
Contents
- Role of Nicotinamide Adenine Dinucleotide and Related Precursors as Therapeutic Targets for Age-Related Degenerative Diseases: Rationale, Biochemistry, Pharmacokinetics, and Outcomes
Role of Nicotinamide Adenine Dinucleotide and Related Precursors as Therapeutic Targets for Age-Related Degenerative Diseases: Rationale, Biochemistry, Pharmacokinetics, and Outcomes
www.ncbi.nlm.nih.gov/pmc/articles/PMC6277084/
2018年11月30日オンライン公開
要旨
意義
ニコチンアミド・アデニン・ジヌクレオチド(NAD+)は、必須のピリジンヌクレオチドであり、酸化的リン酸化および ATP 産生、DNA 修復、エピジェネティックに修飾された遺伝子発現、細胞内カルシウムシグナル伝達、および免疫学的機能に関与する多くの重要な細胞プロセスのための必須の補酵素および基質として機能する。
NAD+枯渇は、フリーラジカルや紫外線攻撃による過剰なDNA損傷、ポリ(ADP-リボース)ポリメラーゼ(PARP)活性化、高ターンオーバー、およびそれに続くNAD+枯渇、および/または慢性免疫活性化および炎症性サイトカイン産生によるCD38活性の促進およびNAD+レベルの低下のいずれかに応答して起こる可能性がある。
最近の研究では、NAD+レベルを高めることで、脳を含む異化組織における酸化細胞損傷を大幅に減少させることができることが示されている。したがって、細胞内NAD+同化の促進は、一般的に加齢に伴う変性疾患の有望な治療戦略を表し、健康的なサーチュイン活性の複数の利点を効果的に実現するために不可欠である。キヌレニン経路は、哺乳類細胞におけるde novo NAD+合成経路を代表する。NAD+は、NAD+サルベージ経路によっても産生され得る。
最近の進歩
このレビューでは、変性疾患状態および生理学的老化におけるNAD+の減少を減衰させることにおける、NAD+前駆体である
- ニコチンアミド(NAM)
- ニコチン酸(NA)
- ニコチンアミドリボシド(NR)
- ニコチンアミドモノヌクレオチド(NMN)
の有効性および利点に関する最近の知見を記述し、議論する。
重要な課題
近年得られた結果は、NAD+前駆体がいくつかの疾患において重要な保護的役割を果たし得ることを示している。しかしながら、いくつかの場合には、これらの前駆体は、NAD+同化経路内の位置を介してNAD+合成を増強する能力が異なることがある。NAD+合成の増加は細胞の保護反応を促進し、NAD+がいくつかの生化学的経路に関連する調節分子であることをさらに示している。
今後の方向性
今後数年のうちに、NAD+前駆体を用いた個別化治療の洗練化と、患者のNAD+レベルに応じた特定のNAD+前駆体の投与を可能にする検出方法の改良が、ヒト疾患におけるNAD+前駆体の治療的役割の理解を深めることにつながるであろう。
キーワード
NAD+、サーチュイン、酸化ストレス、DNA損傷、ナイアシン(ニコチン酸、ニコチン酸アミド/ニコチンアミド)
I. 序論
ペラグラは、必須のピリジンヌクレオチドニコチンアミドアデニンジヌクレオチド(NAD+)の合成前駆体、すなわちナイアシン(ビタミンB3)トリプトファン(75,117,255)の合成前駆体が著しく欠乏した食生活によって引き起こされる症候群である。この致死的な障害は、ニコチン酸(NA)またはニコチンアミド(NAM)の遊離貯蔵がないために、欠乏した食生活を維持していると、60日以内に発症する可能性がある(298)。ペラグラは、病理学的には、はっきりとした濃い色素性の皮膚の発疹と、皮膚炎、下痢、認知症の3つのDによって特徴づけられる(5)。興味深いことに、AIDS認知症複合体(ADC)は、その臨床症状においてペラグラといくつかの神経学的類似性を共有している(55)。
前世紀には、ペラグラは米国南部の貧しい農村部でよく見られた病気であり、未知の感染性病原体に起因するとされていた(299)。しかし、それは1914年にペラグラが食事の欠乏症に起因するものであったという仮説を検討した米国公衆衛生局の博士ジョセフ・ゴールドバーガー、および彼の仲間であった。その後、ペラグラは、これらの集団(1,121)でトウモロコシ、新鮮な牛乳、卵、硬化肉が豊富な食事を使用して防止された。これらの進歩にもかかわらず、それはコンラッドElvehjem、生化学教授は、最初に栄養失調の犬(99,100)に関連する黒舌病にニコチンアミドとニコチン酸抗ペラグラ遺伝的効果を実証した1937年までではなかった。
ペラグラ誘発性認知症と診断された個体は、疾患の初期段階で治療に成功する可能性がある。しかしながら、治療されていないペラグラは、不可逆的な神経学的損傷をもたらし、最終的には死に至る(148)。これは主に、NAD+とそのリン酸化された形態のNADP+が、多くの生物学的プロセスに不可欠な補因子および基質であるため、NAD+の産生および利用可能性が減少したことに起因する(365)。ターンオーバーの増加または合成の減少によるNAD+の利用可能性の局所的な減少もまた、他の条件で見られる病理学の基礎となるかもしれない。ADC患者の率直な病理学の前に、明らかに可逆的な認知症が見られるという観察に合致しているように思われる。現在のところ、ペラグラは、アルコール依存症や食欲不振、あるいは栄養失調の重症例で報告されている稀な状態である(21, 332)。
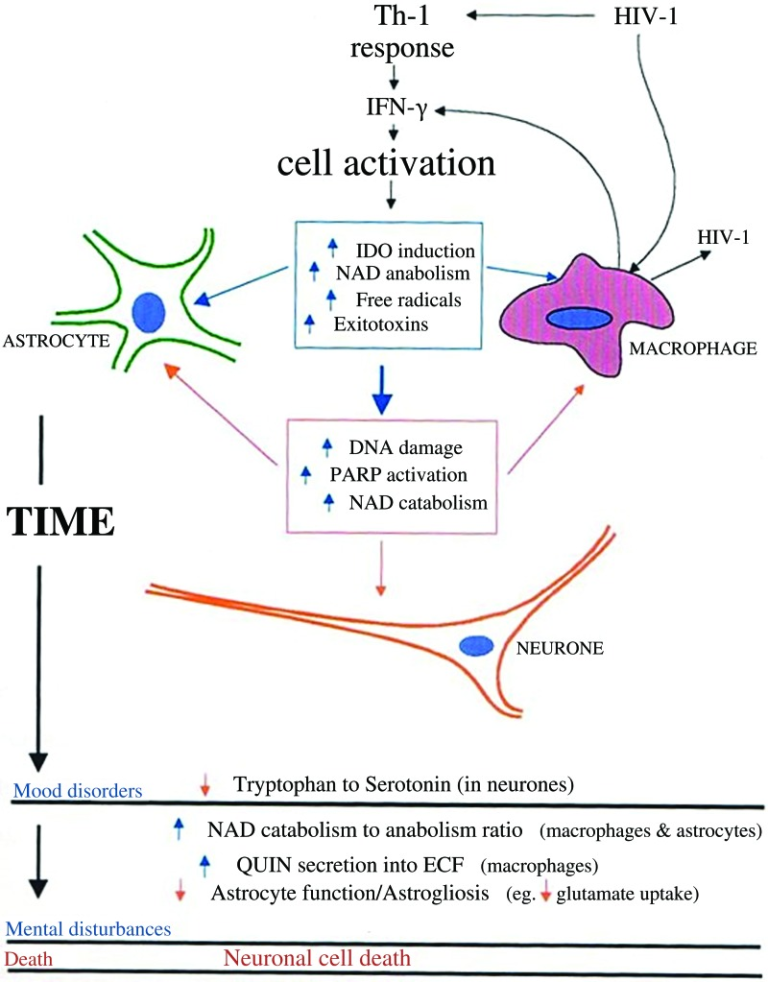
いくつかの生化学的研究では、異化作用が同化作用を上回るNAD+の非効率的な産生が、単に食事によるナイアシンの欠乏によって細胞機能障害をもたらす可能性があることが示されている(27, 325)。それはまた、共置換基依存性キノリン酸ホスホリボシルトランスフェラーゼ(QPRT)の速度制限作用によるものかもしれない(267, 304)。過剰なアミノ酸ロイシンは、ニコチン酸モノヌクレオチド(ニコチン酸モノヌクレオチド(NAMN))へのナイアシンまたはニコチン酸形成を妨げるQPRTを阻害する(189)。トリプトファンの利用可能性の低下は、特に慢性的な免疫活性化の後、またはトリプトファンが豊富な食事(すなわち、大豆、肉、魚、卵、およびピーナッツ)がない場合に、ペラグラの発症と関連している可能性がある(217)。後者はトリプトファンとナイアシンの全身的な欠乏の結果として発症するように、しかし、本質的な違いは、ADCは、特定の、おそらく多数のサイトで増加したトリプトファンとNAD+の異化の結果として発症しながら、ADCとペラグラの間で観察されるかもしれない(図1)。トリプトファン異化の活性化は、ADCでは陽性と陰性の両方の可能性がある。免疫活性化された酸化的トリプトファン異化作用は、脳細胞のNAD+代謝を増加させることで細胞の生存率を積極的に増加させることができる。しかしながら、トリプトファン異化の慢性的な活性化は、増加したNAD+異化に応答して起こり得る。アストロサイトおよび単核食細胞の活性化の増加は、免疫活性化の初期段階でNAD+レベルを維持するためにトリプトファンの異化を刺激する。しかし、免疫活性化が長期化すると、マクロファージの過剰なリクルートと活性化を引き起こし、アストロサイトから神経へのNAD+供給が減少し、ペラグラ様の神経機能障害を引き起こし、短期的には可逆的になる可能性がある(図1)。末期HIVの特徴的な気分障害や抑うつは、セロトニン作動性経路を介したトリプトファン異化のためのトリプトファンの利用可能性の低下につながるトリプトファン異化作用の増加に起因している可能性がある(図1)。
図1 ADC神経病理学におけるトリプトファン異化とde novo NAD+合成の変化との間の仮説的関係
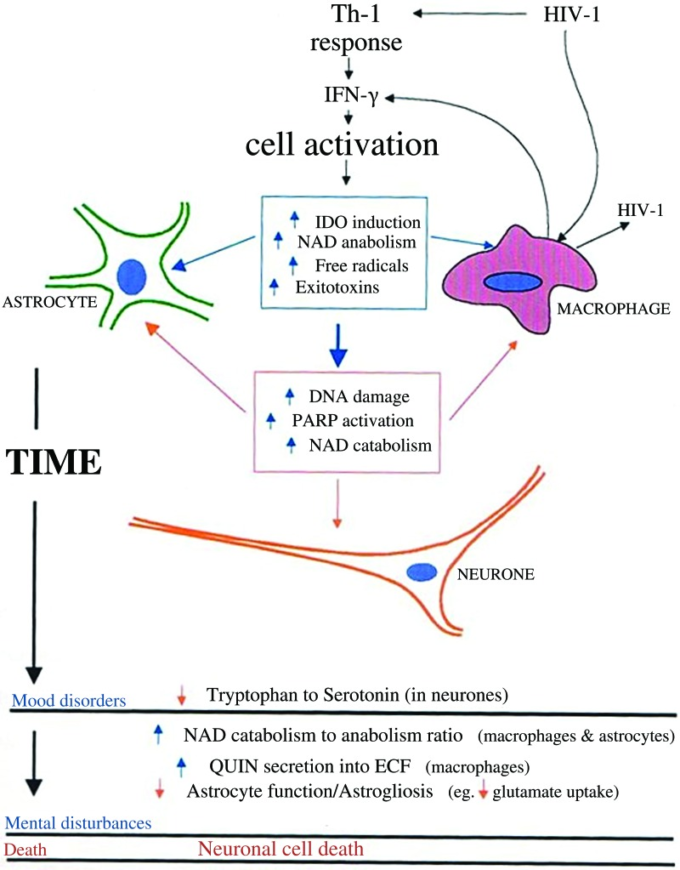
免疫活性化された酸化的l-トリプトファン異化は、アストロサイトおよび単核食細胞におけるNAD+の代謝を増加させることにより、細胞生存率の維持に積極的に寄与することができる。しかし、この経路の慢性的な活性化はまた、QUINや場合によっては3-HKの産生を介して神経細胞の興奮毒性を高める可能性がある。3-HK、3-ヒドロキシキヌレニン、ADC、AIDS認知症複合体、IDO、インドールアミン2,3-ジオキシゲナーゼ、IFN-γ、インターフェロン-γ、NAD+、ニコチンアミドアデニンジヌクレオチド、PARP、ポリ(ADP-リボース)ポリメラーゼ、QUIN、キノリン酸。このイラストをカラーで見るために、読者は、この記事のウェブ版(www.liebertpub.com/ars)を参照してほしい。
NAD+濃度は、エネルギー負荷の低下に関連した条件下で上昇することがよく知られている。これには、絶食、グルコース欠乏、カロリー制限(CR)運動などの活動が含まれる(68)。しかし、ペラグラとは別に、高脂肪食の動物では、NAD+レベルは、老化および細胞老化の間に低下する(293)。NAD+レベルは、寿命または健康寿命が増加した条件下では上昇し、老化が加速された条件下および/または健康寿命が減少した条件下では低下することを考えると、NAD+レベルの低下が老化プロセスの主要な寄与者である可能性があることを示唆している(102)。したがって、NAD+およびその前駆体の補充は、老化プロセス中の炎症および高揮発性活性酸素種(ROS)の蓄積に対する保護を媒介するための潜在的な治療戦略を表す可能性がある。
II. NAD+の生合成経路
私たちの自然食には、いくつかのNAD+前駆体が確認されている。これらには、アミノ酸トリプトファン、ビタミンB3-NA、NAM、ニコチンアミドリボシド(ニコチンアミドリボシド)の3つの形態が含まれる(図2)。キヌレニン経路を介したトリプトファンの異化は、de novo NAD+合成につながる可能性がある(128)。食事中のトリプトファンが制限されている場合、トリプトファンのNAD+への変換効率は、確立された60:1の変換比以下に低下する(107, 164)。NAとニコチンアミドリボシドは基本的な食物連鎖に見られる前駆体である。ニコチン酸は植物および藻類によって産生されるが、ニコチンアミドリボシドは牛乳に存在する(338)。ニコチンアミドはピリジンヌクレオチドの酵素分解の副産物として形成され、動物性食品から吸収可能なビタミンB3の主な形態である。これらのビタミンのNAD+への供給は、腸内マイクロバイオームを含むいくつかの要因によって助けられる(212,213)。生合成遺伝子も概日リズムによって調節される(243)。さらに、NAD+同化に関与する多くの酵素の発現レベルは加齢とともに低下する(236)。
図2 NAD+メタボローム
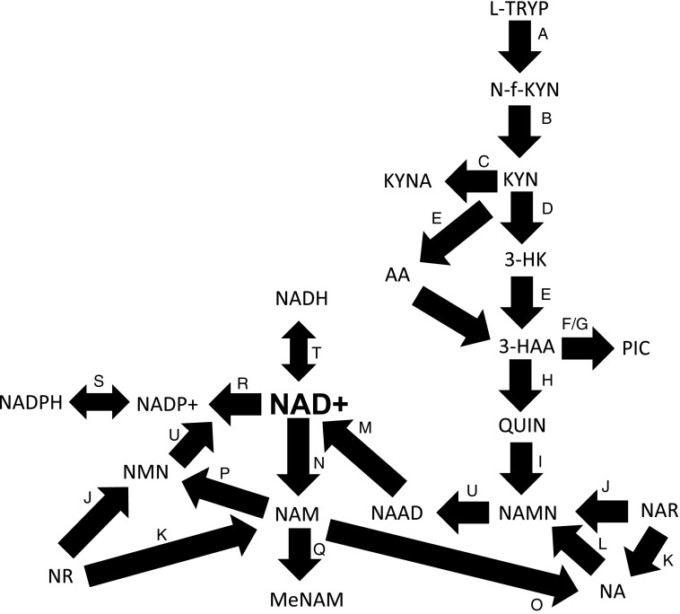
l-TRYP、NA、NAM、NMN、ニコチンアミドリボシド、およびニコチン酸リボシド(NAR)は、NAD+合成の前駆体として使用することができる。
- A) l-TRYPはIDOまたはTDOによってN-ホルミルキヌレニン(N-f-KYN)に異化される。
- B)N-f-KYNはアリルホルミダーゼにより異化されてKYNを形成する。
- C)KATはKYNを異化してKAを形成する。
- D) キヌレニン3-ヒドロキシラーゼはKYNを基質として使用して3-HKを形成する。
- E)キヌレニン分解酵素は、次いで3-HAAを形成し、この生成物は、
- F)3-HAAOによって2-アミノ-3-カルボキシムコン酸セマルアルデヒド(図示せず)に変換される。
- G)次いで、この生成物は、ピコリン酸カルボキシラーゼによりピコリン酸に変換される。
- H)代替的に、セリアルデヒドは、
- I)QPRTによりニコチン酸モノヌクレオチド(NAMN)を形成するQUINを形成するために自然縮合および転位を受ける。
- U)ニコチン酸モノヌクレオチド(NAMN)は、NMNAT1-3によるアデニル化を経てNAADを形成し、これは
- M)グルタミン依存性NAD+合成酵素によってNAD+を形成する。ニコチン酸は、プライス・ハンドラー経路で利用される。
- L)NANは、PRPPPからの5-ホスホリボース基のNAへの付加に続いて、NAPRTによって形成される。
- P)NAMPTは、ニコチンアミドへのホスホリボース基の付加によりNMNを形成する。
- U)NMNはその後、NMNAT1-3の触媒活性を介してNAD+に変換される。
- N)ニコチンアミドはまた、NAD依存性酵素、例えば、PARP、サーチュイン、CD38の副産物として生成される。
- O)ニコチンアミドはまた、細菌のニコチンアミダーゼによってNAに変換され得る。
- J)ニコチンアミドリボシドは、ニコチンアミドリボシドキナーゼ1/ニコチンアミドリボシドキナーゼ2によってNMNを形成するためにリン酸化され、その後、NMNAT1-3によってNAD+に変換される。
- J)ニコチン酸リボシド(NAR)はまた、ニコチンアミドリボシドキナーゼ1/ニコチンアミドリボシドキナーゼ2によってNMNを形成するために、または
- K)プリンヌクレオシドホスホリラーゼによってNAを形成するために利用され得る。
- Q)ニコチンアミドは、NNMMTからMeニコチンアミドにメチル化され、NAD依存性の生物学的プロセスの効率を調節する。T)NAD+は還元されてNADHを形成することができる。
- R)NAD+はまた、NADP+
- S)へのリン酸化を受け、その後、NADPHへの更なる還元を受け得る。
3-HAA、3-ヒドロキシアントラニル酸;3-HAAO、3-ヒドロキシアントラニル酸オキシゲナーゼ;KA、キヌレニン酸;KATs、キヌレニンアミノトランスフェラーゼ;KYN、キヌレニン;l-TRYP、l-トリプトファン。MeNAM、N-メチルニコチンアミド;NA、ニコチン酸;NAAD、ニコチン酸アデニンジヌクレオチド;NAM、ニコチンアミド;ニコチン酸モノヌクレオチド(NAMN)、ニコチン酸モノヌクレオチド;NAMPT、ニコチンアミドホスホリボシルトランスフェラーゼ。NAPRT、ニコチン酸ホスホリボシルトランスフェラーゼ;ニコチン酸リボシド(NAR)、ニコチン酸リボシド;NMNN、ニコチンアミドモノヌクレオチド;NMNAT、ニコチンアミドモノヌクレオチドアデニル転移酵素;NNMMT、ニコチンアミドN-メチルトランスフェラーゼ。ニコチンアミドリボシド、ニコチンアミドリボシド;ニコチンアミドリボシドキナーゼ、ニコチンアミドリボシドキナーゼ;PRPP、5-ホスホリボシル-1-ピロリン酸;QPRT、キノリン酸ホスホリボシルトランスフェラーゼ;TDO、トリプトファン2,3-ジオキシゲナーゼ。
A. キヌレニン経路を介したトリプトファンの異化作用
トリプトファンは、動植物のタンパク質の中で最も少ないアミノ酸であり、タンパク質のアミノ酸含有量の1-1.5%しか占めていない(261)。トリプトファンは、1901年にフレデリック・ゴウランド・ホプキンス卿と彼の学生であるS.W.コールによって最初に単離され(154)、1906年までに成長に必要な最初のアミノ酸として報告された(261)。キヌレニン経路は、1947年にトリプトファン異化の主要経路として初めて記述された(122)。トリプトファン異化のための2つの主要な経路が哺乳類で同定されており、タンパク質の同化とは積極的に独立している。末梢では、キヌレニン経路がトリプトファン代謝の最大95%を占めているが、トリプトファン含有量の約1%のみがインドールアミン経路を介して変換され、神経活性代謝物であるセロトニンおよびメラトニンを形成する(261)。
1. インドールアミン2,3-ジオキシゲナーゼ-1/2とトリプトファン2,3-ジオキシゲナーゼ
キヌレニン経路は、インドールアミン2,3-ジオキシゲナーゼ-1(IDO-1;EC 1.13.11.52)およびそのアイソフォームIDO2またはトリプトファン2,3-ジオキシゲナーゼ(TDO;EC 1.13.11.11)とも呼ばれるトリプトファンピロラーゼ(TDO)のいずれかによるトリプトファンの酸化的切断で進行し、フォルミルキヌレニン(23, 103, 313)を生成する(図2,ステップa)。IDOおよびTDOはともにヘム要求性酵素である。IDOは、主に脳、胎盤、脾臓、肺、腎臓、消化管、精巣上体などの肝外組織に存在する。IDOはトリプトファンアナログの活性化部位を持たず、主にインターフェロン-γ(IFN-γ)などの炎症性サイトカインによって活性化される(109)。
IDOの誘導とIFN-γのフリーラジカル産生は、最初にNAD+生合成を増加させ、NAD+のターンオーバーと需要が増加した環境下での細胞内NAD+レベルの再生に寄与する可能性がある。
このことは、活性化されたマクロファージにおけるトリプトファン異化の増加に対する保護的役割を示唆している(図3)。しかし、TDOは主に哺乳類の肝臓に位置し、空腹時、グルココルチコイド、ヒドロコルチゾン、NA、およびl-トリプトファンを含む多くの要因によって活性化される可能性がある(369)。
図.3. IFN-γによるIDOおよびフリーラジカル生成の同時誘導
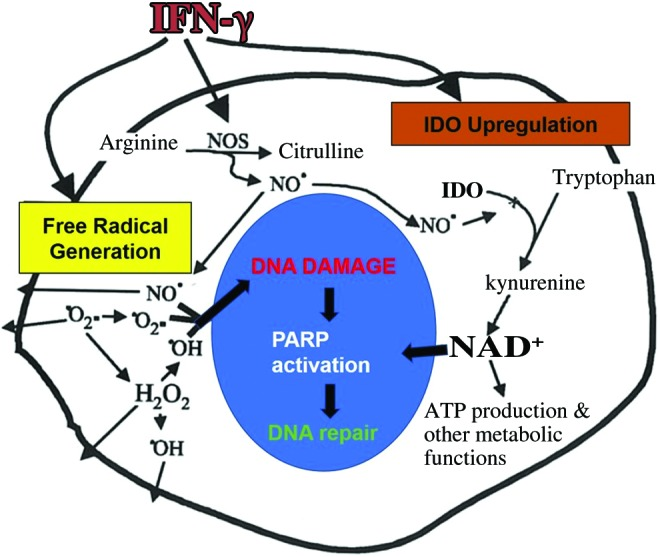
マクロファージおよびアストロサイトの慢性的な免疫活性化は、活性酸素および窒素種の増加をもたらし、グルタミン酸レベルを上昇させる(アストロサイトへの効率的な取り込みがない場合)。IFN-γ刺激によるフリーラジカル産生とIDO誘導との間には、NAD+のde novo合成の増加につながる可能性のある関係が存在する。IFN-γ、インターフェロン-γ。カラーでこのイラストを見るために、読者は www.liebertpub.com/ars でこの記事のウェブ版を参照してほしい。
IDO-1/2およびTDO活性の代謝産物は、不安定な中間代謝物であるN-ホルミルキヌレニン(N-f-YN)(140)であり、これはキヌレニンフォルミラーゼ(EC 3.5.1.9)によって急速に加水分解されてキヌレニンを形成し(図2,ステップb)キヌレニン経路の最初のかなり安定な生成物である。キヌレニンは血液/脳関門(BBB)を横断することができ(240)、3つの異なる酵素、キヌレニナーゼ(EC 3.7.1.3)、キヌレニンアミノトランスフェラーゼ(KAT; EC 2.6.1.7)、およびキヌレニン3-ヒドロキシラーゼ(EC 1.14.13.9)を使用して3つの生成物を合成することができる重要な分岐点を表している(22)。
2. キヌレニン酵素
キヌレニン酵素は、キヌレニンからのアラニン側鎖の切断によってアントラニル酸(AA)を生成する細胞質酵素である(図2,ステップe)。AAは、非特異的ミクロソーム水酸化酵素(184,257,262)を介して、5-または3-ヒドロキシアントラニル酸(5-または3-HAA)への水酸化を受けることができる。AAはまた、受動的拡散を介してBBBを横断することができる。キヌレニナーゼもまた、3-ヒドロキシキヌレニン(3-HK)からの3-HAAの産生において役割を果たす(図2,ステップe)。
トリプトファンからのNAD+の形成は、キヌレニナーゼがキヌレニンからAA、または3-HKから3-HAAへの変換のための補酵素としてピリドキシル-5′-リン酸(ビタミンB6)に依存しているため、ビタミンB6の不十分なレベルによって阻害される(238)。ビタミンB6の低レベルは、心理的苦痛の高レベルと相関することが示されている(172,306)。うつ病におけるB6の関与のメカニズムは、B6がセロトニン生合成の最後のステップを触媒する酵素である5-ヒドロキシトリプトファン脱炭酸酵素の補酵素であるという事実に起因している可能性が高い(126, 233)。
