
Contents
Perspective of SGLT2 Inhibition in Treatment of Conditions Connected to Neuronal Loss: Focus on Alzheimer’s Disease and Ischemia-Related Brain Injury
www.ncbi.nlm.nih.gov/labs/pmc/articles/PMC7697611/
オンラインで2020年11月11日掲載
Michał Wiciński, Eryk Wódkiewicz,* Karol Górski, Maciej Walczak, and Bartosz Malinowski
概要
Sodium-glucose co-transporter 2阻害剤(SGLT2i)は,2型糖尿病の治療薬として承認されている経口血糖降下剤である。SGLT2iは、2型糖尿病の治療薬として承認されている経口血糖降下剤であるが、中枢神経系に存在し、神経保護作用を有する可能性を示唆する報告もある。エンパグリフロジンでSGLT2を阻害すると、APP/PS1xd/dbマウスの大脳皮質におけるアミロイド負荷が減少することが示されている。また、タウ病理や脳萎縮量についても同様の効果が認められた。Empagliflozinは認知機能に有益な効果を示したが、これは脳内脳由来向神経性因子の増加と関連している可能性がある。カナグリフロジンとダパグリフロジンは、アセチルコリンエステラーゼ阻害作用を有しており、アルツハイマー型認知症治療薬に類似している。SGLT2阻害剤は、動脈硬化の危険因子や、脳卒中の急性期と後期の両方に関与する経路に影響を与える可能性がある。SGLT2阻害剤の作用機序は、肝細胞核因子-1α、血管内皮増殖因子-A、および炎症性因子の制限を誘導することに関連していると考えられる。E
mpagliflozinは、糖尿病マウスの神経血管ユニットを維持し、その異常なリモデリングを防止する効果があると考えられる。カナグリフロジンは、ヒトおよびマウスの内皮細胞の増殖を抑制することで、細胞を静める効果があると考えられる。本論文では、アルツハイマー病や脳虚血を中心とした、神経細胞の損傷を伴う状況におけるSGLT-2阻害剤の潜在的なメカニズムを紹介する。
キーワード:薬理学、神経変性、サイトカイン、アルツハイマー病、脳梗塞
1. SGLT2受容体は中枢神経系疾患の治療における潜在的なターゲットとなる可能性がある
ナトリウム-グルコース共輸送体2阻害剤(Sodium-glucose co-transporter 2 inhibitor: SGLT2i)は,2型糖尿病の治療薬として承認されている経口血糖降下剤である[1]。ナトリウム-グルコース共輸送体2(SGLT2)は、主に腎臓の近位畳み込み管のセグメント1および2に発現している。SGLT2は,尿中のグルコースの再吸収に重要な役割を果たしており,尿中グルコースの再吸収はナトリウム濃度勾配に依存している[2]。SGLTファミリーの共輸送体は,14の膜貫通ドメインと1つのN-グリコシル化部位を持つ膜貫通型のモノマー蛋白質を含んでいる。SGLTは、グルコースとガラクトースを濃度勾配に逆らって輸送し、同時にNa+イオンを輸送する[3]。SGLTの主な作用機序は,腎再吸収の低下によるグルコース排泄量の増加である。これらの化合物の利尿作用、造血作用、ナトリウム利尿作用は、尿量とナトリウム量を増加させる[2]。哺乳類の中枢神経系(中枢神経系)にナトリウム-グルコース共輸送体(SGLT)が存在するとする報告が増えている[4,5]。SGLT1は、海馬のCA1,CA3(海馬角の1番と3番)海馬歯状回などに発現しており、SGLT2は、海馬、小脳、血液脳関門(BBB)内皮細胞などに発現していることが確認されている[3,6,7,8]。急性および慢性の脳疾患を治療するための有効な神経保護薬の探索は、長年にわたって行われてきた。この特殊な分布が、神経保護特性を示唆する興味深い証拠の原因となっていると思われる[9]。図1は、SGLT2iの活性化のメカニズムを示している。
