
Contents
www.ncbi.nlm.nih.gov/pmc/articles/PMC8243371/
2021年6月16日オンライン公開 doi: 10.3389/fnins.2021.666958.
概要
アルツハイマー病は、高齢者の進行性認知障害の約65%を占め 2020年には米国で70万人が死亡する、まさに「アンメット・メディカル・ニーズ」の代表的な疾患である。2019年、アルツハイマー病患者の介護にかかる費用は、介護者の精神的・肉体的負担を含めず、244Bドル(約3兆円)に上る。この悲惨な現実にもかかわらず、ADの発症リスクを低減する治療法も、その最も破壊的な症状を長期にわたって緩和させる治療法もないのである。
本総説では、アルツハイマー病の生物学と遺伝学の重要な側面を要約し、ピオグリタゾンがいかにしてADの病態生理学的決定要因の多くを改善するかを説明する。また、前臨床実験、縦断的観察研究、臨床試験の結果もまとめている。動物実験の結果、ピオグリタゾンは保護作用だけでなく矯正作用もあり、その効果は時間および用量依存的に増強されるが、用量-効果関係は単調でもシグモイドでもないことが示唆された。
縦断的コホート研究により、2型糖尿病の既往のある人では認知症の発症を遅らせることが示唆されており、小規模の非盲検パイロット試験で確認されているようである。しかし、プラセボ対照盲検臨床試験の結果はこれを裏付けておらず、これらの矛盾を説明する可能性について議論している。
アルツハイマー病とニーズの定義
アルツハイマー病は、進行性で不可逆的な神経変性疾患であり、その最も恐ろしい臨床症状は認知症であり、ほとんどの治療志向のヒト臨床試験の対象になっている。認知症は、民族や社会経済的な違いに関係なく、無心になる恐怖がつきまといる。
*
アルツハイマー型認知症は、どのような定義にせよ、アンメット・メディカル・ニーズである。認知症の最も一般的な原因であり、現在、がん、心臓病に次ぐ死因の第3位となっている。2000年から2018年の間に、アルツハイマー病に起因する死亡者数は145%以上増加し、心臓病に起因する死亡者数は8%近く減少した(Longhe, 2020)。
2018年には、世界で3,300万人を超える人々がADと共存している。予防的な治療法が開発されなければ、この数は2050年までに世界で1億3200万人に急増すると言われている(Patterson, 2018)。患者の介護にかかる世界の年間負担は現在約1兆米ドルであり 20-30年までに倍増すると予測されている(Patterson, 2018)。
*
このような現実を踏まえると、アルツハイマー病患者の病態生理学的な軌道が少しでも変化すれば、個人と社会の双方に大きな影響を与えることになる。ADの発症を1年遅らせることで 20-30年の経済的影響を1130億ドル(14.7兆円)削減することができる。2050年には、その1年の遅れが2190億ドルを節約し、3年と5年の遅れがそれぞれ4150億ドルと599億ドルを節約することになる(Zissimopoulos et al. 2015年)。
過去10年間の治療研究の失敗率が高かったため、またFDAのガイドラインに沿って、AD臨床研究の焦点は、症状が現れてから治療を開始するのではなく、アルツハイマー病の無症状期における、早期介入に移行している(Sperling et al. 2014)。
アルツハイマー病の創薬への挑戦
アルツハイマー病は、拡大した前臨床期にわたって発症する不均質な疾患である(Sperlingら 2011;Beason-Heldら 2013;Thambisettyら 2013;Neffら 2021)。
早期発症型(EOAD)は、一般的に約65歳以前に発症し、一般的に加齢と関連する「晩期発症型」よりも重度の臨床症状を伴う。早期発症例の約半数は、プレセニリン1(PSEN1)、プレセニリン2(PSEN2)、アミロイド前駆体タンパク質(APP)の3つの遺伝子のいずれかに優性遺伝性の変異があることが原因である。
PSEN1およびPSEN2は、APPを処理するγセクレターゼの活性を調節する。3つの遺伝子に欠陥があると、APPのタンパク質分解産物であるβ-アミロイドペプチドの細胞外への沈着が起こる。
*
遅発性(または散発性)アルツハイマー病(LOAD)は、通常65歳以降に発症し、単一の遺伝子の優性遺伝とは関連がない。アルツハイマー病は、いくつかの遺伝的危険因子を含む多くの危険因子が素因となっている。最も重要な遺伝的危険因子はアポリポ蛋白E(APOE)遺伝子の対立遺伝子変異であり、次いでTREM2(Triggering Receptor ExpressedinMyeloidcell 2)遺伝子のrs75932628(R47H)変異が挙げられている。
TREM2は、ApoEとβ-アミロイドペプチドの両方に結合し、ミクログリア活性化を制御するミエロイド細胞受容体である。また、早期教育の欠如、高血圧、喫煙、肥満、飲酒、糖尿病など、多くの非遺伝的決定要因がアルツハイマー病の素因となる(Zhangら 2021年)。
出生時の生物学的性別は、すべての原因による認知症の重要な危険因子であり、女性は男性よりもAD発症のリスクが高い。APOEがEOADの危険因子であることを含む最近の遺伝学的知見(Geninら 2011)は、EOADとアルツハイマー病の根本的な類似性を裏付けている(Jansenら 2019;Kunkleら 2019;Neunerら 2020)。このレビューの残りの部分では、後期アルツハイマー病を示すためにADという用語を使用し、状況に応じてEOADとLOADを区別することにする。
*
ADを特徴づけるのは複数のホールマークである。すべての症例で検出されるわけではない細胞外のβ-アミロイド沈着(Terry et al., 1999;Monsell et al., 2015;Jack et al., 2019;Sperling et al., 2020)、細胞内の神経原線維変化(NFT、ミスフォールドした高リン酸化タウの不溶性沈着)、神経細胞の酸化ストレス(Nunomura et al., 2001)、神経性炎症(Heneka et al,2015a)、脳インスリン抵抗性(Talbotら 2012)、およびグルコース低代謝(Mosconiら 2008a)、カルシウム過剰(アルツハイマー病協会カルシウム仮説ワークグループ 2017)、ミトコンドリア誤作動(Swerdlow 2018)および再分配(Flannery and Trushina 2019)、シナプス損失(Priceら 2001)および脳萎縮(Jackら 2018)であるという。
これらの要因のいずれかがADリスクまたは疾患の発現に寄与する程度は、生物学的柔軟性およびストレス要因に対する感受性の個人差を反映している(Neffら 2021)。
*
2019年現在、AD治療薬の試験の失敗率は、高度に「検証された」標的であるアミロイドやBACEを含めて99%を超えている(Cummingsら 2014年)。
これらの失敗は、ADを促進および維持するプロセスに関する知識のギャップ、および病原性決定因子に対する感受性が個人間でどのように異なるかを反映している。パノミクスのアプローチにより、基礎疾患や特定の治療法に対する反応に応じて患者の亜集団を層別化し、特定の薬剤に関する安全性の潜在的問題を特定することができる(Roses, 2008)。
がんはすでに「精密医療」アプローチを用いており(Begum, 2019)、他の複雑な疾患についても構想が進行中である(Loscalzo, 2019;Prasad and Groop, 2019;Aletaha, 2020)。
O’Bryantらは、NASID療法に反応するAD被験者を特定するために同様のツールを使用したが(O’Bryantら 2018)、それ以外は、このアプローチはAD臨床研究コミュニティで広く使用されているわけではない。ADの異質な性質の分子基盤の特定(Neffら 2021年)、それを採用するための概念的な推進力を提供するかもしれない。
*
ADの病態生理は複雑であるため、ADリスクを下げるための最も成功する戦略には、他の多因子疾患と同様に、複数の標的を同時に追求する必要がある可能性が高い(米国糖尿病学会 2020;Heidenreichら 2020;Ungerら 2020;)。
しかし、典型的な多剤併用療法には、個々の薬剤では見られなかった新たな副作用の出現や、副作用の相加、効果の減弱など、よく知られた欠点がある。前駆症状のある患者さんや軽度・中等度の認知症の患者さんにとって、複数の薬物療法を継続することは困難な場合がある。
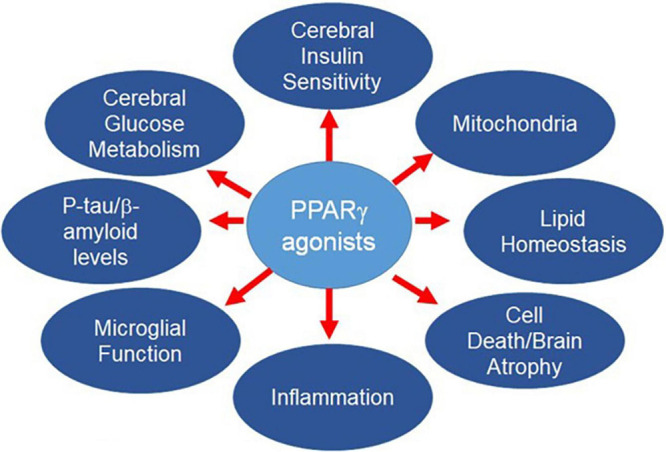
そこで、複数の疾患をターゲットとする単一の薬剤を開発することが考えられる。PPARγアゴニストは、この要望に応えるものである(図1)。
PPARの生物学の概要を説明した後、ADの危険因子とADの病態生理学的決定因子、およびPPARγ作動薬の有用な効果について説明する。GLP-1作動薬のような他の薬剤もまた、AD発症経路の複数のターゲットに影響を与える可能性がある。これらは、今回のレビューの範囲外であるが、他の場所で議論されている(Griecoら 2019;Chengら 2020;Yoonら 2020)。
図1 PPARγは、ADのリスクに寄与する多くの経路を制御している

ADの創薬ターゲットとしてのPparγ
PPARγ受容体は脳に広く分布し(Braissantら、1996;Morenoら 2004;Gofflotら 2007;Sarrufら 2009;Morales-Garciaら 2011)、学習に重要である(Heら 2009;Jahrlingら 2014)。
受容体の活性化は、アストロサイト/ニューロンの代謝的結合を促進し(Dello Russo et al., 2003;Izawa et al., 2009;Cowley et al., 2012)、樹状突起スパインの形成を促進し(Brodbeck et al., 2008)、シナプス不全を修復し(Chen et al,2015;Moosecker et al., 2019)、LTP(長期増強)障害を修正し(Cowley et al., 2012;Chen et al., 2015)、ADの病因の中心であるCNSの炎症性、酸化性の環境を克服する。このトピックは以前にレビューされている(Galimberti and Scarpini, 2017;Cai et al., 2018;Villapol, 2018;Khan et al.)
PPAR は、PPARα、PPARδ、PPARγ という 3 つのリガンド依存性転写因子からなるファミリーで、別々の遺伝子にコードされ、広い範囲に分布しているが、サブタイプには特異的である。PPARは、幅広い代謝および抗炎症活性を有し、脂質異常症(PPARα、Gemfibrozil)、2型糖尿病(PPARγ、Pioglitazone、Rosiglitazone)および肥満(PPARδ)の治療薬として魅力あるターゲットである。
ピオグリタゾンとロシグリタゾンは、PPARγとPPARαの両方に対して高親和性のリガンドであるが、ロシグリタゾンがPPARγに対してより選択的である点、各アゴニストがオーダーメイドのダウンストリーム遺伝子を制御する(Verschuren et al, 2014)、ピオグリタゾンがロシグリタゾンよりも大きく脳に入る(前芝ら、1997;Grommes et al, 2013)点で区別される(Festuccia et al,2008)。
*
PPARは、標的遺伝子の一般的な転写機構をリクルートおよび/または活性を増強し、あるいは他の遺伝子の発現を抑制する。PPARファミリーのメンバーは、構造的および機構的に類似した特徴を有している(図2)。
N末端ドメインには、リガンドに依存しない転写活性化機能であるAF-1があり、PPARサブタイプ選択的な遺伝子発現の主な決定因子である。DNA結合ドメイン(DBD)は、受容体と標的遺伝子のPeroxisome Proliferator Response Element (PPRE) を結合させる。
DBD には、PPAR と他の DNA 結合タンパク質とを区別する 2 本のジンクフィンガーが含まれている。PPREは遺伝子プロモーターまたは近位配列に位置し、コンセンサス配列5′-AGAACA-3′のコピーを1つまたは2つ含んでいる。
DBDに隣接して、転写補因子結合ドメイン(Dサイト)とリガンド結合ドメイン(LBD)があり、受容体とPPREの結合を媒介する。3種のPPARはすべてRXR受容体と義務的なヘテロダイマーを形成する。PPARとRXRのパートナーは、PPREのコンセンサス結合配列のダイレクトリピートの5′と3′の半分に結合している。
図2 PPARγの共有結合修飾

PPARγシグナルは非線形であり、正味の効果はPPARγリガンドの変動、翻訳後修飾の時間的配列と持続時間(図2)、PPARγ転写プログラムと相互作用する下流の遺伝子発現ネットワークの性質に依存する。
Ppar-γとAD関連リスクファクター
導入時のコメント
アルツハイマー型認知症発症の最も重要な危険因子は、潜在的に非修飾性であり、年齢、生物学的性別、第一度近親者のAD歴、遺伝学が含まれる(Gaugler et al. 2019)。AD発症リスクは年齢とともに増加し(Qiu et al., 2009)、女性は男性よりもAD発症リスクが高い(Plassman et al., 2007)。
約30の遺伝的リスク遺伝子座が同定されており(Jansen et al., 2019;Kunkle et al., 2019)、これらはオーバーオールの集団帰属リスクの約65%しか占めていない(Livingston et al., 2017)。リスクの残りは、潜在的に修正可能な併存疾患と関連している(Livingstonら 2020)。
驚くことではないが、生物学的性別とADの遺伝的危険因子の間には根本的な関連がある。第一に、APOEε4は男性よりも女性に重篤な影響を与える(Farrerら、1997;Altmannら 2014;Neuら 2017)。
第二に、最近の調査により、性特異的な常染色体遺伝的影響が明らかになった(Zhouら 2019;Fan C.C.ら 2020;Prokopenkoら 2020)。
いくつかの事例では、一方の性別のリスク遺伝子は他方の性別のリスク遺伝子ではない。例えば、PVRL2のリスクハプロタイプは、女性ではADと有意に関連したが男性では関連せず(Zhouら 2019)、亜鉛フィンガー転写因子をコードするZBTB7Zは、女性ではリスク遺伝子であるが男性では保護的である(Prokopenkoら 2020)。
遺伝的リスクファクター
ADの遺伝的景観は、約30のゲノム遺伝子座からなる(Lambertら 2013;Jansenら 2019;Kunkleら 2019)。PPARγは、これらの遺伝子のうち少なくとも7つの発現を調節するため、この遺伝子ランドスケープの「マスターレギュレーター」と考えられるかもしれない(Barreraら 2018年)。
早期発症のAD
アミロイド沈着とNFTの病理組織学的特徴は、ADの「早期」発症型と「後期」発症型の両方を特徴づけている。APP、PSEN1、PSEN2という3つの遺伝子における原因変異は、早期発症型におけるアミロイド沈着に寄与する(Neunerら 2020年)。
APPは、全身に広く分布する細胞表面分子であり、中枢神経系ではAβペプチドの前駆体分子となる。APPノックアウトマウスは表現型を示さないため、その正確な役割は不明である(O’Brien and Wong, 2011)。
PSEN1とPSEN2は、γセクレターゼ複合体の触媒成分であり、BACE1(βサイトアミロイド前駆体タンパク質切断酵素)と協力してアミロイド前駆体タンパク質を処理し、プラークに見られる凝集しやすいAβペプチドを生成する。
*
ピオグリタゾンは、BACE1を介したAβペプチド産生をいくつかのレベルで制御している。BACE1遺伝子にはPPREが存在し、PPARγはBACE1の発現を制御している。さらに、CDK5は、BACE1の発現を増加させ(Wenら 2008)、リン酸化を介してβセクレターゼ活性を調節することにより、転写および転写後レベルの両方でBACE1を制御する(Songら 2015)。
以下に述べるメカニズムにより、ピオグリタゾンはこれらのCDK5作用を阻害する。細胞ベースおよび生体内試験モデルの両方において、PPARαまたはPPARδではなくPPARγは、BACE1 mRNAおよびタンパク質発現をブロックし、Aβペプチドクリアランスを促進することによって、Aβペプチドの生成および放出(Sastreら 2003、2006;Liuら 2013;Gadら 2016;Quanら 2019b)をブロックした(Camachoら 2004)。
In vivoでは、PPARγの活性化により、βアミロイドプラークが著しく減少した(Henekaら 2005;Escribanoら 2010;O’ReillyおよびLynch 2012;Searcyら 2012;Liuら 2013;Quanら 2019b)。
In vitroでは、RXRリガンドのシス-レチノイン酸単独は、ピオグリタゾンを含むPPARγアゴニスト単独と同等の効果があった(Camachoら 2004年)。
細胞培養では、PPARγアゴニストは、炎症性サイトカインによって誘発されるBACE1発現の増加、Aβペプチドの合成および放出をブロックした(Sastreら 2003)。
逆に、PPARγをノックダウンすると、BACE1の発現が増加した(Sastreら 2006年)。
Aβペプチドおよびフィブリルは、炎症性でCDK5活性化を増加させ(Quan et al., 2019a)、アストログリア症、ミクログリア損傷、神経細胞のアポトーシスを引き起こす(Sastre et al., 2003)。
ピオグリタゾンは、CDK5発現とPPARγリン酸化をダウンレギュレートし、PPARγ発現を増加させ、BACE1発現とAβ産生を抑制した。PPARγアンタゴニストGW9662は、これらのピオグリタゾンの効果をブロックし(Quanら 2019b)、それらがPPARγ受容体によって媒介されることを確認した。
*
PPARγの作用は、共活性化因子であるPGC-1αに依存している。APPを安定的に発現している細胞株でPGC-1αを過剰発現させると、BACE1の発現が減少すると同時に、Aβの産生が抑制された(Katsouri et al.)PPARγの発現をノックアウトすると、PGC-1αの効果は消失した(Katsouri et al.)アルツハイマー病患者の脳抽出液では、認知的に正常な対照群と比較して、PPARγとPGC-1αの両方のレベルが低下している(Sastreら 2006年;Qinら 2009年;Katsouriら 2011年)。
これは、BACE1 PPREへのPPARγの結合が減少し、Aβの産生が上昇することと関連している。PPARγアゴニストのピオグリタゾンは、CDK5や他のシグナルキナーゼの逆調節作用を克服することにより、PPARγの発現を増加させ、BACE1の発現を抑制し、アミロイドプラークの形成を阻害する。
*
ここでNFTを論じるのは、アミロイド斑とユビキタスな関連性があるからだ。神経原線維絡まりは、アミロイド沈着よりも直接的に神経細胞機能障害および脳萎縮と相関する(Brion, 1998;Jackら、 2018)。
