
Pathophysiological Clues to How the Emergent SARS-CoV-2 Can Potentially Increase the Susceptibility to Neurodegeneration
2021年1月8日
pubmed.ncbi.nlm.nih.gov/33417221/
要旨
2019年後半の新型重症急性呼吸器症候群コロナウイルス2(SARS-CoV-2)の出現とともに、脳の構造的変化に関連した無数の神経症状が報告された。本論文では、生存した患者が様々なメカニズムを介して神経変性疾患のリスクを高めている可能性があるという主張を批判的に論じるための証拠を提供する。
このウイルスは、嗅球、末梢神経終末からの逆行性軸索輸送、あるいは血行性ルートやリンパ系ルートを介して脳に直接侵入することができる。感染した神経細胞は末梢白血球の活性化とともに炎症性サイトカインを増加させ、脳の神経変性を引き起こす。また、アンジオテンシン変換酵素2(ACE-2)がウイルスに占有されると、神経保護因子として作用するACE-2活性が低下する可能性がある。
さらに、急性呼吸窮迫症候群(ARDS)や敗血症では低酸素血症や低灌流が誘発され、脳血管の高凝固状態や微小血栓症により局所的に悪化し、酸化ストレスや神経変性を引き起こすと考えられている。COVID-19および神経変性疾患の共通の危険因子、例えば代謝危険因子、遺伝的素因、さらには腸内細菌叢の異常などは、COVID-19生存者における神経変性疾患のより高い発生に寄与し得る。
しかし、感染の重症度、神経症状の程度、およびウイルス感染の結果の持続性が、この関連性の主要な決定因子であることを考慮すべきである。重要なことは,このパンデミックが神経変性疾患の全体的な発症率を増加させるかどうかは明らかではないことだ.
序論
2019年12月、重症急性呼吸器症候群(SARS)を引き起こす新型コロナウイルス(SARSコロナウイルス2(SARS-CoV-2)と短く名付けられた)が中国で出現した。これまで、この大発生は、身体的な健康への負担が大きいだけでなく、多くの社会的、経済的、政治的苦痛を世界中で引き起こしていた[1]。しかし、身体的健康への影響を含むウイルスの長期的な影響は、世界にとってより深刻な脅威となる可能性がある。重要なことに、このウイルス感染は、脳血管事故、意識障害、錯乱、激越、発作、運動失調、頭痛、無感覚、老衰、神経障害、および脳炎または脳症などの神経学的症状を引き起こすか、または提示することができる[2,3,4,5]。さらに、COVID-19に関連した脳構造変化は、生存者および非生存者の両方において、イメージング技術によって確認されている[6,7]。さらに、中枢神経系へのウイルスの侵入の証拠に基づいて、SARS-CoV-2感染で観察される呼吸困難のレベルの違いは、神経系へのウイルスの影響に起因するのではないかと推測されている[8,9,10]。ヒトコロナウイルスが神経細胞株に潜伏したままであることを示す証拠がいくつかあり[11]、COVID-19が脳内の広範な遺伝子発現変化と関連していることが実証されており、その神経機能への長期にわたる広範な影響が強調されている[12]。このように,ウイルス感染の急性期における神経症状とは別に,SARS-CoV-2の長期的な神経学的後遺症,例えば神経変性疾患は非常に重要である[5].