pharmaceuticals-13-00379-g001.jpg である。
図1 ナトリウム-グルコース共輸送体2阻害剤(SGLT2i)の活性化メカニズムの提案
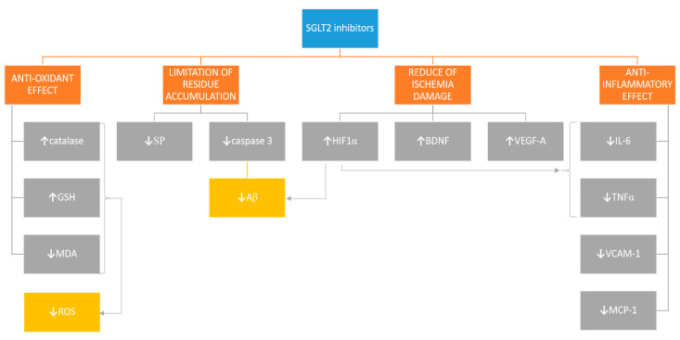
↓還元、↑増加、GSH-グルタチオン、MDA-マロンジアルデヒド、ROS-活性酸素種、SP-セニール・プラーク、Aβ-アミロイドβ、HIF1α-hypoxia-inducible factor 1α。BDNF-脳由来向神経性因子、VEGF-A-血管内皮増殖因子-A、IL-6-インターロイキン6,TNF-α-腫瘍壊死因子α、VCAM-1-血管細胞接着タンパク質、MCP-1-単球走化性タンパク質-1。図1の色の違いは、美観のためにのみ使用されている。
EUでは現在、カナグリフロジン(インボカナ、ボカナメット)ダパグリフロジン(フォルキシガ、ジグドゥオ)エンパグリフロジン(ジャルディアンス、シンジャルディ)の3つのSGLT2iが認可されており、論文でも紹介されている。この3つの化合物は、プラセボと比較して、HbA1c(糖化ヘモグロビン)値の低下に効果があることが示されている。SGLT2阻害剤は、血糖値の低下以外にも、空腹時血糖値(FPG)体重、収縮期および拡張期血圧の改善など、臨床的に有益な代謝効果を示している[10,11]。現在のエビデンスによると、SGLT2阻害剤は、特に心不全患者の心血管イベントのリスクを低減する[12]。エンパグリフロジンは、ダパグリフロジン(1200倍)やカナグリフロジン(250倍)に比べて、SGLT2に対する高い選択性(2500倍)を有することが証明されている[13]。性器感染症は最も一般的な副作用のようである(4倍の増加)[14]。SGLT2阻害剤は浸透圧利尿を誘発するため、体液量の減少につながる可能性がある。副作用の危険因子としては、75歳以上、GFR(糸球体濾過量)が60mL/min/1.73m2未満、ループ利尿薬の使用などが挙げられている[15]。欧州医療機関が行った分析では、2型糖尿病(2型糖尿病)でSGLT2阻害剤(カナグリフロジン、ダパグリフロジン、エンパグリフロジン)の治療を受けている患者で、重篤な糖尿病性ケトアシドーシスが102例発生したことが発表された。興味深いことに、その中には血糖値が中程度に上昇する非典型的なものも発生した[16,17,18,19,20]。残念ながら、この問題に関する現在の情報は、文書化されていないケースに依存しており、適切な評価とリスクグループの特定の可能性が大きく制限されている。
2. SGLT2iはアルツハイマー病の病態に影響を及ぼす可能性のあるもう一つのDM登録薬
アルツハイマー型認知症(AD)は、死亡原因の第6位であり、認知症全体の3分の2以上を占めている。アルツハイマー病の世界的な患者数は 2006年の2,660万人から 2050年には1億680万人になると予想されている[21,22]。このようなデータの多さと、治療法の効果に対する失望感から、広義の認知症の治療には効率的な新しい選択肢が必要とされている。