しかしながら、ピリドキシンはまた、グルタミン酸脱炭酸酵素およびγ-アミノ酪酸(GABA)-トランスアミナーゼ、グルタミン酸からのGABAの合成に必要な2つの酵素を含む、脳神経伝達物質経路のいくつかの反応のための補因子でもある(124,125,368)。ピリドキシン依存性てんかん児において、非効率的なB6レベルは、脳内グルタミン酸のレベルを著しく上昇させる結果となった(141)。さらに、ビタミンB6レベルの低下は、リンパ球の分化と成熟を含む体液性および細胞を介した免疫応答の両方の欠乏と関連している(134)。
キヌレニナーゼ活性の低下は、キヌレニン経路を介した更なるフラックスを減少させ、それによってN-メチル-d-アスパラギン酸(NMDA)受容体アゴニストおよび興奮毒素であるキノリン酸(QUIN)の産生を減少させることが指摘されている(111)。しかし、QUINレベルは炎症時に増加し、キヌレニナーゼ活性は有意に低下しない可能性があることを示唆している。ビタミンB6は、したがって、GABAトランスアミナーゼ(EC 2.6.1.19;GABA産生)5-ヒドロキシトリプトファン脱炭酸酵素(EC 4.1.1.28;セロトニン合成)およびグルタミン酸脱炭酸酵素活性(EC 4.1.1.15)よりもキヌレニナーゼ活性(例えば、QUIN/NAD産生)のために細胞によって優先的に使用される可能性があり、これらの条件下でのde novo NAD+生合成のための細胞の優先順位を示している。神経炎症中の活性化単核食細胞によるQUIN分泌の増加は、これらの細胞におけるNAD+に対する需要の増加を示している可能性があり、その生産は、飽和QUINリボシル化システムによって特定の条件下で制限されている可能性がある(252, 253)。
3. キヌレニンアミノトランスフェラーゼ
KATは、キヌレニンの伝達によりキヌレニン酸を産生する(図2,ステップc)。キヌレニン酸は、グルタミン酸サブタイプであるNMDA受容体において、脳内で非特異的なアンタゴニスト作用を有する安定な化合物である。KATとキヌレニン酵素はともにビタミンB6依存性の酵素である(245)。
4. キヌレニン3-ヒドロキシラーゼ
キヌレニン 3-ヒドロキシラーゼはミトコンドリア酵素であり、芳香環の水酸化によりキヌレニンを 3-HK に変換する(図 2,ステップ d)。3-HKはNADPH依存性の酵素であり、その活性はエストロゲンと甲状腺機能亢進症の状態では減少するように見える(28)。3-HKはまた、BBBを横断し、フリーラジカル産生を刺激し、血管拡張を媒介することができる(104)。
5. 3-ヒドロキシアントラニル酸オキシゲナーゼ
3-HAAの異化は、中間体である2-アミノ-3-カルボキシムコニックセマルアルデヒド(307)を生成するために、細胞質およびシナプトソーム画分の両方に見られる酵素である3-ヒドロキシアントラニル酸オキシゲナーゼ(3-HAAO; EC 1.13.11.6)によって媒介される(図2,ステップf)(図2)。
6. ピコリン酸カルボキシラーゼ
酵素ピコリン酸カルボキシラーゼ(PICAC; EC 4.1.1.45)は、2-アミノ-3-カルボキシムコニックセミアルデヒドを2-アミノムコニックセミアルデヒドに優先的に変換し、それに続く非酵素的なピコリン酸(PIC)(図2,ステップg)への変換(188,230)、金属キレート剤(106,270)、およびNMDA受容体アンタゴニスト、または酵素的な転位が最終的にアセチルCoA(30,79,230)へと導く。2-アミノ-3-カルボキシムコニックセミアリアルデヒドの非酵素的転位は、PICACが基質で飽和されてQUINを生成するときに起こる(図2,ステップh)。PICACの活性は、トリプトファンから合成されたNAD+の量に反比例することが示されている(305)。
7. キノリン酸ホスホリボシルトランスフェラーゼ
QUINは、酵素キノリン酸ホスホリボシルトランスフェラーゼ(QPRT; EC 2.4.2.19)によってニコチン酸モノヌクレオチド(NAMN)に変換される(図2,ステップi)。QPRTは、Mg2+の存在下で5-ホスホリボシル-1-ピロリン酸(PRPP)とQUINの間の反応を触媒し、ニコチン酸モノヌクレオチド(NAMN)を生成する。QPRTの最大酵素速度は、明らかにすべてのキヌレニン経路酵素の中で最も低く、先行する酵素である3-HAAOよりも80倍低い。しかし、3-HAAOとQPRTのマイケル・メントン定数(Km)は同じであると計算されており、これは3-HAAがQUINだけでなくPICの産生にも基質を提供していることに起因していると考えられる。3-HAAから形成されるQUINの相対的な量は、したがって、PICAC活性の速度によって決定される(168,176)。ヒトの脳または他の場所での炎症性条件下でのPICACの挙動は調査されていないようである。しかし、IFN-γはIDOを誘導するだけのように見えるので、PICAC活性は炎症反応中には増加しないと慎重に推測されるかもしれない。したがって、キヌレニン経路を介したフラックスの増加は、比例してQUIN産生を増加させるであろう。
QPRTは、肝臓および脳を含むいくつかの組織に広く分布しており、てんかんおよびハンチントン病を含む神経変性疾患に関連するQUIN誘導毒性に対する神経保護を媒介する上で重要な役割を果たしている可能性がある(58,132,175,229,235,250,333,364)。QUINの生理学的レベルは、低ナノモルの範囲にあると考えられ、QUINのレベルの増加に伴ってQPRT活性が増加する。しかしながら、QUINの高レベル(>500 nM)では、神経細胞のQPRT活性は飽和している(267)。これは、NAD+の産生よりも高い速度でのQUINの産生をもたらし、QUINおよびNMDA介在性興奮毒性の蓄積につながる(254)。
PRPPは、QPRT活性の調節において重要である(図4)(33,161,162,168)。PRPPが合成されて使用される速度は、細胞内での定常状態の濃度を決定し、それによって、PRPPを競合する経路の代謝の進行度が決定される。PRPPは、リボース-5-リン酸とATPを利用した5-ホスホリボースピロホキナーゼまたはPRPP合成酵素(EC 2.7.6.1)によって触媒される反応で細胞内で合成される。PRPP合成酵素は無機リン酸(Pi)を絶対的に必要とし、急速な細胞分裂を受けている細胞で上昇する。PRPP合成酵素の活性は、ADPとATPの増加レベルによって競合的に阻害される。この反応で使用されるリボース5-リン酸は、ヘキソ ース一リン酸シャントを介してグルコース6-リン酸の代謝から、またはホスホリボムターゼ反応を介してリボース1-リン酸(ヌクレオチドのリン酸化によって生成)から生成される(73)。
図4 QPRT活性とNAD+合成に必要な補因子
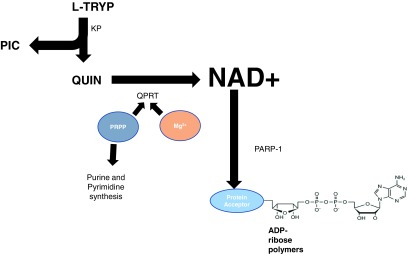
PRPPはQPRT活性の調節に重要である。PIC、ピコリン酸。この図をカラーで見るには、読者はこの記事のウェブ版 www.liebertpub.com/ars を参照してほしい。
PRPP-シンテターゼ活性とNAD)の障害。これは、フリーラジカル誘発性DNA損傷およびアストログリア症の結果として、ADCおよび神経変性疾患で起こり得る。いくつかの神経炎症性疾患で見られるQUINの増加は、したがって、DNA損傷または分裂細胞におけるプリンおよびピリミジン合成のためのPRPPの使用に起因する酵素活性の低下と相まって、キヌレニン経路を介して増加したフラックスの組み合わせである可能性がある。
8. NADピロホスホリラーゼ(NAMモノヌクレオチドアデニル転移酵素
ピリジンヌクレオチドの親分子であるNADの合成につながるさらなる変換は、核内および場合によってはミトコンドリアで起こる。ニコチン酸モノヌクレオチド(NAMN)は、ATPの存在下でNADピロリン酸分解酵素またはニコチンアミドモノヌクレオチドアデニル転移酵素(NMNAT;EC 2.7.7.1)によって触媒され、デサミドNADを生成する(図2,ステップu)。グルタミンの存在下では、デサミド-NADは、親ピリジンヌクレオチドであるNAD図2,ステップm)にアミド化され、キヌレニン経路の最終生成物である(367)。ヒトでは、NMNAT-1(核)NMNAT-2(ゴルジ体)NMNAT-3(ミトコンドリア)という3つのアイソフォームがいくつかの異なる小器官に存在していることが確認されている(31)。これらの酵素の局在が異なることから、これらのタンパク質の小器官特異的な機能と、核、ミトコンドリア、ゴルジ体特異的なNAD+生合成経路が独立していることが示唆される。NAD+合成に好ましい酵素であるNMNAT-1とは異なり(157)。NMNAT活性(および主にNMNAT-1)は、異化組織では高く、非律速制限的であるが、血中ではない(236)。
NAD+生合成とは別に、いくつかの研究では、NMNATアイソフォームが試験管内試験および生体内試験の両方で軸索変性に対して保護することができることが実証されている(80, 183, 211)。NMNATは、熱耐性と酸化ストレスによる寿命の短縮を減衰させるために必要なストレス応答タンパク質として機能することが示されている(11)。さらに、NMNATがヒートショック因子(HSF)と低酸素誘導因子1α(HIF1α)によって転写制御されていることも明らかにした。ヒートショック下では、HSFはNMNATプロモーターに結合し、NMNATの発現を誘導する。しかし、低酸素条件下では、HIF1αはHSFの誘導を介して間接的にNMNATレベルを増強する(11)。さらに、NMNATアイソフォームはタンパク質のシャペロン機能を発揮し、いくつかのショウジョウバエやマウスの神経変性モデルにおいて神経保護作用を発揮する可能性がある(54, 382)。また、NMNAT-1の過剰発現は、慢性タウ症のマウスモデルにおいて、神経細胞の機能を部分的に維持し、生化学的不溶性タウのレベルを低下させることが示されているが、タウのリン酸化、タウの凝集、タウ誘発性炎症および海馬の萎縮には有意な影響を与えないことが示されている()。さらに、NMNAT-3の過剰発現は、網膜神経節細胞株において、p62の発現を減少させ、オートファジーフラックスを増加させることにより、腫瘍壊死因子誘発性および眼圧上昇誘発性視神経変性に対する軸索保護を媒介した(182)。これらの研究は、NAD+生合成のための酵素としての役割に加えて、NMNAT酵素活性を保護する新しいメカニズムの可能性を示唆している。
B. ビタミンナイアシンからのNAD+産生
トリプトファンからのde novo合成に加えて、NAD+は、ビタミンナイアシン(ビタミンB3)の酸、アミド、またはリボシド形態からも合成することができる。
1. ニコチン酸ホスホリボシルトランスフェラーゼ
ニコチン酸は、ATP依存的な方法で、PRPPを共基質として使用する酵素ニコチン酸ホスホリボシルトランスフェラーゼ(NAPRT)(図2,ステップl)(EC 6.3.4.21)により、ニコチン酸モノヌクレオチド(NAMN)に変換される(EC 6.3.4.21)。QUINが酵素QPRTによってニコチン酸モノヌクレオチド(NAMN)に変換されるように、NAD+産生につながるイベントのシーケンスは、いずれかの基質からのニコチン酸モノヌクレオチド(NAMN)形成後に同一である(112)。NAPRTは、結腸、心臓、腎臓、肝臓を含むいくつかの異化組織で発現しているようである(95)。NAD+合成の非脱アミド化ルートは、アミド化ルート(ニコチンアミドホスホリボシルトランスフェラーゼ[NAMPT])と比較して、血液および小腸においてより高い相対割合を示し、肝臓および小腸においてより高い絶対値を示し、これらの組織におけるNAD+合成の前駆体としてのニコチン酸重要性を示唆している()。これは、肝臓、腸、および腎臓においてNAがNAMよりもNAD+合成のためのより好ましい前駆体であることを示したいくつかの摂食研究によって再確認されている(81)。同様に、ニコチン酸は、腎臓細胞株において細胞内NAD+レベルを増加させることが示されている。さらに、NAPRT1の過剰発現は、酸化ストレスが媒介するNAD+枯渇に対する保護を媒介することが示されている(142)。
トリプトファンはニコチンアミドに変換することができるが、de novo合成経路とNAD+図2,ステップo)を発現する脊椎動物の細胞ではNAを生成するために使用することはできない。) しかしながら、ある研究では、細胞内のピロリン酸およびニコチン酸モノヌクレオチド(NAMN)の十分なレベルは、NAを産生するためにNAPRTを誘導することができ、したがって、トリプトファンからのニコチン酸生産を可能にすることが示唆された(212, 214)。この仮説を検証するために、さらなる研究が必要である(そして計画されている)。NAD+の細菌や真菌による分解と直接のNA補給は、血管血流を介して体の残りの部分に分配するために、消化管内のニコチン酸を増加させることもできる(212)。
2. NAMホスホリボシルトランスフェラーゼ
PRPPを共役基質とする酵素NAMPT(EC:2.4.2.12)は、NAMをNMNに変換し(図2,ステップp)ATPの存在下でNADピロホスホリアーゼの作用によりNAD+に変換する(図2,ステップu)(351)。NAD+合成のこのアミド化された経路は、肝臓と腎臓で最も高い速度を示し、血中では最も低い速度を示した(161)。NAMPTの発現は、プレB細胞コロニー増強因子1(PBEF1)遺伝子によってコードされる。PBEFまたはビスファチンとしても知られるNAMPTは、IL-7などの他のサイトカインや幹細胞因子が利用可能な場合にB細胞の成熟を促進するサイトカインとして同定されている。また、インスリン模倣作用を示す(260,374)。細胞内ドメインは、リンパ球を活性化し、NAD+生合成酵素として機能することが示されている(281)。しかし、細胞外ドメインと細胞内ドメインの両方が、有利なホスホリボシル活性を示す。
シスプラチン誘発性急性腎障害(AKI)モデルにおいて、AICARを介したNAMPT発現の薬理学的操作は、腎機能を有意に改善し、尿細管損傷を減少させた。この効果は、ミトコンドリアのサーチュインであるSIRT3のメッセンジャーRNA(mRNA)発現の増加とタンパク質の高アセチル化の減少と関連している(237)。NAMPT経路の阻害はマウスの耐糖能とインスリン分泌を損なう可能性があるが、この効果はその後のNMNの補給によって改善される(275)。これらの知見にもかかわらず、哺乳類細胞でNMNATの基質を同化するNAMPTの阻害は、別の研究ではNMNAT-1を介した軸索保護に有意な影響を及ぼさなかった(289)。
3. NAM N-メチルトランスフェラーゼ
酵素ニコチンアミドN-メチル化酵素(NNNMT;EC:2.1.1.1.1)(図2,ステップq)によるニコチンアミドのメチル化とN-メチルニコチンアミド(MeNAM)へのNAMPTを介したNAD+の生成にサルベージする細胞の能力は、NAD+に依存する生物学的プロセスの効率を調節する(6,263)。N-メチル化はまた、肝臓による特定の薬物および他の外来物質化合物の生物変換および解毒を調節する。NNMTの酵素活性は、S-アデノシルメチオニンをメチル供与体として使用し、S-アデノシル-l-ホモシステインのようなピリジニウムイオンを形成する(287)。この酵素は主に肝臓で発現している。脳や膵臓では検出されなかったが、腎臓、肺、骨格筋、胎盤、心臓、脂肪組織では低い発現が報告されている(287)。NNMTの活性の亢進は、神経毒性を示すN-メチルピリジニウム化合物の産生を促進することが示されており、この化合物が黒質変性に関与している可能性があると考えられている(366)。
4. ニコチンアミドリボシドキナーゼ
ニコチンアミドリボシドまたはニコチン酸リボシド(NAR)は、ニコチンアミドリボシドキナーゼ(ニコチンアミドリボシドキナーゼ; EC 2.7.1.173)経路(図2,ステップj)を介して、またはヌクレオシドホスホリラーゼおよびNAMサルベージ経路(38)の作用によってNAD+に変換され得る、新たに同定された前駆体を表す。ニコチンアミドリボシドキナーゼは真核生物細胞において高度に保存されており、NMrk遺伝子によってコードされている。2つのニコチンアミドリボシドキナーゼ酵素、ニコチンアミドリボシドキナーゼ1とニコチンアミドリボシドキナーゼ2が同定されているが、それらの正確な生理的役割は不明のままである。ニコチンアミドリボシドキナーゼ1は哺乳類の組織ではユビキタスに発現しているが、ニコチンアミドリボシドキナーゼ2は腎臓、肝臓、肺、膵臓、胎盤では発現していない(272)。Nmrk1欠損マウスモデル(ニコチンアミドリボシドキナーゼ1KO)を用いて、ニコチンアミドリボシドキナーゼがニコチンアミドリボシド/MN媒介のNAD+合成の速度制限を行うことが最近明らかになっている(272)。
5. プリンヌクレオシドホスホリラーゼ
第二のニコチンアミドリボシドサルベージ経路はニコチンアミドリボシドキナーゼに依存しない経路で、ニコチンアミドリボシドはリボシル生成物とニコチンアミドに分解され(図2,ステップk)後者はNAMサルベージによりNAD+を産生する。プリンヌクレオシドホスホリラーゼ(PNP; EC 2.4.2.1)は、ニコチン酸リボシド(NAR)をNAに変換することが示されており(図2,ステップk)これはNAPRTの触媒作用によってニコチン酸モノヌクレオチド(NAMN)に変換される(331)。PNP欠損は、デオキシGTPレベルを上昇させることが示されている。これは、デオキシヌクレオチドの形成に必要なリボヌクレオチド還元酵素を阻害する(20, 285, 327)。酵素の欠乏は、リンパ系細胞において毒性を誘発しうる代謝物の蓄積をもたらす(291)。
6. 細胞質5′ヌクレオチダーゼ
最近の研究は、ニコチン酸リボシド(NAR)がヒト細胞によって産生され、細胞内NAD+同化において重要な役割を形成することを示した(190)。この研究は、細胞質の5′-ヌクレオチダーゼ(5′-NT)がニコチン酸モノヌクレオチド(NAMN)を脱リン酸化し、より少ない範囲でNMNを形成することができることを示した。形成されたニコチン酸リボシド(NAR)の量は、非リボシドNAD+前駆体を利用するために必要な機械を欠いている隣接する細胞においてNAD+合成を促進するのに十分であるように見える(190)。
III. NAD+の生物学的役割
NAD+は、酸化的リン酸化とATP産生、コレステロール、脂肪酸、ステロイドの合成など、多くの重要な生物学的プロセスで主要な役割を果たす必須のピリジンヌクレオチドである(224)。NAD+の主な機能は、1936年にWarburgとChristianによって同定された(357)。NAD +は、ミトコンドリアでATPの生産につながる酸化/還元(すなわち、レドックス)反応のための電子の転送を可能にする水素アクセプターとして機能する。ATPは、細胞内の “エネルギー通貨 “を表し、細胞内のNAD +レベルの低下は、エネルギー制限(373)を介して細胞死で絶頂に達する、ATPの減少レベルにつながる。
NAD+とは別に、その密接に関連するリン酸NADP(図2,ステップr)は、脂肪酸およびコレステロール合成(315)などのいくつかの同化プロセスにおける補因子として機能する。NAD+およびNADPの還元形態は、それぞれNADH(図2,ステップt)およびNADPH(図2,ステップs)である。これらのヌクレオチドは、デヒドロゲナーゼ、ヒドロキシラーゼ、および還元酵素が関与する全身の400以上の酵素反応において、水素化物ドナーとして機能する(219)。これらの還元型およびリン酸化型の形態は相互変換することができるが、NAD+のレベルを変えることはない。
