制する効果がより高い。また、シグマ-1受容体に高い親和性を有するパナメシン(EMD 57445)[84]は、抗精神病作用を有し、錐体外運動器系(EPMS)に関連する副作用がない。さらに、中等度から重度のアルツハイマー病[86]で使用が許可されているメマンチン(10μM)[85]は、NG108-15神経芽腫細胞において、ブラジキニンによる細胞内Ca2+の動員を改善することが示されており、PRE-084(1μM)に対するシグマ-1の効果を模倣したものである。シグマ-1に親和性の高いクロルプロマジンやネモナプリドなどの抗精神病薬はまだ多く存在している。
このように、シグマ-1受容体リガンドは、単体でもアジュバントとしても、アルツハイマー病に伴う精神病症状に対して有望な効果を発揮する可能性がある。しかし、その治療効果を確認するためには、大規模な二重盲検無作為化プラセボ対照臨床試験が必要である。
アルツハイマー病におけるシグマ-1受容体の臨床研究と応用
アルツハイマー病発症リスクとシグマ-1受容体遺伝子変異多型との相関に関する研究はあまりない。最初の研究である日本人の症例対照試料[87]では、TT-Pハプロタイプがアルツハイマー病の保護因子であることが示された。しかし、その後のポーランドでの研究では、この知見を検証することはできなかった[88]。最近、A. Feher [89]は、シグマ-1受容体のTT-P遺伝子変異がアルツハイマー病の危険因子であることを発見した。これらの観察結果は一貫性がなく、より大きなサンプルサイズと民族的類似性の高い臨床研究が必要であることを示唆している。
選択的セロトニン再取り込み阻害剤(SSRI)およびシグマ-1受容体に対するフルボキサミンは、Izzo, N.J.によって、アルツハイマー病患者におけるせん妄[90]を緩和するための代替的なアプローチであると考えられている。しかし、うつ病や統合失調症ではいくつかの症例報告があるものの、フルボキサミンがアルツハイマー病患者の認知障害に治療効果があることを示す臨床的証拠はない[91,92]。セルトラリンやパロキセチンを含む他のSSRIとは対照的に、フルボキサミンは強力なシグマ-1受容体アゴニストとして、Akt-1のリン酸化を増加させることでアミロイドβ媒介の神経毒性を減少させる可能性が示唆されている[93]。現在までに、少数のシグマ-1受容体アゴニスト(SA4503およびANAVEX2-73)[22,94,95]のみが、急性/慢性神経変性疾患を対象とした第II相臨床試験を開始している。
したがって、前向きな実験結果が増加しているにもかかわらず、疾患過程の早期にシグマ-1剤を利用することは、依然として見過ごされている治療機会である。
アルツハイマー病におけるシグマ-1受容体の役割をめぐる論争
最近、Tackenbergらは、アミロイドβがNR2B/NR2Aを含むNMDARが関与する経路を介してタウ依存性の神経変性や樹状突起の喪失を誘導することを提唱している[96]。その後、Shaらは、シグマ-1受容体ノックアウトマウスにおいて、NR2Bのリン酸化を減少させることでNMDARの作用がダウンレギュレーションされることを証明した[97]。したがって、アルツハイマー病脳のシグマ-1受容体欠損マウスでは、NR2Bのリン酸化を減少させることで、NMDARを介したアミロイドβ増強Ca2+流入を減少させ、NMDARを介した神経毒性を防ぐことができると予測され、最近Yin, J.らによって証明された。Yin, J.らは、シグマ-1受容体欠損がアミロイドβ25-35誘導海馬ニューロン死および空間認知欠損を減衰させることができることを初めて、生体内試験での証拠を提供している。本研究におけるシグマ-1受容体欠損またはシグマ-1受容体遮断のいずれかが、アミロイドβ25-35誘発性神経細胞死を有意に減少させることができる。逆説的であるが、アミロイドβ25-35/1-42マウスにおけるシグマ-1アゴニストの神経保護効果については膨大な報告がある[98-100]。また、PREGSは培養海馬ニューロンにおいてNMDA誘導性興奮毒性を増幅することが観察されているが[101]、この矛盾は和解するのは困難である。この矛盾した効果を説明するために、シグマ-1受容体の活性化の正確なタイミングが提起された。シグマ-1受容体アンタゴニストをアミロイドβ25-35後48時間以内に投与すると、NMDARを抑制してアミロイドβ神経毒性をブロックすることができる。しかし、アミロイドβ25-35注射の72時間後には、ERK/PI3K-Aktシグナル伝達カスケード[31]を増強したり、酸化ストレス[102]のレベルを低下させたりすることで、シグマ-1の活性化によって神経細胞の損傷を緩和することができる。また、シグマ-1受容体欠損によるPKCのダウンレギュレーションや脂質ラフトの再編成がNR2Bサブユニット含有NMDARを抑制することも提案している[103]。