神経変性疾患は、主に老年期に発生する神経細胞の衰退を特徴とする神経疾患群であり、高い障害率と死亡率により大きな負担となっている[13, 14]。遺伝的素因から代謝・環境リスク因子に至るまでの無数の因子が、酸化ストレスや神経炎症などの様々なメカニズムを介して、誤ったタンパク質の蓄積、オートファジーの障害、ミトコンドリア機能不全、神経細胞のアポトーシス、シナプス障害など、中枢神経系の特定の神経細胞の死滅に寄与し、アルツハイマー病やパーキンソン病(PD)などの神経変性疾患の病態生理を構成している[15, 16]。
中東呼吸器症候群コロナウイルス(MERS-CoV)やSARS-CoVなどのコロナウイルス[17]や、コロナウイルス病2019(COVID-19)[10]などのコロナウイルスは、感染が解消された後に神経障害を引き起こすことが示されている。例えば、ウイルス感染は、一連の錐体外路症状および黒質実質および他の皮質下核における病理学的変化によって特徴づけられる、レザルギカ脳炎に続く後脳性パーキンソニズムとして知られるいくつかの慢性神経学的後遺症と関連し得ることが決定されている。この症状は1918年のインフルエンザ大パンデミック後に初めて報告されたが、コロナウイルス感染が神経変性疾患に関与していることを示す間接的な証拠はあるが、ヒトコロナウイルスの発生ではこのような後遺症の報告はない[18, 19]。さらに,ヒトコロナウイルス(HCoV)感染は,インフルエンザウイルスなどの他の呼吸器ウイルス感染症とともに,中枢神経系,特に側頭領域や海馬に広がり,学習・認知機能の変化を伴うことが示されている[20].この海馬の損傷が神経変性変化を永続させるかどうかは明らかではないが、認知障害に関連したウイルス感染関連の炎症に続くアルツハイマー病特異的なタウ病理は動物実験で確認されている[21]。
中枢神経系へのウイルスの侵入の潜在的な結果を無視しても、COVID-19は炎症性サイトカインの顕著な増加と関連しており、神経変性に寄与する劇症的なメカニズムのカスケードを導くことが示唆されている[22]。ウイルスの神経系への直接的な侵入やウイルス感染による炎症反応に加えて、神経内分泌軸、サイトカインストーム、代謝変化、腸内細菌叢の変化、低灌流などの様々なメカニズムや、SARS-CoV-2に罹患している患者に共通する基礎的な危険因子が神経変性過程の加速に寄与している可能性がある。本論文では、SARS-CoV-2感染者の神経変性、特にアルツハイマー病やパーキンソン病の発症に寄与する可能性のある病態メカニズムを再検討することを目的としている(表1にまとめた)。このことは、将来的にこれらの患者さんの神経変性を予防するための治療介入を設計するための道を開く可能性がある。
表1 COVID-19感染と神経変性疾患との関連メカニズムと危険因子のまとめ
| PD / AD /両方の病態生理学に関与するメカニズム/危険因子 | COVID-19におけるメカニズム/危険因子の証拠 | PD / AD /両方のメカニズム/危険因子の証拠 |
|---|---|---|
| 嗅球の関与 | – COVID-19における香り障害[ 2、23も感染から回復した後] [ 24 ]。 -死後のMRIで検出された嗅球の非対称性[ 6 ]。 -脳皮質及び嗅球[におけるグリアにおけるACE-2及びTMPRSS受容体を介して脳へのウイルスの侵入25、26、27、28 ]。 |
-無嗅覚症及び嗅球の関与は、[AD及びPDの両方の撮像によって明らか29、30、31 ] -嗅球、黒質、及び大脳皮質、PDに関与する領域とADにおけるグリアにおけるACE-2及びTMPRSS受容体の発現を[ 25、26、27、28 ]。 |
| サイトカイン産生 | -髄膜脳炎のCOVID-19患者でIL-6,IL-8,IL-10,およびTNF-αの増加が検出された[ 32 ]。 | -Increased対照と比較してアルツハイマー病患者におけるIL-1β、およびより高いIL-6対照と比較してパーキンソン病患者において、ならびに増加したTNF-αが検出された[ 33、34 ]。 |
| ミクログリアの活性化 | – SARS-CoVの-2オープンリーディングフレーム3a(ORF-3A)タンパク質刺激NLRP3インフラマソームは、それによってミクログリアの活性化[促進する35、36 ]。 -ウイルス感染におけるBBBの分解により、単球はBBBに侵入する可能性がある[ 37 ]。 -死後COVID-19感染における重症ミクログリア活性化[ 38、39 ]。 |