重要なことに、NADPHは活性酸素の還元に必要な必須の補酵素である(29)。チオレドキシン(TXN)は、NADPH依存性のプロセスでチオレドキシン還元酵素によって還元される抗酸化タンパク質である(61)。グルタチオン二硫化物(GSSG)もまた、NADPHを用いてグルタチオン(GSH)に還元するためのグルタチオン還元酵素の基質である。GSHとTXNの生成は、過酸化水素(H2O2)などの活性酸素の除去に極めて重要である(123)。NAD+の同化(または異化作用の増加)の減少によるNADPH産生の減少は、ミトコンドリア機能およびゲノムシグナル伝達と安定性の摂動につながる細胞の酸化還元バランスの障害につながり、その後、壊死およびアポトーシス経路への脆弱性の増加につながる可能性がある。
NAD+は、酸化還元反応における役割とは別に、多くのエビデンスから、DNA修復、エピジェネティックに制御された遺伝子発現、細胞内カルシウム恒常性の維持、免疫学的な役割(52, 118, 119)など、いくつかの重要な酵素反応に必要な基質として関与することが示されている(図5)。
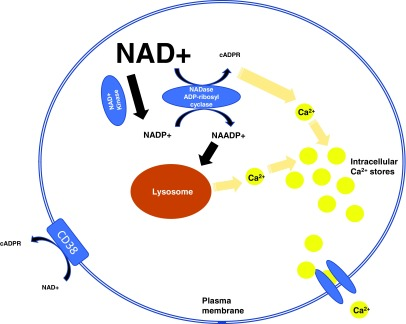
図5 NAD+の細胞内での役割
CD38,PARP、サーチュインなどのNAD+の分解機構。NAD+はNADP+にリン酸化されることがある。また、NAD+からNADHへの酸化反応、NADP+からNADHへの酸化反応もある。CD38はNAD依存性酵素であり、それぞれNAD+およびNADP+からのcADPRの産生を導く。細胞質のcADPRは小胞体上のリアノジン受容体、リソソーム上の一過性受容体電位ムコリピン1を標的とし、小胞体からの細胞内カルシウムシグナル伝達とリソソームを介した細胞内カルシウムシグナル伝達を調節する。 cADPR、cyclic-ADP-リボース。このイラストをカラーで見るには、この記事のウェブ版(www.liebertpub.com/ars)を参照してほしい。
A. ポリ(ADP)リボシル化とDNA修復
DNA鎖切断は、フリーラジカル、紫外線(UV)光、またはアルキル化化学物質に反応して起こることが知られており、酵素PARPを活性化する(図2,ステップn)(320)。グルタミン酸およびQUINの細胞毒性レベルに曝露された神経細胞およびアストログリア細胞は、細胞内酸化ストレスおよびPARP活性の両方の増加を示す(46)。
PARP-1(18個のPARPタンパク質のスーパーファミリーの支配的メンバー)は、そのN末端のジンクフィンガードメイン(312)によってDNA切断の存在を効率的に検出する。PARPのADPリボシル化は、15秒未満でDNA損傷の修復を刺激する鍵となるタンパク質のリクルートを誘発する(85)。重要なことに、PARPがそのADPリボシル化機能を遂行するために、それはその供給のためにNAD+のADPリボース(ADPR)部位を使用する。このようにして、PARPはNAD+を分解してニコチンアミドとADP-リボシル生成物を生成する(図6A)(145)。おそらくDNA鎖切断の結果として、最近の証拠は、ヒストンまたは転写因子のポリ(ADP)リボシル化が核受容体シグナル伝達にも関与している可能性を示唆している。ポリ(ADP-リボース)代謝は、ポリ(ADP-リボース)グリコヒドロラーゼ(295)の作用により、ADP-リボースポリマーの分解が比較的急速に起こる動的なプロセスである(図6A)。
図6 PARP活性の調節
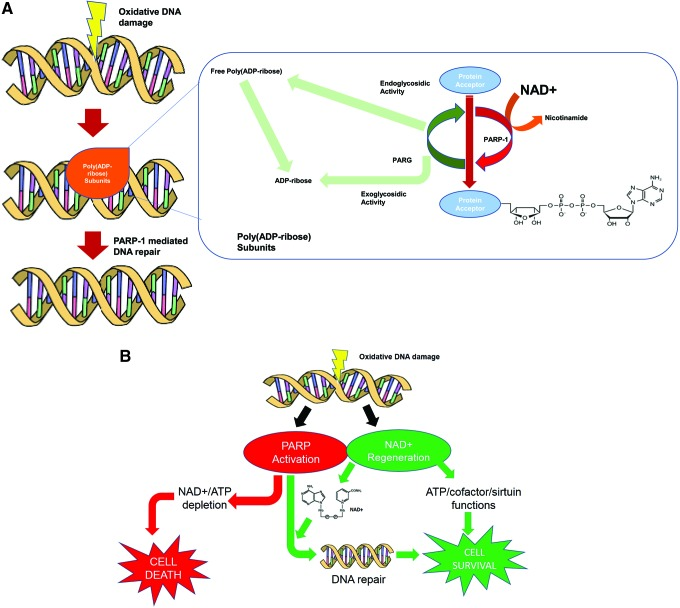
A)PARPおよびPARG酵素活性。PARPはNAD+をニコチンアミドに分解し、PARP-リボースポリマーのADP-リボシル生成物分解がPARPの作用により比較的迅速に起こる。
B)DNA損傷、PARP活性化、NAD+枯渇の関係。通常の生理的条件下では、PARPの活性化は損傷したDNA修復をもたらす。しかし、NAD+の減少をもたらすPARP活性の増加は、ATPを減少させ、細胞の溶解や死を引き起こすことが示されている(45, 203) (B)。PARG、ポリ(ADP-リボース)グリコヒドロラーゼ。カラーでこのイラストを見るために、読者は、www.liebertpub.com/ars でこの記事のウェブ版を参照してほしい。
H2O2,一酸化窒素、HIV感染、または炎症中に曝露された後のDNA鎖切断とPARP活性化の結果として、細胞内NAD+の有意な減少が脳や他の様々な細胞タイプで報告されている(7, 326, 330)。NAD+の減少をもたらすPARP活性の増加は、脳内のATPおよび神経伝達物質レベルを減少させ、細胞溶解および死を引き起こすことが示されている(45, 203)(図6B)。PARP活性を阻害すると、酸化性傷害の後、NAD+とATPレベルが維持され、細胞溶解を防ぐことが示されている(14)が、DNAへの損傷はおそらく防止されていない。PARPの発現を欠いた膵島細胞集団では、DNA鎖切断が同じ程度に発生しているにもかかわらず、酸化剤傷害後にNAD+枯渇は発生しない(68)。このことは、PARPの活性化が、これらの酸化性傷害細胞におけるNAD+枯渇の主な原因であることを示している。フリーラジカル、酸化剤、およびエキサイトトキシンのレベルの上昇は、脳の炎症性媒介性疾患において報告されており、いくつかのケースでは、DNA損傷が実証されている(2,220,221,341,380)。このことは、PARP活性化によるNAD+枯渇が、これらの条件下での中枢神経系(中枢神経系)の機能障害や病理に一役買っている可能性を示唆している(図6)。
最近では、NAD+の減少ではなく、PARPの活性化が遺伝毒性侮辱に曝された後の細胞死に関与している可能性が示唆されている。例えば、ポリ(ADP)リボシル化は解糖酵素であるヘキソキナーゼを直接阻害し、NAD+枯渇前の解糖を大幅に減少させ、ミトコンドリア機能障害や神経細胞死を引き起こすことが示されている(17)。さらに、グリセルアルデヒド3-リン酸デヒドロゲナーゼのポリ(ADP)リボシル化は、虚血性傷害後の腎尿細管における細胞死の主な原因である(94)。これらの研究は、PARP阻害の有益な効果は、病理学的条件下でのNAD+レベルの維持とは独立した代謝効果の変化に起因する可能性があることを示唆している。
PARPはまた、腫瘍抑制タンパク質であるp53のアップレギュレーションに正の役割を果たしているようである。例えば、チャイニーズハムスターV79細胞由来のPARP欠損細胞株は、エトポシドで処理した後、ポリ(ADP)リボシル化を受けず、p53を活性化することができなかった(363)。PARPはまた、リン酸化を介してp53活性を調節するDNA依存性プロテインキナーゼを活性化することができる(318)。したがって、報告されているPARP阻害剤の利点とは逆に、PARP活性の薬理学的阻害は、結果として癌形成のリスクを伴うゲノム不安定性に寄与する可能性がある。
B. CD38/CD39/CD73/CD157と二次メッセンジャーシグナル伝達
免疫関連エクト酵素CD38,CD39,CD73およびCD157は、NAD+を消費する酵素の別のクラスを代表する(155)(図2,ステップn)。これらの酵素は、ADPRを産生するためにNAD+を必要とし、細胞内カルシウム過渡状態を媒介するのに役立つ二次メッセンジャーシグナル伝達分子であるcyclic-ADP-リボース(cADPR)を加水分解する(図7)。CD38はまた、免疫調節の役割も実証されている(135)。例えば、Tリンパ球上のCD38の存在は、抗原提示細胞が抗原特異的T細胞を刺激する能力に影響を与える(256)。CD38発現のアップレギュレーションはまた、炎症性サイトカイン活性化の間に樹状細胞の成熟をシグナルし、樹状細胞とリンパ球との間の調節接着およびシグナル伝達分子として作用する(105)。心筋細胞では、外因性刺激剤が細胞内カルシウムの増加を刺激し、それがCD38の活性化につながることがある(147)。CD38の発現は加齢とともに増加することも示されており(65)、これは循環炎症性サイトカインの加齢に伴う増加に起因している可能性が高く、CD38機能の低下は免疫応答の低下と関連している。
図7 CD38を介したCa2+動員およびNADase活性のストイキオメトリー
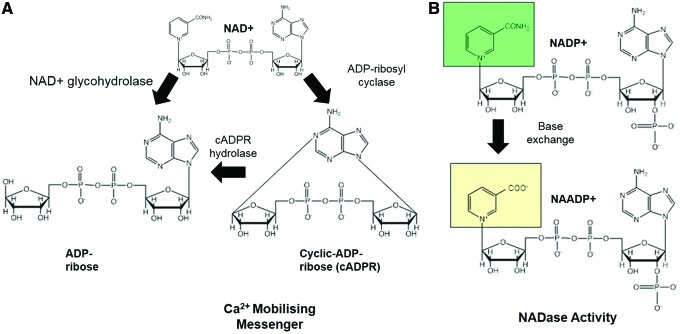
A)CD38は、ADPRを産生し、細胞内カルシウム過渡状態を媒介するのに役立つ二次メッセンジャーシグナル伝達分子cADPRを加水分解するためにNAD+を必要とする。ADPR、ADPリボース。B)CD38はまた、塩基交換を介してNADP+をNAADP+に変換する(CD38のNADase活性)。カラーでこのイラストを見るために、読者は、この記事のウェブ版を参照してほしいwww.liebertpub.com/ars。
1分子のcADPRを生成するためには100分子のNAD+が加水分解されなければならないことを考えると、CD38が細胞内NAD+レベルの主要な調節因子である可能性が高い(76)。したがって、CD38ノックアウトされた神経細胞では、対照細胞と比較してNAD+レベルが5倍に増加していることがわかった(52)。したがって、CD38は非効率な二次メッセンジャー酵素であるだけでなく、NAD+の細胞内レベルとその生理的プロセスを主に調節するNADaseとしても機能している可能性がある(図8)。
図8 CD38が介在する細胞内Ca2+二次メッセンジャーシグナル伝達の模式図
CD38はまた、主にNAD+の細胞内レベルとその生理学的プロセスを制御するNADaseでもある。CD38はまた、NADPとニコチン酸間の塩基交換を触媒し、NAADPの形成につながり、これは加水分解基質としても使用される。NAADP、ニコチン酸アデニンジヌクレオチドリン酸。カラーでこのイラストを見るには、読者は、この記事のWeb版を参照されている www.liebertpub.com/ars
CD38はまた、β-NAD+を基質として使用するが、α-NAD+またはNADHは使用しないことが示されている。CD38はまた、NADPとニコチン酸間の塩基交換を触媒することができ、ニコチン酸アデニンジヌクレオチドリン酸(NAADP)の形成につながるが、これもまた加水分解基質として使用される(90)。また、ニコチンアミドグアニンジヌクレオチド(NGD+)およびニコチンアミドヒポキサンチンジヌクレオチド(NHD+)を含むNAD+のアナログを代謝し、環状化合物(それぞれcGDPRおよびcIDPR)を生成することができる。これらの化合物は蛍光特性を示すが、カルシウム放出性はない(383)。これらの化合物は、ADP-リボシルシクラーゼ活性を調べるための有用な生化学的薬剤である。
心臓ストレス後のCD38の長時間の活性化は、持続的なCa2+放出を誘発し、心臓肥大と不整脈を引き起こすことが示されている(130)。これを裏付ける証拠として、雄性CD38ノックアウトマウスでは心機能の改善が報告されており、ADPRサイクラーゼ阻害剤による治療は複数の試験管内試験モデルや心臓のCa2+過負荷試験で抗不整脈効果をもたらした(131)。同様に、CD73の阻害は、腎ストレス因子に対する保護を媒介することが示されており、CD39活性は、腎虚血性傷害に対する保護を媒介することが示されている(268)。
CD38はまた、その好ましい酵素標的へのNAD+のアクセス性を潜在的に減少させることにより、PARPおよび他のNAD+依存性酵素SIRT1の活性を調節することができる(348)。CD38の触媒活性によって生成されるNAMもまた、SIRT1酵素の内因性代謝物を表す。したがって、CD38 は実際には細胞内 NAD+ レベルと SIRT1 活性の重要な調節因子である可能性があり、その結果、細胞の生体エネルギーの維持、肥満、老化などの SIRT1 機能に影響を与えると考えられている。興味深いことに、ある研究では、CD38ノックアウトマウスでは、野生型動物と比較して、NAD+レベルに有意な影響がないことが報告されている(377)。したがって、CD38の阻害またはアブレーションによる効果の程度については、さらなる調査が必要である。
新しいCD38阻害剤は、最適なNAD+およびNADPHの同化が酸化ストレス侮辱を減衰させるために重要であり、後者はグルタチオンペルオキシダーゼ、ペルオキシレドキシン、およびグルタレドキシンをサポートする究極の電子供与体として機能している。しかしながら、CD38の阻害はまた、免疫学的機能に劇症的な影響を及ぼす可能性がある。CD38/cADPRはまた、多くの社会的行動を制御するオキシトシンの放出をシグナルし、このプロセスを阻害すると、いくつかの形態の精神障害を誘発する可能性がある。さらに、ペラグラ患者で観察されるナイアシン欠乏は、しばしば統合失調症に類似した認知症に進行し(98,264)これは、cADPR形成の障害に起因する可能性がある。
C. サーチュイン活性
もう一つの重要なNAD+依存性機能は、遺伝子転写のサイレント情報調節因子、すなわち酵素のサーチュインファミリーの活性である(図2,ステップn)。サーチュインは、クラスIIIのNAD+依存性ヒストン脱アセチラーゼのファミリーであり、タンパク質リジン脱アセチラーゼ活性、および部分的なADP-リボース転移酵素活性を示す。サーチュインによって媒介される反応では、アセチル修飾されたリジンが標的タンパク質に結合し、特定のポケット内でNAD+が結合する(346)。脱アセチル化は、修飾されたリジン側鎖がNAD+のグリコシド結合の切断に結合することで起こり、副産物として脱アセチル化リジン、アセチル化ADP-リボース、およびニコチンアミドの生成につながる(101)(図9)。
図9 サーチュイン酵素活性
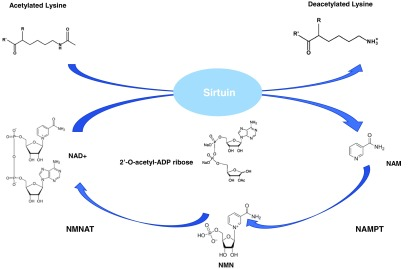
ニコチンアミドは、サーチュイン媒介の脱アセチル化の副産物として生成される。脱アセチル化は、修飾されたリジン側鎖がNAD+のグリコシド結合の切断に結合することで起こり、副産物として脱アセチル化リジン、アセチル化ADP-リボース、およびニコチンアミドが生成される。カラーでこのイラストを見るために、読者は、この記事のWeb版を参照されている www.liebertpub.com/ars
現在、哺乳類細胞では、7つのクラスのサーチュイン(SIRT1-7)が同定されており、それぞれが様々な細胞小器官に局在し、多様な重要な生物学的機能を媒介している(53)(図10)。SIRT1 と SIRT6 は、クロマチン構造の維持、DNA 修復、遺伝子発現に関わる核内タンパク質である。SIRT1 は細胞の長寿を促進する上で重要な役割を果たし、老化表現型の発生を遅らせる鍵を握っている可能性が示唆されている(290)。SIRT1は、代謝調節因子であるペルオキシソーム増殖因子活性化受容体-γ(PPARγ)腫瘍抑制タンパク質(p53)および転写因子の細胞成長連動型FOXOフォークヘッドファミリー(192)を含む、いくつかの重要な転写因子のアセチル化状態に影響を与えることが示されている。しかしながら、いくつかの証拠は、SIRT6もまた、加齢耐性表現型に寄与する可能性があることを示唆している(300)。SIRT2は、細胞質から核へとシャトルする転写因子を脱アセチル化することによって遺伝子発現を制御する細胞質タンパク質である(282)。SIRT3,SIRT4,およびSIRT5は、ミトコンドリアに存在し、マンガンスーパーオキシドジスムターゼ(MnSOD)を含む特定の下流標的の酵素活性を変化させることにより、ミトコンドリアの酸化還元状態の変化に応答する(249)。SIRT7は哺乳類細胞の核小体に局在し、細胞の成長および代謝に関連している(347)。レドックスプロセスにおけるサーチュインの生物学的関連性は、セクションIVでさらに議論される。重要なことに、サーチュイン活性の有益な効果は、NAD+レベルが最適な場合にのみ達成される。
図10 NAD依存性サーチュインと関連する転写因子の機能
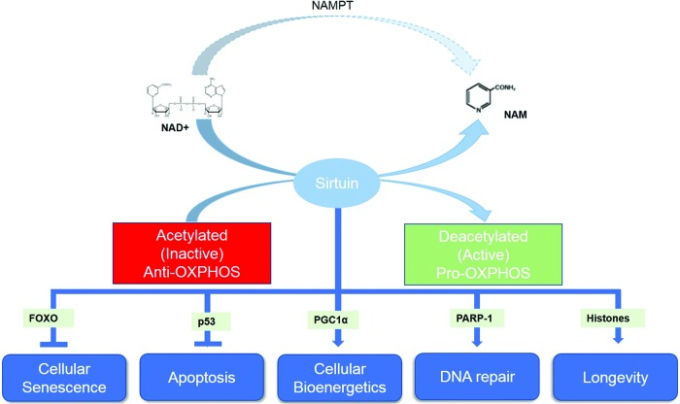 サーチュインが介在する脱アセチル化は、老化や疾患に関連する多数の標的酵素や転写因子に影響を与える。重要なことは、サーチュインの活性がOXPHOSを刺激する一方で、まだ知られていないアセチル化機構が抗OXPHOSを阻害することである。OXPHOS、酸化的リン酸化。このイラストをカラーで見るには、読者はこの記事のウェブ版(www.liebertpub.com/ars)を参照してほしい。
サーチュインが介在する脱アセチル化は、老化や疾患に関連する多数の標的酵素や転写因子に影響を与える。重要なことは、サーチュインの活性がOXPHOSを刺激する一方で、まだ知られていないアセチル化機構が抗OXPHOSを阻害することである。OXPHOS、酸化的リン酸化。このイラストをカラーで見るには、読者はこの記事のウェブ版(www.liebertpub.com/ars)を参照してほしい。
D. NAD+低下の主な原因
NAD+生合成過程での欠乏とは別に、NAD+枯渇が発生する可能性がある条件は主に2つある。
(i)フリーラジカルや紫外線攻撃による過剰なDNA損傷、PARPの過剰活性化につながる。これは最終的にNAD+の高いターンオーバーとその後の枯渇につながる。結果として生じるエネルギー危機と減少したATPの生産は、アポトーシスまたは壊死経路のいずれかを介して細胞死につながることができる
(ii)免疫活性化および炎症性サイトカイン産生の慢性的な増加は、CD38活性を促進し、NAD+の低下に寄与し得る。いくつかの臨床疾患および変性疾患がこれらの基準を満たすことができるが、進行した加齢期における酸化ストレスおよび炎症の慢性的な蓄積は、NAD+低下の主要な推進因子を表す(49)。NAD+前駆体を用いたNAD+同化の促進は、加齢に伴う細胞エネルギーの低下を改善するための臨床的に適切な治療戦略である可能性がある。
IV. サーチュインのレドックス的役割と転写制御
サーチュインは発見されて以来、寿命の延長に関連している。しかし、サーチュインの寿命延長能力はいくつかの小規模なモデルシステムで確立されているが、これらの有益な効果の根底にあるサーチュインの作用様式は不明のままである。経時的なダメージの慢性的な蓄積は、老化プロセスに関連する主な表現型を表している。特に、慢性的な酸化ストレスは、多様な高分子への損傷を誘発し、それらが修復されるメカニズムを乱す可能性がある。最近の証拠は、サーチュインの有益な効果が、酸化還元プロセスを調節する能力によって媒介されている可能性を示唆している。このセクションでは、サーチュインとその酸化還元環境との関連を調査し、サーチュインが介在する脱アセチル化が標的酵素や転写因子にどのように影響するかを検討する。
A. SIRT1
前述したように、腫瘍抑制因子p53は、SIRT1の最初の脱アセチル化基質を表す。転写因子p53は、セストリン、MnSOD、およびグルタチオンペルオキシダーゼ1を含む多数のプロおよび抗酸化遺伝子を活性化することが示されている(284)。Lys382におけるSIRT1によるp53の結合および脱アセチル化は、その転写活性を媒介する(208)。p53のSIRT1脱アセチル化は、酸化ストレスに応答してp53の細胞内局在化に影響を与えることが示されており、抗酸化保護とアポトーシス細胞死の間の代謝スイッチとして機能する可能性がある。例えば、マウス胚性幹細胞では、細胞培養培地中に抗酸化物質が存在しないと p53 のミトコンドリア転座が誘導され、SIRT1 ノックアウト細胞では、酸化ストレスの増加により p53 の核転座が誘導され、抗酸化反応が誘導された(139)。同様に、中膜細胞における SIRT1 のアップレギュレーションは、病理学的濃度の H2O2 に曝露した後の p53 媒介のアポトーシス経路の誘導を減衰させた。しかし、低濃度のH2O2では、SIRT1とp53の相互作用は抗酸化プロセスの誘導をもたらした(191)。
活性酸素ストレスに対するSIRT1の適応的役割は試験管内試験では十分に確立されているが、生きた動物を用いた研究はあまり説得力がない。これは、SIRT1-/-マウスの生産に続く胚性致死率が高いためである。しかし、ヘテロ接合型SIRT1ノックアウトマウスを用いた1つの研究では、腎酸化ストレスに対する脆弱性の増加が報告されており、SIRT1+/-p53+/-の複合体は、p53ハプロインセンス単独の場合と比較して、腫瘍発生に対する感受性が高いことが示されている(146)。