ADの主要な病理学的メカニズムの一つは、βアミロイドからなるプラークと、微小管に結合するタウタンパク質の凝集体である神経原線維変化(NFT)と呼ばれる細胞外および細胞内の残留物の蓄積である[23,24]。最近の研究では、SGLTの阻害がこのプロセスに何らかの影響を与えている可能性が示唆されている。Hierro-Bujalanceら[25]は、SGLT2阻害剤であるエンパグリフロジン(EMP)に関する初めての研究で、APP/PS1xd/dbモデルのEMP投与マウスの大脳皮質および海馬において、老人斑の密度が減少し、可溶性および不溶性のアミロイドβ(Aβ)レベルが全体的に低下したことを報告した。APP/PS1xd/dbマウスでは、病気の進行に伴って脳の萎縮が観察され、実際のADの病態に近いモデルとなっている。アミロイド病理の改善における同様の効果は、代謝制御だけでなく、酸化ストレス、炎症、または血液脳関門(BBB)に対するそれらの効果に基づいて、他の糖尿病(DM)登録された治療法[26,27,28]により観察されている。2種類目のAD特徴的な凝集体についても、EMP投与による制限が現れている。大脳皮質ではタウのリン酸化を抑制する効果があったが、海馬ではインクレチンを用いたDM治療の結果とは逆に統計的有意性が得られていない[29,30]。また、脳萎縮の程度についても、皮質領域でのみ統計的有意性が認められ、同様の矛盾が見られた。さらに、著者らは、EMP治療がAPP/PS1xdb/dbマウスの認知能力にプラスの影響を与え、記憶障害を有意に改善したことから、新しい物体識別課題およびMorris水迷路試験において、認知機能に有益な効果があることを報告した。研究者らは、改善の原因としてレプチンシグナルの変化を除外することはできなかった[25]。Linらの研究[10]では,エンパグリフロジンがdb/dbマウスの認知機能障害を有意に予防し,これは,脳の酸化ストレスの減衰と脳由来向神経性因子(BDNF)の増加と関連していた。BDNFは,神経系における神経細胞の分化,成熟,生存をサポートし[23],グルタミン酸刺激,脳虚血,低血糖症,神経毒性などの条件下で神経保護作用を示す[31,32]。このメカニズムは,グルカゴン様ペプチド1アゴニストであるリラグルチドの神経保護作用の原因として提案されている[33,34]。
FDA(米国食品医薬品局)に登録されているAD治療薬の有効性にはまだ多くの課題があるが、アセチルコリンエステラーゼ阻害剤とN-メチル-D-アスパラギン酸受容体拮抗剤は、苦しんでいる患者のための主要な治療法である。Arafaら[35]のスコポラミン誘発性記憶障害ラットモデルでは、カナグリフロジンという別のSGLT2阻害剤の効果が認められた。この効果は、SGLT阻害剤の付加的な特性であるアセチルコリンエステラーゼ阻害作用によるものと考えられている。この発見は、阻害剤と酵素の間の相互作用の強さを決定するダパグリフロジンとアセチルコリンエステラーゼ(AChE)の19個のアミノ酸残基の結合エネルギーを評価する分子ドッキング技術の研究結果と一致しているようである[36]。阻害剤と酵素との相互作用の強さを決定するアミノ酸残基は19個あり,そのうち最も低い結合エネルギーは,受容体部位または酵素の活性部位における最適なコンフォーマーの印となる[37]。ドッキングされたリガンドは,AChEおよびSGLT2に対して-6.28kcal/molおよび-6.25kcal/molという許容可能な結合エネルギー値を示し[36],ダパグリフロジンの二重阻害剤としての特性の可能性を示唆するとともに,RizviらやShakilらがertugliflozinやsotagliflozinなどの他のSGLTiでも示した結果を再確認することができた[38,39]。
3. SGLT2iは、血管新生と神経発生を促進し、虚血関連の脳障害を予防することが証明された