AD及びPD [における-Microglial活性悪化神経変性40、41 ]。 -NLRP3インフラマソームはADにおけるミクログリアを介したIL-1β放出に役割を果たし[ 42 ]、PDの慢性炎症はNLRP3インフラマソームを介してミトコンドリア機能障害を誘発する可能性がある[ 43 ]。 |
| T細胞浸潤 | – T細胞の軽度の血管周囲浸潤はCOVID-19感染【で死亡した患者の死後中枢神経系分析で認められた38、39 ]。 | -血管に密接に接触している、またはPDのSNのメラニン化したDAニューロンの近くにある中枢神経系浸潤CD4 +およびCD8 +細胞が検出された[ 44 ]。 -T細胞は、CD4 +細胞と比較してCD8 +細胞の数が増加したアルツハイマー病患者の海馬で発見された[ 45 ]。 |
| 酸化ストレス | –SARS-CoV-2は、ARDSを引き起こす可能性があり、ARDSによる低酸素症、ならびに凝固亢進および血栓症は、RONS産生およびその結果としての臓器損傷に関連する酸化ストレスを引き起こす可能性がある[ 5 ]。 | -神経炎症および他のメカニズムを介した、酸化ストレスに応答した、PDにおけるより大きなα-シヌクレイン凝集[ 46 ]、およびADにおけるより高いベータアミロイドペプチド蓄積[ 47 ]の証拠。 |
| ACE軸の不均衡 | -ACE-2はSARS-CoV-2スパイクタンパク質の受容体として機能し、細胞への侵入を可能にする[ 48 ]。SARS-CoV-2がACE-2に結合すると、ACE-2が枯渇し、ACE-2 /アンジオテンシン(1-7)/ Mas軸がダウンレギュレートされることで多臓器障害が悪化する可能性がある[ 49 ]。 | -ACE-2活性はADで低下し、RASの中心的な古典的ACE-1 / Ang II / AT1R軸の重要な調節因子です[ 50 ]。 –ACE-2 / Ang 1-7 / Mas軸は、神経変性メカニズムからニューロンを保護することができる[ 51 ]。 |
| 腸内細菌叢 | -腸microbiomeのdysbiosisとして知られている腸内細菌叢の組成の変化はCOVID-19患者[において検出されている52、53 ]。 | -腸内細菌叢の腸内毒素症障害によって誘発される腸およびBBBの透過性の増加は、神経変性障害の発症に関与している[ 54 ]。 |
| アミロイド-ベータ/タウ/アルファ-シヌクレインの蓄積 | -SARS-CoVスパイクタンパク質は、小胞体のタンパク質機構を乗っ取り、折りたたまれていないタンパク質の応答と誤って折りたたまれたタンパク質の蓄積を促進する可能性がある[ 55 ]。 -ORF-9bの相互作用によるSARS-CoV感染のタンパク質恒常性障害[ 56 ]。 |
-タンパク質恒常性の障害による、ADでのアミロイドベータおよびタウの蓄積やPDでのα-シヌクレインの蓄積など、誤って折りたたまれたタンパク質の蓄積は、神経変性疾患の主力です[ 57 ]。 |
| シナプス機能障害 | -SARS-CoV感染では、IFN-αとIFN-βがウイルス複製の制限に効果的であることが示されている[ 58 ]。 | -IFNはミクログリアを活性化し、炎症誘発性反応を刺激し、シナプス除去を促進する[ 59 ]。 |
| ミトコンドリア機能障害 | -SARS-CoVのSARS-CoVORF-9bは、宿主細胞のオートファジーを誘発するだけでなく、ユビキチン化を誘発し、ミトコンドリアのタンパク質恒常性を損ないます[ 56 ]。 | -ミトコンドリア機能障害、タンパク質恒常性障害、オートファジー、およびリソソーム機能障害は、神経変性疾患の病態生理学に関与している[ 60 ]。 |
| ApoEe4対立遺伝子 | -血清コレステロールはApoE受容体に結合し、細胞表面へのACE-2受容体の輸送を誘導する[ 61 ]。 -アポE e4e4遺伝子型は重篤なSARS-CoVの-2感染〔の危険因子として作用する62、63 ]。 |
-ApoE e4は、ベータアミロイドおよびタウ凝集体の形成と沈着を増加させ、樹状突起棘形成とシナプス可塑性を破壊することにより、ADのリスクをほぼ14倍増加させる[ 64 ]。 |