ピオグリタゾンは、細胞ベースのタウオパシーモデル、および前臨床マウスモデル(Escribanoら 2010;Searcyら 2012)において、タウのリン酸化(Choら 2013;Hamanoら 2016;Mooseckerら 2019)およびオリゴマー化(Hamanoら 2016)を阻害した。また、試験管内試験で樹状突起スパインへのタウのミスルーティングをブロックした(Moosecker et al. 2019)。
PPARγ特異的拮抗薬GW9662はこれらの効果をブロックし、PPARγ受容体依存性であることが確認された。ロシグリタゾンは、マウスで同様の効果を示した(Escribano et al.、2010)。
PPARγはシナプスを維持し、これはタウのミスソーティングの修正によると考えられる(Moosecker et al.)また、ピオグリタゾンは、3xTgマウスADモデルにおいて、タウのリン酸化を抑制した(Searcy et al.)タウのリン酸化および凝集に対する効果は、ピオグリタゾンが介在するCDK5の直接阻害の結果であると考えられる(Hanger et al.、1998)。
さらに、PPARγは、炎症反応への影響を通じて間接的にCDK5を調節する可能性がある。 p35はCDK5を活性化する調節タンパク質であり、ADの発症過程で神経細胞に生じる細胞質Ca2+の上昇に応じたカルパイン触媒によるp35の切断は、CDK5を過活性化してタウリン酸化を増加させるp25を形成する(Kimura et al., 2014;Seo et al., 2017).IL-6はp35 – から – p25への変換を促進することでCDK5活性を高め(Quintanilla et al., 2004)、PPARγはIL-6の放出を抑制する(Jiang et al., 1998)。
遅発性AD
晩発性アルツハイマー病に関連する遺伝子のおよそ50%は自然免疫系に関与するタンパク質をコードしており、残りの遺伝子の多くは脂質代謝に関与するタンパク質をコードしている(Jones et al. 2010)。
ADの最も重要で高度に再現された遺伝的危険因子であるアポリポ蛋白Eε4(APOEε4)の両方(Corderら、1993;Saundersら、1993;Lambertら、.2013)、および2番目に大きな効果量を有するTREM2R47H多型(Guerreiro et al., 2013;Jonsson et al., 2013)は、自然免疫および脂質代謝に影響を与える(Shi and Holtzman, 2018;Nugent et al., 2020)。
メタボロミクス研究は一貫して、脂質代謝の顕著な変化をADの初期マーカーとして指摘している(Han 2005年、2010年)。
*
APOEは、Chr19q13.32ゲノム領域に存在するADリスクに影響する遺伝子群の一つであり、PVRL2、TOMM40、APOC1も含まれる。APOEには、112位と158位のアミノ酸の同一性によって区別される3つの一般的な型があり、APOE遺伝子内の2つの密接に関連したSNP:rs429358とrs7412によって決まり、3つの代替タンパク質アイソフォームの発現につながる。
両位置にシステイン残基を持つAPOE ε2、システイン112とアルギニン158を持つAPOE ε3、そして両位置にアルギニン残基を持つAPOE ε4である。APOEε4はAD発症リスクを用量依存的に増加させ、発症年齢も低下させる(Corderら、 1993;Roses, 1994;Frisoniら、 1995;Farrerら、 1997)。
一方、APOEε2はADに対して保護的であり、APOEε3は中程度のリスクを有する(Corderら、 1994;Farrerら、 1997)。脳はすべてのApoEを局所で産生し、肝臓とマクロファージは末梢でApoEを産生する。脳におけるApoE産生の大部分はグリア細胞が担っている。ApoEは、アストロサイトやミクログリアと神経細胞との間で、HDL様リポ蛋白質粒子を介したコレステロールやリン脂質の授受を仲介している。
これは、低密度リポタンパク質受容体ファミリーのメンバーによって取り込まれるこれらの粒子の主要なリポタンパク質成分である(Holtzmanら 2012)。ストレス条件下では、神経細胞もAPOEを発現する(Han et al, 1994a,b)。
APOE ε4とLOADの関連が最初に報告されて以来(Saundersら、1993)、神経突起伸長の障害(Holtzmanら、1995)、可塑性(Weeberら 2002)、修復(Ignoberら 2002)など、LOADの発症にAPOEε4が寄与するさまざまな潜在的メカニズムが明らかにされつつある。
2002)および修復(Ignatiusら、1987)、Aβクリアランスの欠陥(Vergheseら 2013;Kanekiyoら 2014;Mouchardら 2019)、ミトコンドリア機能障害(Chenら 2011)およびエンドソーム-リゾーム輸送障害(Nurielら 2017;Zhaoら 2017)へと至るまで。
APOEはADの最も重要な遺伝的危険因子であるが、chr19q13.32ゲノム領域に起因するリスクを完全に説明するものではない。APOEに近接する少なくとも3つの追加遺伝子、PVLR2、APOC1、TOMM40(Takei et al., 2009;Roses, 2010;Zhou et al., 2019;Bussies et al., 2020;Fan K.H. et al., 2020;Squillario et al., 2020)が独立して寄与している。
TOMM40はミトコンドリアタンパク質輸入チャネルをコードし、ミトコンドリアの恒常性維持(Baker et al., 1990;Taylor et al., 2003)および生命維持に不可欠である(Zeh, 2013)。
TOMM40の複数のSNPは、APOE遺伝子とは独立してADリスクと関連しており、rs7259620(Takei et al., 2009;Nazarian et al., 2019)、rs760136(Marioni et al,2018)、rs2075650(Heら 2016;Huangら 2016;Bussiesら 2020;Soyalら 2020;Squillarioら 2020)、およびrS10524523(Roses 2010;Liら 2013;Yuら 2017a、b)である。
APOC1も PVRL2も、脂質や免疫に関連するというパターンに当てはまる。ApoC1は、ApoE濃縮リポタンパク質粒子の低密度リポタンパク質受容体への結合を阻害することにより、CNSにおけるApoE介在のコレステロールおよびリン脂質の取り込みを妨害し(Kowalら、1990;Weisgraberら、1990;SehayekおよびEisenberg、1991)、APOE ε3-およびAPOE ε4-濃縮粒子の結合も同様にブロックする(Kowalら、1990)。
APOC1リスクハプロタイプは、Aβ40の血漿レベルと関連していた(Zhouら 2019年)。PVRL2はヘルペスウイルスの取り込みを媒介する(Warnerら、1998)。ヘルペスウイルスがADの病因に寄与するという不朽の憶測があるが(Itzhaki et al., 2016;Readhead et al., 2018)、この説は議論の余地がある(Rizzo, 2020)。PCRL2のリスクハプロタイプは、認知パフォーマンスの悪化、脳および海馬の総体積の減少、および血清Aβ42の総量と関連していた(Zhou et al.、2019)。
さらに、PVRL2、APOE、APOC1は、この連鎖不平衡領域の遺伝子の発現に調節的な役割を担っている。APOEε4は、脳内のTOMM40、APOEおよびAPOC1の転写を抑制し、PVRL2およびAPOC1のリスクハプロタイプは、APOE遺伝子型にかかわらず、脳内APOE発現を増加させる(Zhou et al.、2019)。
TOMM40プロモーターのメチル化は、TOMM40の発現を減少させ、APOE発現を増加させる(Shaoら 2018)。これらの研究を合わせると、19番染色体上で連鎖不平衡にある4つの遺伝子、PVRL2、TOMM40、APOE、APOC1が独立して脳の構造、神経エネルギー、認知パフォーマンス、およびADのリスクに影響を与えることが示された。
*
chr 19q13.32ゲノム領域はPPARγ結合部位に富んでおり(Subramanianら 2017)、内因性PPARリガンドのほとんどが脂質または脂質誘導体であり、この領域はリポタンパク質またはそれと相互作用するタンパク質に富んでいるので驚くには当たらない(Zhouら 2019)。
PPARγアゴニストは、この領域の4つの遺伝子のうち3つの遺伝子の発現に影響を与える。TOMM40、APOE、APOC1であり、PVRL2発現への影響はこれまで検討されていない。ピオグリタゾンは、マクロファージにおけるAPOE発現を増加させ(Ricoteら 2004年のレビュー)、脳においても増加させる(Mandrekar-Colucciら 2012年)。
一方、Subramanianらは、ヒト肝癌株HepG2におけるPPARγの発現を低下させると、逆説的にTOMM40、APOEおよびAPOC1の発現が増加することを示した。これらの結果と一致するように、低濃度(nM)のピオグリタゾンは、APOEと APOC1の両方の発現を抑制したが、TOMM40の発現には検出できない影響を及ぼした。
他の研究者は、高濃度(μM)のPPARγアゴニストであるシグリタゾンと15d-PGJ2が、APOEの発現を強く増加させ、APOC1の発現を適度に抑制すると報告している(Dahabreh and Medh, 2012)。これらの対照的な結果は、二相性のPPARγ用量効果曲線が報告されていることから、使用されたそれぞれの薬物濃度を反映していると考えられる(Wadaら 2006年;Miglioら 2009年;Moonら 2012年)。
ADにより適切なSKNMC細胞株を用いて、ピオグリタゾンがTom40タンパク質発現を増加させることを見出した(Charalambous et al. 2016;図3)。

脂質代謝
ADの病態は、脂質代謝の広範な変化と絡み合っており(Foley, 2010)、脳だけでなくCSFや血漿でも検出される(Wood, 2012;Trushina et al, 2013;Varma et al., 2018)。この話題は最近レビューされているので(Penke et al., 2018;Kao et al., 2020)、ここでは厳選したトピックに限定して解説することにする。
エタノールアミンプラズマローゲン(PIEtn)は、ヒト灰白質および白質中の神経細胞膜画分に含まれる全リン脂質のそれぞれ60〜90 mol%、シナプス小胞中のホスファチジルエタノールアミン全体の60 mol%以上を占める(Han,2010)。
プラズマローゲンは、sn-1位にビニルエーテル脂肪アルコール(-O-CH = CH-R)を置換基とするグリセロリン脂質である。プラズマローゲンは、多価不飽和ジアシルリン脂質に対する酸化的損傷から保護し(Reissら、1997)、膜融合を促進する(GlaserとGross、1994)。
プラズマローゲンの欠乏は、ADの初期に検出される(Han et al.)ハンチントン病やパーキンソン病では検出されない(Ginsberg et al., 1995;Farooqui et al., 1997)。欠損は灰白質でも白質でも検出されるが、白質ではADの病態が進行するにつれて悪化するのみである(Hanら 2001)。循環血中PIEtn濃度は、機能状態の悪化の程度と正の相関がある(Woodら 2010)。
ADの後期には超長鎖脂肪酸が増加し、脂質毒性を引き起こし(Schönfeld and Reiser, 2016)、神経細胞障害を悪化させる。ペルオキシソームは、PIEtnの合成と超長鎖脂肪酸の酸化の両方を司るが、ADでは欠損または機能不全に陥る(Grimm et al. 2011;Kou et al. 2011)。
ピオグリタゾンは、いくつかの異なるメカニズムによって、これらの欠損を修正する。部分的なPPARαアゴニストとして(Sakamotoら 2000;Orasanuら 2008)、それはペルオキシソーム生合成(Hoivikら 2004)および関連する脂質代謝(Kersten 2014)を促進する。
また、ピオグリタゾンは、脂肪酸結合タンパク質5を介してドコサヘキサエン酸を含む前駆体脂肪酸の取り込みを促進することにより、PIsEtn合成を促進する(Pan et al. 2015)。FABP5ノックアウトマウスは、ワーキングメモリおよび短期記憶の障害を示し(Panら 2016)、ピオグリタゾンはFABP5の発現を増加させる(Lowら 2020)。
また、ピオグリタゾンは、APPのアミロイド形成処理を阻害することにより、PIsEtn合成を亢進させる。APP細胞内ドメインは、PIsEtn合成の律速酵素であるアルキルジヒドロキシアセトンリン酸合成酵素の発現を促進する(Grimmら 2011年)。APPのプロセシングがAβ経路に転換されると、これは失敗するが、ピオグリタゾンはこれを阻止し、PlsEtnの合成を救済する。
*
スフィンゴ糖脂質はミエリン鞘の主要成分であり、MRIではMCI期に脱髄が起こることが示されている(Bouhrara et al. 2018)。同様に、発症の初期段階では、スルファチドスフィンゴ糖脂質レベルは、脳領域にかかわらず、灰白質で約92mol%、小脳で35mol%、側頭皮で58mol%減少している。
ロシグリタゾンは、齧歯類モデルにおいてミエリンの構造的損傷を回復させた(Cowley et al.)セラミドレベルは、年齢をマッチさせた対照群と比較して、MCI被験者のすべての脳領域の白質で約3倍高い。また、セラミド合成の基盤となる広範な遺伝子ネットワークの発現も、ADの初期段階で増加している(Katsel et al. 2007)。
セラミドの増加は、ミトコンドリア障害を引き起こし、アポトーシスを増加させることによってADの病態に寄与し(Yuら 2000)、PIEtn-PLA2の刺激を介してPIEtnの枯渇に貢献する(Farooqui 2010;Ongら 2010)。セラミドの代謝産物であるスフィンゴシン-1-リン酸塩(S-1-P)は、一般にセラミドの効果を打ち消す(優れたレビューについては、Wang and Bieberich(2018)およびCzubowiczら(2019)参照)。
PPARγ(Parhamら 2015)は、S-1-P効果を媒介し、T-リンパ球の恒常性表現型を維持するいくつかの表面および細胞内受容体の1つであり、この役割はミクログリアでは調査されていない。
自然免疫
かなりの割合で、ADは自然免疫系の疾患である(Zhangら 2013;Jonesら 2015;Kanら 2015)。AD GWASのアットリスク遺伝子多型の大部分(約60%)は、Simsら(2017)、またはミクログリアに濃縮されている遺伝子またはその調節要素の近くにある(Tanseyら 2018;Jansenら 2019;Kunkleら 2019;Nottら 2019)。
APOEε4の発現と免疫制御遺伝子の発現は正の相関がある(Keren-Shaulら、 2017;Mathysら、 2019)。さらに、ミクログリアの発達に極めて重要な遺伝子であるPU.1の発現低下に関連するハプロタイプは、ADの発症年齢を遅らせる(Huangら 2017)。
*
ミクログリアは、CNSの常駐自然免疫系細胞である(Ginhouxら 2010;Schulz C.ら 2012)。その主な機能は、ニューロンの健康と接続性を保証することである[Streit and Kincaid-Colton (1995)andNayak et al. (2014)and references therein]。
「静止」状態では、ミクログリアは、ramified extensionsを通じて、近隣のニューロンやアストロサイトとの直接的なコミュニケーションを含む、局所環境を調査する。周囲の細胞が傷害されることで発生する特定のシグナルを検出すると、ミクログリアが活性化され、形態変化や特定の生化学的・遺伝学的プログラムが誘発される。
活性化状態を維持するために、生体エネルギー代謝は酸化的リン酸化から解糖に切り替えられ、ATPだけでなく、NADPHやペントースリン酸シャントの他の中間体を含む重要な代謝中間体も供給する(Lauro and Limatola, 2020)。
*
ミクログリアは、その局所環境の逸脱に絶妙に敏感であり、ミクログリアのトランスクリプトミクス、形態または行動(食作用)の変化は、しばしば病理学の最初の兆候である(Boza-Serrano et al. 2018)。
そのプログラムされた転写反応は、異なる刺激に対してオーダーメイドであり、食作用の増加、インターフェロンおよび細胞毒性サイトカイン、ケモカイン、細胞外プロテアーゼおよび活性酸素種の生成、ならびに抗炎症サイトカインおよび組織修復および細胞外マトリックスのリモデリングを促進する因子をサポートする(Porcherayら 2005;Stoutら 2005)。これらの炎症性及び免疫抑制性の表現型は、反応のスペクトルの両極を表している(Coltonら 2006;Colton 2009;Grayら 2020)。
マウスADモデルから単離されたミクログリアの縦断的な遺伝子転写プロファイルは、インターフェロン関連、増殖関連および神経変性関連の表現型を反映する、AD脳における活性化ミクログリアの複数の離散集団があることを明らかにした(Keren-Shaulら 2017;Friedmanら 2018;Mathysら 2019)。
*
活性化したミクログリアは、アミロイドの代謝において2つの異なる役割を担っている。一方では、インターフェロンを介したIFITM3(インターフェロン誘導型膜貫通タンパク質3)の誘導を介して、Aβペプチドの生成を促進する。IFITM3は、以前は抗ウイルス活性が認められていたが(Baileyら 2014)、γセクレターゼ複合体と会合し、アミロイド形成性APPプロセッシングを促進する(Hurら 2020)。
この役割は、Aβペプチドが自然免疫系の抗感染レパートリーの一部であるという説と一致する(Eimerら 2018)。活性化したミクログリアもアミロイド沈着物のクリアランスに参加し、βアミロイドプラークに隣接した-時には取り囲んだ-クラスターを形成する(Condelloら 2015)。
プラークに関連する一部のミクログリアは、疾患が進行するにつれて、疲弊によって、または隣接するミクログリアの無制限の炎症性活性による付随的な損傷を介して異形化し、プラークが一部を飲み込む(Streitら 2009年、2018)。
*
APOEを含む炎症関連遺伝子の発現が増加し、恒常性維持遺伝子の発現が減少していることが、活性化したミクログリアを特徴づけている。APOEは活性化反応に必要なのかもしれない(Ulrichら 2018)。APOEの発現はプラークに近いほど高いが(Krasemannら 2017)、勾配シグナルは不明である。
ApoEは、NF-κBシグナルを活性化することにより、恒常性維持に関わるミクログリアの遺伝子発現を抑制し、炎症性遺伝子発現を強化する(Krasemannら 2017)(Ophirら 2005;Maezawaら 2006)。NF-κBは、自然免疫系および炎症反応のマスターレギュレータである(Liu T. et al., 2017)。APOEε4は、おそらく免疫抑制相への分化を阻害することによって、これらの効果を悪化させる(Brownら 2002年;Vitekら 2009年;Zhuら 2012)。