脳虚血性脳卒中は、世界における死亡および長期障害の主要原因のひとつである[40]。血流障害は,病態生理学的な変化を引き起こし,最終的には組織の壊死や神経細胞の喪失につながる[41].Abdel-Latifら[42]は,低用量および高用量のエンパグリフロジンが,頸動脈の閉塞を利用した虚血モデルのラットにおいて,神経学的障害を軽減することを明らかにした。なお、エンパグリフロジンの効果は用量に依存しており、高用量投与群では神経学的スコアが高くなっていた。エンパグリフロジンは、対照群と比較して、低酸素誘導因子1α(HIF-1α)のレベルと血管内皮増殖因子A(VEGF-A)タンパク質の発現を増加させ、カスパーゼ-3の発現を減少させ、梗塞体積を制限した。虚血の悪影響を防止または軽減するために、低酸素症の影響を受けた細胞ではシグナル伝達経路や因子が活性化される。その代表的なものがHIF-1αである。解糖、赤血球生成、血管新生を誘導することで、酸素のホメオスタシスの回復をサポートする[43]。Xingら[44]が得た結果によると、HIF-1αの活性化は、ラットによる全身虚血において、インターロイキン-6/インターロイキン-6受容体(IL-6/IL-6R)経路、腫瘍壊死因子α/腫瘍壊死因子受容体1(TNF-α/TNFR1)の発現上昇を有意に弱め、カスパーゼ-3の発現上昇を減少させた。著者らは、炎症性サイトカインの発現低下を介したHIF-1αが、有害な脳虚血の影響を抑えることに貢献していると示唆している[44]。前項に関連して、カスパーゼ-3の過剰発現は、アミロイドーシス[45]、NFT形成[46]、神経細胞のアポトーシス[47]など、ADの病因にいくつかの役割を果たしていると考えられている。
DM2(2型糖尿病)におけるSGLT2発現の変化のメカニズムはよくわかっていないが、肝細胞核因子1α(HNF-1α)および肝細胞核因子3β(HNF-3β)の活性の変化が、SGLT2発現の変化に寄与していることが報告されている[48]。HNF-1αは、マウスおよびヒトのSGLT2の発現を直接制御しているようである[49]。研究者らは、HNF-1αの発現および溶質キャリアファミリー5メンバー2のプロモーターにおける結合活性の上昇が、糖尿病によって誘発されるSGLT2の過剰発現につながることを示した[50]。上述のEMPによるHIF-1αの上昇の例は、SGLT-2受容体とHNF-1αの発現を含むある種のフィードバックループの前提となっていると仮定するのが妥当であろう。
低酸素の影響から細胞を保護する役割を果たし、EMP投与中に増加したもう一つの因子はVEGF-Aである。VEGFは、血管新生および神経発生の重要な調節因子であることが知られている[51]。脳虚血イベントの結果,VEGF-Aとその受容体である血管内皮増殖因子受容体1および血管内皮増殖因子受容体2がアップレギュレートされている[52]。齧歯類のHeterocephalus glaberの脳では,VEGF-Aの発現が著しく上昇しており,このことが低酸素に対する彼らの並外れた固有の耐性に寄与していると思われる[53].一方、脳卒中の急性期にVEGF-Aが活性化されると、BBBが破壊され、ホメオスタシスが損なわれ、その結果、浮腫が発生する[54]。VEGF-A治療の効果を評価する上で、VEGF-Aレベルが上昇する瞬間が重要であると考えられる。残念ながら、脳卒中におけるVEGF-Aの役割に関する知識の現状は、主に動物モデルを用いた研究に基づいている。
脳微小血管系は、脳実質と密接な構造的・機能的関係にあり、神経血管ユニット(NVU)と呼ばれる生物学的システムの制御下にある[55]。NVUは、神経細胞、グリア細胞、血管細胞、および細胞外マトリックスからなる統合的な生物学的システムである。Haydenらは,db/dbマウスの大脳皮質灰白質および移行皮質下白質のNVU内で,同じ背景を持つ年齢・性別の非糖尿病野生型マウスと比較して,認知機能障害,脳組織の酸化ストレス,超微細構造(US)のリモデリングを報告した[56,57].さらに彼らは,USリモデリングを伴う皮質灰白質NVU,神経膠,ミエリンの損傷を観察した。