SIRT1はまた、酸化ストレスにさらされた後にFOXO3aを脱アセチル化し、活性化することも示されている(57)。FOXO3aは、内因性抗酸化タンパク質MnSODの産生をコードするSOD2遺伝子の重要な転写活性化因子であるようである。フリーラジカルに直接作用するカタラーゼ酵素は、主にペルオキシソームに局在しており、FOXO3aのもう一つの標的である(185)。SIRT1とp53の間の関係に従って、低レベルのH2O2は、FOXO3a媒介のカタラーゼ誘導を媒介することができ、一方、細胞毒性レベルのH2O2は、FOXO3a媒介のアポトーシスを誘導することができる(144)。心血管疾患では、酸化ストレスの増加は、SIRT1発現をアップレギュレートし、カタラーゼおよびMnSOD発現を刺激し得る。しかし、SIRT1 のレベルが高いと、アポトーシス経路を介して心肥大と細胞死を引き起こす可能性がある(9)。これらの研究を総合すると、SIRT1 は活性酸素センサーとして機能し、低レベルのストレスでは保護を誘導し、厳しいストレスレベルではアポトーシスを誘導することが可能であることが示唆されている。
最近では、細胞内の NAD+:NADH 比の調節に関与するメカニズムが、細胞のエネルギー恒常性を制御するために不可欠な AMP-activated kinase (AMPK) を介して SIRT1 の機能にも影響を与えることが明らかになっている。いくつかの研究では、筋芽細胞で利用可能なグルコースの減少が AMPK の活性化と NAMPT のアップレギュレーションを誘導し、細胞内 NAD+ レベルの上昇と活性化 SIRT1 の活性化につながり、FOXO タンパク質や PGC-1α を含むいくつかの転写メディエーターの活性化につながることが示されており、これにより異化とミトコンドリアの生合成が促進されることが示されている (110)。さらに、AMPKの活性化は、別の研究でSIRT1の下流の転写活性を刺激した(66)。SIRT1はまた、正のフィードバック機構を介してAMPKを活性化することができる。例えば、低栄養下でリン酸化してAMPKを活性化する肝キナーゼB1(LKB1)は、SIRT1の刺激または過剰発現によって(直接的または間接的に)脱アセチル化され得る。これは、核から細胞質へのLKB1の転座を促進し、AMPKをさらにリン酸化する(192)。このように、AMPK-SIRT1軸を介して発生する代謝調節には複数のレベルがあるようであり、これらのステップの多くはさらなる解明が必要である。
AMPK-SIRT1軸とは別に、SIRT1は、PGC-1αと相互作用し、脱アセチル化することもできる。PGC-1αは、ミトコンドリアの生合成を刺激し、組織依存的な方法で間接的にミトコンドリアのダイナミクスも刺激する重要な転写コアクチベーターである。例えば、空腹時に増加した肝内SIRT1はPGC-1αを脱アセチル化し、解糖遺伝子の阻害とグルコース新生に関連する遺伝子の発現増加の両方をもたらす(276)。別の研究では、SIRT1が副腎PC12細胞においてPGC-1αと直接相互作用し、PGC-1αの転写活性を低下させ、ミトコンドリアの酸化的代謝に関係するPGC-1αを脱アセチル化する可能性があることが示された(246)。しかし、骨格筋では、SIRT1活性の増加とPGC-1αの脱アセチル化は、ミトコンドリアの脂肪酸酸化の増加をもたらした(116)。PGC-1α活性の低下は、ミトコンドリア抗酸化タンパク質MnSODの発現低下と関連しており、酸化還元ストレス因子の制御におけるSIRT1の役割をさらに支持している(207)。
最近の研究では、カロリー制限および植物化学物質レスベラトロールは、SIRT1を活性化することが知られており、内皮一酸化窒素合成酵素(eNOS)の発現を増強し、PGC-1αなどの転写因子をアップレギュレートすることによってミトコンドリアの生合成を促進することができることが示されている(78)。同様に、SIRT1は、生体内でeNOSを脱アセチル化し、eNOS活性および細胞内NO産生の増加につながることが示されている(225)。したがって、SIRT1は、eNOSに依存する血管緊張の重要な調節因子を表している。
さらに、低酸素に対する転写応答は、主にHIFファミリーのタンパク質によって制御されることがよく確立されており、そのうちHIF1αおよびHIF2αはよく特徴づけられている[Majmundarら(216)にレビューされている]。HIF1αとHIF2αの両方が、2つの異なるメカニズムでSIRT1によって脱アセチル化されることが実証されている。正常な生理学的条件下では、SIRT1はHIF1αに結合して脱アセチル化し、HIF1αが転写コアクチベーターp300と相互作用するのを防ぎ、その転写活性を阻害することができる(200)。しかし、低酸素環境下では、酸素濃度の低下により、NAD+:NADH比が低下し、最適なSIRT1活性のために利用可能なNAD+が減少するため、HIF1αはアセチル化されたままとなり、低酸素転写活性が阻害される(200)。HIF1αに対するその効果とは逆に、SIRT1はまた、低酸素条件下でSIRT1と複合体を形成することができ、カルボキシ末端の3つのリジン残基(K385,K685,およびK741)で脱アセチル化され、HIF2αおよび関連タンパク質、特にエリスロポエチンの転写活性の増加を導く(92)。
B. SIRT2
また、SIRT2 の発現は、酸化ストレスなどの細胞性ストレス因子に応答して、mRNA とタンパク質の両方のレベルでアップレギュレーションされることが示されている。数多くの研究で、酸化ストレス後の SIRT2 発現の増加は、プロアポトーシスタンパク質 Bim の誘導を介して細胞のアポトーシスにつながることが実証されている (350)。SIRT2 の過剰発現は神経変性を促進することも示されているが、正確なメカニズムは不明である(322)。SIRT2遺伝子が存在しない場合、細胞質シャペロン14-3-3ζのアップレギュレーションは、プロアポトーシスミトコンドリアタンパク質BADを細胞質に隔離し、無酸素・再酸素化誘導細胞死からの保護を媒介する(210)。SIRT2阻害剤は、パーキンソン病(PD)の細胞モデルにおいて、α-シヌクレイン媒介毒性を改善することが示されている(210)。しかしながら、低ストレス条件下では、SIRT2は、FOXO3aの脱アセチル化を介してミトコンドリアMnSODをアップレギュレートし、活性酸素のレベルの低下を導く。
C. SIRT3
イソクエン酸デヒドロゲナーゼ2(IDH2)は、ミトコンドリアのサーチュインであるSIRT3のもう一つの主要な標的を表している。IDH2は、NADPHを使用して還元GSHを生成し、抗酸化作用を媒介する。Schlickerらは、SIRT3はSIRT5ではなく、SIRT3がK211およびK212残基でIDH2を脱アセチル化し、その活性を促進し得ることを示した(294)。CR 後の肝臓、脳、内耳では、SIRT3 依存的な方法で GSH:GSSG 比と NADPH のレベルが増加することが示されている (311)。また、SIRT3 は IDH2 を直接脱アセチル化して活性を阻害し、SIRT3 の過剰発現は NADPH レベルを増加させ、酸化ストレスを媒介とする細胞死を減少させる (311)。これらの研究から、CR、SIRT3,IDH2は加齢性難聴の管理と治療のための重要なターゲットであり、細胞内のNAD+レベルの維持は変性に対する細胞応答を調節することが示唆されている。
IDH2 と同様に、SIRT3 はミトコンドリアの FOXO3a 活性を調節することで SOD2 活性を媒介することが示されているが、正確なメカニズムは不明のままである。マウス胚性線維芽細胞における SIRT3 の過剰発現を用いたある研究では、SOD2 依存的な方法で活性酸素のレベルが劇的に減少したことが示されている(266)。同様に、SIRT3欠損マウスにおけるSOD2のハイパーアセチル化は、SOD2活性の低下と活性酸素産生のアップレギュレーションをもたらした(329)。しかしながら、SIRT3によるSOD2の部位特異的な調節の違いが報告されており、これは細胞タイプ、種、またはストレス条件の違いによるものと考えられる。
SIRT3およびSIRT5の追加のミトコンドリア標的も最近同定されており、これらは酸化ストレスを調節することができる。SIRT3 は、複合体 I、II、III、IV とグルタミン酸デヒドロゲナーゼを脱アセチル化し、グルタミン酸の酸化ストレスを制御して NADPH を生成し、SIRT3 によって脱アセチル化される (そして SIRT4 媒介の ADP リボシル化によって拮抗される) (205)。SIRT5はまた、チトクロムcを脱アセチル化することができる(294)。同様に、SIRT3とSIRT5は共に、尿素サイクルのミトコンドリア局在化反応を制御することができる。より具体的には、SIRT3はオルニチントランスカルバモイルラーゼを脱アセチル化することができ、一方、SIRT5はカルバモイルリン酸合成酵素1に作用して尿素サイクルの機能を高め、酸化ストレスを促進するアンモニウムのクリアランスを促進する(137)。
SIRT3は、正常な心機能の調節および心疾患に対する保護に重要であることが最近示されている。SIRT3のノックアウトは、ミトコンドリアタンパク質の過アセチル化を増加させ、加齢に伴う自発的な心肥大とATPレベルの50%以上の低下をもたらすことが示されている(319)。SIRT3発現の低下および心臓ミトコンドリア酵素の過アセチル化は、心疾患のマウスモデル、および貧弱なヒト心臓においても報告されている(156)。また、SIRT3ノックアウトマウスでは、アシル-CoAデヒドロゲナーゼや脂肪酸酸化に関与する他の酵素(FAO)の活性の増加も報告されている(13)。しかし、別の研究では、絶食動物の心臓におけるFAOの割合が減少したことが報告されている(151)。これらの違いは、タンパク質アセチル化の活性に影響を与えうるストレス因子の種類の違いに起因すると考えられる。
腎ストレスは、SIRT3の発現を低下させることが示されている。例えば、遊離脂肪酸関連の尿細管間質性炎症のモデルでは、SIRT3 mRNA 発現が減少することが示されており、これは、年齢をマッチさせた対照動物と比較して、活性酸素や炎症のマーカーのレベルの増加と並行して発生した(187)。興味深いことに、SIRT3のレトロウイルス過剰発現はこれらの変化を減衰させ、最適なSIRT3機能が腎機能に必要であることを示唆している(370)。同様に、高グルコースレベルでは SIRT3 の mRNA およびタンパク質発現が低下することが示され、NAD+ の補給は、ゲノムレベルとタンパク質レベルの両方で、高グルコース誘発性のメサンジア ル肥大と SIRT3 発現を改善した(390)。これらの知見は、SIRT3が糖尿病性腎症における腎変性から保護できることを示唆している。
D. SIRT4
SIRT3と同様に、SIRT4は脳、心臓、肝臓、腎臓などの異化組織で高発現しているようである(136)。SIRT4は、心筋芽細胞における低酸素誘発性アポトーシスから保護することが示されている(202)。しかし、SIRT4のノックアウトは、マウスのアンジオテンシンII誘導心肥大および線維化に対して保護され、SIRT4が心血管系疾患の病態に直接関与している可能性を示唆している(209)。どちらの研究も、心機能における SIRT4 の正確な役割については見解の相違を示唆しているが、これらの効果は、細胞の酸化ストレスレベルに対する SIRT4 の調節的な役割によるものである可能性が高いと考えられる。
また、腎機能、SIRT4 レベル、NAD+ メタボロームの間には強い相関関係がある。例えば、シスプラチンと植物化学物質クルクミンの共処置は、NAD+レベルを回復させ、シスプラチン誘発性腎毒性によるNAMPT、SIRT1,SIRT3,およびSIRT4発現の低下を減衰させた(342)。しかし、NAMPT、SIRT1,およびSIRT3のレベルも影響を受けたので、これらの影響がSIRT4に直接反応しているとは考えにくい。脳、心臓、腎臓の変性疾患、およびNAD+枯渇に関連する他の加齢に関連した状態におけるSIRT4の役割および作用様式を評価するためには、さらなる研究が必要である。
E. SIRT5
正常な細胞恒常性の維持におけるSIRT5の正確な役割はよくわかっていない。ある研究では、高脂肪食に暴露したSIRT5ノックアウトマウスでは、心臓の体重、心拍数、収縮期血圧に有意な差は見られなかった(379)。しかし、別の研究では、タンパク質のサクシニル化がSIRT5ノックアウトマウスで特異的に上昇していることが示された(248)。これらのタンパク質には、脂肪酸代謝、アミノ酸異化、TCAサイクル、酸化的リン酸化、およびケトンおよびピルビン酸代謝に関与するものが含まれる(41)。心臓虚血にさらされたマウスでは、SIRT5ノックアウトマウスの心臓では、野生型のコントロールと比較して、梗塞容積の増加と酸化ストレスの上昇が報告されている(41)。これらの変化は線維化の増加を伴い、短縮率と駆出率が減少した。SIRT5ノックアウトマウスではコハク酸脱水素酵素(SDH)活性の亢進も報告されており、SDH阻害薬は梗塞サイズを「正常」レベルまで減少させた(41)。このことは、SIRT5の保護効果がSDHのデスキニル化によって媒介されている可能性を示唆している。
同様に、SIRT5のノックアウトはまた、ミトコンドリアタンパク質の過剰スクシニル化、および腎臓におけるマロニル化およびグルタリル化の翻訳後修飾をもたらした(198)。さらに、SIRT5はカルバモイルリン酸合成酵素1(CPS1)を脱アセチル化し、CPS1の活性を高め、血漿中尿素濃度を低下させることが示されている(242)。SIRT5ノックアウトマウスでは、年齢をマッチさせた野生型コントロールと比較して、血中アンモニア濃度の上昇が報告されている。これらの知見は、アンモニアの調節におけるSIRT5の役割に重要な役割を提供している。
F. SIRT6
現在の文献では、SIRT6 機能に対する酸化還元ストレス要因の影響はいまだに不明であるが、ある研究では、SIRT6 のノックダウンは、退行性特徴の発現、テロメア長の短縮、寿命の短縮によって証明されるように、老化を加速させることが示されている(239)。興味深いことに、HIF1αはSIRT6を欠損した細胞でアップレギュレーションされ、グルコース取り込みの増加と解糖の改善につながることが示されている(384)。正常なマウス胚性線維芽細胞では、SIRT6はH3K9ヒストン脱アセチル化酵素として機能し、複数の解糖遺伝子のHIF1α依存性転写を阻害し、HIF1αのコアプレッサーとして作用する。
G. SIRT7
サーチュインファミリーの中で、SIRT7 は最も研究が進んでいない。ある研究では、SIRT7をノックダウンするとp53のアセチル化が促進され、遺伝毒性侮辱に対する脆弱性が高まることが示された(241)。また、SIRT7は、高酸化ストレスレベルに曝露した後の細胞増殖を抑制することも示されている(344)。
H. NAD+前駆体による活性化
NAD+同化のアップレギュレーションがサーチュインによって制御されるプロセスに影響を与えうることを示唆する証拠が増えている。したがって、これらの経路は、NAD+またはNAD+前駆体、またはNAD+生合成経路を操作する他の手段によってアップレギュレートされ得る。SIRT3およびSIRT5のKmがミトコンドリアのNAD+レベルよりも有意に低いことが示されており、これらのサーチュインの活性がミトコンドリアのNAD+レベルの利用可能性によって速度制限されていることを示唆している(150)。現在の証拠は、NAD+の有益な効果を調節する上でSIRT1とSIRT3が重要であることを示唆しており、他のサーチュインに対するNAD+補給の効果については不明な点が多い。他のサーチュインの活性がNAD+療法によって影響を受けるかどうかを調べることは、新たな研究分野となっている。NAD+の補給は、サーチュインファミリーの複数のメンバーを活性化し、複数の生物学的プロセスに多様な効果をもたらし、その結果、さまざまなストレス要因の下で脳、心臓、および腎臓の機能を改善する可能性がある。
V. NAD+メタボロームの分布
NAD+(特にNAD+/NADH比)が細胞のバイオエネルギーのマスターレギュレーターであることはよく知られている。全細胞内NAD+含有量は,0.2〜0.5 mMの範囲であると推定されている(388)。この濃度は、NAD+に対するPARP(0.02〜0.08 mM)(15)およびSIRT1(0.56 mM)の推定NAD+ Km値の範囲内である(278)。このことは、必須基質であるNAD+の利用可能性が、PARPsおよびSIRT1にとって速度制限的であることを意味する。例えば、PARP活性の増加によるNAD+レベルの低さは、SIRT1活性の低下につながり、一方、NAD+レベルの高さは、PARPおよびSIRT1活性を増強する。我々のグループの研究では、慢性的な酸化ストレスとPARPの過剰活性化によるNAD+レベルの低下が、サーチュイン活性の有意な低下と関連していることが実証されている(51, 223)。
最近、一晩絶食したヒトの末梢血単核細胞(PBMCs)血漿、尿中のNAD+メタボロームのメタボロームプロファイリングが発表された(337)。この研究では、リン酸化された NAD+ 代謝産物である ニコチン酸モノヌクレオチド(NAMN)、ニコチン酸アデニンジヌクレオチド (NAAD)、NADP+、NMN、および ADPR は、血球のみに存在し、血漿や尿には存在しないことが示された。NA、NAM、およびニコチンアミドリボシドのレベルは、正常な空腹時血中ではかなり低い(337)。これらの NAD+ 代謝物の正確な測定には限界があるため、これらの NAD+ 代謝物のレベルを調べた研究はほとんどない。ガスクロマトグラフィー質量分析法を用いたある研究では、空腹時血中の NAM の濃度は約 300 nM であり、NA のレベルは 30 nM であったと報告されている (72)。これは、肝外組織へのナイアシンの好ましい形態としてのニコチンアミドの生理学的重要性を示す証拠を提供する。NAおよびニコチンアミドの両方の血中濃度は、ビタミンB3の補充に続いて有意に増加させることができる。これらの薬理学的用量は、ニコチン酸またはニコチンアミドの1〜3gの間である。一方、ナイアシンが豊富な食事には、NAとニコチンアミドの混合物からなるビタミンB3が約10g含まれており、その濃度は植物性および動物性食品中の含有量によって異なる(231, 232)。
これまでに、少量のニコチンアミドが腸や肝臓でNAD+に変換され、ニコチンアミドが全身血中に検出されない場合があることが示されている。さらに、小腸または肝臓におけるNAD+グリコヒドロラーゼの触媒活性またはADPリボシル化は、血流中へのニコチンアミドの放出を誘導することができる(274)。食事からのニコチンアミドはまた、小腸および肝臓でNAD+を形成するために使用され、血流に放出されることもある。肝性NNMTの発現は、NAMからのMeニコチンアミドの形成を導き、従って肝臓でのSIRT1活性を維持する(152)。
ニコチンアミドが、ニコチンアミドが豊富な食事の後に血流中に蓄積することができるか、または必要に応じてNAD+を生成するために複数の組織に貯蔵され、後に血流中に放出されて血流中の閾値レベルを維持することができるかどうかは、依然として不明である。しかしながら、以前のある研究では、ナイアシン欠乏症のラットモデルでは、赤血球において、総NAD+レベルの最大60%が枯渇していることが示された。残りの40%は枯渇に抵抗性があるように見えた(274)。逆に、肝臓でのNAD+のレベルはかなり高く、欠乏の間はより遅い速度で枯渇した(274)。NAD+の短期または長期の貯蔵は、肝臓および赤血球で行われ、ここでは、ナイアシン欠乏の期間、例えば空腹時などの間、血中NAMレベルを調節する。通常の生理学的条件下では、低〜中程度のナノモル濃度で血流中に存在するNAおよびニコチンアミドの肝外組織への移行を促進するために、高親和性トランスポーターが必要である。これらの前駆体間の相互作用を理解することは、NAとニコチンアミドの適切な薬理学的用量を明らかにすることにつながる。