| メタボリックシンドローム/要因 | -肥満とメタボリックシンドロームは、この感染症に苦しむ可能性を高める[ 65 ]。 -SARS-CoV後の代謝結果が報告されている[ 66 ]。 |
-肥満、メタボリックシンドローム、脂質代謝の変化、糖尿病は、複数のメカニズムを通じて神経変性疾患を発症するリスクを高める[ 67 ]。 |
| HPA軸異常 | – SARS-CoVの-2の急性期におけるサイトカイン産生は、潜在的にHPA軸【刺激することができる68、69 ]。 | -コルチゾールレベルが高いと、タウの過剰リン酸化、アポトーシス、シナプス喪失、ミトコンドリア機能障害が増強される可能性がある[ 70 ]。 |
| 自己免疫反応の遅延 | -SARS-CoV-2はニューロンに潜伏したままである可能性があり[ 11 ]、SARS-CoV-2に対する自己免疫反応は自己免疫神経学的実体を引き起こす可能性がある[ 10 ] [ 71 ]。 | -自己免疫メカニズムは神経炎症を促進する可能性があり、パーキンソン病患者の脳脊髄液で抗CoV抗体が同定されている[ 72 ]。 |
中枢神経系への直接または間接的な侵入:神経侵襲の可能性
SARS-CoV-2は、ウイルスS1スパイクタンパク質と複数の脳組織に豊富に発現しているアンジオテンシン変換酵素2(ACE-2)との相互作用を介してヒト細胞に侵入できることが明らかにされている[73]。以前、ACE-2と同様に結合するSARS-CoV mRNAが、このウイルスに感染した患者の脳組織に見られることが示された[74]。全体として、呼吸困難のレベルの差、一部の患者で観察された神経学的症状、および患者の脳の構造的変化は、ウイルスの神経侵襲性の手がかりとして示唆されている [3, 6, 7, 8, 9, 10, 75] が、脳脊髄液 分析および神経画像学的証拠に基づく研究では、神経侵襲が脳症の原因となる可能性は低いことが示唆されている [37]。
SARS-CoV-2の中枢神経系への侵入には複数の経路が提案されている [2, 76]。
1)SARS-CoV-2と毛細血管内皮のACE-2受容体との相互作用は、内皮炎を引き起こす可能性があり[77]、それによって破壊された血液脳関門(BBB)を介した中枢神経系へのウイルスの侵入が促進される可能性がある。この仮説と一致するように、COVID-19感染者の脳脊髄液(脳脊髄液)中のウイルスRNAおよび髄腔内炎症のマーカーが検出されている [78, 79]。
2)もう一つのウイルス侵入経路として、ウイルスの血行性伝播、またはリンパ管を介したウイルスの伝播が提案されている[80]。最近まで、脳には特徴的なリンパドレナージがないというのが一般的な意見であったが、(Loveau et al 2015)は、SARS-CoV-2の脳内侵入の適切な経路となり得るリンパ管が脳内にいくつか存在することを示している[23]。実際、死後の研究では、様々な組織の内皮細胞がウイルスに感染し、結果として内皮炎がリンパ管や血管を介したウイルスの拡散を促進し、髄膜炎・脳炎を引き起こす可能性があることが明らかになっている[77]。
3)SARS-CoV-2に感染した患者で観察された高率の嗅覚異常と老衰、および一部の患者では感染から回復した後も持続することから、SARS-CoV-2は嗅球を介して神経系に侵入し、さらに軸索輸送を介して脳内に拡散することが示唆されている[2, 81]。さらに、COVID-19患者を対象とした死後の脳MRI研究では、嗅球の非対称性の証拠が観察されたが[6]、最近の研究では、COVID-19患者の嗅覚異常は一過性の浮腫によるものであることが示唆されている[82]。ウイルスの受容体と考えられるACE-2と膜貫通型セリンプロテアーゼ2(TRPMSS2)は嗅上皮に広く発現していることから、ヒト嗅神経細胞を対象とした最近の研究では、ウイルスに感染した嗅神経細胞でTRPMSS2受容体の発現が上昇していることから、嗅神経細胞へのウイルスの侵入はTRPMSS2受容体を介して行われることが示唆されている[29]。一方、パーキンソン病(PD)やアルツハイマー病などの神経変性疾患では、嗅覚機能障害や嗅覚関連領域の構造変化が早期に観察されている[24, 30, 31]。重要なことは、COVID-19からの回復後も持続的な低酸素血症が、本感染症患者、特に高齢者に多く見られることである[25]。