脳卒中後の回復には、このユニットの再構築が重要であると考えられている[58,59,60]。EMPを用いたSGLT2の阻害は、ECのタイトジャンクションやBBBのアドヘントジャンクションの減少または消失、ECや大脳皮質物質を含む様々な異常からなる、NVUの細胞やミエリンのUSリモデリングを防止した[61]。EMPや他のSGLT2がBBBを通過するという証明はないが、SGLT2阻害剤は脂溶性なので、BBBを通過するはずである[62]。さらに、脳卒中の急性期には、血液脳関門(BBB)の完全性と機能が損なわれているという報告がある[63]。エンパグリフロジンは、破壊されたBBBを通過することで、神経保護効果を発揮するという仮説を立てている研究者もいる[64]。これを裏付けるように、Haydenら[61]は、エンパグリフロジンが糖尿病マウスの脳の神経血管ユニットと神経膠の超微細構造のリモデリングを改善することを示し、エンパグリフロジンがBBBの完全性が失われたこれらの領域に入ることができることを強調した。他の研究者は、SGLT阻害剤(この場合はダパグリフロジン)がGLP-1濃度を上昇させ、GLP-1が血液脳関門を通過してコルチコステロン濃度を低下させ、神経保護効果をもたらすと考える方がより妥当であると主張している[65]。
4. SGLT2iの抗炎症作用は、動脈形成を遅らせ、酸化ストレスに関連した神経細胞の損失を防ぐ可能性がある
脳卒中は、頸動脈のアテローム性動脈硬化を含む危険因子を排除することで予防することができる[66]。これは、プラーク形成とそれに続く動脈の狭窄を引き起こす血管の慢性的な炎症である[67]。炎症に関連した動脈硬化の誘導には、腫瘍壊死因子α、IL-6,単球走化性タンパク質-1(MCP-1)などの様々なサイトカインが関与しており、また、メディアによる発現誘導や細胞接着分子1(VCAM-1)の発現誘導もある[68]。全身性の炎症は、血液脳関門の完全性を妨げ、炎症誘発物質の中枢神経系への移動を引き起こす可能性がある[69]。その結果、慢性的な低グレードの炎症が、神経細胞の損失を促進することが証明されている[70,71,72,73]。
脳虚血イベントの主な原因の1つであるアテローム性動脈硬化症の発症を遅らせることに、SGLT2阻害が関連している可能性があるという報告がある。Hanら[74]は、ApoE-/マウスにおいて、エンパグリフロジンが、グリメピリドを投与した対照群と比較して、大動脈弓部および弁部のアテローム性動脈硬化プラークの面積を制限することを見出した。エンパグリフロジン投与後、TNF-α、IL-6,MCP-1の濃度が低下し、プラークの大きさとの間に有意な相関関係が認められた。
IL-6とTNF-αは炎症性因子であり,特に脳卒中時にその濃度の上昇が観察される[75,76]。TNF-αとIL-6は、脳卒中のリスクを高める可能性があるという報告がある[77,78]。これは、Cuiら[79]が中国人集団で得たメタアナリシスデータで支持されているが、Jefferisら[80]は、イギリス人集団ではそのような依存性がないことを示している。Pennigら[81]も同様の結果を得ており、エンパグリフロジンを投与したマウスのアテローム性動脈硬化プラークは有意に小さくなり、同時に脂質が減少し、構造中のコラーゲン含量が増加した。Dimitriadisら[82]は、エンパグリフロジンがコレステロール値を低下させ、HDL(高密度リポタンパク質)コレステロール値を増加させ、動脈硬化病変の形成と炎症分子VCAM-1およびMCP-1の発現を減少させることを発表した。Ganbaatarら[83]は、エンパグリフロジンの投与により、大動脈弓部の動脈硬化病変の大きさが有意に減少し、脂質の沈着とマクロファージの蓄積が減少し、MCP-1の発現が低下したことを示した。前出のHierro-Bujalanceら[25]も、EMPを投与したdb/dbおよびAPP/PS1xdb/dbマウスの実質的なミクログリアの負担が減少したことを確認している。しかし、この評価は、Iba1免疫染色という限られた範囲で行われたものであり、この現象の複雑さ全体をカバーするものではない。