最近、ニコチンアミドリボシドは、マウスおよびヒトにおいて特異的かつ経口的に生物学的に利用可能なNAD+前駆体ビタミンとして同定されている(337)。血中 NAD+ 濃度は、ニコチンアミドリボシド(1000 mg)を 1 日 1 回 7 日間投与すると 2.7 倍に増加し、同時に PBMC において NAAD が最大 45 倍に増加することが示されている。ニコチンアミドリボシド の経口投与がどのようにして NAAD レベルを上昇させるかは不明であるが、ニコチンアミドリボシド は NAD+ サルベージ経路を介して部分的に NAM に変換される可能性が示唆されている(337)。このような変換は、細菌によるニコチンアミドのNAへの加水分解を刺激し、NAAD中間体を用いたNAD+の産生に至る可能性がある。別の研究では、NMNは細胞外で代謝されてニコチンアミドリボシドを生成し、その後、細胞内でNAD+に変換されることが示された(272)。したがって、ニコチンアミドリボシドとNMNは、NAD+同化を強化するための収束性のあるサプリメント戦略を表している。
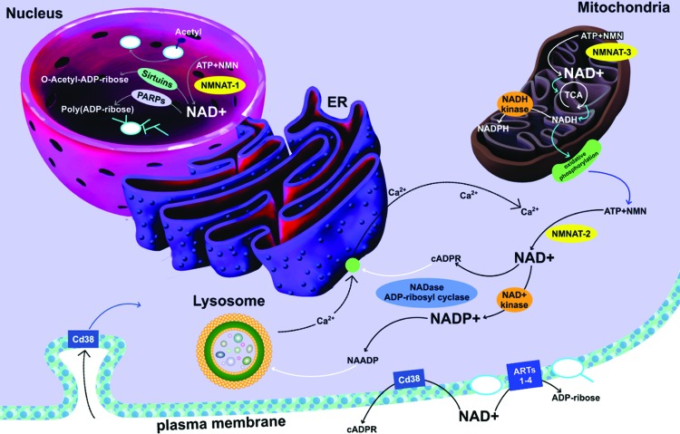
VI. NAD+の細胞内区画化
従来、NAD+は核内に分布していると考えられていたが、核内に起源を持つNMNATは1つの形態のみであることが確認された(296)。したがって、核内のNAD+はポリADPリボースの形成を触媒するために利用可能であったが、核内の細孔を介してサイトゾル内で平衡化することも可能であった(32)。最近まで、ミトコンドリアのNAD+の重要性は不明であり、NAD+はそのままの形でミトコンドリアに輸送されると考えられていた(138)。しかし、現在では、細胞内には核、細胞質、ミトコンドリアの3つのNAD+コンパートメントがあることがわかってきている(31)。NAD+の細胞内コンパートメント化は、ナイアシン枯渇後に重要な役割を果たすと考えられている。細胞内のNAD+レベルが低下すると、細胞内に蓄積されたNAD+は、NAD+の消費に依存する生化学的プロセス間の競争に影響を与え、組織病理に関与する代謝経路に重大な変化をもたらす可能性がある。
最近、核(NMNAT-1)ミトコンドリア(NMNAT-2)ゴルジ装置(NMNAT-3)に局在する3つの異なるNMNAT酵素が発見された(31)。NMNAT-1とNMNAT-2の触媒活性に必要なNMNのレベルは非常に近いが、NMNAT-2の活性にはより多くの量のNMNが必要である(121)。異なる細胞内コンパートメントでのNMNAT酵素の異なる発現は、様々な細胞における最適な代謝機能を促進するための複数の役割、またはストレスへの適応応答のための付加的なメカニズムを示唆している。例えば、ある研究では、ナイアシン欠乏とノルモキシアを併用すると、フィッシャー344ラットの肺NAD+レベルが40%減少することが示された(273)。興味深いことに、慢性的な低酸素状態に曝露しても、肺組織でポリ(ADP-リボース)形成が誘導されたが、肺のNAD+含量は減少せず、むしろナイアシン欠乏の肺組織ではNAD+レベルはほぼコントロールされていないレベルにとどまった。
NMNAT-1は、ADP-リボシル化を担当する主要な酵素、PARP-1だけでなく、PARP-2および3,タンクラーゼ、およびサーチュインを含む近接した位置にNAD+合成を媒介する上で重要な役割を果たしている。核内のNAD+は特定の核孔を介して細胞質に入る可能性があるが、核内でのNAD+の形成にはさらなる利点もある。NMNAT-1の過剰発現は、ワレリアン変性として知られる軸索変性からニューロンを救うことが示されている(381)。同様に、NMNAT-1の不活性変異体もまた、シャペロン効果に起因する可能性があり、神経損失に対して有益な効果を示した(54, 382)。NMNAT-1は、NMNAT-1とポリ(ADP-リボース)との非共有結合を介して、NAD+合成を自動修飾PARP-1の活性部位に向けることができる(32)。
さらに、ミトコンドリアは、TCAサイクルやATP産生のための酸化的リン酸化などの重要な酸化還元反応の主要な部位でもある。また、ミトコンドリアのポリADPリボース代謝やSIRT3-5活性の拠点でもある(247)。これらの基本的なプロセスは、細胞内ADP-リボシル化の増加およびナイアシン欠乏によるNAD+の低下の存在下でも、可能であれば維持される必要がある。NAD+は、アポトーシスまたは壊死の条件下では、特定の透過性遷移孔を介して、ミトコンドリアから細胞質および核に放出され得る(89,149)。したがって、最適な酸化還元機能を維持するためには、細胞質レベルよりも一桁大きいNAD+のミトコンドリアレベルの高い開始レベルが必要である。
一方、ゴルジ装置は、他の小器官への多量栄養素の輸送や細胞外への排出に関与している。ゴルジ装置は、他の小器官のNAD+レベルを制御している可能性があるが、これについては不明である。NAD+はゴルジ装置から細胞質に排泄されるか、または細胞外空間に放出され、通常は大量のNAD+にアクセスできない重要なエクトモノ(ADP-リボシル)トランスフェラーゼおよび/またはADP-リボシルシクラーゼの基質として作用する可能性がある(31)。
細胞内分布におけるNAD+前駆体の効果は依然として不確実であり、いくつかの疑問が未解答のままである。それはポリ(ADP)リボシル化とDNA損傷の修復を調節する最大の能力を持っているので、ビタミンB3の高レベルの治療に続いて、核内のNAD+がより利用可能になるのであろうか、脳の環状ADP-リボースレベルが増加することができることを考えると、細胞質のNAD+の増加もあるのであろうか、そしてミトコンドリアのNAD+プール上のビタミンB3の高レベルの影響は何か?興味深いことに、ニコチン酸NAD+への変換を担う酵素であるNAPRTは細胞質に存在する(142)。したがって、高レベルのニコチン酸補充は、NAD+の細胞質下の内容を変化させる可能性がある。
VII. カロリー制限によるNAD+代謝の変調
マウスやラットを含む短命種の長寿を促進し、霊長類の健康で平均的な寿命を維持するためには、CRが最も効果的な介入であることはよく知られている。CRは、栄養失調や重要なビタミンや栄養素の減少を招くことなく、栄養補助給餌と比較してカロリー摂取量を20%減少させるものと定義されている(222)。CRの分子基盤はまだ明らかになっていないが、CRは脂肪および炭水化物代謝を調節し、酸化ストレスおよび炎症を改善し、インスリンおよびインスリン様シグナル伝達(ILS)ラパマイシンのアミノシグナル伝達標的(TOR)-S6キナーゼ経路、およびグルコースシグナル伝達のRas-タンパク質キナーゼA(PKA)経路をダウンレギュレートするストレス誘発ホルモン応答を活性化すると考えられている(36)。高分子消費量の調節は減食への直接的な反応であると考えられており、ホルミシスおよびTORおよびPKAのダウンレギュレーションはCRの分子的側面である可能性が高い。
いくつかの研究は、モデル生物の様々なCRの効果を検討していた。酵母では、亜致死ストレス条件に曝露するとニコチンアミダーゼの発現が増加し、その結果、NAD+代謝が変化し、哺乳類SIRT2の酵母ホモログであるSir2の活性が増強される(16)。これは、年齢に関連した染色体外リボソームDNAサークルの抑制によって証明されている(309)。TORおよびPKAのダウンレギュレーションもまた、細胞生存研究で報告されているように、CRの寿命に対する有益な効果を媒介している(361)。逆に、ワームの長寿は、ILSまたはフォークヘッドFoxO転写因子daf-16の不活性化によって媒介される(25)。さらなるメカニズムが哺乳類のCRに起因する可能性があるが、NAD+メタボロームの変化とサーチュイン活性の増加は、CR食後の脳と肝臓で報告されている健康上の利点を媒介する上で重要な役割を果たしている可能性がある。
げっ歯類では、CRを投与したマウスでは脳の総NADレベルが上昇し、同時にNAMレベルが低下したことが報告されている(265)。これらの観察は、神経細胞のSIRT1活性の増加と並行して行われ、アルツハイマー病に関連する神経病理を低下させた。別の研究では、絶食マウスで肝内の NAD レベルが上昇し、その変化は SIRT1 活性化、PGC1α脱アセチル化、ミトコンドリア生合成の亢進を伴っていた(276)(図 11)。CR後のNAD+メタボロームにおけるこれらの変化を説明するために、3つのメカニズムが開発されていた:(i) l-トリプトファンとニコチン酸利用可能性の増加は食事の利用可能性に依存しているため、NAD+前駆体であるニコチンアミドとニコチンアミドリボシドの全身的な動員が増加する;(ii) PARPsやCD38などの主要なNAD消費酵素がCRによってネガティブに調節されている場合、NAD+の異化が減少する;(iii) ニコチンアミドリボシドおよび/またはニコチン酸リボシド(NAR)経路のCRが介在するネガティブな調節は、脳と肝臓のNAD+レベルを増加させる可能性がある。
図11 マウスおよびヒトにおけるカロリー制限によるNAD+およびNAD依存性経路の変調
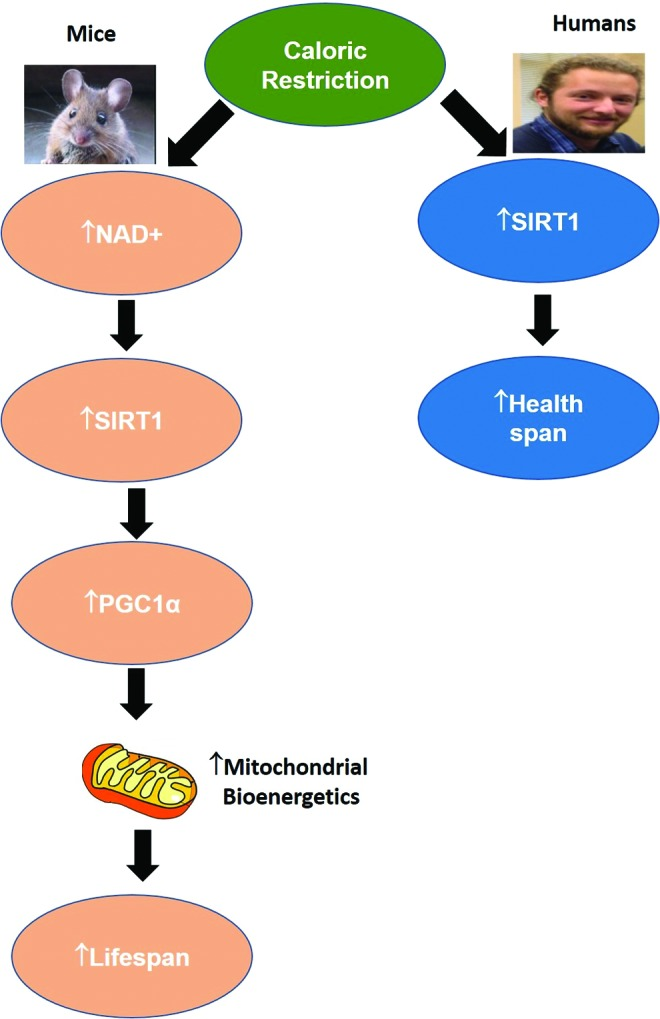
カロリー制限は、ヒトでは神経細胞のSIRT1活性を増加させることが示されている。マウスでは、絶食マウスで肝総NAD+レベルが上昇し、これらの変化は、SIRT1活性化、PGC1α脱アセチル化の増加、およびミトコンドリア生合成の増加を伴っていた。SIRT、サーチュイン。この図をカラーで見るために、読者は、www.liebertpub.com/ars でこの記事のウェブ版を参照してほしい。
実験的な小型モデル生物から得られた情報から、CRや老化の分子基盤についての知見が得られていた。しかし、より大きな動物を用いた研究にはまだ矛盾がある。CRは、ヒトに最も近い実験的モデル生物であるアカゲザルを用いて、制御された環境下で研究されてきた(226)。国立老化研究所(NIA)が行ったある研究では、寿命の有意な改善は報告されていない。しかし、加齢に伴う変性疾患の発症を遅らせる正の傾向が観察された(227)。対照的に、ウィスコンシン国立霊長類研究センター(WNPRC)による別の研究では、寿命と健康寿命の両方で有意な改善が見られた(82,83)。これらの相違は、対象者の食事組成の違いや遺伝的背景の不均一さに起因していると考えられている(図11)。
それにもかかわらず、CRの有益な効果は、国立衛生研究所(NIH)が実施したCALERIE(Comprehensive Assessment of Assessment of Long-term Effects of Reducing Intake of Energy)研究で文書化されている。この研究では、2年間の25%のCRレジメンは、炎症性マーカーや心代謝リスク因子の減少など、肥満ではないヒトにおいて有意な健康上の利益をもたらしたことが示された(280)。しかし、アカゲザルで観察された結果を考えると、ヒトの寿命および正常な生理機能に対するCRの潜在的な効果を検証するために、より大きなサンプルサイズでより長期の研究を実施する必要がある。
VIII. NAD+前駆体の有益な効果
哺乳類細胞におけるNAD+の同化は、2つの主要な経路(de novo経路とサルベージ経路)を介して起こることが知られている。組織、臓器、および細胞におけるNAD+レベルの維持による栄養的および治療的利益を決定するために、NAD+およびその還元型NADHまたはその前駆体のいずれかを補充することは、老化プロセスを遅らせ、および/または加齢に関連する変性疾患の管理を改善するための潜在的な治療戦略を表している。NAD+およびNADHの経口サプリメントは、NAD+の血漿または組織レベルの有意な上昇を示していないが、これは潜在的に、腸を介したNAD+の代謝が非効率であるために、バイオアベイラビリティーの低下につながる可能性がある(177)。さらに、経口NADHは、体内でNAD+に酸化されず、消化管系で効率的に吸収されず、または吸収前にNAMを得ることができない生成物に変換される可能性がある(34,35)。現在のところ、NAD+の静脈内注入は、臨床的に全身のNAD+レベルを増加させる唯一の有効な手段として認識されている。しかし、NA、NAM、NMN、ニコチンアミドリボシド、およびニコチン酸リボシド(NAR)(図12)を含む代替的なNAD+前駆体のいくつかは、いくつかの利点を提供する可能性があると予想される。
図12. NAD+前駆体の化学構造
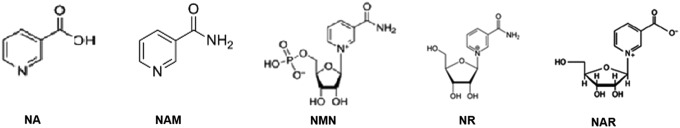
A. ニコチン酸
ニコチン酸はナイアシンの酸性形態を表する。高脂血症の治療のために一般的に臨床的に処方されている。1日1~3gの摂取で血中中性脂肪レベルと低密度リポタンパク質(LDL)を低下させる一方、高密度リポタンパク質(HDL)のレベルを上昇させ、LDL:HDL比を有利に調節することが報告されている(133, 343)。我々の研究グループは、外因性のNAが脳細胞の細胞内NAD+レベルを効率的に増加させることを初めて示した(127)。しかし、NA療法は、大多数の人に有意な皮膚紅潮を誘発するため、臨床での使用は制限されている。50mgの経口NAに暴露された患者では軽度の皮膚紅潮が報告されており、米国およびカナダではニコチン酸許容上限は成人の1日あたり35mgに設定されている(314)。ニコチン酸脂質低下効果は、HM74AまたはGPR109Aとして知られるGタンパク質共役型受容体の細胞表面へのニコチン酸結合によって媒介されると考えられている(314)。脂肪細胞におけるこの結合は、トリグリセリドの脂肪分解を抑制し、循環脂肪酸の減少をもたらし、肝臓の超LDL形成と循環LDL-コレステロールの減少をもたらす(314)(図13)。
図13. 脂質異常症におけるニコチン酸作用機序
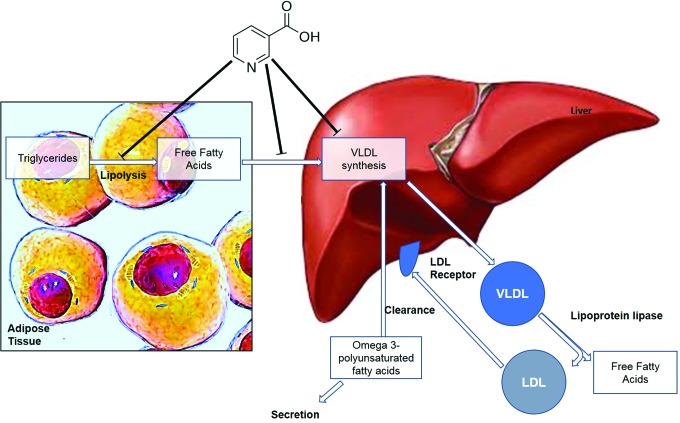
ニコチン酸脂質低下作用は、HM74AまたはGPR109Aとして知られるGタンパク質共役型受容体の細胞表面へのニコチン酸結合によって媒介されると考えられている。脂肪細胞におけるこの関連性は、循環脂肪酸の減少で絶頂に達する、トリグリセリドの脂肪分解を抑制し、肝臓の非常にLDL形成と循環LDL-コレステロールを減少させる。LDL、低密度リポタンパク質。カラーでこのイラストを見るには、読者は、この記事のWeb版を参照されている www.liebertpub.com/ars
この不快な副作用は、皮膚免疫細胞の一部でHM74AがNAを介して刺激されることにより、オメガ6の代謝物であるアラキドン酸がプロスタグランジンE2に変換され、皮膚毛細血管の血管拡張を刺激し、皮膚のフラッシュを引き起こすために発生する(314)(図14)。HM74A とはアミノ酸が 1 つだけ異なる受容体である RUP25 が同定されている。RUP25は、HM74AよりもNAに対してより大きな親和性を示すことが示されており、一部の人では極端な皮膚紅潮反応と関連している(314)。このしばしば劇的で歓迎されない副作用は、したがって、ニコチン酸用途を本質的に治療抵抗性の脂質低下療法に限定している(170)。
図14 NAによる治療後の皮膚紅潮の分子機構の模式図
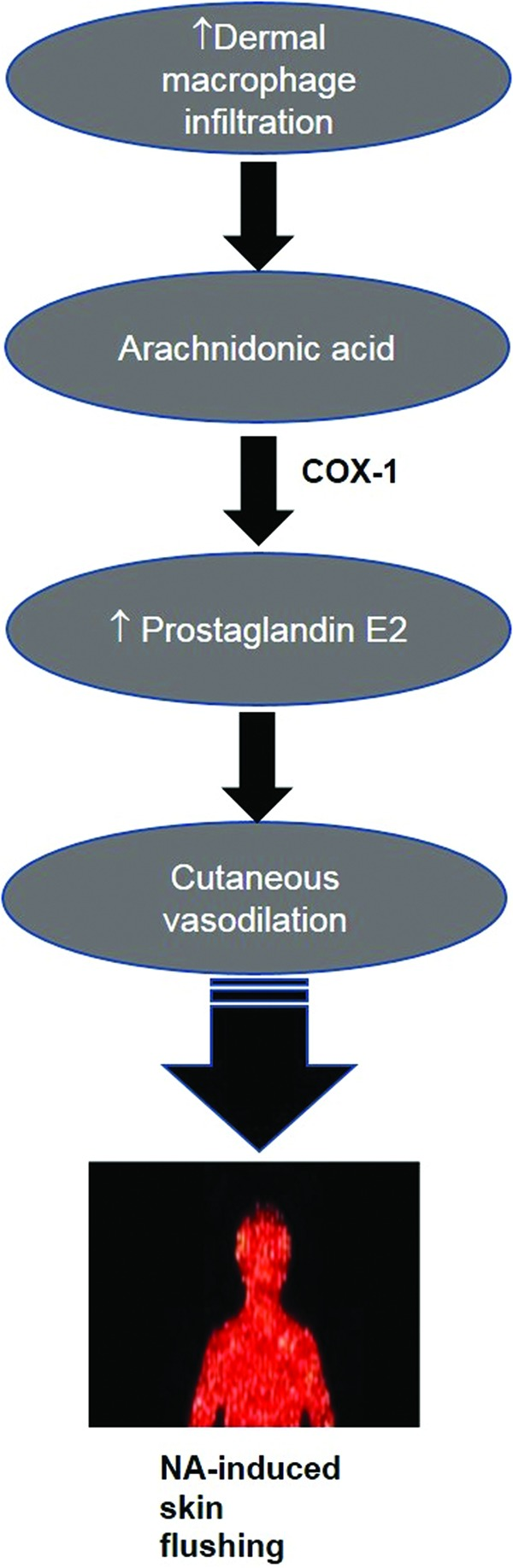
いくつかの皮膚免疫細胞におけるHM74AのNAによる刺激は、オメガ6代謝物AAのプロスタグランジンE2への変換をもたらし、皮膚毛細血管の血管拡張を刺激し、皮膚の炎症を引き起こす。カラーでこのイラストを見るために、読者は www.liebertpub.com/ars でこの記事のウェブ版を参照されている。
さらに、ケトン体であるβ-ヒドロキシ酪酸は、空腹時に産生されるHM74Aの天然リガンドである(314)。ニコチン酸は、β-ヒドロキシ酪酸(半減期に必要な700 nM)と比較してHM74A(半減期に必要な100 nM)に対するより大きな親和性を実証したが、内因性のニコチン酸は、ケトン体が必要なレベルで循環している間、この受容体を活性化するために必要な濃度に達していない(314)。しかしながら、脂質異常症に対するニコチン酸効果を説明するために、他のメカニズムが規定されている。これらには、肝臓ジアシルグリセロールアセチルトランスフェラーゼの阻害、肝臓におけるHDLのクリアランスに関連する経路の阻害(74,345)および肝外組織からのPPAR媒介コレステロール輸送の活性化(84,169,171)が含まれるが、これらに限定されるものではない。より最近では、SIRT1は肝臓X受容体(LXR)の陽性調節因子であることが示されている。保存されたリジン残基でのSIRT1媒介によるLXRの脱アセチル化は、コレステロールレベル、HDL生合成、および脂質恒常性を調節するLXRの活性化につながり得る(199)。
同様に、ニコチン酸レベルの上昇は、小核の頻度を減少させることによりゲノムの完全性を改善することが示されており、ニコチン酸欠乏は染色体の不安定性をもたらす(178-181)。NAによる治療は発がんを遅らせ、マウスメラノーマ細胞およびヒトPBMCsにおけるγ線およびX線照射後の修復効率を高め、低酸素障害後の神経細胞機能を改善することが報告されている(244, 362)。ニコチン酸はまた、NADP+およびGSHの内皮レベルを増加させることで内皮保護を強化することも示されている(114)。しかし、これらの研究は、培養液中の250〜1000μMの範囲の濃度を用いて行われたが、これはヒトにおける生理学的濃度を超えている。
B. ニコチンアミド
ビタミンB3のアミド形態であるニコチンアミドは、SIRT媒介の脱アセチル化、PARP媒介のADPリボシル化、およびCD38 NADaseおよびADPリボシル化活性の副産物としても生成され、サルベージ経路を介してNAD+に変換される。高レベルのニコチンアミドは、腫瘍内の微小血管の流れを促進することにより、放射線治療を増強したり、固形腫瘍を化学増感させたりするために使用されてきた(3,4)。臨床レジメンでは、95%酸素/5%二酸化炭素の吸入と組み合わせて、全身血中濃度を700μM以上に上昇させることを目的とした経口投与(3-6g)が記載されている(358,359)。これは、改善された腫瘍血流および酸素生成をもたらし、したがって、ミオシン軽鎖キナーゼ(MLCK)を阻害することによって放射線の効果を増強する。MLCKのリン酸化の減少は、血管平滑筋の収縮を阻害し、血管拡張を促進する(283)。しかし、試験管内試験で使用される濃度は臨床的な全身レベルよりも桁違いに高く、大血管効果はNAD+産生に対する効果とは独立しているようである。
ニコチンアミドはまた、動物モデルにおいて、いくつかのタイプの糖尿病の進行を予防または遅らせることが示されているが、この効果は、ランダム化対照試験において、グラム量のNAMを使用して再現可能ではなかった(10, 113, 215, 323, 371, 386, 387)。ニコチンアミドはまた、いくつかの動物モデルにおいて、低酸素および/または化学毒性の侮辱に対する脳および他の組織の血管損傷および虚血を制限するための潜在的な治療戦略としても使用されており、いくつかの成功を収めている(91,301,302,334,352)。