したがって、ウイルス感染後の低汗症のメカニズムは十分に解明されていないが [25]、黒質、脳室、大脳皮質のオリゴデンドロサイト前駆細胞およびアストロサイト、および嗅球周囲細胞においてACE2およびTMPRSS2の両方が発現していることから [26,27,28,83]、パーキンソン病またはアルツハイマー病の初期徴候である可能性が示唆されている [2]。臨床的には、この事実は、COVID-19と診断された無症状の患者における無呼吸症の鑑別診断に注意を払う必要があるかもしれない。嗅球がウイルスの侵入経路となる可能性、神経変性疾患と関連した神経炎症過程や神経細胞の蛋白質機械系の変化などの病態生理学的変化、およびこれらの病態との関連性については、今後の研究が必要であると考えられる。
4)感覚ニューロンおよび後根神経節(DRG)ニューロンは、ACE-2だけでなく、TMPRSS2およびFURINなどの他のウイルス受容体を発現することができる[84]。また、SARS-CoV-2は、皮膚の外層または腔内臓器の上皮にある遊離神経末端を介して脳に到達する可能性がある[85]。腸管神経系および迷走神経もまた、患者で観察される消化器症状と相まって、ウイルスの侵入経路の可能性があると考えられている[86]。同様に、パーキンソン病のα-シヌクレイン病理は、腸神経系から始まる同じ進行パターンに従うことが示唆されている[87]。
ウイルスが神経系に入ると、脳組織で広く発現しているACE-2と結合し、脳全体に播種することができる。例として、(Paniz-Monodolfi et al 2020)がCOVID-19を有する患者に対して行った死後電子顕微鏡検査では、前頭葉におけるウイルスの存在が明らかにされている[88]。重要なことに、SARS-CoVは、脳炎を引き起こすことなく、感染したマウスの神経細胞の死につながることが示されている[89]。また、最近では、COVID-19に関連した広範な脳の転写変化が検出されている[12]。前述したように、HCoV感染は他の呼吸器感染と同様に脳、特に側頭部と海馬領域に伝播する可能性があり、ウイルス感染動物モデルでは海馬領域におけるアルツハイマー病特異的病理学的な証拠があるが、それが炎症の結果なのか、ウイルスの直接的な浸潤の結果なのかは明らかではない[20, 21]。さらに、パーキンソン病におけるBraak仮説と病期分類によれば、パーキンソン病患者の脳領域における疾患特異的な病理学的変化は、腸神経系および/または嗅球から始まり、大脳辺縁系および大脳皮質へと拡大するという特徴的な順序で起こるが、このパターンは普遍的なものではなく、これについてはいくつかの議論がある[87]が、これはウイルスの侵入のメカニズムとして提案されているものとほぼ一致している。
サイトカインストーム 神経免疫機構
脳脊髄液中のSARS-CoV-2検出に関する報告はあるが[78]、重症SARS-CoV-2髄膜脳炎の2例ではウイルスRNAが検出されなかったことから、中枢神経系における一過性または未検出のウイルスRNA量を示唆している可能性がある[90, 91]。一方、サイトカインストーム(サイトカインの循環レベルの顕著な増加)は、HCoVファミリーの他のメンバーと同様に、SARS-CoV-2によって誘発された肺への損傷と死を引き起こす主要なメカニズムの1つと考えられている。インターロイキン-6(IL-6)を含むプロ炎症性サイトカインのレベルのアップレギュレーションは、COVID-19患者、特に重症化した患者で検出されている[32, 92]。この観察は、重度の髄膜脳炎または他の神経学的症状を引き起こすのは、ウイルスの直接的な浸潤ではなく、末梢の炎症ではないかという疑問を提起するかもしれない。一貫して、インテルキン-6(IL-6)IL-8,IL-10,および腫瘍壊死因子α(TNF-α)などの高レベルのサイトカインおよび陽性の抗S1 IgMが、COVID-19髄膜脳炎の3例で検出されている [93]。実際、高レベルの末梢性サイトカインはBBBを直接通過して神経炎症反応を開始したり、BBBの完全性を損なうことがあり、ウイルス感染した末梢性白血球、特に単球の透過を可能にし、サイトカインの制御不能な産生、微小グリアの活性化および神経炎症につながる [33, 94]。一方、免疫応答の調節障害は、神経変性疾患の病態生理に関与する主なメカニズムとして認識されており、IL-1βやIL-6などの炎症性サイトカインのレベルの上昇が観察されている[34, 95]。