TREM2は、環境の変化をモニタリングし、その変化に対するミクログリアの反応(増殖、移動、活性化など)を制御するためのミクログリアモニタリングシステムの一部である。TREM2の変異は、遅発性ADの遺伝的リスクを2〜4倍増加させ、これはAPOEε4の効果の大きさに次ぐものである(Jonssonら 2013;Guerreiro and Hardy 2014)。
TREM2は、損傷関連分子シグネチャー(DAMPS)(Dawsら 2003)、リポタンパク質およびリポタンパク質粒子、細胞損傷によって露出したアニオン性脂質およびスフィンゴミエリン、β-アミロイドペプチド(Wangら 2015;Yehら 2016;Song W. et al 2017)などに結合する単一パス受容体である。
リガンド結合は、TREM2受容体とアダプタータンパク質DAP12(TYROBP)との間の会合を促進し、静電相互作用を介して会合し、リガンド結合による生存と増殖、食作用および炎症への影響を媒介する細胞内シグナル伝達カスケードを活性化する(Wang et al.、2015)。
マウスADモデルにおいて、TREM2は、β-アミロイドプラーク周辺のミクログリアのクラスタリングと、貪食および「炎症性」応答の活性化を仲介し(Jayら 2015;Wangら 2015;Ulrichら 2018;Zhaoら 2018;Zhongら 2018)、すべてのミクログリアモジュールにわたるAβ病理に対する応答の完全発現に必要であった(Friedmanら 2018)。
APOEは TREM2のリガンドであり(Atagiら 2015;Baileyら 2015;Jendresenら 2017)、TREM2はAPOE以外のコア神経変性ビンのほぼすべての遺伝子の発現を調節し(Friedmanら 2018)、Trem2-/-およびApoe-/-マウスは表現型(Ullandら 2017;Ulrichら 2018)なので、APOEおよびTREM2が同一の分子経路で動作する可能性がある。
*
TREM2タンパク質のR47Hスイッチは、ADと関連する最も一般的なTREM2変異株である。これは、CSFにおける総タウの増加と関連しているが、CSF Aβペプチドレベルには影響しない(Lillら 2015)。
In vitroでは、APOEは、野生型受容体よりも低い親和性でこのTREM2変種に結合し(APOEアイソフォームの区別なく)(Atagi et al., 2015)、R47H変種は単球由来マクロファージによるAβ-リポタンパク質複合体の取り込みを減少させた(Yeh et al., 2016)。
*
化学的解剖により、ADおよび関連するタウパチーにおけるミクログリアの役割にさらなる光が当てられている。TGF-1βおよびCSF-1シグナルはミクログリアを維持し、脳から常駐ミクログリアを枯渇させるCSFR1拮抗薬またはcFMS阻害剤(Dagherら 2015;Spangenbergら 2016、2019;Sosnaら 2018)が分子メスとして使用されてきた。
予想に反して、ミクログリアを排除すると、βアミロイド斑の発生と神経細胞内アミロイドの蓄積がブロックされ、神経細胞とシナプスの喪失を防ぎ、記憶と学習が改善された。これらの有益な効果は、阻害剤を早期に添加して長期間維持しても(Sosnaら 2018;Spangenbergら 2019)、プラーク形成が進行した後に添加しても(Spangenbergら 2016)起こった。
CSFRを自己リン酸化し活性化するcFMSキナーゼの阻害剤でミクログリアの増殖を阻害すると、同様にアミロイド斑の形成を防ぎ、記憶と行動を改善し、脳環境を免疫抑制相に移行させた(Olmos-Alonso et al.,2016)。
したがって、予想に反して、アミロイド斑の形成にはミクログリアが必要であることは明らかである。さらに、ミクログリアの不在やミクログリア機能の低下は、学習や記憶にとって有害ではない。Shiらは、タウを介した神経変性におけるミクログリアの役割を知るために、同じ戦略を用いた。彼らは、タウを介した毒性ではなく、ミクログリアが介在する損傷が、マウスのタウオパシーモデルにおける神経変性の原因であることを実証した(Shi et al. 2019)。
したがって、ミクログリアの増殖および/または活性化が、アルツハイマー病の2つの主要な病理学的特徴である神経原線維変化およびβアミロイド斑に共通して関連する神経変性に関与していると思われる。ミクログリアが介在する炎症反応、あるいはミクログリアの免疫抑制反応の失敗が、認知機能低下や認知症につながる障害を引き起こしている可能性がある。ミクログリアは神経変性疾患の標的になっている(Dong et al., 2019)。
PPARγと自然免疫
PPARγアゴニストは、骨髄系細胞が免疫抑制刺激に反応するように促し、骨髄系細胞の免疫抑制状態への分化を促進する(Bouhlelら 2007年)。PPARγは、ミクログリアを含むマウスおよびヒトの脳に広く分布しており(Wardenら、2016)、炎症性刺激で骨髄系細胞を活性化するとPPARγのmRNAおよびタンパク質発現が増加する(Fakhfouriら 2012;Song J. et al 2017)。
PPARγ活性化剤もPPARγのmRNAとタンパク質の発現を増加させる。骨髄系細胞では、PPARγの結合部位はマクロファージ/ミクリア特異的標的のPU.1部位に隣接し(Lefterova et al. 2010)、PU.1応答性遺伝子の発現を制御する(Lefterova et al.、2010)。
PU.1は、炎症反応に直接関与するサイトカインやサイトカイン受容体の制御に加え、M-CSF(Macrophage-specific CSF)(Zhang et al., 1994) など、骨髄系やリンパ系細胞の発生に必要な因子の発現を制御する(Turkistany and DeKoter, 2011)。PPARγは、M-CSFの発現をブロックし(Bonfieldら 2008)、転写因子AP-1、STAT3、NF-kBを阻害する(Ricoteら、1998)。
PU.1に対する効果とともに、正味の結果として、炎症性活性化の抑制と免疫抑制表現型への分化のためのミクログリアの感作がもたらされる。ピオグリタゾンは、IL-1、TNF-α、IL-6、iNOS、COX2、MMP9、カスパーゼ3などの炎症性分子の合成を阻害し(Kapadiaら 2008)、Arg1、IL-4、IL-10、TGFb、カタラーゼ、SOD、および関連遺伝子などの免疫抑制関連の分子の合成を促進する(Bouhlelら 2007)。
*
In vitroの研究では、PPARγがAβやLPSなどのAD関連の病原性トリガーに対する細胞応答を制御することが確認された(Combsら 2000年;Henekaら 2000年;Hunterら 2008年)。
これらの効果は、単に炎症性分子や抗炎症性分子の発現を調節することにとどまらない。PPARγは、ミクログリアが免疫抑制表現型に移行するのを妨げる、病因に関連した発生ブロックを克服する。病原性TREM2R47H変異をヘテロ接合で持つiPSC由来のミクログリアは、解糖能力が不足しており、ミクログリアの免疫抑制表現型への分化を支える代謝スイッチを実行できない(Piersら 2020);結果として、食作用が欠損している。
ピオグリタゾンは、解糖の欠損を修正し、代謝シフトのブロックを逆転させ、Aβ42の貪食を回復させた(Piers et al. 2020年)。ピオグリタゾンは、p38-MAPKのリン酸化と活性化を増加させ、MAPK2をリン酸化・活性化し、さらに、解糖の重要な調節ステップである6-ホスホフルクト-2-キナーゼ/フルクトース-2,6-ビスホスファターゼ3(PFKFB3)をリン酸化・活性化させることでこれを実現させた。
*
ピオグリタゾンの自然免疫系に対する顕著な効果は、外傷性脳損傷の前臨床モデル(Dengら 2020)、うつ病(Zhaoら、…)における免疫抑制状態への移行とも相関している。2016)、軸索損傷(Wenら 2018)、神経炎症(KielianとDrew 2003)および脳卒中(Tureyenら 2007;Caiら 2018)、およびパーキンソン病(Swansonら 2011;CartaとPisanu 2013)である。
脳内グルコースホメオスタシス
脳は、必要なエネルギーのほとんどをグルコースに依存しており、体内の1日のグルコース負荷の25%を消費する。エネルギー生産に加えて、グルコースはアセチルコリン、アスパラギン酸、グルタミン酸、GABAなどの神経伝達物質の合成に寄与している。
摂食状態では、神経細胞はグルコースを直接消費し、グリア細胞(主にアストロサイト)はグルコースをグリコーゲンとして貯蔵する。酸化ストレス下では、ミトコンドリアの生体エネルギーが低下し、ニューロンはアセチル-CoAを脂肪酸に転換し、アストロサイトはAPOE依存的にこれを取り込んで脂質滴として貯蔵する(Liuら 2015;Liu T.ら 2017)。神経細胞からアストロサイトへの脂質の移動に欠陥があると神経変性が起こるため、これらの飛沫は神経細胞の健康にとって不可欠である(Liu L. et al. 2017)。
ドロップレットは、必須のエネルギー貯蔵量を表している可能性がある。通常の絶食期間中、アストロサイトはグリコーゲン分解および解糖を介して貯蔵グリコーゲンを乳酸に変換し、これはニューロンによって消費される(Calì et al. 2019)。同様に、アストロサイトは、脂質滴に貯蔵された脂肪酸をケトン体に変換し、ニューロンによって消費される可能性がある。グルコース不足下では、神経細胞はケトン体を消費する(Ding et al. 2013)。
ヒトアルツハイマー病患者およびADモデルマウスの遺伝子発現解析から、疾患の進行に伴い、グルコース消費量の減少に伴い、脂質代謝への依存度が高まることが明らかになった(Yaoら 2011;Demarestら 2020)。あるいは、貯蔵されたトリグリセリドは、貪食をサポートする膜の合成に、あるいはストレスに応答して使用されるかもしれない(Martínez et al. 2020)。
最後に、細胞内脂質滴は、病原性微生物を引き寄せ、抗菌ペプチドの貯蔵庫として、またRSAD2を含む他の免疫タンパク質の核形成部位として機能することにより、感染性物質に対するグリアベースの反応を調整するセンターとなるかもしれない(Boschら 2020)。また、これらは相互に排他的な選択肢でもない。[脂質滴の最近のレビューについては、Welte and Gould (2017)を参照〕。
*
グルコース代謝の低下は、アルツハイマー病に特徴的な現象である。これは、18F-deoxyglucose-positronemission tomography (FDG-PET)(Minoshima et al., 1995,1997;Herholz, 2010) や、15O-PET(Beason-Held et al., 2013) により局所血流が日常的に測定され、高い相関性がある。
ナトリウムに鈍感なGLUT1およびGLUT3トランスポーターは、血液からのグルコース抽出の大部分を占め、アルツハイマー病患者では、脳内のこれらのトランスポーターのレベルは、AD症状の発症の数十年前に低下し始める(シンプソンら、1994;Patching 2017)。
GLUT1は、血液脳関門(BBB)およびアストロサイトにおける優勢なグルコーストランスポーターであり、脳で消費されるすべてのグルコースの全身循環からの取り込みを担っている。高親和性、高容量のGLUT3トランスポーターは、神経細胞のグルコース取り込みを担っている。
脳はまた、小脳、大脳皮質、海馬、視床下部など、インスリン受容体が高度に発現している領域で、インスリン感受性GLUT4トランスポーターを低レベルで発現している(McEwen and Reagan, 2004;Alquier et al, 2006)。ピオグリタゾンは、神経刺激による大脳のグルコース取り込みを促進する。
*
ADにおける脳内グルコース利用の減少は、解糖および解糖後経路の障害を反映する解糖中間体のCSFレベルの低下(Bergauら 2019)と関連している(Anら 2018)。
それは、Aβ42およびAβ40レベル(Venziら 2017)、または脳萎縮(Smithら、1992;Ibáñezら、1998)または脳構造における他の変化(Smallら、1995、2000;Minoshimaら、1997;Reimanら、,2004;Samuraki et al., 2007;Beason-Held et al . , 2013)、臨床症状が現れる数十年前に出現する(Cutler, 1986;Kennedy et al., 1995;Small et al., 1995;Reiman et al.)AD病態に最も脆弱な脳領域におけるエネルギー代謝遺伝子の発現変化と関連しており(Xuら 2006;Brooksら 2007;Wangら 2007、2010;Liangら 2008a、b;Bossersら 2010)、正常老化とは異なる局所性側頭葉代謝低下の出現が認められる(Kuhlら、1982;de Leonら、1983;Duaraら、1984)。
大脳低代謝はタウのリン酸化の増加(Planelら、 2004)やアミロイドの蓄積(Gabuzdaら、 1994)につながる。逆に、EP2受容体の阻害によって恒常的な骨髄細胞のグルコース代謝を再確立すると、加齢に伴う認知機能低下が逆転した(Minhasら 2021)。したがって、脳内グルコース代謝の欠陥は、ADの神経変性の早期、一貫した、特異的なマーカーであり、ADの病理学に結果的に先行するものである。
Pioglitazoneと脳内血糖代謝の関係
In vivoにおいて、ピオグリタゾンは、脳血流および脳内グルコースの取り込みと消費を改善し(Nicolakakisら 2008;Satoら 2011;Papadopoulosら 2013)、一部はGLUT4トランスポーターの発現増強を介して(Sandoukら、1993;OlefskyおよびSaltiel 2000)、一部はミトコンドリア機能および生起の改善によって、改善される。
ピオグリタゾンはまた、PGE2合成を抑制し、EP2によって引き起こされるPKAシグナル伝達を阻害することによって、グルコース代謝を正常化する(Subbaramaiahら 2012年)。
脳内インスリン抵抗性
インスリン抵抗性(Craft 2005;Benedictら 2012;Willetteら 2013、2015a;Ferreiraら 2018)および2型糖尿病(DM2)(ChatterjeeおよびMudher 2018;Barbiellini Amideiら 2021)は関連するが独立したADのリスクファクターである。
DM2と認知機能障害の両方は、炎症およびPI3K-Aktシグナルに関与する遺伝子に富む遺伝子発現ネットワークを共有しており(Potashkinら 2019)、死後脳サンプルの直接解析により、ヒトおよびマウスAD脳サンプルにおいてインスリンおよびIGF1トリガーシグナルの障害が明らかになった(Bomfimら 2012;Talbotら 2012)。
*
インスリン、インスリン様成長因子1、およびそれぞれのmRNAは、脳全体に存在する(Blázquez et al. 2014)。インスリン受容体は、脳全体のすべての細胞型にマッピングされており、特に海馬と視床下部での濃度が高く、全身のエネルギー恒常性(Chen et al., 2017)、バランスと運動(Zhao et al., 2004)、記憶の形成と定着(McNay et al., 2010;McNay and Recknagel, 2011;Kullmann et al., 2016)など多様なプロセスにおけるその役割もよく確立されている。
*
脳と末梢のインスリン受容体(IR)は同じ遺伝子でコード化されている。しかし、脳と末梢の受容体は3つの重要な点で異なっている。脳内受容体はIR遺伝子の交互スプライシングによって生じ、末梢型IRよりも小さい。末梢IRとは異なり、脳内IRにインスリンが結合しても、その内在化は促進されない。
最後に、脳内IRは末梢IRよりも容易にIGF1受容体(IGF1R)とのハイブリッド受容体を形成する。IRとIGF1R受容体は同じ受容体チロシンキナーゼファミリーに属している。どちらもα2β2ヘテロテトラマーであり、2本の細胞外リガンド結合α鎖が、チロシンキナーゼ活性を持つ膜貫通型βサブユニットにジスルフィド結合している構造である。
また、α鎖は互いにジスルフィド結合している。一般に、IRシグナルは、トランスポーターの内部貯蔵庫から細胞表面への移動を含む代謝反応を引き起こし、グルコースとアミノ酸の取り込みと代謝を促進する。一方、IGF1Rは、主にタンパク質合成と細胞増殖に影響を与える。
両受容体の活性化は、遺伝子発現の変化を引き起こす。ハイブリッド型IR-IGF1R受容体は末梢よりも脳に多く存在する。本稿執筆時点では、CNSにおけるこれら3種類の受容体の具体的な役割は解明されていない。
*
インスリンは、末梢において、主にグルコースと脂質のホメオスタシスを促進する。その鍵となるのが、インスリンによって刺激されたグルコーストランスポーターGLUT4の細胞内プールから脂肪細胞や筋肉の表面膜への移動である。シグナル伝達アダプタータンパク質とキナーゼのカスケードによって、リン酸化の連鎖を介して、小胞移動、タンパク質合成、代謝経路の活性化を仲介する標的機能タンパク質とキナーゼが次々とつながるのである。
リガンドが結合すると、受容体のチロシン残基の自己リン酸化が活性化され、アダプタータンパク質であるIRS(IR、主に)またはShc(IGF1R、主に)の結合部位が形成される。これらはシグナル伝達カスケードのハブを形成する。PI3K-Akt経路は、細胞内トランスポーターの細胞表面への移動を促進し、インスリンの代謝作用に関与している。
MAPK経路はIRSとShcの両方が制御しており、遺伝子発現に対するインスリン(およびIGF1)の効果を媒介する。両経路は協力して、細胞の成長、分化、修復を制御している(Boucher et al. 2014)。
*
脳では、インスリン伝達経路は、GLUT4のトラフィッキングだけでなく、高親和性コリントランスポーターとAMPA、NMDA、GABA受容体のトラフィッキングも促進する(Zhaoら 2004;Fishwick and Rylett 2015;Spinelliら 2019)。これらのシナプス後作用に加えて、インスリンは樹状突起棘とシナプスの形成を促進する(Leeら 2011)。
BBBの主要なグルコーストランスポーターであるGLUT1、および脳内の主要なグルコーストランスポーターであるGLUT3はインスリンに反応しないため、インスリンは脳のグルコース消費の大部分を調節しない(Simpsonら 2008年)。
しかし、インスリンは、GLUT4トランスポーターを発現する数少ない脳領域の一つである海馬でのグルコース利用を刺激する。記憶の形成と回復に関連する高いエネルギー需要を満たすために、GLUT4トランスポーターのトランスロケーションは、膜電位の制御下でAMPKによっても媒介される(Ashrafiら 2017)。
IRとGLUT4は共にシナプスに集中しており、インスリン刺激、GLUT4を介したグルコースの取り込みは、持続的なシナプス小胞リサイクルをサポートし(Ashrafiら 2017)、記憶形成に不可欠である(Pearson-Leary and McNay, 2016;Pearson-Leary et al.)