また、NAM外用製剤は、酒さ、自己免疫性水疱性皮膚炎、およびにきびを含む炎症性皮膚疾患の治療にも成功している(251)。また、ニコチンアミドは、皮膚の完全性の維持、皮脂レベルの低下、および色素沈着斑および赤みの低減にも以前に使用されている(328)。ニコチンアミドはまた、炎症性メディエーターIL-6およびTNF-α、ならびにDNA損傷マーカーであるシクロブタンピリミジン二量体および8-オキソ-7,8-ジヒドロ-2-デオキシグアノシンの発現を抑制することにより、紫外線誘発性皮膚損傷の急性および慢性的な影響を減少させることが示されている(234)。同様に、ニコチンアミドは、げっ歯類モデルおよびヒトの研究において、紫外線誘発免疫抑制および光発がんを改善することが示されている(115)。同様に、ヒト臨床試験では、経口ニコチンアミドがプラセボと比較してアクチン性角化症を有意に減少させることが示されており、非メラノーマ皮膚癌の予防にも有用である可能性が高い(321)。
しかしながら、ニコチンアミドは、NAD+異化の副産物として、NAD依存性酵素の天然フィードバック阻害剤としても機能することが十分に確立されている(図15)。例えば、NAM濃度が上昇するとPARP、サーチュイン、CD38の活性が比例して阻害され、これがヒトにおけるニコチンアミドの抗糖尿病効果のメカニズムであると推測されている。NAD+濃度は上昇しているが、重要なNAD依存性機能(例えばSIRT1活性)は阻害される。さらに、NAM補給は、コリン欠乏ラットモデルにおいて肝脂肪蓄積を悪化させ、この効果は、ポリADP-リボースの蓄積、およびNAM排泄におけるメチル基の利用によるエピジェネティックメチル化の減少に起因していた(19, 173)。したがって、外因性ニコチンアミドは、NAD+に変換することができるが、それは再び、その酵素阻害とメチル枯渇の可能性のために、特に中長期的には、理想的なサプリメントとは考えられていない。
図15 NAM の作用機序と NAD+ メタボロームへの影響
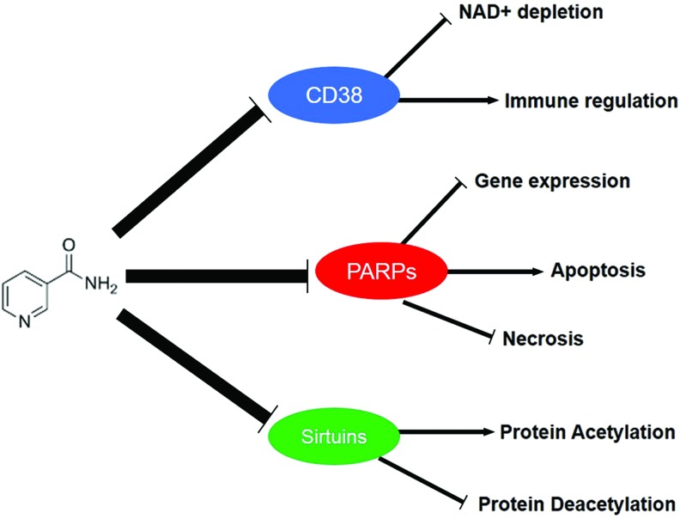
ニコチンアミドはまた、NAD依存性酵素の自然なフィードバック阻害剤としても機能する。例えば、PARP、サーチュイン、CD38活性はNAM濃度が上昇すると比例して阻害され、これがニコチンアミドのヒトにおける抗糖尿病効果のメカニズムであると推測されている。NAD+レベルはまだ上昇しているが、重要なNAD依存性機能(例えば、SIRT1活性)は阻害される。この図をカラーで見るために、読者は、この記事のウェブ版を参照してほしいwww.liebertpub.com/ars
C. ニコチンアミドモノヌクレオチド(NMN)
NMNはNAMからのNAD+合成のための重要な前駆体である。NMNの補給は、最も可能性の高い膵β細胞への作用を介してインスリンレベルに正の効果を持つことが示されている(316)。NMNの補給はまた、いくつかの試験管内試験および生体内試験モデルで肥満と血管障害を軽減することが示されている(87,355)。また、NMNは脳のミトコンドリア呼吸障害を増加させ、アミロイドβ(Aβ)オリゴマー誘発毒性と認知障害から保護し、反応性グリア誘発運動ニューロン喪失を改善し、神経幹細胞と前駆細胞を維持することで中枢神経系機能を改善することも報告されている(356)。また、NMNは脳虚血誘発性アポトーシスから保護し、脳損傷後の神経新生を促進することも示されている(360)。また、NMN治療は、Nrf2およびHP-1タンパク質の発現をアップレギュレートし、脳虚血後の神経炎症後にそのトランザクティベーションのためのNrf2核転座を促進し、二次的な神経学的損傷を減衰させることが示されている(360)。同様に、NMNの同化に必要な酵素であるNAMPTの過剰発現は、脳卒中において神経保護効果があるようである(353, 354)。
最近の研究では、APPswe/PS1dE9 ADトランスジェニック(tg)マウスにおいて、NMN治療により、認知障害、神経炎症、Aβ沈着、シナプス欠損を含むアルツハイマー病(AD)の主要な病理学的特徴が有意に改善されたことが示された(372)。別の研究では、NMN治療は、AD-tgマウスのJNKの活性化とAPP-leavage secretaseによるAPP媒介アミロイド原性処理を阻害することも発見された(349, 356)。したがって、NMNはADや他の加齢性変性疾患の治療や管理のための新たな治療標的となる可能性がある。
心臓特異的複合体I欠損マウスモデルであるNDUSF4KOマウスでは、NAD+/NADH比の低下、ミトコンドリア透過性遷移孔(mPTP)を含むミトコンドリアタンパク質の過剰アセチル化、酸化的リン酸化の障害、および心臓ストレスに対する脆弱性の増加を示した(174)。NMNを投与すると、NAD+の細胞内レベルが部分的に回復し、mPTPの過リン酸化が抑制され、慢性ストレスにさらされた後の心不全が改善された。さらに、これらのマウスはまた、SIRT3の活性化の増加を示し、NMN治療後のNAD+レベルの増加に続く活性の増加によるmPTPを含む主要なタンパク質の脱アセチル化もまた、NDUFS4KOにおけるNAD+の有益な効果を説明する可能性がある(174)。
NMN処理のこれらの報告された有益性にもかかわらず、証拠はまた、NMNが細胞膜内に効果的に含まれており、高い拡散勾配を受けないことを示唆している。このことは、NMNがほとんどの細胞間を効果的に行き来することができるかどうかという問題を提起している。興味深いことに、細胞外NMNは外因性NAD+の直接代謝から積極的に産生される可能性がある(388)。しかし、NMNがヒトにおいて有益であることを証明する条件の範囲を確立するためには、さらなる研究が必要である。
D. ニコチンアミドリボシド
ニコチンアミドリボシドは、もともと新鮮な牛乳から分離されたNAD+の天然に存在する前駆体である(338)。ニコチンアミドリボシドを外因性に投与すると、さまざまな細胞株において細胞内NAD+レベルが上昇することが示されている。ニコチンアミドリボシドの補給は、ニコチンアミドリボシドキナーゼ2遺伝子の転写誘導が関与するメカニズムを介して、マウス後根神経節ニューロンを軸索障害から保護した(288)。この効果はニコチン酸またはニコチンアミドによって再現可能ではないので、ニコチンアミドリボシドは、キヌレニン経路によるNAD+のde novo合成が障害されたときに、中枢神経系における主要な前駆体を表している。ニコチンアミドリボシド は、NA とは対照的に皮膚紅潮を起こさず、NAM とは対照的に肝障害を起こさずに NAD+ レベルを効率的に増加させることが示されている (337)。ニコチンアミドリボシドはまた、肥満マウスにおいてコレステロール低下剤としての役割を果たすことも示されている(67)。最近の研究では、ニコチンアミドリボシドはミトコンドリアで好まれるNAD+前駆体であり、ニコチンアミドリボシドの有益な生体内試験効果は、ミトコンドリアのサーチュイン活性だけでなく、PARP、サーチュイン、CD38,NAD依存性酸化還元酵素、およびNADPH依存性活性酸素解毒酵素を含む核細胞質標的の調節に起因していることが示されている。ニコチンアミドリボシド の投与はまた、SOD2 および NADH ユビキノン酸化還元酵素サブユニット A9 を含む SIRT3 のいくつかのタンパク質標的のアセチル化状態を減少させることが示されており、ニコチンアミドリボシド は薬理学的に SIRT3 を活性化するために使用される可能性があることを示唆している (67)。さらに、ニコチンアミドリボシドの投与により、NAD+-SIRT3経路による騒音曝露後の神経細胞の変性が遅くなった(56)。
ナイアシンの排他的な供給源としてのニコチンアミドリボシドの代謝健康への影響はまだ明らかになっていない。仮定的なヒト癌遺伝子である型破りなプレフォルジンRPB5インタラクター(URI)を過剰発現させたマウスにおける食事中のニコチンアミドリボシドの補給は、形成不全病変を減少させ、腫瘍の発生を防止し、したがって、肝細胞癌の治療および管理のための新規なアプローチとしてのNAD+補給の証拠を提供する(340)。ヒト URI の過剰発現は、アリル炭化水素受容体とエストロゲン受容体(OR)およびキヌレニン経路の代謝障害が関与するメカニズムを介して、肝細胞における異形成の発生を促進する(340)。hURI過剰発現マウスの末期腫瘍は、ニコチンアミドリボシドを補充したマウスではアポトーシスが増加して退行した(320, 340)。
ニコチンアミドリボシド補給の有益な効果にもかかわらず、有益な効果をもたらすためにしばしば使用される用量は、現在市販されているサプリメント(6-500mg/kg体重/日)と比較して著しく高い(400mg ニコチンアミドリボシド/kg体重/日)。ある研究では、ニコチンアミドリボシド(300mg/kg体重/日)はラットの運動パフォーマンスを低下させることが示され、以前に報告されたニコチンアミドリボシドのエルゴジェニック効果は確認できなかった(186)。この観察を説明するために2つの仮説が立てられている。(i) NAとニコチンアミドの類似した効果に基づいて、ニコチンアミドリボシドは運動中にFAOも減少させ、疲労の早期化をもたらす可能性がある;および/または(ii)ニコチンアミドリボシドはNAD+とNADP+の酸化還元特性を変化させ、非最適な還元状態に導く可能性がある(186)。運動パフォーマンスに対するニコチンアミドリボシドの効果を調べるためには、さらなる研究が必要である。
最近、軽度の肥満誘発性(40%脂肪)食に曝露したマウスを用いて、代謝柔軟性および精巣上体白色脂肪組織の遺伝子発現に対する幅広い範囲の食事性ニコチンアミドリボシド濃度の影響を調べた(303)。この研究では、30mg ニコチンアミドリボシド/kgの食事は、代謝柔軟性、および脂肪形成のマスターレギュレーターであるPPARγ、および2つの抗酸化遺伝子であるSOD2およびPRDX3の発現の増加に関して、代謝健康の改善に最も有益であることが示された(303)。この研究では、30 mg ニコチンアミドリボシド/kg の食事が代謝健康を増強するための最適な濃度であると結論づけられた。
E. ニコチン酸リボシド
NAD+前駆体の中で最も検討されていないニコチン酸リボシド(NAR)は、細胞質の5′-NTによるNMNとニコチン酸モノヌクレオチド(NAMN)の脱リン酸化を介してヒト細胞で産生されることが示されている(38, 190)。この代謝物は、NAD+生成の重要な前駆体となることが予想されている。低マイクロモル濃度のニコチン酸リボシド(NAR)は、細胞の生存率を維持するのに十分な量のNAD+を生成することがすでに実証されている。ある研究では、ニコチン酸リボシド(NAR)は生理学的に十分なレベルで細胞によって産生され、送達されることが示されている(190)。他の細胞タイプがニコチン酸リボシド(NAR)を利用して、お互いの間でニコチン酸リボシド(NAR)を輸送している可能性がある。IX. NAD+前駆体の薬物動態
前駆体分子を利用してNAD+プールを増強することは、複数の健康上の利点と多様な治療上の意味合いを持つ可能性がある。NA、NAM、NMN、およびニコチンアミドリボシドは、強力なNAD+ブースターとして公表されている。NMNおよびニコチンアミドリボシドはまた、NAおよびニコチンアミドに対して有害な反応を示す患者において、一般的なサプリメントとして使用され得る。NAおよびニコチンアミドの薬物動態は広範囲に調査されているが、ニコチンアミドリボシドの薬物動態特性は、マウスおよび中年のヒト被験者において最近決定されたばかりである(337)。しかし、NMNやニコチン酸リボシド(NAR)の薬物動態効果は、ヒトモデルやマウスモデルではまだ十分に検討されていない。
A. ニコチン酸
ニコチン酸薬物動態は、これまでにニコチン酸薬理学的用量といくつかの徐放製剤を用いて検討されてきた。非盲検、用量漸増、クロスオーバー試験では、12名のヒト被験者に溶液状のNA 2000mgを徐放性(ナイアシン水溶液25mgを80用量で10分ごとに投与)中間性(ナイアシン水溶液50mgを40用量で10分ごとに投与)または速放性(ナイアシン水溶液100mgを20用量で10分ごとに投与)で投与した(232)。ピークニコチン酸は10μM(徐放)と240μM(速放)の間で変化した。興味深いことに、徐放製剤の曲線下面積(AUC)は、速放製剤の対応するものよりも25倍低かった。徐放製剤中のニコチン酸は、腸と肝臓に取り込まれ、NAD+を形成し、ニコチンアミドとして循環に放出される(232)。重要なことに、10μMの濃度は、血中のニコチンアミドの生理的レベルよりも約30倍高いと推定される。ニコチン酸速放製剤は、より高いピークレベルとAUCをもたらすだけでなく、それはまた、16μMにピークのNAMレベルを上昇させる。ニコチン酸は優先的にニコチンアミドのための4時間の半減期と比較して、1時間の半減期を持つ高レベルで循環から除去される(231,232)。したがって、ニコチン酸高用量はまた、ニコチンアミドのレベルを超生理学的レベルまで上昇させた。したがって、脂質に対するニコチン酸効果は、ニコチンアミドの保護効果とNAD+同化の増加によるものである可能性もある。
経口投与後のマウスにおけるNAD+前駆体のNAD+生成効率を比較した最近の研究では、ニコチン酸は最も低いレベルのNAD+を生成した(337)。しかし、肝NAD+蓄積の速度論は、NAMまたはニコチンアミドリボシドのいずれかよりも4-6時間速かった。ニコチン酸経口投与は、NAAD(中間体)のレベルを増加させることにより肝NAD+を2倍(1から2mM)に増加させ、Meニコチンアミドのレベルの増加によって報告されているようにNAD+の異化を促進した(337)。肝臓は、十分なNAが利用可能である限り、NAD+の同化を促進し、一方で、副産物としてNAMを生成するいくつかのNAD依存性プロセスの活性を増加させた(337)。MeNAMレベルの増加によるNNMT発現の増加は、マウスおよびヒトにおいて肝SIRT1タンパク質を安定化し、脂質レベルを調節する(152, 194, 336)。
別の研究では、2-3週間の給餌間隔で経口補給を行うと、NAD+のレベルが有意に変化したことが報告されている(26)。以前は、NAD+レベルはプラトーに達する前に時間に応じて増加し続けると考えられていた。NA(30および40000mg/kg)を補充したラットでは、40000mg/kgを給餌した動物では骨髄NAD+が有意に増加した(26)。しかし、摂取が慢性化するにつれてNAD+レベルは低下し、この効果がニコチン酸ニコチンアミドへの変換に関連したニコチン酸取り込みのみによるものか、骨髄におけるNAD+の異化作用の変化によるものかは不明であった。したがって、NAD+前駆体の長期補充に対する薬理学的反応は、時間の経過とともに変化する可能性があると推測されている(26)。このことはまた、より高いNAD+レベルが細胞機能への悪影響を誘発する可能性があるかどうかという重要な疑問を提起し、それによって適応応答を刺激する。
B. ニコチンアミド(NAM)
ヒトにおける経口NAM 3~6gの薬物動態は以前に調査されている(93)。高用量では、吐き気および嘔吐を含む副作用が生じやすい。ニコチンアミドの血中濃度のピークは1~2mMであった。これは、循環レベルの3000倍以上と推定される(93)。この数値はまた、放射線感受性に関連する最小濃度をはるかに上回っている。ニコチンアミドの半減期は4-5時間であり、これはカルボゲン呼吸および放射線治療を促進するのに十分な時間を提供する。MLCKがニコチンアミドの標的となる可能性が高い場合、抑制濃度を細胞内に蓄積するためにMLCKが必要であることが示唆されている。ニコチンアミドの細胞内トランスポーターは以前に同定されているが、高濃度のニコチンアミドに対する細胞応答のメカニズムは明らかにされていない。ある研究では、ニコチンアミドの循環レベルが低下しても放射線感受性は維持されたことが示されており、ニコチンアミドの有益な効果は、NAD+生成の増加(279)、および/またはポリADP-リボースおよびDNA損傷誘発性アポトーシスの阻害(153)を含む追加の下流効果に関連している可能性が示唆されている。
高用量のニコチン酸はNAMレベルを増加させることが示されているが、ニコチン酸に対するNAM増加の効果は依然として不明である。現在のところ、NAMからNAへの酵素的変換は同定されていない。しかし、経口および腸内細菌叢を介したニコチンアミドの脱アミド化は可能である(212)。ある研究では、多量の唾液中のニコチンアミドがNAに変換されたが、ニコチンアミドの大量経口投与後のニコチン酸循環レベルは検出されなかったままであった(317)。この研究で使用された定量法は、NAD+メタボロームの変化を明確に区別するのに十分な感度を持っていなかったと思われる。NA治療後の一般的な副作用である皮膚潮紅もNAMで報告されており、高用量のNAM投与後にNAが増加する可能性があることを示唆している(167)。
ボーラスで提供される3種類のNAD+前駆体ビタミンを同等の経口投与量で比較した最近の研究でも、肝NMN、NAAD、NAD+、およびNADP+レベルの上昇、ならびにMeNAMおよびADP-リボースの上昇によって証明されるNAD+異化活性の上昇が示された(337)。しかしながら、ニコチンアミドによるNAD+上昇のAUCはNAと比較して50%の有益性を示したが、この研究では、NAM媒介のADP-リボース蓄積はNAと比較して50%の欠損を示した(337)。これらの研究はまた、NAADがNAD+合成の増加のためのバイオマーカーを表す可能性があり、l-トリプトファンおよびNAなどの従来のNAAD前駆体とは独立していることを示唆している(337)。NAとは異なり、ニコチンアミドは強力なコレステロール低下剤ではなく、高レベルのニコチンアミドはPARPおよびサーチュイン活性を阻害する可能性がある。
高用量のニコチンアミドに続く潜在的な適応応答の証拠は以前に報告されている。高用量のNAM(4 g/kg)を補給したラットでは、脳内のNAD+とADP-リボースのレベルが上昇した。興味深いことに、これらの変化は、海馬依存性の空間学習テスト(197)で障害されたパフォーマンスによって報告されるように、認知障害を伴っていた。NAD+の増加は、最終的にPARP、サーチュイン、CD38/CD157を含む多様な酵素の活性の増加につながる可能性がある。これは、順番に、細胞機能における動的な効果を有する可能性がある。
C. ニコチンアミドモノヌクレオチド(NMN)
血中のNMNの検出は依然として困難である。血漿中のNMN濃度は約50μMであることが報告されているが(275)別の研究ではNMN濃度は検出できなかった(272)。これらの違いは、NMNの検出技術の違いに起因する可能性がある。例えば、高速液体クロマトグラフィー(HPLC)ベースの方法を用いて、NMNとNAD+の細胞内濃度は、500 mg/kgのNMNを腹腔内注射してから15分後に、白色脂肪および膵臓組織で最大500 pmol/mgおよび50 pmol/mgまで上昇することが報告されている(376)。しかし、肝NMNおよびNAD+レベルは、185mg/kgを経口投与した6時間後に、それぞれ10および4000pmol/mgに達することが報告されている(337)。同様に、NMNのレベルは、腫瘍では組織で1.5pmol/mg程度、腹水では80nM程度であることが報告されている(310)。
NMNは、10%ウシ胎児血清(FBS)を補充した血漿および細胞培地中で安定であるようであり、1時間のインキュベーション後にNAMレベルの増加は報告されていない(272)。しかし、NMNの注射は血漿中のNAMレベルの有意な増加をもたらし、これはNMNが腹腔内注射後に部分的にニコチンアミドに変換される可能性を示唆している。NMN注射後のマウス血漿中のニコチンアミドの存在は、NMNが最初にニコチンアミドリボシドに変換される可能性を示唆している(272)。