神経炎症が神経変性過程の引き金となるかどうかは明らかではないが,少なくとも前臨床段階の患者では,これらのメカニズムを促進し,神経変性の促進につながることが示されている[95].さらに、高レベルのサイトカインレベルによって特徴づけられる全身性炎症は、重症敗血症で観察されるように、その後の海馬の萎縮に寄与することが明らかにされている[96, 97]。実際、プロ炎症性サイトカインレベルの上昇は、サイトカインおよびウイルスに感染/活性化された末梢性白血球の両方に対するBBB透過性を増加させ、これが常駐する脳ミクログリアを活性化させ、M1神経毒性表現型への成熟を促進する[40]。この持続的なミクログリアの活性化は、他のミクログリアをさらに活性化し、酸化ストレスを促進し、タウの高リン酸化とタンパク質の凝集を促進し、ミトコンドリアの機能不全とアポトーシスを引き起こし、シナプス可塑性と神経伝達を損なう可能性がある[41, 98]。さらに、感染に起因するサイトカインは、脳に到達することでドーパミンの合成を変化させ、パーキンソン病の感受性を高める可能性がある [58]。SARS-CoV-2の症例では、IFN-γを含むインターフェロンを産生するCD8 T細胞の増加が報告されており[93]、SARS-CoV感染では、IFN-αおよびIFN-βがウイルスの繁殖を制限するのに有効であることが示されている[59]。インターフェロンは、他のサイトカインと同様に、ミクログリアを活性化することによってシナプスの喪失を増強することができ[99]、βアミロイドの存在下では、これらのペプチドは宿主免疫系の一部として作用し、ウイルス粒子を巻き込むことができ、インターフェロンによって誘導されるウイルス後の炎症反応を促進する[35]。
サイトカインレベルの上昇は、少なくとも部分的には、コロナウイルスオープンリーディングフレーム3a(ORF3a)タンパク質に起因し、関連する急性肺炎(ALI)によって悪化させることができ、これらのタンパク質は、NOD様受容体タンパク質3(NLRP3)の炎症ソーム活性を誘導し、サイトカイン産生、すなわちIL-1βを促進することが認識されている[36, 100]。一方、全身性の炎症は、NLRP3 インフラマソームの活性化を介して脳の免疫恒常性を損ない、炎症性サイトカイン、特に IL-1β の産生を促進し、病原性フィブリルの形でペプチドの凝集を促進するとともに、ミトコンドリア機能障害やアポトーシスを引き起こし、アルツハイマー病 や PD と同様に神経変性を引き起こす [42, 43, 71] 。このような証拠はすべて、NLRP3 インフラマソームが、SARS-CoV-2 感染による神経変性の影響を引き起こす可能性のあるクロスロードとして機能していることを強調している。
提起されるであろう重要な疑問は、SARS-CoV-2感染が長期的に影響を及ぼすかどうか、そしてその結果として生じるサイトカインストームが持続し、神経変性疾患に関連した神経病理学的変化を引き起こす可能性があるかどうかである。実際、この質問に対する答えは、炎症の基礎となる基盤がどの程度あるか、末梢白血球の脳への潜在的な神経浸潤や移動の程度、あるいはウイルス感染後の分子模倣のようなメカニズムによる自己免疫反応の有無など、複数の要因に依存しているかもしれない。これに関して、COVID-19は、頭蓋神経障害[85]、多発性硬化症[101]、ギラン・バレー症候群[10]、または脳脊髄炎[72]を引き起こす分子模倣機構による自己免疫反応を引き起こすことが知られている。これに伴い、パーキンソン病患者の脳脊髄液サンプルから高レベルの抗コロナウイルス抗体が検出されており[102]、免疫反応による糖タンパク質(S)の変異など、ウイルスの特異的な変異がグルタミン酸興奮毒性を促進し、それによって神経変性に寄与することが明らかにされている[103]。さらに、炎症性条件下、特に高齢者ではBBBの完全性が損なわれ、ウイルスに感染したミエロイド細胞が中枢神経系に移動し、サイトカインの産生やミクログリアの活性化を促すことで神経炎症を持続させることが明らかになっている[104, 105]。時間の経過とともに、これらの白血球はHCoVに感染したままになり、脳内で持続的な炎症の原因として作用し、神経変性環境に寄与することがある [106]。
養子性免疫系と細胞性免疫系 脳内の免疫細胞