グルコースセンサーN-アセチルグルコサミンO-トランスフェラーゼによるミトコンドリアの翻訳後修飾は、IRおよびGLUT4と同じ細胞領域内にミトコンドリアを局在させる(Pekkurnaz et al, 2014)。O-GlcNAcylationはまた、ミトコンドリアATP合成酵素の完全な活性に必要である(Chaら 2015)。
GLUT4トランスポーターおよびミトコンドリアとインスリン受容体の共局在化は、シナプス小胞輸送および活動電位のエネルギー的要件を支えるためのインスリン制御グルコース取り込みおよび代謝の重要性を強調する。タウはまた、恒常性条件下でO-GlcNAc化されるが、インスリン抵抗性はO-GlcNAc循環を擾乱し、タウの過リン酸化に寄与する(Liuら 2009、2011;Bourréら 2018)。
*
解糖とピルビン酸脱水素酵素の順次作用によるアセチルCoA生成とコリンの取り込みという2つのレベルでのアセチルコリン合成の促進から、神経伝達物質の放出と取り込みを操作し、神経新生と修復を支援し、タウリンのリン酸化を調節することまで、インスリンは認知を支えるプロセスに大きな影響を与えることが明らかである。
*
インスリンが脳生理学において重要な役割を果たすことを考えると、末梢インスリン抵抗性に起因する脳低インスリン血症、または脳インスリン抵抗性自体が、アルツハイマー病を含む神経変性疾患の重要な危険因子であることは驚くべきことではない(Baura et al, 1996;Matsuzaki et al, 2010;Willette et al, 2015b,c;Ekblad et al, 2017;Kong et al, 2018)。
中年期のインスリン抵抗性は、晩年期の認知症を予測する(Ekbladら 2017,2018;Lutskiら 2017;Tortelliら 2017;Kongら 2018)。代謝制御を(Ryanら 2006)または薬理学的介入(Naorら、1997)なしで増加させると、ワーキングメモリが改善される。膵臓機能不全と結合した全身的なインスリン抵抗性を反映するDM2は、ADの危険因子である(Ottら、1999;Schrijversら 2010;Livingstonら 2020)。メタボリックシンドロームは、脂質および心臓の併存疾患を伴う全身性インスリン抵抗性を反映しており、APOE遺伝子型とは無関係にADと関連している(Kuusistoら、1997年)。
これらの関係は全身のインスリン抵抗性を反映しているが、死亡時にDM2、肥満、メタボリックシンドロームなどのインスリン抵抗性を特徴とする他の併存疾患がない被験者では、ADにおいて脳組織自体がインスリン抵抗性であることに注目することが重要である。
ADに関連した脳のインスリン抵抗性は、死亡時に認知障害のなかった高リスク者において、人生の早い時期に検出可能である。インスリンおよびIGF-1シグナルの減少は、in situでのインスリンシグナル関連タンパク質の阻害性リン酸化、インスリンシグナルカスケードのキナーゼの活性低下、ex vivoでのインスリンシグナルカスケードの活性化障害によって検出できる(Steenら…2005;Bomfimら,..2005)。2005;Bomfim et al., 2012;Talbot et al., 2012)、およびインスリン/IGF1シグナル伝達経路のメンバーをコードする遺伝子の調節された発現異常によってである(Katsel et al. 2018)。
インスリンシグナル伝達の欠損は相加的であり、ADとDM2の両方を患った個体でより大きかった(Liuら 2011)。マウスEOADモデルもまた、インスリンシグナル伝達の欠損を示す(Takedaら 2010;Bomfimら 2012)。脳のインスリン抵抗性とアルツハイマー病との関連についての優れた最近のレビューについては、Ferreiraら(2018)、de la Monte(2019)を参照のこと。
*
ADに寄与する決定要因の多くは、脳インスリン抵抗性を誘発し、持続させる。IL-1βおよびTNF-αを含む炎症性サイトカイン(Gengら、1996;Morrisonら 2010;Bomfimら 2012;Kitanakaら 2019)は、インスリン/IGF1-PI3K-Aktシグナル伝達を妨害するERK2、JNKおよびPKCζ/λ(20)などの「逆調節」キナーゼを活性化させる。
これらは、IR/IGFRシグナル伝達カスケードのタンパク質を、正常なドッキングやキナーゼ活性を妨げる部位でリン酸化する。β-アミロイド線維やミトコンドリア機能不全に起因する酸化ストレスもまた、これらのキナーゼを活性化する(Okazawa and Estus, 2002;Persiyantseva et al.)炎症と酸化ストレスは、同様に全身のインスリン抵抗性を説明する(Czech, 2017)。
ピオグリタゾンが脳内インスリン抵抗性を克服する
ピオグリタゾンは、脳内インスリン抵抗性を克服し、血流とグルコースの取り込み・利用を促進することにより、ADのリスクを低減させることができる。ピオグリタゾンは、脳内インスリン抵抗性を克服する。
インスリンシグナル伝達経路の遺伝子の正常な発現を回復し(Katselら 2018)、脆弱なニューロンにおけるグルコースの取り込みを促進し、炎症を抑え、免疫抑制を促進し(Zhangら 2008;原口ら 2008;Swansonら 2011;Kaplanら、,2014)、酸化ストレスを改善し(Gumieniczek, 2003;Wang et al., 2014;Paciello et al., 2018)、Aβペプチドの合成を阻害し(Liu et al., 2013;Quan et al., 2019b)、脳からの迅速なクリアランスを促す(Mandrekar-Colucci et al., 2012)。
糖尿病でない被験者を含む全身のインスリン抵抗性を克服することにより、ピオグリタゾンは脳低インスリン血症も緩和する(Baura et al, 1996;Miyazaki et al, 2002;Kernan et al, 2003)。
ミトコンドリア機能障害
バイオエナジェティクス
ミトコンドリア機能障害は、ADにおける脳内エネルギー代謝異常の主要な要因である。酸化的損傷とミトコンドリアストレスは、ヒトのAD(Hirai et al .変化したミトコンドリア形態は、樹状突起プロファイル、スパインおよびシナプス末端、ならびに脳全体のアストロサイトにおいて明白であり(Baloyannis 2011)、生体エネルギー欠損を裏切る(Parkerら、1994a;Vallaら 2001、2010;Yaoら、,2009)、核にコードされたミトコンドリア遺伝子の発現の減少およびmtDNA欠損の誤った修復(Lovellら 2000;Weissmanら 2007;Sykoraら 2015)に一部起因し、ダイナミクス(Manczakら 2011)、およびプロテオスタシス(Alikhaniら 2011;Westerlundら 2011)が損なわれていると考えられる。
ミトコンドリアのエネルギー産生の阻害は、APPのアミロイド原性プロセシングを誘発し(Gabuzdaら、1994)、その結果、ミトコンドリア機能を悪化させる(Manczakら 2006;Ceniniら 2016)。Swerdlow (2018); Wang et al. (2020)ほか(Lin and Beal,2006;Reddy and Beal, 2008;Gibson et al., 2010;Moreira et al., 2010;Swerdlow et al., 2010,2014) は、ミトコンドリア障害のアルツハイマー病の病態への寄与を徹底的に検討しており、ここにそのハイライトを記した。
*
ミトコンドリアは独自のゲノムを持つが、約1500種類あるミトコンドリアタンパク質のうち、13種類しかコードしていない。残りは核ゲノムによってコードされており、これらの核ゲノムにコードされたミトコンドリア遺伝子の多くは、AD初期に発現が制御されなくなる。
そのパターンは、代謝低下を示す脳領域の勾配とほぼ平行であり(Liang et al., 2008b)、重度の影響を受ける後帯状皮質(PCC)から、代謝異常を比較的免れる中側頭回、海馬、内嗅皮質、視覚皮質、上前頭回に及ぶ(Minoshima et al., 1997;Mosconi et al., 2008b;Herholz, 2010)。
ADでは、ミトコンドリア遺伝子に加え、解糖系やTCA経路の遺伝子もダウンレギュレートされている(Brooks et al.)TOMMおよびTIMMは、核にコードされたミトコンドリアタンパク質の輸入を触媒するミトコンドリア外膜および内膜複合体の成分をそれぞれコードしている(Wiedemannら 2004年)。
ミトコンドリアの健康に対する重要性に加え(Zeh, 2013)、輸入が阻害されるとミスフォールドしたタンパク質の細胞質への過剰な蓄積につながるため、細胞質プロテオスタシスの制御にも重要である(Liu W. et al., 2018)。
ADではミトコンドリアタンパク質の輸入が制御不能であり(Anandatheerthavarada et al., 2003;Devi and Anandatheerthavarada, 2010;Devi and Ohno, 2012;Cenini et al., 2016;Sorrentino et al., 2017)、この制御不能はOXPHOS遺伝子と同じ領域パターンに従っている。ADでは、後帯状皮質でTOM複合タンパクの50%、TIMタンパクの27%が発現低下していたが、視覚野ではそれぞれ17%、0%しか発現低下していなかった(Liang et al. 2008b)。
*
OXPHOSシステムを構成する5つの複合体それぞれのサブユニットの発現もADでは抑制されており、各脳領域での発現低下の程度はそれぞれのmRNAの発現パターンと一致した。これらの研究は、AD脳におけるCOX(Parker et al., 1994a;Gonzalez-Lima et al., 1997;Bosetti et al., 2002)、α-ケトグルタル酸脱水素酵素(Gibson et al., 1988;Bubber et al., 2005) およびピルビン酸脱水素酵素(Sorbi et al., 1983;Rex Sheu et al., 1985) の活性低下という以前の報告を補完するものであった。
COX活性はまた、ADの血小板において減少する(Parkerら、1994b;Bosettiら 2002;Vallaら 2006)ので、ミトコンドリア関連のAD病態生理決定因子が脳に限定されないことが示唆される。
*
これらのデータは、ADに関連した脳代謝低下の生化学的根拠を与える一方で、観察されたミトコンドリア欠損がADに関連した障害に起因する可能性もある。Vallaらは、AD発症リスクのある若年成人のAPOEε4キャリアと、APOEε4を持たない年齢をマッチさせた対照者を用いてこの仮説を検証した(Vallaら、 2010)。
どちらのグループにも、組織学的なβ-アミロイド沈着、神経原線維変化、可溶性Aβ42は見られず、不溶性Aβ42と可溶性Aβ40についても一致した。しかし、COXの活性とタンパク質のレベルは、リスクのある集団で低く、ミトコンドリアの損傷が、ADの病理学的徴候が検出される前に起こることが確認された。
*
ADにおけるミトコンドリア機能障害を要約したデータおよびその概念図は、剖検標本に基づいている。なぜなら、生きた被験者のミトコンドリアを調べることは不可能だったからである。むしろ、生体内のミトコンドリア機能は、解糖を直接測定するFDG-PET解析から推測されてきた。
今回、塚田らのグループは、ミトコンドリアOXPHOS複合体Iのロテノン阻害部位に結合するPETリガンドを導入し、生体脳における複合体Iの稼働率を直接測定できるようにした(原田ら、 2013;寺田ら、 2020)。彼らは、ADの海馬傍で複合体Iの消失がFDG-PETで検出される代謝低下に先行することを発見し(Terada et al., 2020)、ミトコンドリア機能障害がAD発症の初期イベントであることを確認した。
CSF中のミトコンドリア関連マーカーは、脳のミトコンドリアの健康状態を直接プローブするための追加のツールを提供する。Podlesniyらは、CSF中のmtDNAレベルが、AD発症リスクのある無症状の被験者およびアルツハイマー病患者で、認知的に正常で年齢を合わせた対照群と比較して低いことを示した(Podlesniyら 2013年)。この欠陥は、アルツハイマー病の病態に関連する前頭側頭葉変性症(FTLD)の被験者では観察されなかった(Podlesniyら 2013)。
以前、AD脳におけるmtDNAの減少は、全脳の免疫組織化学(Hiraiら 2001)、およびレーザー捕捉マイクロダイセクションによって分離された単一細胞におけるqPCR(Riceら 2014)により検出された。これらの死後のデータは、CSF mtDNAの定量をミトコンドリアの健康状態の生体内試験の指標として使用することを支持するものである。
ミトコンドリアダイナミクス
シナプスの損失は、AD発症の初期に明らかになり、AD関連の認知障害の重症度と高い相関がある(DeKosky and Scheff, 1990;Terry et al.、1991)。
ミトコンドリアは非常に動的なオルガネラであり、特定の地域のニーズを満たすために細胞内で絶えず再分配し、機能的なミトコンドリアを維持するために必要な形状変化および連続的かつ同時の分裂と融合のラウンドを受ける。健康な神経細胞では、ミトコンドリアは細胞体とシナプスの両方で神経細胞全体に均一に分布し、シグナル伝達(Werner and Werb, 2002;Yang et al., 2009)、ATP生成(Rangaraju et al., 2014;Sobieski et al., 2017)および Ca2+ -buffering(Contreras他, 2010;Tarasov他, 2012)の役割を介してシナプス形成とシナプス機能をサポートしている。
対照的に、AD被験者の神経細胞では、ミトコンドリアは細胞体に大きく制限されている(Baloyannisら 2004;Wangら 2009;Pickettら 2018)。APPとタウの両方が、ミトコンドリアの分布の乱れに寄与している。APP/PS1ニューロン、およびPS1およびTg2576APPマウスのニューロンでは、ミトコンドリアの軸索輸送が遅延した(Calkinsら 2011;Trushinaら 2012)。
APP過剰発現マウスから単離したニューロンを培養下でAβ42にさらすと、ミトコンドリア輸送が阻害され、軸索のミトコンドリア密度が減少した(Duら 2010年)。コントロールのC57Bl/6マウスから得た海馬ニューロンをAβ25-32ペプチドに暴露しても、同様の結果が得られた(Calkins and Reddy, 2011)。
高リン酸化タウおよびP301L変異を含む疾患関連タウ変異は、微小管とミトコンドリアを含むカーゴとの相互作用を破壊し、正常な輸送を阻害する(Kopeikinaら 2011;Schulz K.L. et al 2012;Shahpasandら 2012;Rodríguez-Martínら 2016)。
タウの枯渇は、Aβ誘発性のミトコンドリアトラフィッキングの欠損から保護する(Vosselら 2010)。これは、タウノックダウンAPPマウスで観察される神経細胞機能不全の緩和に寄与すると考えられる(Roberson Scearce-Levie et al., 2007;Ittner et al., 2010)。インスリン抵抗性およびミトコンドリアのシナプス結合に必要なO-GlcNAc合成の阻害(Pekkurnazら 2014)も、ニューロンにおけるミトコンドリアの分布を乱す可能性がある。
*
ミトコンドリアダイナミクスの障害は、ミトコンドリア機能欠損にも寄与している。これは、ミトコンドリア分裂遺伝子DRP1およびFIS1、ならびにミトコンドリア融合遺伝子MFN1、MFN2およびOPA1の欠陥発現と関連している(Wangら 2009;Manczakら 2011)。ヒトAD被験者の脳サンプルでは、認知的に正常で年齢をマッチさせた対照群と比較して、FIS1の発現が亢進し、MFN1、MFN2およびOPA1の発現が抑制されていることがわかった。
しかし、Manczakら(2011)がADサンプルにおけるDRP1の発現上昇を報告したのに対し、WangらはADにおいてそのmRNA発現に変化はなくDrptタンパク質の発現低下を報告している(Wangら 2009)。
これらの違いは、これらの研究チームが使用したADサンプル(BraakステージやAPOEステータスを含む)の違いを反映している可能性がある。分裂タンパク質と融合タンパク質の分布は、健常者とAD症例の脳におけるミトコンドリアの分布とそれぞれ一致した(Wang et al. 2009)。
Drp1のGTPase活性は、S616のリン酸化によって増強される(Taguchi et al. 2007)。ウェスタンブロット解析により、アルツハイマー病患者のミトコンドリアおよび細胞質画分の両方において、年齢をマッチさせた認知機能正常対照者よりもDrp1のS616リン酸化が大きいことが明らかになった(Wang et al. 2009)。
アミロイドペプチドやオリゴマーはDrp1を活性化する可能性がある。Manczakらは、共免疫沈降法とIHC法により、単量体およびオリゴマーAβとDrp1が物理的に結合し、それが疾患の重症度が増すにつれて増加することを明らかにした。神経芽腫細胞または初代培養神経細胞におけるAPPswe変異の過剰発現は、ミトコンドリアの断片化と核周囲の分布を増加させ、これはBACE阻害剤によって逆転した(Wangら 2008)。
続いてWangらは、培養神経芽細胞腫細胞をオリゴマーAβに暴露すると、Drp1のリン酸化とミトコンドリア画分への蓄積、およびミトコンドリアの断片化が増加することを示した(Wangら 2009)。タウはまた、Drp1のGTPase活性を増加させるような形で、Drp1と物理的に相互作用している。この相互作用は、ヒトのAD前頭葉皮質で検出されたが、対照群では検出されなかった。また、APP、APP/PS1、3×Tgマウスの皮質サンプルで確認されたが、年齢を合わせた同腹の対照群では確認されなかった(Manczak and Reddy, 2012)。
*
これらの結果は、Drp1活性、またはアミロイドおよび/またはタウとの結合が、ADを遅延または治療するための魅力的な標的であるかもしれないことを示唆している。Kuruvaらは、分子ドッキングシミュレーションを使用して、Drp1とアミロイドの結合をブロックするDDQを設計した(Kuruvaら 2017)。
培養神経芽腫細胞において、DDQはAβとDrp1の会合をブロックし、ミトコンドリアの断片化と酸化ストレスを防ぎ、ミトコンドリア生合成とシナプス形成を促進した(Kuruva et al.、2017)。我々は、ピオグリタゾンが、Aβ生成およびタウリン酸化に対する効果によって、Aβおよびタウを介したDrp1活性化もチェックすることを提案する。
*
細胞は、タンパク質の合成、フォールディング、輸送を制御するために、すべての主要なオルガネラ間のクロストークを含む一連の統合経路を使用している。どの区画のどの枝が機能不全に陥っても、それはシステム全体に影響を及ぼす。凝集しやすい多くの細胞質タンパク質は、ミトコンドリアに輸入され分解される(Ruan et al.)Aβペプチドは、TOMM20を輸入「受容体」として、TOM複合体(Hansson Petersenら 2008)を介してミトコンドリアに輸入されることができる(Huら 2018)。