もう1つのSGLT2阻害剤であるカナグリフロジンは,内皮細胞(EC)の増殖とDNA合成を阻害することが証明されている[84]。ECの増殖を制御するプロセスは、アテローム発生の抑制に極めて重要な役割を果たすことが示されている[85]。カナグリフロジンの抗増殖作用は,マウスの動脈循環に加えて,ヒトの動脈循環および静脈循環に由来するECでも観察された。乳酸脱水素酵素活性の測定結果から、カナグリフロジンの作用機序は、細胞毒性というよりもむしろ細胞静菌作用であると考えられる。この作用は濃度依存性であり、この薬剤で治療を受けた患者の血漿中で到達可能な濃度で現れるようである[86]。さらに、この結果は、マウス由来のECよりもヒトの細胞の方がカナグリフロジンの増殖抑制作用に敏感である可能性を示唆している。興味深いことに、この特定の効果は化合物特異的な効果である可能性があり、したがって、エンパグリフロジンやダパグリフロジンなどの他のSGLT2阻害剤の薬理学的に適切な濃度では、ECの増殖に影響を与えなかった[87,88]。
Aminら[64]は、ラット中枢神経系におけるSGLT2治療と酸化ストレス制限に関する結果を発表した。グリクラジドと比較して、エンパグリフロジンの投与は、ラットの脳組織において、脳の酸化ストレス、炎症、アポトーシスのマーカーを抑制するとともに、梗塞体積の減少をもたらした。著者らは、エンパグリフロジンの神経保護作用は、主に酸化ストレスの軽減によるものと推測している。EMPは、脳組織において、マロンジアルデヒド(MDA)を減少させ、カタラーゼの活性を高め、グルタチオン(GSH)濃度を上昇させた。MDAは、酸化ストレスのマーカーである[89]。カタラーゼは,過酸化水素(活性酸素種(ROS)の原因)を水と酸素に分解する酵素であり[90],GSHは,活性酸素による細胞成分の損傷を防ぐ抗酸化物質である[91]。本稿で紹介した実験研究の有望な結果にもかかわらず,脳虚血イベントにおけるSGLT2iの臨床的有効性については,依然として議論の余地がある。心血管リスクの高い2型糖尿病患者を対象としたZinmanらのメタ解析[92]では、プラセボと比較してエンパグリフロジン投与後の脳卒中を含む脳血管リスクに有意な差は認められなかった。EMPA-REG OUTCOME試験では、脳卒中リスクの上昇傾向がわずかに認められたが、統計的には有意ではなく(HR, 1.18; 95% CI, 0.89 × 101.56; P¼ 0.26)これはヘマトクリット値の上昇と関連している可能性がある[93]。カナグリフロジンのCANVAS試験では逆の傾向が得られている(HR, 0.87; 95% CI, 0.69 × 101.09)が、統計的有意性には再び達していない[94]。本論文で紹介した研究結果の概要を表1に示する。
表1 レビュー結果のまとめ
| 著者 | 研究対象 | SGLT2iの種類 | SGLT2iの投与量 | 結果 |
|---|---|---|---|---|
| Abdel-Latif etal。[ 29 ] | ウィスターラット | エンパグリフロジン | 1および10mg / kg、1時間および24時間の再灌流後に2回経口投与 | ↓神経学的欠陥、↑HIF-1α、↑VEGF-A、↓カスパーゼ-3、↓骨折量 |
| アミン等。[ 41 ] | ウィスターラット | エンパグリフロジン | 1および10mg / kg、1時間および24時間の再灌流後に2回経口投与 | ↓グリクラジドと比較したインフラクトボリューム、↓MDA、↑カタラーゼ、↑GSH |