最近の研究では、NMNのニコチンアミドリボシドへの脱リン酸化は、酵母においてNAD+を産生するために必要であり、哺乳類細胞における外因性NAD+前駆体としての主要なステップを表すことが実証された(272)。細胞外受容体CD73がニコチンアミドリボシド放出酵素として作用すると考えられている。CD73はピロホスファターゼと5′NT活性の両方を持ち、細胞外NAD+とNMNのニコチンアミドリボシドへの変換を促進し、その結果、さらなるNAD+合成を刺激するために使用することができる。これは、CD73の遺伝子サイレンシングが潜在的なNAD+前駆体としてのNMNの使用を阻害することを示した別の研究によって支持されている(310)。さらに、ニコチンアミドリボシドキナーゼ1は最近、NAD+合成のためのNMNからニコチンアミドリボシドへの変換のための重要な律速酵素として同定されており(272)、NMNとニコチンアミドリボシドについて報告されている重複する効果を説明するための信頼できる説明を提供する。
D. ニコチンアミドリボシド(NR)
哺乳類組織におけるニコチンアミドリボシドの薬物動態を評価するためには,生体試料中のニコチンアミドリボシドの感度や検出性が悪いため,その評価には限界がある。また、健康なヒトにおけるニコチンアミドリボシドの薬物動態を調べることを目的とした臨床試験の結果はまだ得られていない。しかし、ニコチンアミドリボシドはマウス血漿中で培養すると急速に分解され(10分後には約10%、1時間後には約66%が分解される)NAMも同等に増加した。これらの結果は、ニコチンアミドリボシドをニコチンアミドに分解する血漿因子の存在を示唆している(272)。ニコチンアミドリボシドの分解に続いてニコチンアミドの検出も、10%FBSを含む細胞培地で観察されている(272)。重要なことに、ニコチンアミドリボシドは、1週間の潜在的な寿命を持つ乳中のタンパク質分画中で安定である(338)。ニコチンアミドリボシドはまた、細胞結合型で数時間循環する可能性がある。
ある研究では、ニコチンアミドリボシドはマウスとヒトにおいて安全で経口的に生物学的に利用可能であり、副作用は報告されていないことが示された(337)。しかし、将来の研究では、ニコチンアミドリボシドが潮紅エピソードと関連しているかどうかを評価するために、検証された潮紅症状の質問票を組み込む必要がある。NMN、ニコチン酸モノヌクレオチド(NAMN)、NAM、NAM、NAAD、NAD+およびNADP+のレベルは、経口ニコチンアミドと比較してニコチンアミドリボシドの経口投与後に高レベルであった(337)。さらに、ADP-リボースレベルは、NAおよびニコチンアミドと比較してより有意に上昇しており、ニコチンアミドリボシドは、モル当量のNAMおよびNAよりもNAD+を消費する酵素の活性を高めることができることを示唆している(337)。さらに最近、ランダム化二重盲検プラセボ対照試験では、ブルーベリーに含まれる天然由来のフィトケミカルであるプテロスチルベンと組み合わせたニコチンアミドリボシドは、8週間の試験全体を通して全血中の溶解液中で用量依存的にNAD+を増加させることが示された(88)。このことは、ニコチンアミドリボシドが、ナイアシンのアミド化および酸性形態よりもNAD+合成およびNAD依存性活性のより強力な前駆体であることを示唆している。
E. ニコチン酸リボシド(NAR)
私たちの知る限りでは、ニコチン酸リボシド(NAR)の薬物動態は文献に報告されていない。また、ニコチン酸リボシド(NAR)は生理的にサブマイクロモル濃度が低いため、薬理学的な理解は困難なままでした。1H-核磁気共鳴(NMR)法は、細胞培養液中のニコチン酸リボシド(NAR)の化学シフトが観察されることから、ニコチン酸リボシド(NAR)の検出に成功している(190)。また、検出法の感度が低いため、長時間にわたるスペクトルが必要である。HEK293細胞におけるNAPRTの過剰発現は、細胞培養液中のニコチン酸リボシド(NAR)の検出につながり、この効果は、ニコチン酸異化作用の増加によるものである(190)。HeLa細胞では、ニコチン酸リボシド(NAR)ではなくニコチン酸リボシド(NAR)は、NAD+生合成および細胞生存を維持するのに十分な量で放出される(190)。
F. ニコチン酸アデニンジヌクレオチド-NAD+代謝上昇のバイオマーカー
上述したように、NA、NAM、およびニコチンアミドリボシドは、中間体であるNAADのレベルを上昇させることが示されている。しかし、NAADのレベルの増加は、ニコチン酸摂取後に最も低かった(337)。この知見は、NAD+前駆体の補給によるNAD+同化の増加は、NAD+異化の副産物(ADP-リボースおよびMeNAMなど)の蓄積を増加させるだけでなく、NAADおよびニコチン酸モノヌクレオチド(NAMN)の逆行性合成を刺激することを示唆している。NAD+の同化速度が増加すると、NAD+は脱アミド化されてNAADを形成し、まだ知られていないメカニズムによってNAADを形成する。同様に、NMNの脱アミド化は、ニコチン酸モノヌクレオチド(NAMN)およびNAADのレベルの増加につながる可能性もある。脱アミド化反応はまだ検証されていないが、このとらえどころのない生化学的反応を引き起こすメカニズムはまだ特定されていないが、NAADもNAADPから形成される可能性がある。
X. NAD+前駆体のNAD依存性プロセスへの影響
NAD+前駆体は、NAD+およびNADP依存反応やADP-リボシル化などの正常な細胞機能に対する有益な効果とは別に、NAD+同化のためのこれらの基質は、複数の細胞区画における細胞内NAD+レベルを増加させるという共通の効果を共有している。これまで、ビタミンB3の1日の推奨摂取量に応じて、通常の代謝過程が満たされる可能性があり、また、高用量では代替メカニズムが誘導される可能性があると考えられていた。しかし、NAD+前駆体の生理的役割は、食事中に自然に存在する用量とは異なる可能性があることが明らかになっている。
A. ニコチンアミドとPARPs
ニコチンアミドがPARP-1活性を阻害することができるかどうかは、依然として議論の余地がある。ニコチンアミドが媒介するPARP-1の阻害のためのKi値は、無細胞系では30〜200μMの範囲である。培養細胞でのKi値は、無細胞系でのKi値よりも3倍大きかった(271)。このことは、ニコチンアミドのNAD+への取り込みおよび/または変換が細胞培養において制限されている可能性があることを示唆している。哺乳類では、経口ニコチンアミドは血流に入る前に小腸および肝臓で代謝される。ニコチンアミドは、少量ではあるが肝外組織に取り込まれ、直ちにNAD+に変換される。さらに、血液の量は組織の量よりも著しく少ない。したがって、組織のNAMレベルがPARP-1活性を阻害するのに必要な濃度に達する可能性は低い。
逆に、基質の増加によるニコチンアミドのNAD+への変換は、ポリ(ADP-リボース)レベルを促進することが示されている。例えば、NAM(1g/kg)の補給は肝NAD+レベルを50%増加させたが、基質のポリ(ADP-リボース)レベルは2倍増加した(163)。このことは、ニコチンアミドがPARP-1阻害を媒介するよりも基質プールの増強に効果的であったことを示唆している。しかしながら、PARP-1活性を増強するために同じ動物を肝癌原体に曝露した場合、ポリ(ADP-リボース)含量は同じであった。ニコチン酸はまた、より高いレベルのポリ(ADP-リボース)形成を促進した(163)。まとめると、基底PARP-1活性はDNA損傷誘発PARP-1活性とは異なる形で調節されている可能性があり、ニコチンアミドはPARP-1活性の後者の形を阻害することでより効果的であるかもしれない。
別の研究では、NAM(4 g/kg)の経口投与に応答して肝外組織においてPARP-1活性が増加したことが報告されている。その研究では、骨髄のNAD+レベルが2.5倍に増加し、基底のポリADP-リボースレベルが5倍に増加し、DNA損傷誘発ポリADP-リボースが2倍に増加した(42, 43)。同様に、放射線感作モデルにおける研究では、ニコチンアミドによる放射線感受性は、PARP活性およびDNA修復過程の阻害とは独立したメカニズムに起因することが示された(279)。
B. ニコチンアミドとサーチュイン
哺乳類のサーチュインは低いNAD+結合親和性を有しているため、細胞内のNAD+濃度のわずかな変化によってその脱アセチル化酵素活性を効率的に調節することができ、強力なNAD+センサーとして機能している。加齢に伴い細胞内のNAD+濃度が低下すると、サーチュイン活性とSIRT1を介したp53の脱アセチル化が抑制される可能性がある(51)。逆に、CRまたはNAD+補給による細胞内NAD+レベルの増加は、サーチュイン活性をアップレギュレートする可能性がある。植物由来のスチルベンであるレスベラトロールは、相対的にはSIRT1のみを活性化するが、NAD+の補給は哺乳類の7つの形態のほとんどすべてのサーチュインを活性化することができる。例えば、細胞内NAD+レベルによるSIRT3の調節は、アポトーシスに対する細胞の回復力の主要な決定因子であることが実証されている(143)。
生物学的系におけるニコチンアミドの増加がPARP-1活性を阻害することができるならば、それはまた、サーチュインのような他のNAD+依存性プロセスを阻害する可能性がある。高レベルのニコチンアミドは、NAM切断反応を阻害するか、またはNAD+結合部位での競合的阻害を媒介して、機能またはサーチュイン酵素の変化をもたらし、最終的にNAD+のレベルを高める可能性がある。ニコチンアミドの生理学的レベルは、いくつかのサーチュインのIC50と同じ範囲内である(159)ことから、サーチュインはNAD+センサーとしてだけでなく、NAMセンサーとしても機能する可能性があることが示唆されている。
ニコチンアミドはサーチュインの触媒部位の特定の保存領域に結合し、脱アセチル化ではなく中間体との逆塩基交換反応を誘発し、サーチュインの脱アセチル化酵素活性を阻害することが示されている(129)。基底交換平衡定数は、SIRT1では約20であると推定されている(37)。このことは、任意のNAM濃度での塩基交換反応の完全阻害によるSIRT1の最大活性化は、酵母ではSir2よりも大きいことを意味する。最近、ニコチンアミドの合成アナログであるイソニコチンアミド(isoNAM)が、触媒部位での結合のためにニコチンアミドと競合することが示されている(228)。しかし、ニコチンアミドとは異なり、isoニコチンアミドは中間体と実質的に反応せず、Sir2活性の増加につながる。
ニコチンアミドはサーチュインの生理的阻害剤を表す。細菌Sir2,酵母Sir2,マウスSir2,SIRT1,SIRT2,SIRT3,およびSIRT5の阻害のためのIC50値は、それぞれ26,120,160,50,100,36.7μM、および1.6mMであると測定された(129)。酵母核内のニコチンアミドの濃度は10〜150μMと推定されており、ニコチンアミドが生体内でのSir2活性の調節因子であることが示唆されている(292)。酵母および細菌のサーチュインは、哺乳類のサーチュインと比較して、NAD+に対するKmsおよびKdsが低いため、細胞内NAD+濃度の変化に対して哺乳類のサーチュインよりも感受性が低い可能性がある(68)。したがって、NAD+レベルの増加は、哺乳類サーチュインの活性化をもたらす可能性が高い。哺乳類細胞では、いくつかのラット組織で低レベルのニコチンアミドが報告されている(142)。これは、NAD+および関連するピリジンヌクレオチドの産生のためのニコチンアミドの迅速な異化によるものと考えられる。しかしながら、高濃度のNAM(最大300μM)がTg2576マウスの脳で報告されており(265)ニコチンアミドが哺乳類組織におけるサーチュイン活性の調節因子としての追加の証拠を提供している。さらに、細胞サブコンパートメントにおけるNAD+とニコチンアミドの比率は加齢とともに低下し、サーチュイン活性を低下させることがある(223)。この効果は、PARPによるNAD+の利用の増加と、NAMPTを介したサルベージ経路によるNAMからのNAD+同化の減少によるものと考えられる(160)。
これらの生理的役割に応じて、いくつかの他のメカニズムが、ニコチンアミドによるサーチュインの調節を説明するために帰結されている。例えば、ニコチンアミドはSIRT1の完全な阻害剤であるのに対し、SIR2AF2(Archaeon Archaeglobus fulgidus由来のSIRT2ホモログ)を部分的にしか阻害できない(129)。同様に、ニコチンアミドは、他のサーチュインについて報告されている非競合的阻害とは対照的に、SIRT3の競合的阻害剤である(129)。哺乳類のSIRT3は、腫瘍抑制効果が実証され、解糖代謝を調節するミトコンドリアのサーチュインである(18)。ニコチンアミドによるSIRT3の阻害は、解糖代謝を増加させ、好気性解糖を阻害することで、がん細胞のバイオマスを減少させ、慢性的な組織障害を改善する可能性がある。
サーチュイン活性の前方反応の速度を増加させることが知られている塩基交換とは別に、SIRT1と比較してSIRT3の阻害速度におけるニコチンアミドとNAD+の間の競争の程度が大きいことを説明する別のメカニズムとして、直接的な競争が提案されている(129)。これは、ニコチンアミドの存在下でNAD+が触媒部位に結合することに関係している(129)。SIRT3および他のサーチュインに対するニコチンアミドの作用機序を解明することは、より効率的なサーチュインの阻害剤および活性化剤を開発するのに役立ち、臨床への応用が期待される。
C. CD38を介したプロセス
CD38が必須基質NAD+、ニコチンアミドのSIRT1酵素への利用可能性を調節することで、SIRT1活性を調節できることが、我々のグループや他の研究者によって提案されている(52)。これは、肥満、代謝障害、細胞エネルギーのホメオスタシスや細胞の老化、老化の調節に大きな影響を与える可能性がある。細胞内の NAD+ 同化を促進しながら NAM レベルを低下させることにより、CD38 の阻害は SIRT1 活性を増加させることができる。
いくつかのCD38阻害剤が同定されている。これらには、NAMおよびNA、アラビオン-NADなどのNAD+アナログ、およびジチオスレイトールを含む還元剤が含まれる(76)。CD38は、カルシウム生成に対する効果に起因して、平滑筋収縮、細胞死およびアポトーシス、神経およびホルモンシグナル伝達、ならびに卵の受精の重要なメディエーターとしても機能する(76)。したがって、CD38の阻害は、高血圧、心臓虚血、喘息、機能不全性陣痛など、カルシウムの恒常性が損なわれる病理学的条件の下で有用である可能性がある。
しかしながら、CD38はまた、母体および社会的行動を調節するオキシトシンおよび副腎皮質刺激ホルモンなどのホルモンの放出にも関与している(166)。これらの状態でCD38を阻害すると、心理機能に重大な悪影響を及ぼす可能性がある。また、CD38は免疫系において重要な役割を果たしており、CD38のノックアウトは細菌感染に対する感受性を高めることが示されている(201)。したがって、NAD+前駆体の効果およびNAD+メタボロームへの変化は、CD38活性およびNAD依存性プロセスのこれまで知られていなかった効果を有する可能性があり、適切な用量レジメンが考案され、個々の患者の要件を満たすように適応される場合、代謝性および炎症性疾患の治療のための重要な治療戦略として機能する可能性がある。
D. 酸化還元反応
真核細胞では、ATPの生成は、ミトコンドリアの酸化的リン酸化によって主に達成される。このプロセスでは、炭素基質の分解に続いて放出された自由エネルギーは、ATPの生産(308)につながる電子輸送チェーン(ETC)を介して電子供与体と電子受容体との間の交換によって捕捉される。NAD+は電子受容体として機能し、その還元は、その後、NAD+を生成するためにETCの複合体Iによって酸化されることができるNADHの生成につながる。NAD+/NADH比は、いくつかの酸化還元酵素の重要な指標となる。NADHのレベルが高くなると、NAD依存性のプロセスが阻害される可能性がある。酸化的リン酸化における代謝的不均衡は、いくつかの心臓、神経、および腎臓の病理学的疾患と関連している(206)。ETCへの変化は、ATP産生の大幅な減少、細胞内Ca2+流入とフリーラジカル産生の増加、およびNAD+/NADH比の低下につながる可能性がある。いくつかの心臓ストレス因子に応答して嫌気性代謝に酸化的な間のスイッチは、酸化的損傷を低減し、ATPレベルを維持することが示されている。しかし、この代償メカニズムは、ミトコンドリアのNAD+プールを制限しながら、酸化的リン酸化を損なう(97)。
同様に、NAD+/NADH比は心臓と腎臓で重要な役割を果たしているようであり、NAD+前駆体の補給は、心臓ストレス因子およびAKI誘発腎障害による酸化的リン酸化の障害から保護することが示されている(150)。PGC1α欠損マウスでは、ニコチンアミドの投与はFAO、ATP生成、およびNAD+/NADH比を増加させ、AKI毒性から保護することが示された(339)。したがって、酸化的リン酸化の障害やNAD+の低下につながる他の異常を伴う変性状態では、NAD+前駆体を介したNAD+同化のアップレギュレーションが酸化還元機能を改善する可能性がある。
NAD+/NADH比の効果については、ヒトの生体内試験試験ではまだ未熟である。しかし、二光子顕微鏡を用いて表皮皮膚層のNADHおよびNADPHの定量化を行ったある研究では、動脈閉塞後のNADH蛍光の有意な増加が報告されており、NADHからNAD+への酸化のための電子供与の必要性の低下による酸化的リン酸化の減少があることが示唆されている(24)。同様に、若年者と比較して高齢女性の顔面皮膚では、減少したNADPH蛍光発光が報告されている(286)。これらの研究は、細胞のバイオエネルギーの調節におけるNAD+の役割を支持する証拠を提供している。
ポリ(ADP)リボシル化の障害は、ペラグラとして知られるナイアシン欠乏症のヒト疾患で報告されている皮膚病変の基礎となるDNA損傷に対する感受性の増加と関連している(274)。さらに、細胞内カルシウムレベルを調節する環状ADPリボースの形成障害は、ペラグラ性認知症で観察される神経細胞の損失に寄与している可能性がある(378)。しかし、NAD+/NADHの比率に対応する酸化還元反応は、ADP-リボシル化反応のようにNAD+レベルの変化による影響を受けにくい。NAD+/NADP+は、多数の酸化/還元反応において可溶性の補酵素として機能する。触媒酵素は、リボフラビンをベースとしたヌクレオチドをプロステック基の供給源として利用する。他のものは電子移動を容易にするために鉄を含んでいる。ポリおよびモノ(ADP-リボシル)アチオン反応とは異なり、鉄およびリボフラビンの欠乏は、皮膚の日光過敏症、およびペラグラの2つの主要な特徴である認知症を誘発することは知られていない。
ある研究では、健康な高齢者から採取したリンパ球の増殖、サイトカイン放出、細胞の酸化還元状態に対するNAD+の還元型であるNADHの影響を調べた(40)。NADH(500μM/L)に曝露した細胞では、GSHやカタラーゼ活性の増加が見られたが、マロンジアルデヒドやカルボニル蛋白質は著しく減少しており(40)酸化ストレスの低下が示唆された。最近では、1mM NADHで処理すると、SIRT2の機能を増加させることで、核内Nrf2の発現、カタラーゼ活性、総GSHが増加することが示されている(69)。
同様に、ナイアシン欠乏が内因性抗酸化防御機構、NADPH:NADP+、およびGSH:GSSGレドックスカップルに及ぼす影響は依然として不明である。2つの研究では、ナイアシン欠乏は酸化ストレスのマーカーを増加させるが、NADPHまたはGSHの低下を誘発しないことが示された(27, 325)。このことは、ナイアシン欠乏はポリADP-リボースの蓄積を障害したが、GSH防御を維持しながら、さらなる組織損傷を刺激しなかったことを示唆している。ナイアシン欠乏の期間中の酸化還元反応の維持を説明するために、いくつかのメカニズムが仮定されてきた。これらのメカニズムには、NAD+に対する基質親和性の変化、酵素や補酵素の細胞内局在化、酵素活性/発現レベルの直接的な調節などが含まれる。