Aβペプチドでミトコンドリアプロテオスタシスシステムを圧倒することは、以下を含む多くの有害な結果をもたらす:脂肪酸短鎖デヒドロゲナーゼ/還元酵素の阻害に続いて生じるROSの生成を介した酸化ストレスの増加(Lustbader et al,2004)およびOXPHOS複合体Iの阻害(Bobbaら 2013)、ミトコンドリア輸送の阻害および軸索ミトコンドリア密度の減少(Duら 2010;CalkinsおよびReddy 2011)、ならびにミトコンドリア断片化の増加(Baloyannis 2006)。
Aβのミトコンドリア内分解が障害された場合(Lautenschlägerら 2020)、またはミトコンドリアへの輸入が阻害された場合(Liu Y. et al 2018)、細胞質プロテオスタシスが阻害され、細胞質へのタンパク質蓄積および凝集が生じる(Lautenschäger and Schierle, 2019に総説あり)。
ミトコンドリアタンパク質のホメオスタシスに関与する2つのプロテアーゼ、PreP(Alikhani et al., 2011)とHtr2(Westerlund et al., 2011)がADで減少すること、Htr2の多型がADと関連しているという示唆に富む証拠は(Westerlund et al., 2011)、このモデルと一致している。
しかしながら、ミトコンドリアAβ蓄積とミトコンドリア外タンパク質凝集の関係は、Aβによる生体エネルギーの阻害とAPPのプロセシングがフィードバックサイクルによって関連しており、Aβによる生体エネルギーの阻害は、APPプロセシングと追加のβ-アミロイドの生成を停滞させる(Wilkins and Swerdlow, 2017)ため非線形であると考えられる。
それにもかかわらず、β-アミロイドとホスホタウの両方の細胞内蓄積とミトコンドリア局在は、アルツハイマー病を特徴付けるミトコンドリア機能障害と異常なトラフィッキングおよびダイナミクスに寄与する。ピオグリタゾンは、Aβペプチドの生成とタウのリン酸化を阻害することで、これらの作用をブロックする。
ミトコンドリアと細胞カルシウムの調節障害
ADではカルシウムのホメオスタシスが乱れ(Khachaturian, 1994)、病気の初期と後期(Bezprozvanny and Mattson, 2008;Calvo-Rodriguez et al, 2020)の両方の段階に寄与すると考えられている。
カルシウムは、ミトコンドリア機能に加えて、神経形成やシナプス形成、シナプス伝達、シナプス可塑性など、複数の神経細胞活動に必須である。カルシウムのホメオスタシスの変化は、アルツハイマー病および他の神経変性疾患の主要な特徴であり(Mattson, 2007;Tong et al., 2018)、β-アミロイド蓄積などの外因性要因(Bezprozvanny and Mattson, 2008)に加えて、内因性要因によるところもある。
電位依存性カルシウムチャネルを介した流入の増加およびERからのカルシウム放出の増大は、再吸収の鈍化と相まって(Popugaeva and Bezprozvanny, 2013)、細胞質カルシウムを上昇させる(Thibault et al. 2007)。これらは、ミトコンドリアカルシウムの過負荷(Calvo-Rodriguezら 2020)、過剰なROS生成、ミトコンドリアエネルギー産生およびアポトーシスの障害に寄与する(CeniniとVoos 2019)。
また、細胞質Ca2+の上昇は、カルパインを介してCDK5を活性化し、高活性p25調節サブユニットの生成につながる(Kimura et al. 2014;Seo et al. 2017)。神経細胞のカルシウムホメオスタシスの破綻は、カルシウム調節に重要な遺伝子の発現にまで及ぶ(Emilsson et al. 2006)。
酸化ストレスおよび脂質過酸化(Mattson, 1998)、ミトコンドリア-ER膜(MAM)の摂動(Hedskogら、 2013;Area-Gomezら、 2018)およびβ-アミロイドペプチドの蓄積(Mattsonら、 1992)は、ADにおけるカルシウムの恒常性の変化に寄与している。
*
形態学的解析と遺伝子発現、プロテオミクス、機能的データを総合すると、ミトコンドリア機能障害は、Aβプラークやタウタンゲルの蓄積などAD病態の検出可能な段階の前に早期に存在し、AD発症に寄与しているという結論が導き出された。
ピオグリタゾンとミトコンドリア機能障害
PPARγアゴニストは、上述のAβペプチド産生の阻害、マイトビオジェネシスの誘発(Strumら 2007;Miglioら 2009)、およびミトコンドリア膜電位の改善(Wangら 2002;Pipatpiboonら 2012)によりAD関連のミトコンドリア機能障害を改善させる。
また、複合体Iによる活性酸素の生成を抑制し(Brunmairら 2004;Ghoshら 2007)、グルタチオンや抗酸化物質SODおよびカタラーゼの発現を増加させることにより、酸化ストレスによるダメージを制限する(Collinoら 2006;AleshinおよびReiser 2013)。
PPARγアゴニストは、ADにおいて減少しているGLUT3の発現を刺激することによって(Garcia-Buenoら 2006;Wangら 2012)、神経細胞のエネルギーバランスにさらなる正の効果を及ぼす(Simpsonら、,1994)、GLUT4を介したグルコースの取り込みを刺激することによって、およびインスリン刺激Akt活性化の増強(Karwiら、2020)およびPDHキナーゼ活性の阻害(Wayら 2001)を介して神経細胞の乳酸酸化(Izawaら 2009)およびピルビン酸フラックス(Rossiら 2020)の促進によって、神経細胞のエネルギーバランスにさらなる正の効果を与える。
PioglitazoneとRosiglitazoneの前臨床試験における有効性
表1は、PPARgアゴニストのロシグリタゾンとピオグリタゾンを用いた代表的な前臨床試験をまとめたものである。
これは網羅的なリストではなく、いくつかの点を説明するために選択したものである。まず、両薬剤は少なくともいくつかのAD関連表現型に対して生体内試験で有効性を示すが、すべての研究結果が再現されるわけではない。
一般に、PPARγアゴニストは、酸化的損傷から保護し、シナプスの回復を促進し、学習と記憶を改善し、脳血流とグルコース取り込みを促進し、コルチコステロンレベルとアミロイド沈着、Aβペプチドレベルと反応性アストロサイトとミクログリアを減らし、ミクログリア貪食を促進することがわかった。これらの研究から、ヒト試験の適切なデザインを考える上で重要な2つの一般論が導き出される。
表 1 アルツハイマー病の前臨床モデルに対するPPARγアゴニストの作用のまとめ
| モデル/リファレンス | 投与量 | 人間換算値 ドセア | コメント | 結果 |
| 治療パラダイム | ||||
| Tg2576(Pedersenら 2006) | Rosiglitazone, 4 mg/kg, po (chow), vs vehicle control, 4 months. | 19.51mg/d | 男性、治療開始時9ヶ月、治療開始時にアミロイドの沈着、海馬の樹状突起のスパインの損失、空間学習の欠陥が明らかであった。 | ロシグリタゾンは、記憶力の改善、学習障害(放射状腕迷路)の軽減、不溶性Aβ42レベルの低下、コルチコステロンレベルの低下と統計的に関連した。 |
| Tg2576、Nenov 2014,2015(Nenovら 2014、2015) | Rosiglitazone, 30 mg/kg, po (chow), vs vehicle control, for 30 days. | 146.35mg/d | 同数の雌雄、治療開始時8ヶ月齢。 | 学習・記憶の改善は、自発的シナプス活動および短期可塑性の改善と相関する。ERK経路の関与とシナプスタンパク質の発現、成熟DG:未熟DG顆粒細胞比の回復、Nav媒介電流の正常化。 |
| APP(V717I)、(Henekaら 2005) | ピオグリタゾン, 40 mg/kg/d; po (chow), vs vehicle control, 7 日間 | 195.12mg/d | 男女同数、治療開始時10ヶ月齢、アミロイド病変あり。 | BACE1の減少、アミロイドプラークの沈着、可溶性Aβ42レベル、反応性ミクログリアの減少。 |
| APP (Swe/PS1)Δ9,(Mandrekar-Colucci et al., 2012;Skerrett et al., 2015) | ピオグリタゾン、80 mg/kd/d、po(経口投与) vs ビークルコントロール、9日間 | 390.24mg/d | 雌雄同数、投与開始時に6ヶ月齢と12ヶ月齢で病態負荷が異なるマウスでも同様の結果が得られた。 | 記憶保持の改善アミロイドプラーク沈着量の減少、6ヶ月齢マウスにおける可溶性および不溶性Aβ40およびAβ42の減少、12ヶ月齢マウスにおける不溶性Aβ42およびAβ40および可溶性Aβ40の減少、反応性ミクログリアおよびアストロサイトの減少、ミクログリア貪食の促進、IL-1β、TNF-α、Tm1、Fizz1、Arg1発現量の上昇を示した。 |
| APP/PSI(Chen et al., 2015) | Pioglitazone, 10 mg/kg/d, ip, vs. vehicle control, 7 and 10 days. | 48.78mg/d | 男女同数、治療開始時12ヶ月齢、病態あり。 | 7日間の投与でLTPが、10日間の投与で水迷路の成績が改善された。タウのリン酸化の代用となるCDK5の発現と活性を低下させた。 |
| APP(Swe/PS1)Δ9(Toba et al.) | Pioglitazone, 80 mg/kg/d, po (chow), vs vehicle control, 9 days. | 390.25mg/d | 同数の雌雄マウス、投与開始時5~6ヶ月齢、病態のエマージェンシー・ステージ。 | 運動協調性、LTPの増加、CDK5制御タンパク質(p25 & p35)の発現低下。 |
| J20 (PDGFプロモーター下V717F)(Escribano et al., 2010) | Rosiglitazone, 5 mg/kg/d, po (gavage) vs vehicle control, 1ヶ月および4ヶ月間投与した。 | 24.39mg/d | 雌雄同数、投与開始時生後10ヶ月。 | 1ヶ月後に物体認識力が向上し、空間記憶(モリス水迷路)が徐々に改善した。アミロイドプラーク、不溶性Aβ42およびAβ40レベル、リン酸化タウが減少し、抗炎症、食作用のあるミクログリア表現型が促進された。 |
| J20(Nicolakakis et al., 2008) | ピオグリタゾン、20mg/kg/d、po(chow)、対ビヒクルコントロール、1.5~2ヶ月間。 | 97.56mg/d | 男女同数、投与開始時生後14ヶ月、アミロイドーシス、神経細胞喪失がよく確認された。 | 水迷路の成績、アミロイド沈着、可溶性・不溶性Aβ42およびAβ40のレベルには影響なし。
脳血流およびグルコース取り込みの改善,脳血管機能の回復,皮質コリン作動性刺激の改善傾向,アストログリオーシスの減少,脳酸化ストレスの回復. |
| 3xTg、検索(Searcy et al.) | ピオグリタゾン、18 mg/kg, po (chow)、対ビヒクルコントロール、3.5ヶ月間 | 87.81mg/d | 雌マウス、治療開始時11-12ヶ月齢、アミロイド沈着は十分に確立され、タウ凝集塊も存在する。 | 能動的回避課題における学習の改善、LTPの増強、CA1におけるアミロイド沈着および高リン酸化タウの減少。 |
| 予防のパラダイム | ||||
| J20(Escribano et al., 2009) | Rosiglitazone, 5 mg/kg/d, po (gavage) vs vehicle control. | 24.39mg/d | 予防 vs. 救援試験。1.5ヶ月齢の雌雄同数を2.5ヶ月間投与(予防)し、9ヶ月齢の同数のマウスを1ヶ月間投与(救済)した。 | 両コホートで物体認識力を向上させた。高齢のマウスでは、コルチコステロンレベルを下げ、グルココルチコイド受容体のダウンレギュレーションをブロックした。 |
| J20(Badhwarら 2013)。 | ピオグリタゾン、20 mg/kg/日、po(チャウ) vs 対照群 | 97.56mg/d | 3ヶ月齢のマウスで14週間投与開始、小コホートで3日間投与。 | 幼若マウスで開始した14週間の投与は、統計的に空間学習の改善と関連し、記憶力の改善傾向も認められた。3日間投与で脳血流が回復し、その効果は長期投与マウスでも持続した。 |
| Tg2576(Rodriguez-Riveraら 2011年)。 | Rosiglitazone, 30 mg/kg, po(chow), vs vehicle control, for 4, 8 or 12 months. | 146.16mg/d | 同数の雌雄、投与開始時1ヶ月齢。 | 9ヶ月齢の動物(8ヶ月間摂取)の連合学習および記憶障害を回復させたが、5ヶ月齢(4ヶ月間摂取)および13ヶ月齢(12ヶ月間摂取)のマウスでは回復させなかった。 |
| SCAMP8(Seok et al., 2019) SCAMP8は、アミロイドとタウの病理、ニューロンと樹状突起スパインの損失、およびCNS酸化ストレスを示す自然発症の「アルツハイマー様」マウスモデルである(Armbrecht et al., 2014;Cheng et al., 2014).。 | Pioglitazone、2または5 mg/kg/d、po(経口投与)対ビヒクル対照、7週間。 | 9.76 又は 24.39 mg/日 | 同数の雌雄マウス、投与開始時9ヶ月齢。 | 水迷路の成績向上、アミロイド沈着と可溶性Aβ40の減少、LRP1発現の増加。5mg/kg/dと2mg/kg/dの比較では、すべての反応が減衰した。 |
| 脳血管モデル | ||||
| J20/TGFβ1(Papadopoulosら 2013) | ピオグリタゾン, 20 mg/kg/d, po (chow), vs ビークルコントロール, 6ヶ月間 | 97.56mg/d | 治療対予防研究」でもある。男女同数で実施。投与開始時に生後6ヶ月と12ヶ月の2つのコホート。成体マウスは6ヶ月間投与、老齢マウスは3ヶ月間投与。 | 空間学習や記憶には影響なし。成体マウスでは反転学習が改善されたが、老化マウスでは改善されなかった。成体および老化マウスにおいて、脳血流および脳内グルコース取り込みの改善,海馬ではなく大脳皮質でのアストログリアの抑制,海馬でのミクログリア活性化の抑制がみられた。アミロイド病変や脳血管反応性には両群とも影響なし。 |
| TGFβ1(Lacombe et al., 2004) | ピオグリタゾン、18 mg/kg/d、po(chow)、対ビヒクル対照、2ヶ月。 | 87.81mg/d | 同数の雌雄マウス、投与開始時2ヶ月齢。 | Aβ42量とグリアの活性化が減少し、水頭症が増加した。 |
| TGFβ1(Galea et al., 2006) | ピオグリタゾン、18 mg/kg/d、po(chow)、対ビヒクル対照、2ヶ月。 | 87.81mg/d | 同数の雌雄マウス、投与開始時2ヶ月齢。 | ピオグリタゾンは、コントロール(非トランスジェニック同胞)マウスの脳内グルコース取り込みを抑制し、トランスジェニックマウスではTGF1bによる抑制を逆転させることができなかった。 |
| 糖尿病モデル | ||||
| ICR マウス(Jiang et al., 2012) ICR 系統は、汎用マウス系統である。
10週齢のマウスに高脂肪食(脂肪60%、炭水化物20%、タンパク質20%)を1ヶ月間与えて末梢性のインスリン抵抗性と高血糖を誘発し、その後ストレプトゾトシン(100 mg/kg)を注射してインスリン欠乏と脳低インスリン血症を引き起こすことにより糖尿病を発症させた。 |
Pioglitazone, 18 mg/kg/d, po (chow) or 9 mg/kg/d vs vehicle control, for 6 weeks. | 87.81mg/d または 43.9mg/d | 投与開始時に16~18週齢で、体重および高血糖の程度が同程度の雌雄同数のマウスを、無作為に等しい大きさの投与群に割り付けた。非糖尿病対照群も同様に治療群に分けた。 | HFD/strep-糖尿病は記憶障害と関連していた。ピオグリタゾン投与は学習と記憶を改善し、可溶性Aβ42とAβ40、BACE1、NF-κB、RAGEの減少を認めた。 |
| Sprague-Dawley ラット(Gao et al., 2017)12週齢のラットに60%脂肪食を20週間与えた後、ストレプトゾトシン(27mg/kg)を注射した。 | Pioglitazone, 10 mg/kg/d, po (chow) vs vehicle control, for 10 weeks. | 48.78mg/d | 20週齢の雌雄同数のマウスを、ICRマウスの場合と同様に、処理群に分けた。 | HFD/strepラットは、対照群および糖尿病+ピオグリタゾン群に対して記憶障害を示したが、両群に差はなかった。ピオグリタゾンは、高血糖によるERK1/2 mRNAおよびタンパク質の発現障害を改善した。 |
| APOEモデル | ||||
| APOETRマウス(Toら 2011年) 3ヶ月齢の雄マウスに60%脂肪食を32週間与えた。 | ピオグリタゾン、20 mg/k/d、po(経口投与) vs ビークルコントロール、3週間。 | 97.56mg/d | 40週齢の雄性HFまたはLFマウスを対照群およびピオグリタゾン投与群に分け、3週間投与した。 | 高脂肪食はインスリン抵抗性と耐糖能異常を引き起こし、すべてのマウスですべてのphospho-tauエピトープを減少させた。
食事とピオグリタゾンのいずれもAPP代謝に影響を与えなかった。HFマウスにおいて、ピオグリタゾンはAPOEε3マウスにおけるAT8 p-tauの減少、APOEε4マウスにおけるAT8 p-tauの増加と関連していた。 |
| 加齢に伴う神経病理学的変化 | ||||
| Wistar ラットにおける老化現象の逆転(Cowley et al., 2012) | Rosiglitazone, 3 mg/k/g, po (chow), vs vehicle control, 56 days. | 14.63mg/d | 投与開始時、22ヶ月齢 vs 3ヶ月齢(対照)の雌雄同数のラット。 | ロシグリタゾンはT1緩和時間を改善し、LTPのシナプス後成分を改善し、アストログリアシスおよびRANTES発現を減少させたが、これはロシグリタゾンによる内皮細胞-アストロサイト相互作用の増強が介在していた。ミクログリア活性化には影響なし。 |
| Wistar ラットにおける老化防止効果(Wang et al., 2012) | Rosiglitazone, 6 mg/kg/d, po (chow), vs vehicle control, 40 days. | 29.27mg/d | 投与開始時に12~14ヶ月齢(中)の雌雄同数のラットと1ヶ月齢の対照群を比較。 | Rosiglitazoneは水迷路学習を改善し、シナプス可塑性、場所細胞の活性を高め、LTPのシナプス後成分を改善し、海馬のGLUT3発現を回復させた。 |
| BOLDイメージング ファーマコダイナミクス | ||||