| アラファら [ 22 ] | ウィスターラット | カナグリフロジン | 10mg / kg経口1回 | ↓記憶障害 |
| Behnammanesh etal。[ 63 ] | HUVEC、HAEC、MAEC | カナグリフロジン、エンパグリフロジン、ダパグリフロジン | 1〜50μM | ↓ECの増殖 |
| ガンバタール他 [ 62 ] | ApoE-/-マウス | エンパグリフロジン | 20mg / kg /日で8週間または12週間 | ↓アテローム性動脈硬化症病変のサイズ、↓脂質沈着、↓マクロファージ蓄積、↓MCP-1 |
| ハンら。[ 53 ] | ApoE-/-マウス | エンパグリフロジン | 3mg / kg /日で8週間 | ↓TNF-α、↓IL-6、↓MCP-1、↓アテローム性動脈硬化症プラーク |
| ヘイデンら。[ 75 ] | db / dbマウス | エンパグリフロジン | 10mg / kg /日で10週間 | NVUの細胞とミエリンの米国の改造を防いだ |
| Hierro-Bujalance etal。[ 11 ] | APP / PS1xdb / dbマウス | エンパグリフロジン | 10mg / kg /日で22週間 | ↓SP、↓Aβ、↓NOD、↑MWM、↓ミクログリア負荷 |
| リンら。[ 17 ] | db / dbマウス | エンパグリフロジン | 0.03%のエンパグリフロジンを含む食事を7日間または10週間 | 認知機能障害の予防、↑BDNF |
| Pennig etal。[ 60 ] | C57BL / 6Jマウス | エンパグリフロジン | 35mg / kg /日で3週間 | ↓アテローム硬化性プラークのサイズ、↓脂質および↑アテローム硬化性プラーク構造のコラーゲン含有量 |
備考 ↓減少、↑増加、GSH-グルタチオン、MDA-マロンジアルデヒド、SP-セニール・プラーク、Aβ-アミロイドβ、HIF-1α-hypoxia-inducible factor 1α、BDNF-脳由来向神経性因子、VEGF-A-血管内皮増殖因子-A。IL-6-インターロイキン6,TNF-α-腫瘍壊死因子α、VCAM-1-血管細胞接着タンパク質、MCP-1-単球走化性タンパク質-1,ECs-内皮細胞、NVU-神経血管ユニット、NOD-新物体弁別、MWM-モリス水迷路。
5. 結論
文献で得られる情報の範囲は残念ながら限られている。実験モデルでの結果は有望であり、ADや、主にあるいは二次的に神経変性を背景とする他の疾患の治療ソリューションの分野における研究の発展のための新たな方向性を示している。とはいえ、この分野ではさらなる研究の必要性がある。SGLT2の伝達に影響を与える薬剤が、中枢神経系疾患の治療に効果的に役立つかどうかは、臨床効果の評価によってのみ明らかになる。今回収集したデータは、この多因子による神経保護プロセスの完全な理解に向けて、一歩前進したことになる。
著者の寄稿
M.W. (Michał Wiciński), E.W., M.W. (Maciej Walczak)は、データ解析、知見の解釈、論文の作成に貢献した。K.G.とB.M.は、データ収集、論文の重要な修正、最終承認に参加した。すべての著者は、本稿の公開版を読み、同意した。
資金調達
本研究は,Nicolaus Copernicus University, Toruń, PolandのCollegium Medicum in Bydgoszcz, Faculty of Medicine, Department of Pharmacology and Therapeuticsの支援を受けて実施した。
利害の衝突
著者は利益相反を表明していない。