しかし、シグマ-1受容体がアルツハイマー病に及ぼす悪影響についてのエビデンスは十分ではない。また、伝統的に両刃の剣とされてきたNMDARを介した応答は、この数十年の間に、シグマ-1受容体との相互作用について議論の的となっていた。例えば、ハロペリドール、(+)-ペンタゾシン、(+)-SKF-10,047,DTGなどのシグマリガンドは、Xenopus卵母細胞のNMDAR電流を抑制する[104]。また、久米らは、NMDARに親和性を持つシグマ-1受容体リガンドが、NMDARを介したCa2+の流入を抑制することで、ニューロンプロテクターとして作用する可能性を示唆している[105]。しかし、神経ステロイドであるPREGはシグマ-1アゴニストとして、NMDARの機能を双方向に制御している。その結果、PREGSはNMDARを介してNR2Bチロシンのリン酸化とCa2+の流入を抑制し、シナプス可塑性に関与するNMDAR依存性長期増強(LTP)に重要なERK/CREB経路をカスケードしていることが示唆された[106]。上記の結果とは逆に、シグマ-1 受容体は NMDAR 応答の促進因子であると考えられる多くの証拠がある。シグマ-1受容体は、嗅球摘出術によって誘発されたNMDA障害行動を逆転させることが早くから実証されている[107]。さらに、シグマ-1受容体は、背側海馬CA3領域の錐体ニューロンをNMDAに誘導(興奮性活性化)し[29]、NMDA依存性CA1シナプスを増強し[30]、学習能力や空間記憶の記憶に不可欠であると考えられている。プロゲステロンやテストステロンのような非ステロイドホルモンは、シグマ-1のアンタゴニストとして作用し、結果としてNMDAを介した応答のアンタゴニストとして作用する[1]。一般的に、シグマ-1受容体はNMDARの機能に対して二方向の制御を行っている。しかし、シグマ-1受容体がどのようにしてNMDARを介した応答に作用するのか、そのメカニズムはまだ解明されなかった。Martinaは、ラット海馬の小コンダクタンスCa2+活性化K+電流(SKチャネル)を遮断することで、シグマ-1がNMDARのシナプス伝達と可塑性を調節していると考えている[108]。一方で、これはサブユニットの構成の複雑さや局在の変化に起因するNMDARの応答のさらなる多様性に起因している可能性がある。シナプス外NMDARは、サブユニットNR2A>NR2B [109]の数を持ち、核内カルシウムシグナル伝達経路を活性化してシナプス可塑性を促進し、神経興奮性を向上させることができる。その代わり、サブユニットNR2A<NR2Bの数を持つシナプス外NMDARの活性化は、神経細胞にカルシウム過負荷を引き起こし、アポトーシスまたは死の経路を開始する[110,111]。したがって、NMDAR自体については、この2つの異なる経路のアンバランスが神経変性疾患、特にアルツハイマー病の病態を引き起こす可能性がある。
したがって、NMDARに関わる場合、シグマ-1受容体の活性化は、アルツハイマー病に対して2つの全く異なる影響をもたらす可能性があることを提案する。
- (a) NMDAR依存性の学習・記憶の調節による認知改善の促進、
- (b) アポトーシスや死の経路におけるNR2Bサブユニットを含むNMDARによるアミロイドβ介在性神経毒性の悪化、
である。アルツハイマー病とNMDARサブタイプの両方の応答に対するその二重効果の正確なプロセスを評価するために、さらなる研究が緊急に必要である。
おわりに
シグマ受容体は、脳の細胞分化、神経保護、神経可塑性、抗健忘症に関与していることが明らかになってきており、認知障害の治療への可能性が示唆されている。しかし、この受容体に関するいくつかの疑問は未だに残されている。現在、臨床応用されているシグマ受容体リガンドがない。神経変性疾患の第Ⅱ相臨床試験に入ったシグマ-1アゴニストはわずかである。
アルツハイマー病とシグマ受容体遺伝子多型の変異との相関研究の結果はまだ統一されていない。NMDAR反応に関しては、シグマ受容体がNMDAR依存性の神経アポトーシスを悪化させるかどうかについては、さらなる研究が必要とされている。さらに、アルツハイマー病の初期段階でシグマ-1受容体の密度が低くなる原因はまだ不明である。
アミロイドβがどのようにしてシグマ受容体の発現に作用しているかについてはほとんど知られていないため、シグマ受容体に関する既存の研究は、内因性リガンドと外因性リガンドに限定されている。また、特定の内因性リガンドについては、深く研究されていない。
しかし、シグマ受容体リガンドは、単体でもアジュバント剤としても、中枢神経系の変性疾患、特にアルツハイマー病関連の認知機能障害に有効であり、現在入手可能なAchEIs/メマンチンに代わる薬剤として期待されている。アミロイドβがどのようにしてシグマ受容体の発現に作用するのか、そのメカニズムはアルツハイマー病の発症に向けて新たな方向性を示している。