| Wistar ラット若齢成体(Crenshaw et al.) | ピオグリタゾン、0.04, 0.08, 0.16, 0.32 mg/kg/d、po(経口投与)対ビヒクル対照、2日間および7日間。 | 0.195, 0.39, 0.78, 1.56 mg/日 | 投与量は(TOMMORROW)で使用した用量(0.8mg/日)に等尺性を持たせて選択された。比較対象が多いため、試験パワーが不足。 | 安静時の機能的結合は、0.08mg/kg/day投与2日後に2領域間で増加し、7日後には5用量群すべてにおいてベースラインに対して17個の結合が変化した。7日目には、CA1と視床腹部の間の結合は、すべてのピオグリタゾンの用量で増加したが、0.32mg/kg/dayで最も弱かった。 |
第一に、治療のタイミングと期間が、疾患関連の表現型ごとに固有の重要な変数であるということだ。疾患の自然史における治療時期が極めて重要な意味を持つことがある。刺激結合型脳血流やグルコース取り込みなどの一部のパラメータは、成体マウス(治療開始時6カ月齢、プラーク病理の目に見える兆候や学習・記憶における著しい欠損の前に)および老化マウス(治療開始時15〜18カ月齢)の両方で正常化し、反転学習などの他のパラメータは成体マウスで改善したが、老化マウスでは改善しなかった(Papadopoulos et al., 2013).また、空間学習などの他の形質は、若いマウスを長期間治療した場合にのみ改善された(Badhwar et al. 2013)。
第二に、短期間の治療に反応するパラメータもあれば、反応しないパラメータもある。脳血流は短期間(3日間)の薬物曝露で正常化したが、その期間では学習の改善は見られなかった(Badhwar et al. 2013)。
第三に、有効性は治療時間の経過とともに顕著になる(Escribano et al. 2010;Chen et al. 2015)。
*
第二の教訓は、投与量が重要であるということである。これらの代表的な研究では、幅広い薬物濃度が採用されている。ピオグリタゾンの場合、80〜0.04mg/kg/dayの範囲であり、アロメトリックスケーリングの結果、390mg/day、つまり60kgのヒトで0.8mg/dayとなる。
比較のため、DM2治療のためのピオグリタゾンの推奨開始用量は15〜30mg/日である。このモードは約20mg/kg/day/マウスで、体重60kgのヒトでは約97mg/dayに相当する。rosiglitazoneの推奨開始用量は4mgであるが、生体内試験のAD関連試験に使用されるヒト等価用量は29~58mg/animal/dayである。
PD/PKタイプの実験は、これらの用量の選択を正当化するようなものは行われていないようで、オフターゲット効果の危険性に加えて、このような高用量の使用は、多くの(もしあれば)読み出される用量反応特性を十分に理解できていない点で問題があると言えるだろう。
例えば、ヒト(Knodtら 2019)およびラット(Crenshawら 2015)の両研究は、逆U字型fMRI BOLDピオグリタゾン用量反応曲線を明らかにした。
さらに、Seokらは、2mg/kg/日のピオグリタゾンが、9ヶ月齢のSAMP8マウスにおいて、モリス水迷路試験における学習および記憶の統計的に有意な改善と、IHC検出可能および可溶性海馬Aβ40沈着の減少に関連したが、5mg/kg/日ではこれらのパラメータすべてに対する効果が少なかった(Seokら 2019)ことを明らかにした。また、ラットのBOLD研究の結果は、低用量が高用量と同じかそれ以上に有効であることを示唆している(Crenshawら 2015)。
これらの結果は、ミトコンドリア生合成とAβクリアランスに関する細胞培養実験(例えば、Miglioら 2009;Moonら 2012)を彷彿とさせるものである。この用量反応パターンに関する合意された説明はなく、どの疾患表現型がこのようにPPARγアゴニストに反応するかを予測することもできない。したがって、ADの治療または発症を遅らせるためのPPARγ作動薬の評価は、測定されるパラメータに対する薬物濃度の影響を考慮する必要がある。
*
アルツハイマー病に関連するPD/PK試験が、前臨床モデルまたはヒトにおいて、脳内標的関与の測定を含めて行われていないことは、信頼できるヒト試験を計画することを困難にし、観察された反応に関する薬物の作用メカニズムについて検証可能な理論の開発を阻害する一因になっている。実際、表1にまとめたどの試験でも、これらの治療レジメンのもとで、薬物がネズミの脳に入り込んだことは確認されていない。
ロシグリタゾンは基本的にBBB伝染性がなく(GSK、未発表)、ピオグリタゾンはBBB伝染性が低い(Maeshiba et al.、1997)。しかし、0.04から0.32mg/kg/dのピオグリタゾンは、Wistar系若齢成体ラットのCA1領域と腹側視床の間の機能的結合性を増加させたが、最高用量で結合性が低下した(Crenshaw et al. 2015)。
これは、(Longhe, 2020)高濃度のピオグリタゾンは、少なくともいくつかの反応には不要であるか、(Patterson, 2018)いくつかのプロセスはBBBを伝染する少量に反応し、(質量作用によって)輸送障壁を乗り越えるために大量投与が必要であるか、(Zissimopoulos et al, 2015)いくつかのプロセスはBBB外の薬物の作用に反応していると思われる。
ある調査では、ロシグリタゾンは、BBB内皮細胞とアストロサイトの相互作用を調節することを介して、生体内試験で間接的にアストロサイトの挙動を調節すると結論付けた(Cowleyら 2012)。
我々の知る限りでは、これらの観察結果はフォローアップされていない。アストロサイトとミクログリアの間のクロストークの可能性を考えると(Clarkら 2021)、このような間接的な経路もPPARγアゴニストによるミクログリア機能の生体内試験制御に寄与しているのかもしれない。
In vivoの有効性及びMOA試験を設計し、ヒト臨床試験のガイダンスを提供するためには、脳及びBBB、並びに関連する末梢細胞における薬物物質及び標的関与の定量化を含む詳細なPD/PK試験の実施を通じて、用量及び治療時間を最適化する必要がある。
人間研究
観察型コホート研究
いくつかの縦断的観察コホート研究により、ピオグリタゾンが2型糖尿病における認知症のリスクを低減し、発症を遅らせることが示されている。これらの研究は、国民健康保険記録から抽出したデータを用い、指標日に2型糖尿病(DM2)と診断され、認知症(国際疾病分類第9版(ICD-9)または第10版(ICD-10)によりコードされる)のない被験者を対象に行われた。
指標日は、薬剤の初回処方日とした。ほとんどの場合、5年後または被験者が認知症と診断された時点で観察が終了した。ロシグリタゾンは中立的な効果を示した(Tseng, 2019)。これらの観察研究のメタアナリシスが発表されている(Yeら 2016;Zhouら 2020)。
*
Millerらは、退役軍人省(VA)の記録を用いて、糖尿病を患っているが2年前にADの診断記録がない米国退役軍人の解析を行った(Miller et al. 2011)。彼らの分析では、ロシグリタゾンまたはピオグリタゾンを処方された被験者が対象となった。研究対象者は、白人(79%)、男性(98%)の2型糖尿病患者が多く、薬剤開始時からICD-9コードを用いて行われた最初のAD診断まで追跡調査された。
この集団において、チアゾリジンジオン(TZD)のみの投与とインスリンのみの投与のハザード比(HR)は、0.81(95%CI, 0.73 – 0.89)であった。血糖コントロールを改善するためにインスリンとTZDの使用を併用した場合、インスリンに続いてTZDを使用した場合のHRは0.63(95%CI, 0.53 – 0.74)、TZDに続いてインスリンの場合は0.72(95% CI, 0.61. – 0.84)であった。
*
Henekaらは、ドイツの公的医療保険会社の記録を用いて、インスリンを投与しておらず、ピオグリタゾンを初めて使用した日である指標日の2年前から認知症でない被験者のみを考慮し、5年間被験者を追跡した(Henekaら、 2015b)。
彼らが追跡した集団は、60歳以上、試験開始時に認知症がない、ピオグリタゾンを服用していない糖尿病患者、ピオグリタゾンを服用中の糖尿病患者(服用期間別に分類)、および非糖尿病患者であった。ピオグリタゾンの長期使用者(ピオグリタゾンの処方を受けている期間>8四半期)は、非糖尿病患者と比べて認知症のリスクが低かった[相対リスク(RR)、0.53(95%CI、0.301〜0.936、P=0.029)]が、短期使用者(<8四半期)はRR ∼ 非糖尿病患者となり(RR, 1.16, P=0.317)。
ピオグリタゾンを処方されていない糖尿病患者の場合,相対リスクは1.23であった(P < 0.0001)。このデータセットでは、ロシグリタゾンとメトホルミンの使用はいずれもリスクを変化させなかった。インスリン使用のRRは1.608(95%CI、1.459 – 1.773、P < 0.001)であった。
*
Chouらは、台湾人を対象に、ピオグリタゾンがDM2患者の認知症リスクを低減することを確認した(Chou et al. 2017)。彼らは、台湾の国民健康保険研究データベース(NHI)の縦断的健康保険データベースサブセットから、「ピオグリタゾン使用経験者」対「使用経験なし」のデータを抽出した。コホートは、指標日に認知症がなく、5年間のフォローアップ期間を組み込んだ。
ピオグリタゾンコホートは、年齢、性別、指標日をピオグリタゾンコホートと一致させた比較コホートと比較して、脳卒中と高血圧の有病率が高かった。彼らは、ピオグリタゾンの1日の使用量を定量化するために、WHOが推奨する「定義された1日量」(DDD、30mg/日)を使用した。
全体として、認知症のリスクは、比較群に対してピオグリタゾン使用群で23%低かった(HR = 0.77(95%CI, 0.62 – 0.95, P = 0.015))。ピオグリタゾンの効果は時間および用量依存的であった。ハザード比は、高累積使用者群(>444定義日用量)で0.50(95%CI, 0.34 – 0.75, P = 0.001)、長期使用者群(>536日)で0.53(95%CI, 0.36 – 0.77, P < 0.001 )、高平均日用量使用者群(>平均日用量)では0.66(95% CI, 0.49 – 0.90, P = 0.009 )であった。
*
Tsengも台湾のNHIの被験者を追跡調査したが、解析は2年間の追跡期間に限定した(Tseng, 2018)。Chouらとは異なり、Tsengは、高血圧、脂質異常症、虚血性心疾患、末梢動脈疾患、パーキンソン病、スタチン使用、その他の血糖コントロール薬などの併存疾患についてピオグリタゾン-と比較-コホートをマッチングさせた。非対照コホートとマッチングコホートの解析では、ピオグリタゾンの使用は認知症リスクの有意な低下と関連していることが確認された。
メトホルミンもこの研究で保護作用を示したが、ピオグリタゾンの保護作用はメトホルミンとは無関係であった。ピオグリタゾンの効果は、メトホルミンを服用したことがない患者と20ヵ月以上ピオグリタゾンを服用した患者で最大であった。マッチドコホートでは、メトホルミンを使用したことがない患者において、ピオグリタゾンを使用したことがある場合とない場合のハザード比は次の通りであった:<11ヵ月。
11ヵ月未満:0.588(95%CI、0.272 – 1.273、P = 0.1778)、11 – 19.6ヵ月。0.265(95% CI, 0.102 – 0.688, P = 0.0064)であった。一方、全患者の場合、ピオグリタゾン使用歴のある患者とない患者のハザード比は0.716(95%CI、0.545 – 0.940, P = 0.163)、110ヵ月未満、11 – 19.6ヶ月、11~19.6ヶ月、19.6ヶ月以上の使用に対するハザード比は0.806(95%CI, 0.544 – 1.193, P = 0.2809), 0.654(95% CI, 0.430 – 0.994, P = 0.0467) 及び 0.694(95% CI, 0.469 – 1.026, P = 0.067)だった。
*
メトホルミンが保護的であるというTsengの知見は、メトホルミン使用者に利益がないことを観察したHenekaらと相反するものである。興味深いことに、Bohlkenらもドイツのコホートにおいてメトホルミン使用は認知症の発症を減少させないことを報告している(Bohlkenら 2018年)。
彼らの研究は、一般診療所(神経内科を含むすべての健康事例源とは異なる)を代表するGerman Disease Analyzerデータベース(IQVIA)に依拠し、指標日の少なくとも1年前から認知症でなかった疾患コホートを対象としている。
また、年齢、性別、併存疾患をマッチングさせた「ever」と「never」のコホートも用意された。グリタゾン(ピオグリタゾンまたはロシグリタゾン)服用者の認知症発症のオッズ比は0.80(95%CI, 0.68 – 0.95, P = 0.011)、メトフォルミンのオッズ比は0.96(95%CI, 0.88 – 1.04, P = 0.153)であった。
ドイツと台湾のメトホルミンの結果の不一致は、認知症のサブタイプの違い(Neffら 2021)、それぞれのデータベースに表れた民族グループの他の遺伝的背景の違い、それぞれの研究における「経験あり」「経験なし」グループの併存疾患、糖尿病の重症度、またはデータ収集と分析における方法論の違いを反映していると考えられる。
*
インスリン、グリコシダーゼ阻害剤、メトホルミン、スルホニル尿素、DPP-4阻害剤など他の血糖降下剤についても多数の観察研究が行われ、その結果が2つのメタアナリシスで検討・まとめられている(Yeら 2016;Zhouら 2020)。一般に、DPP-4阻害剤が最も認知症リスクが低く、次いでメトホルミン、ピオグリタゾンとロシグリタゾンをひとまとめにしたチアゾリジン系薬剤が多かった(Zhou et al.、2020)。
また、別の研究では、チアゾリジン系やメトホルミンなどの血糖降下剤はADの発症を遅らせないことが示されている(Imfeldら 2012)。ロシグリタゾンの効果は中立であるため(Tseng, 2019)、ピオグリタゾンと一緒にすると、これらのメタアナリシスで交絡する結果が生じる。
DPP-4阻害剤は、ミクログリア機能を調節するインクレチンホルモン(GIP、GLP-1)の分解を阻害する(Spielmanら 2017年)。インクレチンはPPARγの発現を誘発するため(Svegliati-Baroniら 2011;Onumaら 2014)、これらのデータで明らかなDPP-4効果に対するPPARγおよびインクレチンの寄与を分離することは不可能である。
*
全体として、縦断的コホート研究は、成人発症糖尿病患者の集団において、ピオグリタゾンの使用が認知症リスクの低下と関連し、その効果は時間および用量依存的であることを実証している。これらの結果は、DM2症例を用いて実施されたパイロット臨床研究(羽生ら 2009、2010;佐藤ら 2011)と一致している。
これらの縦断的コホート研究の長所は、年齢、性別、合併症をマッチさせ、現実的な追跡期間(2-5)を持ったプラセボ対照臨床試験に近いものであることである。しかし、これらの研究はすべて認知症の定義にICD診断コードを用いており、認知症の発症と診断の間にしばしばタイムラグがあった。
Henekaらは、診断に多層的なアプローチを採用することによって、この欠点を克服しようとした。さらに、これらの研究はいずれもAPOEやその他の遺伝的危険因子、生活習慣の要因、糖尿病の重症度やインスリン抵抗性の程度、血糖コントロールの程度を説明するものではなかった。
これらの観察研究やDM2を用いた小規模な臨床試験(10.2節)の困難を克服するために、アルツハイマー病の発症リスクが高い成人発症糖尿病患者を対象とした大規模な盲検プラセボ対照臨床試験が必要であると思われる。
臨床研究
表2は、チアゾリジンジオン系PPARγアゴニストをアルツハイマー病の治療薬として評価したデータを報告した12の臨床試験の要約である。
ここでは、重要なポイントを強調した研究に焦点を当てる。脳内エネルギーの測定値を読み出した薬力学的研究2件、Aβペプチド濃度に対する薬効を測定した小規模の研究2件、インスリン低下に対する薬効を相関させた2件、DM2既往のあるボランティアにおけるピオグリタゾンの有効性を評価した小規模の研究3件を含む。
このグループによる同様の結果を報告した追加研究は表には含まなかったが、以下で議論することにする。DM2患者を対象とした試験を除き、すべての試験は、糖尿病の既往のある人、またはグルコースコントロールのための薬剤を服用している人を除外している。
2つの試験は、登録時に認知症でない被験者を対象とした予防試験であり、前臨床試験の重要な教訓の1つである、AD関連の病態が発現する前に治療を開始すると学習・記憶が維持されるという結果を反映していた(Badhwarら 2013年)。
残りは、当初、軽度から中等度のアルツハイマー病または軽度認知機能障害を有する被験者を対象とした治療研究であった。 を除き、and, すべての試験は、正常認知からMCIまたは軽度ADへの転換率に対して、パワー不足または実施期間が短すぎた可能性がある。
表 2 アルツハイマー型認知症に対するPPARγ作動薬の臨床試験結果の概要
| 研究内容 | 治療法 | 研究デザイン | 人口 | 結果 |
| A.ロシグリタゾン | ||||
| A.1.フェーズ2介入 | ||||
| ロシグリタゾン投与中の初期アルツハイマー病および健常軽度認知障害患者における認知機能の維持(Watson et al.) | Rosiglitazone 4 mg/日、vs プラセボ。 | 早期ADおよびAmnestic MCIを対象とした24週間のプラセボ対照二重盲検並行群間比較試験。アウトカム評価項目:認知機能、血漿インスリン、血漿Aβ。 | プラセボ、N=10、Rosiglitazone、N=20。平均年齢73歳,女性70%,白人100%。ベースラインのインスリンは8.1μU/mL、MMSEは平均23。グルコースコントロールのための薬物を服用している被験者は除外された。 | ロシグリタゾンは、4ヶ月と6ヶ月の遅延記憶、6ヶ月の選択的注意、血漿Aβ42、Aβ40、Aβ42/Aβ40比の安定と統計的に関連していた。 |
| 遺伝学的に定義された軽度から中等度のアルツハイマー病患者集団におけるrosiglitazoneの有効性(Risner et al.) | Rosiglitazone 2, 4, 8 mg/日 vs プラセボ。 | 軽度から中等度のアルツハイマー病患者を対象とした24週間のプラセボ対照二重盲検並行群間パイロット試験(第2相試験)。アウトカムメジャーADAS-Cog、CIBIC+。 | 平均N=128、平均年齢70.7歳、60%女性、100%白人。APOEε4でバランス。ベースラインのインスリンは14.2μU/mL。ベースラインのMMSE平均値は21.3。1型糖尿病または2型糖尿病の既往のある者、空腹時血糖値7mM以上またはHbA1c8.5%以上の者は除外した。 | 全体:アウトカム指標に対するロシグリタゾンの統計学的有意な効果は認められなかった。APOEε4非キャリアでは、ロシグリタゾン8mg投与は、ADAS-Cogの改善と統計学的に関連した。 |