XI. NAD+と関連する前駆体はホルミシスを示すか?
NAD+の減少が老化プロセスの主な原因であり、心臓、脳、肝臓、腎臓、皮膚に影響を与えるいくつかの加齢性変性疾患の病態に関与している可能性があることを示唆する証拠が増えている。これらの結果は、NAD+単独でもNAD+前駆体を用いても、加齢や関連する病態から保護するためのNAD+補給の可能性を強調している。このような可能性は臨床的には重要な意味を持つが、ヒトの老化におけるNAD+とその調節の役割はまだ部分的にしか理解されていない。特に、「非常に高い」NAD+レベルを有することの影響については、ほとんど理解されていない。我々は、NAD+レベルの調節が、多くの臨床結果を混乱させる可能性のあるホルミシス用量/反応を誘導する可能性があることを示唆している。
ホルミシスという用語は、1943年にSouthamとEhrlich(314a)によって、木材腐朽菌に対するレッドスギの木の抽出物の効果を説明するために、最初に生物医学的な文脈に組み込まれた(62)。この研究では、様々な種類の真菌が低用量の刺激と高用量の細胞代謝抑制効果を示すことが示された。21世紀になって、ホルミシスは、化学的または物理的な薬剤への曝露後に起こる二相性の用量/反応、または細胞毒性のある侮辱に対する過補的な反応として定義するために使われるようになった(63)。サーチュインの活性化剤であるレスベラトロールは、最近、乳房、前立腺、結腸、肺、子宮、白血病腫瘍細胞株を含む様々な生物学的モデルにおいて、ホルモン性の用量/反応を誘導することが示されている(64)。これらの研究では、レスベラトロールの濃度が低いほど腫瘍細胞の増殖が促進された。しかし、高濃度では、レスベラトロールは抑制効果を誘導した。例えば、レスベラトロールは、ビタミンD受容体の活性を増加させ、4μMまでのT47D乳癌細胞の増殖を促進し、それ以上の濃度では、腫瘍細胞株の増殖を減少させた(64)。
他の研究では、レスベラトロールは5~25μMの濃度で培養海馬ニューロンを酸化ストレスから保護できることが示されている(108)。レスベラトロールはまた、COX-2の阻害により、培養腫瘍細胞の炎症および酸化ストレスを改善する可能性がある(391)。しかし、これらの細胞が酸素およびグルコースの利用可能性が低下した条件下にある場合、レスベラトロールはアポトーシス細胞死を誘導することができる(64)。
これらの知見に照らすと、NAD+同化のアップレギュレーションもホルミシスの二相性の用量/反応に適合している可能性がある。例えば、神経細胞が虚血性障害やグルタミン酸などのサイトトキシン、Aβ凝集体などの細胞ストレスにさらされている場合、NAD+レベルの上昇は、細胞障害性刺激剤と比較して投与量や投与期間に依存して、有益な効果と破壊的な効果の両方をもたらす可能性がある。例えば、ナイーブT細胞をNAD+でインキュベートするとアポトーシスが誘導されたが、活性化されたT細胞をNAD+でインキュベートしてもアポトーシスの兆候は見られなかった(204)。ADPリボシル化の基質としてのエクトNADはナイーブT細胞には作用するが、活性化T細胞には作用しないことが示唆された(297)。このことは、NAD+の多くの効果が、好ましい応答をもたらすと思われる環境因子に依存していることを示している。
NAD+を消費する酵素間の競争もまた、ホルミシスを示す。例えば、先に述べたように、PARP1の活性は、酸化的DNA損傷の蓄積により、また、高エネルギー摂取に応答して、加齢とともに増加する。NAD+、PARP1,およびSIRT1のKmは比較的類似しているので、PARP1活性化後のNAD+レベルの低下は、SIRT1活性の低下も誘発し得る。したがって、PARP1活性の低レベルは、軽度の酸化ストレスレベルにさらされた後にDNA損傷を修復することができるが、PARP1活性の増加は、SIRT1活性の低下とエネルギー制限を介して細胞死につながる可能性があり、したがって、疾患の進行を悪化させる(269)。したがって、ヒトにおけるNAD+の用量/反応効果の性質を評価するためには、厳密な二重盲検およびプラセボ対照臨床試験が必要である。老化や加齢に伴う疾患に対するNAD+同化作用の役割をさらに理解するためには、さらなる研究が必要である。
XII. 体外・生体内研究の限界
人間の健康や病気に対するNAD+代謝の重要性にもかかわらず、NAD+のレベルを決定することは依然として課題である。また、臨床でのNAD+前駆体の補充には臨床的な意義があるが、そのような補充でNAD+レベルが上昇することを示す証拠は限られている。細胞培養や動物モデルは研究研究によく使用されているが、人間の生理を正確に表しているわけではない。さらに、組織サンプルまたは細胞ホモジネートを分析するために現在使用されている生化学的アッセイまたは分析方法は、pH、温度、光、および化学剤または緩衝液の変化に対して脆弱である。したがって、より正確で信頼性の高い定量化と生体内試験条件への外挿が必要とされている。
A. 細胞培養系
蓄積されたエビデンスは、いくつかの変性疾患における酸化ストレス、炎症、およびl-トリプトファン異化の増加が関与していることを示唆している。このことは、フリーラジカル損傷の基本的なメカニズムの研究と、フリーラジカル損傷から身を守るための治療戦略としてのNAD+メタボロームの調節への道を開いていた。例えば、マウスやヒトの初代脳細胞培養や不死化細胞株は、酸化的損傷や適応細胞応答の影響を調べるモデルとして非常に有用である。中枢神経系の酸化ストレスおよび変化したキヌレニン経路代謝をモデル化するための最も一般的なアプローチは、初代グリア細胞およびニューロン細胞を有害な条件に曝露し、外因性のプロオキシダントおよび神経保護剤を添加することである(44, 46, 48, 50, 71, 195, 333)。細胞培養モデルはまた、ナイアシン欠乏の効果、およびNAD依存性プロセスの阻害(PARP、サーチュイン、およびCD38阻害など)の、いくつかの試験管内試験疾患モデルにおける細胞機能への影響を調べるために使用されてきた(46,47,52,119,267)。しかしながら、これらの研究の基礎となるほとんどの細胞培養成分は、培養中の細胞増殖および生存率を最大化することを目的としており、生体内試験での自然な生物学的プロセスを完全に再現しているわけではない。また、培養系におけるNAD+前駆体の有益な効果が、非生理学的メカニズムを介して発生する可能性があるという強い証拠もある。
ビタミンB3は、そのアミドおよび酸性形態の両方で細胞培養中に存在する。しかしながら、ニコチンアミドは最高濃度で存在し、これは血流中のナイアシンの主要な形態としてのニコチンアミドの意義を反映している。一般的に使用される細胞培養培地(例えば、MEM、Williams、RPMI、BME、L-15,およびDulbecco’s)は、1〜4 mg/LのNAMを含有するが、より特殊な培地(MCDB 131およびBGjB)は、6〜20 mg/Lを含有することがある。4mg/Lの等価モル濃度は、∼33μMである。この量は、血漿中の平均的なNAM濃度の約300倍である。これらの濃度は、細胞内のNAD+貯蔵、cyclic-ADP-およびモノリボシル化、ならびに阻害剤の研究に深い影響を及ぼす可能性がある。他の細胞培養培地は、4μMまでの濃度でNAMおよびニコチン酸等しい内容物を含む。同様に、その濃度は、全身循環におけるニコチン酸生理的濃度をはるかに上回る。さらに、これらの濃度は、これらが培養中の細胞で発現している場合、HM74A受容体を活性化するのに必要な量よりも有意に大きい。
B. 生体内試験モデル
ヒトの寿命は、小型の哺乳類よりもはるかに長いため、ヒトの正常な老化の間のNAD+代謝の影響を完全に特徴づけることは困難である。そのため、従来の生体内試験研究では、再現性のある結果を得るために、表現型的に老化が加速されているか、または寿命が延長されている動物、単一遺伝子の役割に焦点を当てたトランスジェニックモデル、変異体モデル、およびノックアウトモデルを用いて行われてきた。寿命が短いため、寿命に対する内因性因子と外因性因子の影響を調べるためのモデルとして、近交実験用げっ歯類、特にラットとマウス(例えば、老化促進マウス)が使用されている(324)。しかし、これらの近交系モデルでは、ヒトと比較するための有意な遺伝的多様性が得られず、ヒトの条件との相関性が低いため、これはかなり限定的である。これまでのところ、重篤な患者の炎症を抑えるための臨床試験候補は、不十分な動物モデルに過度に依存したために、150以上もの臨床試験が失敗に終わっている。
我々は、老化した雌のWistarラットから収集した生化学データを概念的に翻訳することで、NAD+代謝の役割やヒトの「正常な」老化の一部として起こる他の分子変化の理解をさらに深めることに取り組んできた(51-53)。生理的に老化したウィスターラットは、少数の個体の中に有意な遺伝的多様性を示す外交系モデルである。この多様性交配ラットモデルは自然集団をより代表するものであり、したがって、これらの薬理学的戦略の有効性を評価するための遺伝的基盤を明らかにするための強力なツールであり、以前に交配された動物ではそうでなければ初期段階にある第一選択治療試験における副作用を特定するための強力なツールである。この動物モデルで以前に実証された老化の追加的な影響には、アストロサイト/神経細胞比の著しい減少、「血管の安定性」に影響を及ぼすペリサイト/内皮関係の変化、著しい炎症性カスケード、中枢神経系の新生血管化および血液/網膜バリアの破壊、および防御関連Fos発現の低下が含まれる(158, 218)。
C. 検出方法
NAD+およびその関連代謝物は、これまでに様々な方法を用いて測定されてきた。例えば、間接的な測定を提供する酵素アッセイや比色アッセイは、信頼性に影響を与える固有の困難性を有しており、温度やpHのわずかな違いによる代謝物レベルの大きな変動の影響を受けやすく、低ピコモルレベルを検出することができない。さらに、緩衝塩やイオンペアリング剤を含む移動相に依存する逆相HPLCは、感度を高めるために使用されているが、依然として低ピコモルレベルの検出に限られている(70)。
対照的に、液体クロマトグラフィー/タンデム質量分析法は、高い特異性と感度で、異なる生物学的サンプル中のNAD+代謝物の微量レベルをより強固に定量することができる。これは、NAD+ メタボロミクスのゴールドスタンダードである。しかし、NMR とは異なり、複雑なサンプリングプロセスが必要となる(335)。NAD+代謝物(すなわち、遊離塩基、モノおよびジヌクレオチド)の多様性は、それらの同時差分分析を大きな課題としている(335)。私たちは最近、脳や生殖細胞を含む生物学的サンプル全体の NAD+ メタボロームとアデノシンリン酸塩を定量するための改良された方法を開発した。この方法の主な特徴は、分解能が向上したことと、アミノ相カラム上で 17 種類の分析物を同時に定量できることで、2 つの別々のグラジエント(アルカリ性と酸性のクロマトグラフィーグラジエント)の必要性を回避できることである (60)。
NAD+メタボロームの非破壊的検出・定量法の開発は、無傷のヒトや動物の体内における細胞内NAD+レベルや酸化還元状態を解明するために望まれている。最近、生きた動物の脳内のNAD+分子の内因性31P MR信号を決定するための新しい磁気共鳴イメージング(MRI)が開発された(389)。この技術は、与えられた磁場強度における特定の分光学的特性を利用することにより、NADHのMRI信号をNAD+のMRI信号から分離することができる。このアプローチには9.4Tと16.4Tの超高磁場が必要である。この非侵襲的手法は、7TのヒトMRスキャナを用いて、健康なヒトの脳における細胞内NAD+とNADHの含有量とNAD+/NADHの酸化還元状態を測定するためにさらに使用されてきた(389)。我々は、サーチュイン活性の必須基質である細胞内NAD+レベルが、ヒトおよび生理学的に高齢化したラットでは加齢とともに低下することを初めて示した(51, 52, 223)。MRIを用いて、健康なヒトの脳の細胞内NADHの年齢依存的な増加とNAD+、総NAD含量、NAD+/NADH酸化還元電位の年齢依存的な減少を再確認した。
NAD+メタボロームを定量する方法の改善により、異なる研究室間でのNAD+研究の標準化や、前臨床研究の臨床への翻訳に伴う課題の克服が期待されている。
XIII. NAD+前駆体の臨床応用の展望
ペラグラは、ニコチン酸またはl-トリプトファンのいずれかが欠乏している食事によって引き起こされる症候群であり、精神病性の症状は、必須アミノ酸であるl-トリプトファンの神経細胞を枯渇させることができるIDOのアップレギュレーションのために、おそらく老年期の認知症につながる可能性があり、神経変性を引き起こす可能性がある。NAD+前駆体であるニコチン酸またはニコチンアミドの投与は、1930年代に認知症患者の神経学的状態を改善したことがある。ニコチン酸またはニコチンアミドのいずれかの薬理学的用量はまた、動物モデルおよび臨床環境の両方において、脂質異常恒常性障害、関節リウマチ、I型糖尿病、大腸炎、多発性硬化症(MS)および統合失調症を含む他の疾患に対して劇的な治療上の利益を提供してきた。これらの前駆体の中で、ニコチン酸はGタンパク質共役型受容体であるGPR109を特異的に活性化し、プロスタグランジンであるPGE2とPGD2の放出を導くようである(314)。これらのプロスタグランジンは、内因性のシグナル伝達機構を介して強力な抗炎症効果を発揮する。ニコチンアミドは動物モデルにおいてMSを予防することができるが、サーチュインの阻害剤でもあるため、長期的な細胞生存および長寿に有害であることが証明される可能性がある(258, 259)。
NAD+投与は、複数の酸化ストレス誘発性変性疾患における細胞傷害をも減少させる可能性があることを示唆する証拠が増えてきている。NAD+治療は、PARP1誘導性アストロサイト死を減少させることが示されている(7)。PARP1は、糖尿病、AD、およびPDを含むいくつかの疾患の病因に関与している(196,221)。NAD+の補充は、PARP1が媒介する細胞死から保護することができるので、NAD+の投与は、少なくとも部分的にPARP1の毒性を改善することによって、これらの疾患における細胞生存率を改善する可能性がある。試験管内試験での研究では、NAD+は、PARP1活性化後3〜4時間で投与しても保護されたままであることが示されており、NAD+投与は、細胞傷害を減少させるための長いウィンドウ期間を有することを示唆している(8)。さらに、NAD+はまた、サーチュイン活性を増強し、および/またはエネルギー代謝を改善することにより、細胞の生存性を改善する可能性がある。
エネルギー代謝およびミトコンドリア機能におけるNAD+代謝経路の潜在的な関与はかなり前から知られてたが、DNA修復および長寿におけるNAD+の関与の示唆は、過去10年間で急速に増加している。NAD+合成経路の特性評価は、これらの進歩を可能にしただけでなく、細胞生物学におけるピリジンヌクレオチドの多様な役割の理解に大きく貢献していた。しかしながら、神経変性や老化におけるNAD+の基本的な役割に関する情報はまだ限られている。この分野でのさらなる研究が必要とされている。
今回のレビューでは、細胞変性におけるPARP1に焦点を当てたが、タンキラーゼのような他のPARPが細胞機能に果たす役割についてはほとんど知られていない。NAD依存性タンキラーゼはテロメラーゼ活性の主要な媒介者であるため、NAD+がタンキラーゼ活性の調節を通じて老化過程にも影響を与える可能性が高い(385)。したがって、加齢脳に関連する可能性のある神経発生を含む特定の生物学的機能に対するタンキラーゼおよびテロメラーゼに対するNAD+前駆体の効果を研究することは興味をそそられる。
さらに、NAD+は寿命を制御する多様な経路を制御している。NAD+の重要性は、NAD+合成に必要ないくつかの経路が存在することを示す遺伝的証拠を提供する最近の研究によって、さらに強調されている。例えば、新たに同定されたNAD+前駆体ニコチンアミドリボシドは、酵母Saccharomyces cerevisiaeにおいて少なくとも2つのユニークな経路によってNAD+合成に寄与し、マウスおよびヒトにおいて細胞内NAD+レベルをアップレギュレートすることが示されている(338)。どちらの経路も、NAD+合成のための以前に確立された経路に入るためにNAMリングを必要とする。今後の研究では、ヒトの健康と疾患におけるニコチンアミドリボシドの重要性、およびADなどの加齢関連疾患で低下したNAD+レベルを補充するためにニコチンアミドリボシドを効果的に使用できるかどうかを検討する必要がある。NAおよびニコチンアミドの高用量使用に関連する副作用を考えると、ニコチンアミドリボシドはNAD+レベルを高めるための代替的な前駆体である可能性がある。
同様に、NADHレベル、NAD+酸化還元電位、およびNAD+レベルの変化は、他の病理学的状態に存在する可能性が高く、疾患の進行と関連している可能性がある。特に、AD、PD、ADC、および筋萎縮性側索硬化症(ALS)における酸化ストレスおよび免疫活性化の増加は、これらの分子の利用可能な濃度に影響を及ぼす可能性がある。これらおよび他の変性疾患におけるNAD+の代謝および生物学的機能についての将来の研究は、これらの疾患における治療のための補助療法としてNAD+前駆体を使用することに関係する基本的な特性を明らかにする可能性があり、おそらく加齢に関連した疾患プロセスを遅らせるのに役立つ可能性がある。
レスベラトロールは、「アンチエイジング」酵素SIRT1を直接活性化することで作用すると考えられている、健康上の大きなメリットを持つポリフェノールである。しかし、最近の報告では、この「直接活性化」仮説に疑問を呈しており、レスベラトロールがSIRT1機能を増加させるメカニズムはまだ不明であることが示唆されている(39, 86)。我々のグループの以前の研究では、レスベラトロールがNMNAT-1活性の用量依存的な増加を誘導することが初めて示された。SIRT1はその遺伝子サイレンシング機能を実行するために基質としてNAD+を必要とするため、NAD+レベルが高いとSIRT1活性が増強されることになる。この知見は、レスベラトロールが、直接的な活性化を必要とせずに全細胞系でNAD+合成を促進することで、SIRT1の機能を促進する可能性を示唆している。レスベラトロールがNMNATに作用してヒト初代脳細胞のNAD+レベルを増加させるという我々の観察結果は、緑茶ポリフェノールのQUIN介在性興奮毒性に対する神経保護効果(47)と相まって、ポリフェノールが特に神経変性疾患の治療に大きな可能性を秘めているという見解を支持するものである。NMNATは、すべての6つの基質、QUIN、ニコチンアミドリボシド、NMN、NA、ニコチン酸リボシド(NAR)、およびNAMからのNAD+合成を促進することができるように、レスベラトロールによるNMNATの活性化は、NAD+レベルを補充するための理想的な自然療法を表すかもしれない。細胞内のNAD+を高く維持することは、SIRT1活性と他のNAD+依存性経路を強化し、細胞の生存率と寿命にプラスの影響を与える。
最後に、コラーゲン誘発性関節炎のマウスモデルでは、NAMPTの増加が血清中および関節炎足の両方で報告されている(59)。NAMPTの阻害は、エタネルセプトに匹敵する関節炎の重症度を減少させ、影響を受けた関節におけるサイトカイン放出のレベルを有意に低下させ、炎症細胞の細胞内NAD+濃度を低下させた(59)。したがって、NAMPTは、白血球からのサイトカイン分泌に関連する炎症性疾患の間に重要な役割を果たす可能性がある。したがって、NAD+濃度の増加は、炎症性疾患の状態では劇症的である可能性があり、NAMPT活性の増加および免疫細胞でのNAD+の使用のために疾患を悪化させる可能性がある。これは、NAD+の 「単独 」使用に対して、さらなる潜在的なマイナス要因となる可能性を示している。
本論文で検討されたエビデンスのほとんどは、NAD+のターンオーバーが高い状態および/または濃度が低下している状態でNAD+レベルを増加させるための戦略を調査し、開発するという現在の熱意を強く支持しているが、細胞内のNAD+レベルがすでに十分な状態でNAD+を増強する治療法を使用することは賢明ではないかもしれない。NAD+および関連する代謝物によって影響を受ける生化学的システムの複雑さを考えると、NAD+治療薬への単純論的な、ワンサイズフィットオールのアプローチは、おそらくNAD+治療の真の可能性を制限し、実際にはいくつかの状況下で害を引き起こす可能性がある。また、いくつかのNAD+前駆体が最近同定され、いくつかのモデルで検討されているが、これらの前駆体を並べて比較することは、現在の文献ではまだ始まったばかりである。これらの前駆体は、様々な病理学的障害において、その効果に重要な違いを示すことが予想される(375)。
これを回避するためには、NAD+を増加させる効率的な方法を特定することに焦点を当てた多くの研究に加えて、「健康的な」NAD+レベルとは何かについての明確な理解を確立するために、組織および細胞外液中のNAD+レベルを測定し、細胞および臓器の健康と相関させる費用対効果の高い方法の開発にも力を入れなければならない。この知識があれば、臨床家はNAD+レベルを適切に評価した後、自信を持ってNAD+治療を行うことができ、各患者のケースで治療が有効であるかどうかを判断することができる。
XIV. おわりに
NAD+の研究は、過去20年の間に多くの発見をもたらした。細胞呼吸とエネルギー生産における補因子としてのNAD+の重要な役割が明らかになった後、多数のNAD+生合成経路が発見された。近年では、NAD+がタンパク質の脱アセチル化を介してDNA修復やエピジェネティック制御にユニークな役割を果たすことが示されている。
NAD+が、酸化ストレスや免疫活性化、エネルギー代謝、エピジェネティック制御、細胞生存率などの重要な生化学的・細胞的プロセスと関連して果たす重要な役割を解明することは、変性疾患の効果的な治療法の進歩に大きく貢献すると考えられている。
NAD+は、NAD+、NADH、NADP+、NADPHの代謝と機能の中心分子であることに変わりはない。これらの4つの分子のうち、NAD+のみがキヌレニン経路を介して新規に合成され、NADH、NADP+、NADPHの生成には元の前駆体としてのNAD+が必要である。細胞内のNAD+レベルの維持は、DNA修復、ストレス抵抗性、および細胞死の調節にとって極めて重要であり、キヌレニン経路および/またはサルベージ経路を介したNAD+合成は、加齢に関連する変性疾患における治療的介入のための魅力的なターゲットであることを示唆している。
ニコチンアミドリボシド、さらにはNAおよびニコチンアミドのような薬剤は、ワレリアン変性の動物モデルにおいて、切断された軸索を変性から保護し、小動物の寿命を延ばすことが示されている。しかし、細胞内のNAD+同化を効率的に促進するために、特定のNAD+前駆体を使用すべき条件を明らかにするためには、さらなる研究が必要である。
そのためには、健康モデルや疾患モデルでの薬物動態、安全性、有効性を評価し、変性過程を改善し、健康寿命の維持・向上に寄与する標的治療法を開発する必要がある。
NAD+療法だけでは、神話のような「生命の万能薬」ではないことはほぼ確実に証明されるであろうが、細胞のエネルギー、核シグナル伝達、生存能力におけるNAD+の基礎的な役割は、NAD+が重要な成分である可能性を示唆している。
使用している略語
- 3-HAA 3-ヒドロキシアントラニル酸
- 3-HAAO 3-ヒドロキシアントラニル酸オキシゲナーゼ
- 3-HK 3-ヒドロキシキヌレニン
- 5′-NT 5′-ヌクレオチダーゼ
- AAアントラニル酸
- Aβアミロイドベータ
- AD アルツハイマー病
- ADC エイズ認知症コンプレックス
- ADPR ADPリボース
- AKI急性腎障害
- AMPK AMP活性化キナーゼ
- 曲線下面積
- BBB血液・脳関門
- cADPR 環状ADPリボース
- 中枢神経系
- CPS1 カルバモイルリン酸合成酵素1
- CRカロリー制限
- eNOS内皮一酸化窒素合成酵素
- ETC電子輸送鎖
- FAO脂肪酸の酸化
- エフビーエスウシ胎児血清
- GABAγ-アミノ酪酸
- GSHグルタチオン
- GSSGグルタチオン二硫化物
- 過酸化水素
- HDL 高密度リポ蛋白質
- HIF1α 低酸素誘導性因子1α
- HPLC高速液体クロマトグラフィー
- HSFヒートショックファクター
- IDH2 イソクエン酸デヒドロゲナーゼ2
- IDO インドールアミン2,3-ジオキシゲナーゼ
- IFN-γインターフェロンγ
- ILS インスリン様シグナル伝達
- イソナムイソニコチンアミド
- KAT キヌレニンアミノトランスフェラーゼ
- LDL低密度リポ蛋白質
- LKB1 肝キナーゼB1
- LXR 肝臓X受容体
- MeNAM N-メチルニコチンアミド
- MLCK ミオシン軽鎖キナーゼ
- MnSODマンガンスーパーオキシドジスムターゼ
- mPTPミトコンドリア透過性遷移孔
- エムアールアイ
- mRNAメッセンジャーRNA
- 多発性硬化症
- NAニコチン酸
- NAADNAアデニンジヌクレオチド
- NAADPニコチン酸アデニンジヌクレオチドリン酸塩
- NAD+ニコチンアミドアデニンジヌクレオチド
- NAM
- (NAMN)ニコチン酸モノヌクレオチド
- NAMPTニコチンアミドホスホリボシルトランスフェラーゼ
- NAPRTニコチン酸ホスホリボシルトランスフェラーゼ
- ニコチン酸リボシド(NAR)ニコチン酸リボシド
- NMDA N-メチル-D-アスパラギン酸
- NMNATニコチンアミドモノヌクレオチドアデニル転移酵素
- 核に局在するNMNATのNMNAT-1アイソフォーム
- NMNAT-2 ゴルジ体複合体アイソフォーム
- ミトコンドリアに局在するNMNATのNMNAT-3アイソフォーム
- NMR核磁気共鳴
- NNMTニコチンアミドN-メチル転移酵素
- ニコチンアミドリボシドニコチンアミドリボシド
- ニコチンアミドリボシドキナーゼニコチンアミドリボシドキナーゼ
- PARPポリADPリボースポリメラーゼ
- プレB細胞コロニー増強因子
- 末梢血単核球
- PDパーキンソン病
- ピコリン酸
- PICAC ピコリン酸カルボキシラーゼ
- PKAプロテインキナーゼA
- PNPプリンヌクレオシドホスホリラーゼ
- PPARペルオキシソーム増殖因子活性化受容体
- PRPP 5-ホスホリボシル-1-ピロリン酸塩
- QPRTキノリン酸ホスホリボシルトランスフェラーゼ
- クインキノリン酸
- 活性酸素種
- SDHコハク酸脱水素酵素
- サーチュイン
- TDO トリプトファン2,3-ジオキシゲナーゼ
- TG形質転換体
- ラパマイシンのTORターゲット
- TXNチオレドキシン
- URI 型破りなプレフォルジン RPB5 インタラクタ
- UV 紫外線