| {軽度から中等度のアルツハイマー病の被験者における認知および脳内グルコース利用に対するロジグリタゾンの効果(Tzimopoulouら 2010年)。 | Rosiglitazone 4 mg/日を1ヶ月間投与し、残りの期間は8 mg/日に増量。 | 52 週間の並行群間二重盲検法、第 2 相試験。アウトカムメジャー12ヶ月間の脳糖代謝率変化、脳容積、ADAS-Cog、CIBIC+の変化。 | 平均N=31人、平均年齢71.25歳、女性46.2%、白人94.9%が研究を完了した。APOEε4でバランスをとった。1型糖尿病または2型糖尿病の既往のある者、またはグルコースコントロールのための薬物*を服用している者は除外した。 | 総グルコース代謝量、局所グルコース代謝量、脳容積に持続的な効果は認められず、ADAS-Cog、CIBIC+にも影響はない。 |
| A.2.フェーズ2 – 糖尿病 | ||||
| 2型糖尿病およびMCIを有する高齢者におけるロシグリタゾンと認知機能の安定性(Abbatecola et al, 2010) | ロシグリタゾン4mg/日、対メトホルミン500mg/日、対ロシグリタゾン+メトホルミン、対食事療法。 | 2型糖尿病を合併した軽度~中等度アルツハイマー病患者を対象とした36週間の前向き無作為化非対照試験。アウトカム評価項目:神経心理学的テストスコアおよび代謝制御パラメータ(FIRI、FPG、HbA1C)の変化。 | 平均N=32.2,平均年齢76歳,女性45%;FPG平均ベースライン,8.44mmol/L;FIRI,148pmol/L;HbA1Cベースライン平均,7.5%;MMSEベースライン,24;TMT-Aベースライン平均,67.6;TMT-Bベースライン,161.1;DIFFBAベースライン平均,101.2;RAVLTベースライン平均,24.5 | メトホルミンとロシグリタゾンの併用により、すべての神経心理学的検査が安定した。メトホルミンはMMSE、TMT-A、TMT-Bを安定化させ、食事療法はMMSE、TMT-Aを安定化させた。線形固定効果モデルでは、FIRI x 時間とメトホルミン/ロシグリタゾン併用RAVLTに相関がみられた。 |
| A.3.フェーズ3介入 | ||||
| {ロシグリタゾン(徐放錠)の単剤療法として軽度から中等度のアルツハイマー病を対象に実施(Gold et al., 2010) | Rosiglitazone 2 mg または 8 mg extended release, daily vs プラセボ(REFLECT-1)。 | 軽度から中等度のアルツハイマー病患者を対象に、APOEε4の有無で層別化した24週間の二重盲検、二重ダミー、無作為化、並行群間第3相試験。アウトカムメジャーADAS-Cog、CIBIC+。 | 平均N=159、平均年齢72.3歳、女性37%、白人72%、APOEε4でバランス。ADAS-Cog平均ベースライン、19.1。1型糖尿病または2型糖尿病の既往のある者、またはグルコースコントロールのための薬物*を服用している者は除外した。 | 全例でロシグリタゾンの投与量に統計学的または臨床的に有意な効果は認められず、ロシグリタゾンによる治療とAPOE遺伝子型との間に有意な相互作用の証拠もない。 |
| ロシグリタゾン(徐放錠)の軽度から中等度のアルツハイマー病の被験者に対する補助療法(Harrington et al.,2011) | Rosiglitazone 2または8 mg徐放、donepezilとの併用(REFLECT-2)、または任意のAChEIとの併用(REFLECT-3)。 | 軽度から中等度のアルツハイマー病患者を対象とし、APOEε4の有無で層別化した48週間の二重盲検無作為化プラセボ対照並行群間比較試験。アウトカムメジャーADAS-Cog、CDR-SB。 | (REFLECT-2):平均N=464,平均年齢74.1歳,女性60%,白人90.67%,APOEε4でバランス。
ADAS-Cogの平均ベースラインは25.3、MMSEの平均ベースラインは19.46。(REFLECT-3):平均N=476.3,平均73.9歳,女性55.6%,白人91.76%,APOEε4でバランス。 ADAS-Cogの平均ベースラインは24.1、MMSEの平均ベースラインは19.7。1型糖尿病の既往のある被験者、または2型糖尿病でグルコースコントロールのための薬物*を服用している被験者は除外された。 |
REFLECT-2 または REFLECT-3 では、Rosiglitazone の投与量と APOE 遺伝子型との間に有意な相互作用は認められなかった。 |
| B.ピオグリタゾン | ||||
| B.1.フェーズ1 – 投与量測定 | ||||
| 認知的に健康な高齢被験者におけるピオグリタゾンの連日投与の脳血行動態への影響を評価する試験(Knodt et al., 2019) | ピオグリタゾン、0.6mg、2.1mg、3.9mg、6.0mg/日、 vs プラセボ | 2 週間、複数回投与、単盲検、無作為化、並行デザイン、プラセボ対照、第 1 相用量設定試験。アウトカム評価:エピソード記憶に関連する海馬の活動を、血中酸素濃度依存性(BOLD)機能的磁気共鳴画像法(FMS)で測定。 | 平均N=11、平均年齢=66.08歳、71%F、85.45%White。CERAD-WLMの平均ベースラインは8.02、TMT-Bの平均ベースラインは97.58。血糖値をコントロールする薬を服用している糖尿病患者*、またはHbA1Cが6%を超える者は除外した。 | ピオグリタゾン0.6mg/日投与は、ベースライン時と比較して、7日目および14日目に新規顔名ペアのエンコーディング時の右海馬の活性化を統計的に関連付けた。2.1,3.9,6.0 mg/dayでは、統計的に有意な改善はみられなかった。 |
| B.2.パイロット版 – 糖尿病 | ||||
| アルツハイマー病におけるペルオキシソーム増殖因子受容体γアゴニストpioglitazone治療後の認知機能改善における腫瘍壊死因子αの役割(Hanyu et al.) | ピオグリタゾン、15mg/日 vs. 無し。 | 2型糖尿病を合併した軽度から中等度のアルツハイマー病患者を対象とした24週間の前向き無作為化非対照試験。アウトカムメジャーADAS-JCog、MMSE、TNF-α、IL-6、C反応性蛋白。 | 両群ともN=17、平均年齢78.7歳、女性50%、白人100%、APOEε4とドネペジル使用で均衡、MMSE平均ベースライン21.85、ADAS-JCog平均ベースライン15.65、TNF-α平均ベースライン1.38 pg/mL、IL-6平均ベースライン2.62 pg/mL、C反応蛋白ベースライン 0.08 mg/dL。 | ピオグリタゾンは、ADAS-JCogおよびTNF-αの改善と統計的に関連しており、ADAS-JCogの変化はTNF-αの変化と相関していた。 |
| PPARγ作動薬pioglitazoneの軽度アルツハイマー病に対する有効性(佐藤ら 2011) | ピオグリタゾン、1日15mgまたは30mg vs. 無し。 | 2型糖尿病を合併した軽度から中等度のアルツハイマー病患者を対象とした24週間の前向き無作為化非対照試験。アウトカムメジャーADAS-JCog、MMSE、WMS-R、rCBF、血漿Aβ40およびAβ42、HOMA-R、HbA1c、FIRI。 | 両群ともN=21、平均年齢77.5歳、女性52%、白人100%、APOEε4でバランス、他の血糖降下剤、donepezilでバランス。 | ピオグリタゾンは、MMSE、ADAS-JCog、WMS-Rの改善、頭頂葉の血流改善、代謝性因子の改善と統計的に関連していた。血漿Aβ40/Aβ42比はピオグリタゾン群では変化せず、対照群では増加した。ADAS-JCogは対照群で有意に悪化した。 |
| B.3.フェーズ2介入 | ||||
| {Pioglitazone in Alzheimer’s disease safety trial(Geldmacher et al., 2011):アルツハイマー病におけるPioglitazoneの臨床試験。 | ピオグリタゾン、1日15mg、毎週45mgまで漸増 vs プラセボ。 | 軽度から中等度の確率のアルツハイマー病を対象とした72週間の二重盲検無作為化プラセボ対照群間比較試験 アウトカム尺度(3ヶ月間隔で収集)。
(Longhe, 2020)。 ADAS-Cog、CDR-SBを含む認知機能の測定。(Patterson, 2018)。効果量計算のための推定値。 |
平均N=14.5,平均年齢70.95歳,女性62%,MMSE平均ベースライン21,ADAS-Cog平均ベースライン21,CDR-SB平均ベースライン5.8。 | ピオグリタゾンは、統計学的に認知機能の改善と関連しなかった。ADAS-Cogの1ヶ月あたりの調整平均値は、ピオグリタゾン群で低かったが、統計学的な有意差はなかった。
α=0.05、検出力=0.80の場合、ADAS-Cog(-0.746)およびCDR-SB(-0.354)に対するピオグリタゾン効果の推定回帰係数が有意であるためにはそれぞれ340名(pio170名、プラセボ170名)および155名(pio78名、プラセボ77名)の被験者サイズが必要であったと思われる。 |
| MCIおよびメタボリックシンドロームの高齢者に対するPoem(Pioglitazoneおよび運動効果)(Hildrethら 2015年)。 | 試験概要】 Pioglitazone(15mg/日、1ヵ月後に45mg/日に漸増)、プラセボ、または45~75分の運動トレーニングを3回/週、現状維持の運動と比較した。運動負荷は最大心拍数50~60%で開始し、試験期間中に最大心拍数80~85%に増加させた。 | MCIおよび中心性肥満の座りがちな成人を対象とした24週間の二重盲検無作為プラセボ対照パイロット試験。アウトカムメジャー認知機能、インスリン抵抗性、体組成、代謝および炎症マーカーに関するベースラインからの変化。 | 平均N=22、平均年齢65.6歳、女性51.8%、白人87.8%、APOEε4でバランス、平均コンプライアンス、ピオグリタゾン76%、プラセボ89%、グルコース平均ベースライン、101mg/dL、インスリン平均ベースライン、16.1μU/mL。
1μU/mL、CRPベースライン、3mg/L、IL-6ベースライン、1.7pg/mL、TNF-αベースライン、1.56pg/mL、MMSEベースライン、28.6、ADAS-Cogベースライン、6.5pg/mL。 |
ピオグリタゾンは認知機能の改善と統計学的な関連は認められなかった;視覚再生テストの成績;ピオグリタゾン群対プラセボ群でスコアが悪化した;ADAS-Cogは運動により改善した(-1.3 EX vs -0.3 CON;P = 0.05)。グルコース排出速度と認知能力の間に統計学的に有意な相関は認められなかった。 |
| B.4.第3段階 予防 | ||||
| ADによるMCI発症リスク予後のバイオマーカー・アルゴリズムの適格性と認知正常者におけるADによるMCI発症遅延に対するピオグリタゾンの安全性と有効性を同時に試験(Alexander et al,2019;Burns et al., 2019)注:本研究を記述した完全な出版物は、今回の論文投稿時に審査中であった。 | ピオグリタゾン、0.8 mg エクステンド・リリース・デイリー、 vs プラセボ。 | AD予備軍である認知機能正常成人(APOE、TOMM40遺伝子型、年齢)を対象としたイベントドリブン(予想5)、二重盲検、無作為、並行群プラセボ対照の第3相予防試験。アウトカムメジャー年齢や遺伝的リスク因子によりリスクが高まっている健常者において、MCI発症を遅らせる。 | N = 433低リスクプラセボ,1516高リスクプラセボ,1545高リスクpioglitazone。平均年齢73.1歳、女性56.16%、白人96.6%、高リスク群におけるAPOEε4キャリッジ平均92.45%、MMSE平均ベースライン28.56。アウトカム評価:高リスク層におけるピオグリタゾン投与群とプラセボ投与群のADによるMCI診断までの時間。事前に設定した無益性の閾値、40%の治療差が検出される条件付き確率は30%。 | 無益性解析のため試験終了。1278日後、プラセボの総イベント数は46、ピオグリタゾンの総イベント数は39。ピオグリタゾンとプラセボのリスク比は0.8(95%CI、0.45 – 1.4)、P = 0.307。ポストホックのサブグループ解析では男性にピオグリタゾンが有効である可能性が示唆された。 |
ADAS-Cog, Alzheimer’s Disease Assessment Scale-Cognitive Subscale; ADAS-JCog, ADAS-Cog 日本語版; ATP III, Adult Treatment Panel III criteria for central obesity; rCBF, regional cerebral blood flow; CDR-SB, Clinical Dementia Rating scale-Sum of Boxes; CIBIC+, Clinician’s interview-based impression of change with caregiver input; DIFFBA, TMT-A minus TMT-B – a measure of cognitive efficiency.I ICRT, TMT-2;FIRI:空腹時免疫反応性インスリン、FPG:空腹時血糖値、HOMA-R:インスリン抵抗性の恒常性モデル評価、MCI:軽度認知障害、MMSE:ミニメンタルステート検査、RAVLT:レイ聴覚言語学習テスト、1型糖尿病:1型糖尿病、2型糖尿病:2型糖尿病、TNF-α:腫瘍壊死因子α、TMT-A、TMT-B:トレイルマークテストA、トレイルマークテストB、それぞれを指す。*インスリン、スルホニル尿素、PPARγアゴニスト、またはグリチニド。
いくつかの小規模なパイロット試験では、代謝危険因子であるインスリン抵抗性および2型糖尿病との関連において、ロシグリタゾンおよびピオグリタゾンの有効性を評価した(Luchsingerら 2004;Biesselsら 2006;Mullerら 2007;Xuら 2009;Willetteら 2015a、b;Wuら 2016)。
Watsonらは、ロシグリタゾンを用いた24週間の試験に、早期ADおよび記憶障害性MCIのボランティアを登録した(Watsonら 2005年)。これらのボランティアは、報告されたベースラインのインスリンおよびグルコース平均値から計算されたHOMA-IRスコア(計算値2.0;スコア>1.9は軽度インスリン抵抗性)に基づく軽度インスリン抵抗性でもあった(Matthewsら、1985年)。
ベースラインの認知スコアからの変化に加えて、これらの著者らは、血漿インスリンと、我々が見つけた他のほとんどの研究とは異なり、血漿バイオマーカーAβ42とAβ40を定量化した。6ヵ月の試験期間中、ロシグリタゾンは遅延記憶のスコアと選択的注意力を維持したが、対照群では記憶力が悪化した(Watson et al.)Aβ42/Aβ40比はプラセボ群では低下したが、ロシグリタゾンでは安定した。
ロシグリタゾンはまた、末梢インスリンのわずかだが統計的に有意な低下を惹起し、記憶の保存の程度と干渉テストの誤り率は、血漿インスリンレベルの変化と逆相関していた。Risnerらは、インスリンレベルの変化と認知の間の相互作用の示唆的な証拠も見出した(Risner et al. 2006)。
この効果は、末梢性高インスリン血症が引き起こす中枢性低インスリン血症の是正と関連している可能性がある(Bauraら、1996)。グルコースレベルは上昇したが、その変化は統計的な有意差には至らなかった。
APOEε4は、晩発性アルツハイマー病の最も重要な遺伝的危険因子であり、いくつかの研究では、APOEε4保有とロシグリタゾンまたはピオグリタゾンの認知機能に対する効果の相互作用があるかどうかが検討された。
Risnerらは、24週間の小規模な用量反応試験を行った(平均N=128)(Risnerら 2006年)。全体として、ロシグリタゾンは認知機能に有意な影響を与えなかったが、ADAS-CogスコアとAPOEステータスの相互作用の検定は有意であった。APOEε4陰性者では、ロシグリタゾンの最高用量(8mg)において認知機能が改善した。
注目すべきは、APOEε4陰性者は、少なくとも1つのAPOE ε4対立遺伝子を有する被験者よりも、ロシグリタゾン8 mg投与によって誘発される血漿インスリンの低下も大きかったことである。インスリン低下と認知機能改善との関係は、Watsonら(2005)を想起させるが、後者の研究とは異なり、ここではその相互作用は正式には解析されていない。
*
グラクソ・スミスクライン社は、APOEステータスおよびロシグリタゾンの有効性の相互作用をさらに大規模な2つの用量反応試験で検討し、AChEIの補助療法として、APOEステータスで層別化した試験集団における低用量(2 mg)および高用量(8 mg)のロシグリタゾンの有効性を比較した(Goldら 2010年;Harringtonら 2011年)。{は24週間実施し、平均N = 124人の被験者(Gold et al,2010)、及びand「は、臨床試験である。
{NCT00348140試験は、48週間実施され、平均N=464人の被験者(Harrington et al.,2011).ロシグリタゾンは、いずれの試験においても、APOEε4+またはAPOEε4-の被験者において、いずれの用量においても、認知機能に統計的に有意な影響を与えなかった。
HbA1c値は、Goldら(2010年)では2つのロシグリタゾン濃度間で基本的に差がなく、Harringtonら(2011)ではロシグリタゾンの用量増加とともに増加した。なお、これらの変化と認知に関するスコアとの統計的な相互作用は報告されていない。なお、空腹時血糖値および空腹時インスリン値は、いずれの試験においても測定されていない。
*
DM2のボランティア(Sato et al., 2011)やADのマウスモデル(Nicolakakis et al., 2008;Papadopoulos et al., 2013)で実証されたように、PPARγアゴニストは中枢の糖代謝を促進する。
Tzimopoulouらは、rosiglitazoneの中枢作用の薬力学的マーカーとして、脳糖代謝率(CMRglu)および脳萎縮を測定した。この研究のボランティアは、軽度から中等度のアルツハイマー病であり、52週間の試験で認知的に正常な対照者と年齢および性別をマッチさせた(Tzimopoulou et al. 2010)。
ロシグリタゾン(8mg徐放製剤/日)は、投与開始後1カ月間、プラセボの4.7%減少に対し、統計的に緩やかな(1.5%)グルコース利用率の増加と関連していた。しかし、この直後の増加は強固なものではなく、試験の残りの11ヶ月間、CMRglu率の平均値は両群とも減少した。
CMRgluの減少率はプラセボ群よりロシグリタゾン群で低かったが、その傾向は示唆的であり、ロシグリタゾンが脳容積や認知力の変化に影響を与えるという証拠はなかった。APOEε4保有はいかなる結果にも影響を与えなかった。REFLECT試験と同様に、空腹時インスリン値やグルコース値が記録されていないため、インスリン値やインスリン抵抗性の変化と認知やこれらの薬力学的マーカーとの相互作用を評価することはできない。
*
羽生らのグループは、DM2症例を対象とした一連の小規模パイロット試験において、ピオグリタゾン(15〜30mg/日)が6ヵ月後に2型糖尿病患者の認知機能を改善したと報告した。同剤を服用したDM2症例ではADAS-JCogスコアが改善し、服用しなかった対照糖尿病患者では悪化した(羽生ら 2009、2010、佐藤ら 2011年)。局所脳血流もピオグリタゾンで改善し(佐藤ら 2011)、末梢のTNF-α値も改善した(羽生ら 2010)。ピオグリタゾンは、対照群で低下していたAb42/Ab40比を安定化させた(Sato et al.)
羽生氏のグループは、ピオグリタゾンの認知機能低下遅延効果を評価するために、既存のDM2症例をモニターしたのに対し、Abbatecolaらは、PPARγ作動薬が既存のMCIを改善できるかどうかを調べるという、異なるアプローチをとった。
もう一つの違いは、メトホルミンにロシグリタゾンを加えた併用療法と、メトホルミン単剤または食事療法による血糖コントロールとの比較検討である。ロシグリタゾンとメトホルミンの併用は、メトホルミン単独や食事療法単独よりも認知機能の低下を遅らせることに優れていた(Abbatecola et al. 2010)。これらのデータは、メトホルミン使用者において、ロシグリタゾン追加後の最初の12カ月で保護作用が高まる傾向を示した縦断コホート研究と一致する(Tseng, 2019)。
*
残りの3つの研究のうち、1つは薬力学的研究であり、2つは予防研究であった。Hildrethらは、遺伝的リスクの低い集団において、代謝的危険因子であるインスリン抵抗性が認知障害に及ぼす役割を、ピオグリタゾンの高薬理学的用量を用いて直接的に検討したものであった。
TOMMORROW試験では、測定「NCT01931566″}}NCT01931566)は、代謝的に強固であるが遺伝的に晩発性アルツハイマー病の発症リスクがある集団において、認知障害の発症を遅らせるためのピオグリタゾンの有効性を測定したもので、PD試験の助けにより確立された非常に低いピオグリタゾン投与量が用いられている。
*
Knodtらは、BOLD fMRIを薬力学ツールとして、海馬機能に対するピオグリタゾンの効果を決定し、TOMMORROW試験 )で用量選択を知らせるツールとして用いた(Knot et al. 2019)。健康で認知的に正常なボランティアは、14日間毎日、車両(プラセボ)、または0.6 mg、2.1 mg、3.9 mg、または6.0 mgのピオグリタゾンを投与された。DM2の治療開始用量は15mgで、その0.4〜40%の範囲で投与された。
全体として、正しく想起された顔名対の反応時間は、符号化中の右および左の海馬の活動と負の相関があった。0.6mgのピオグリタゾンは、ベースラインから7日目まで、およびベースラインから14日目までの右海馬の活性化に関連していた。
プラセボ群では、7日目から14日目にかけて右海馬の活性化が減少した。これらのデータは、ピオグリタゾンが意識下のヒトにおいて脳機能に影響を及ぼすことを支持し、さらに、CA1領域と視床下部および腹側視床との相互作用に関するラットBOLD試験で観察されたものと同様の、海馬機能に対するホルモン的用量反応効果を示唆している(Crenshawら 2015年)。
上記の羽生グループの知見や、臨床レベルのピオグリタゾンを用いた観察コホート研究(Henekaら 2015b;Chouら 2017;Tseng 2018)とともに、これらの結果は、認知症のリスクに対するピオグリタゾンの全体的な有用な効果は、複数のターゲットを介して、それぞれが固有のピオグリタゾン濃度範囲に応答して仲介される可能性を示唆している。
*
POEM研究では、認知スコアの変化と代謝パラメータに関する運動とピオグリタゾンの効能を評価・比較した(Hildrethら 2015年)。さらに、炎症の循環マーカー(CRP、IL-6、TNF-α)を測定した。Watsonら(2005)と同様に、本研究の参加者はベースラインでインスリン抵抗性であったが(HOMA-IRスコア、4.0)、Watsonらと異なり、認知的には正常であった(ベースラインの平均MMSE、28.6)。
ピオグリタゾンと運動のいずれも循環炎症マーカーに影響を与えなかった。ピオグリタゾン群では、空腹時インスリンと高血糖クランプによるインスリン抵抗性が改善したが、運動とピオグリタゾンは認知能力に影響を与えず、糖代謝率の改善と認知能力のどの領域にも相互作用はみられなかった。
APOEε4陰性者では認知能力が改善されたが、その変化は統計学的に有意ではなかった。Hildrethらが指摘したように、ベースライン時の参加者に認知機能障害があったとしても、それは非常に軽度であった。平均22名の被験者をわずか6ヵ月間追跡調査しただけで、プラセボ群では正常な認知機能からMCIへの転換が少なすぎて、Pioglitazoneの効果の可能性を検出することはできなかったと思われる。
*
TOMMOROW試験) は、遺伝的な危険因子により認知機能および代謝機能が正常な被験者において、低用量(0 … 続きを読む8mg/日、徐放性)により、アルツハイマー病による軽度臨床障害の発症を遅らせることができるかどうかを検討するために、遺伝的リスク因子により5年以内にアルツハイマー病によるMDIを発症するリスクの高い、認知・代謝的に正常な被験者を対象に実施された。
対象は65歳〜83歳の3500人。対象者は、入所時の年齢とAPOEおよびTOMM40’523遺伝子座の遺伝子型によってリスクを層別化し、その後5年以内にADによるMCIを発症するリスクが低いか高いかのいずれかに割り付けられた。
高リスクの被験者は、そのほとんどが少なくとも1つのAPOEε4対立遺伝子を持っており、プラセボ群または治療群のいずれかに割り付けられた(平均N = 1530)。ベースラインの平均MMSEスコアは28.56であり、糖尿病の既往がある、あるいは血糖値に影響を与える薬剤を服用しているボランティアは除外された。
研究成果はMCI発症の遅延であり、仮にMCIが発症していたとしても、予想される5年間におけるベースラインからの変化量の30%の差を検出するのに十分な検出力があった。しかし、試験開始後、無益性の基準が40%の差に変更され、無益性解析により、被験者の大半が3年未満の薬剤曝露期間であったため、試験は早期に終了した。
高リスクのNon-Hispanic/Latino Caucasian被験者に対するPioglitazoneの効果は、プラセボと比較して統計的に有意ではなかった(39/1430 [2⋅7%]vs46/1406 [3⋅3%]; HR 0⋅80; 99% CI, 0⋅45 – 1⋅40; p = 0⋅307).統計的に有意ではなかったが、事前に特定した性別サブグループ解析により、男性被験者における潜在的な差が明らかになった(ピオグリタゾン HR 0⋅56; 95% CI, 0⋅30-1⋅06; p =0⋅074)(Alexander et al. 2019)。
血漿Aβペプチドレベルを含む薬力学的測定は収集されなかった。HbA1c以外の代謝パラメータは収集されておらず、認知バッテリーのパフォーマンスの変化と空腹時インスリンまたはインスリン抵抗性の変化との間に考えられる相互作用の解析は不可能である。
表2にまとめたPPARγ作動薬の臨床試験に加えて、現在、PPARδ/PPARγのデュアルアゴニストが、軽度から中等度のアルツハイマー病患者における認知症発症リスクへの影響を評価中である。
T3D-959は、PPARδに対してPPARγよりも15倍強い活性を有している。PPARδアゴニストは、グルコースおよび脂肪酸の利用を調節し、抗酸化および抗炎症シグナル伝達を強化するため、ADのリスクを低減すると仮定されている(Liu Y. et al. 2018)。
探索的な第IIb相試験において、T3D-959は脳内グルコース利用を増加させ、認知評価において示唆的な改善をもたらした(Chamberlainら 2020)。
現在、二重盲検プラセボ対照の第2相用量設定試験が進行中である。その主要アウトカムは、認知およびグローバル機能に対する効果であり、探索的な測定には、血漿Aβ42/40比、Nflおよびタウ、および脳グルコース利用が含まれる(Clinicaltrials.gov )(Didsbury et al., 2020).
*
全体として、ピオグリタゾンとロシグリタゾンは、DM2でないボランティアにおいて、認知機能の回復やアルツハイマー病によるMCIの発症を遅らせることには効果がないことが示された。しかし、これを決着した問題と考える前に、いくつかの注意点を考慮する必要がある。
まず、これらの研究のほとんどは、検出力が不十分であるか、統計的信頼性をもって変化を検出するのに十分な期間実施されていない(Watson et al., 2005;Hanyu et al., 2009,2010;Abbatecola et al., 2010;Tzimopoulou et al., 2010;Geldmacher et al., 2011;Sato et al., 2011;Hildreth et al., 2015;Knodt et al., 2019)。
例えば、Geldmacherらは、アルツハイマー病のボランティアを対象に、18ヶ月間の第2相ピオグリタゾン(当初15mg/日、1ヶ月後に45mg/日に漸増)の安全性試験を実施し、効果量の算出も可能とした(Geldmacherら 2011年)。
各コホートには平均14.5人のボランティアが登録され、MMSEのベースライン・スコアは平均21点であった。彼らは、6ヵ月ごとに5つの認知テストを実施し、各テストについてマルチレベルモデルの回帰係数を算出した。ADAS-Cogのパラメータ(-0.746)は有意ではなく、また、他のテストでも有意ではなかった。
Geldmacherらは、彼らのデータから、観察されたADAS-Cogパラメータが有意であるためには、平均コホートサイズが約170でなければならないと推定し、α=0.05、検出力=0.8としている。この研究を他の研究と直接比較することはできないが、同様の計算を他の研究に適用した場合、どのような結果になるのか疑問が残る。
*
第二に、WatsonらとRisnerらは、空腹時インスリンの変化を考慮した上で、ロシグリタゾンが認知機能低下に正の効果を示すことを示し、Risnerらは、空腹時インスリンの変化がAPOEε4対立遺伝子を持っていない人により広範囲であることも明らかにした。
末梢性の高インスリン血症は中枢性の低インスリン血症を引き起こし(Bauraら、1996)、この効果の根底にあるのは、末梢性の高インスリン血症である。いくつかのグループ(Watson et al., 2005;Gold et al., 2010;Tzimopoulou et al., 2010;Harrington et al., 2011)が空腹時血糖値および/またはHbA1cを測定したが、これらの測定値は有用ではなかった。
インスリン自体は、PPARγ作用の臨床試験で使用するには、グルコースやその代替物よりも良い共変量である。しかし、Risnerら(2006)以降の研究のほとんどは、空腹時インスリンに対する薬効を考慮に入れていない。
*
第三に、Watsonら(2005)およびSatoら(2011)における認知に対するロシグリタゾンの効果およびAβ42/Aβ40比の一致は、Aβペプチドが、この種の臨床試験においてPPARγ-介在効果をモニターするための有用なバイオマーカーであり、特にAPP処理およびAβ放出に対するPPARγ効果の基礎をなす機構が十分に理解されており、標準的血液試験が臨床使用において可能であるので、それを示唆するものであった。
PPARγアゴニストはタウのリン酸化も抑制し、血漿p-tau181はADに伴う代謝障害や認知障害と関連しているため(Lussierら 2021)、p-tau181の標準化された高感度検査(Karikariら 2020)を組み込むことも有用であると思われる。
ADにおけるPPARγアゴニスト効果の薬力学的マーカーとして脳内糖代謝を用いる試み(Tzimopoulouら 2010)は、研究の限界により不成功に終わった。Tzimopoulouらは、グルコース代謝に対するロシグリタゾンの持続的な保護効果を明らかにしたが(Tzimopoulouら 2010)、この研究は、統計的または臨床的に意味のある結論を導き出すのに十分な期間行われなかった。
PDマーカーとしてのBOLD fMRIも、Knodtら(2019)に要約された理論的正当性があるが、BOLDシグナルに対するピオグリタゾンの効果に関連する前臨床研究は1件しか発表されておらず(Crenshawら 2015)、より確立したバイオマーカーに対するその適合性は確立されていない。
これらのPDバイオマーカーの難しさ、およびAβレベルの変化の情報性を考慮すると、血漿Aβ42およびAβ40測定が、ADによる認知症の発症を遅らせるPPARγアゴニストの有効性を試験する臨床試験においてより広く取り入れられなかったことは残念なことであった。
*
パイロット試験および第2相試験で記録された認知スコアの変化と空腹時血漿インスリンの変化、Aβ42およびAβ40ペプチド濃度の変化との関係が、単なるタイプIエラーであったかどうかを知ることは、アルツハイマー病の基礎的な病態生理の理解および薬剤開発目的の双方にとって有益であると思われる。
このレビューで示したように、これらの各パラメーターとPPARγ MOAとの関係は経験的に正当化されるので、PPARγ作動薬またはPPARγ/δ二重作動薬T3D-959の将来の大規模AD薬剤試験には、共変量として空腹時インスリン、Aβペプチドおよびp-tau 181の測定を組み込むよう推奨する。
結論
ピオグリタゾンは、「錠剤の中の多剤併用療法」と呼ばれ、炎症、酸化ストレス、ミクログリアの欠陥、アミロイド斑や神経原線維変化、脳内グルコース消費障害、ミトコンドリア機能障害(生体エネルギーの抑制とダイナミックスの阻害)など、アルツハイマー病の複数の病理学的決定因子を改善することが示されている。
前臨床試験において、pioglitazoneは学習・記憶を改善し、それはシナプス活動の改善、アミロイドおよびタウ病変の減少と相関しており、AD病変の発症前に治療を開始した場合に、より良い効果が得られることが示されている。縦断的コホート研究により、ピオグリタゾンは、DM2患者において、時間および用量依存的な保護因子であることが示されている。
これらの結果は、ピオグリタゾンが脳血流を増加させ、痴呆の発症を遅らせることを示したDM2症例における小規模のパイロット試験と一致している。これまで実施された臨床試験のほとんどは、小規模で検出力が十分でないか、あるいは決定的な結果を得るには十分な期間実施されていないものであった。しかし、これらの研究は、ピオグリタゾンの認知機能に対する作用が、DM2のない症例においても、インスリン低下作用と相互作用することを示唆するものである。
利益相反
ASはZinfandel Pharmaceuticals, Inc.の社長兼CEOである。DBは、ジンファンデル・ファーマシューティカルズのシニアバイスプレジデント兼COOである。WGは、ジンファンデル・ファーマシューティカルズ・インクからコンサルティング料を受け取っている。