COVID-19感染における細胞性免疫系の役割は、最近の研究で強調されている。実際、SARSコロナウイルスの構造抗原および非構造抗原は、感染の重症度に比例してCD4およびCD8 T細胞応答を活性化することが明らかにされている[107, 108]。軽度の場合には、ウイルスに対するCD8 T細胞応答が優勢であるが、重度の場合には、体液性免疫応答とCD4 T細胞が抗ウイルス免疫応答をより顕著に引き継ぎ、それ自体が免疫病理学に寄与している可能性さえある[108]。さらに、ウイルス感染によって誘導される全身性炎症反応は、抗炎症状態を炎症状態に有利に変化させる可能性がある[108]。末梢での細胞性および養子性免疫系の活性化が中枢神経系に影響を与えるかどうかは明らかではないが、ウイルス感染では、BBBの破壊により、白血球がさらにBBBに侵入し、脳内で感染を永続させ、神経炎症過程に至ることが明らかにされている[76]。COVID-19感染では、サンプルサイズが小さい研究では、脳脊髄液サンプル中の白血球の増加などの炎症の細胞マーカーの上昇は観察されなかったが[79]、いくつかのケースでは脳脊髄液サンプル中の多血球症が報告されている[37, 38]。COVID-19感染で死亡した患者の死後病理組織学的解析では、中枢神経系では重度のミクログリア活性化と軽度のT細胞の血管周囲浸潤が認められた [39, 45]。このことは、軽度や中等度の感染症では顕著な免疫細胞の活性化は見られないが、重度の感染症では免疫系の細胞成分が中枢神経系に浸潤していることを示唆しているのかもしれない。先に述べたように、中枢神経系におけるミクログリアの浸潤は、サイトカインの産生異常、神経伝達の障害、さらには海馬の萎縮[97]や神経変性メカニズムの伝播、アポトーシス[41, 98]と関連している。さらに、中枢神経系におけるT細胞の浸潤は、アルツハイマー病 [44]とPD [109]の両方で検出されている。このような免疫細胞の炎症性、細胞毒性の分極化は、COVID-19感染の高齢者ではより起こりやすく、神経変性メカニズムをさらに増殖させ、加齢によるタンパク質の誤集積によってすでに引き金となっている可能性がある。全体として、感染の異なる段階で、また軽度または重度の感染形態では、細胞免疫応答が可変であるため[108]、神経変性過程に対するこのような変化の影響は、特に長期的には明確に予測することができない。
ウイルスタンパク質の相互作用 タンパク質の凝集とミトコンドリア機能不全
α-シヌクレインを発現するドーパミン神経細胞をH1N1などの呼吸器ウイルスに感染させると、α-シヌクレイン凝集体の形成とオートファジーの抑制が起こり、これがパーキンソン病における神経変性の促進に関与する主なメカニズムとなっている[111]。一方、SARS-CoV-2タンパク質は、ユビキチン化、ミトコンドリア活性、RNAプロセシング、小胞輸送などの老化に関連するタンパク質を含むヒトタンパク質と相互作用することができる[55]。SARS-CoV感染は、ウイルスタンパク質合成のための小胞体(ER)機械を乗っ取ることができ、ERシャペロンと相互作用することでスパイクタンパク質がPERK経路を活性化し、その結果、グルコース制御タンパク質78(GRP78)やGRP94を刺激してタンパク質のアンフォールド応答(UPR)を誘導し、タンパク質のミスフォールディングを促進することが明らかにされた[56]。一方、SARS-CoVのオープンリーディングフレーム9b(ORF-9b)は、宿主細胞のオートファジーを誘導するとともに、ユビキチン化を誘導し、ミトコンドリアのプロテオスタシスを阻害する可能性がある[112]。さらに、SARS-CoVタンパク質であるORF-3a、ORF-3b、ORF-6,ORF-7aは、カスパーゼ-3やERストレス、ヤヌスキナーゼ(JNK)経路を介して宿主細胞のアポトーシスを誘導することが知られている[113]。SARS-CoV-2ではまだそのようなエビデンスは得られていないが、これらのウイルスの構造的・機構的挙動の類似性から、ERストレスを標的としたSARS-CoV-2の治療法が示唆されている[114, 115]。したがって、COVID-19の感染は、タンパク質の機械を乗っ取ることで神経細胞に入り込み、ERとミトコンドリアの機能を破壊し、ミスフォールディングされたタンパク質の蓄積を増加させることで、ミトコンドリアの酸化ストレスを誘発し、神経細胞のアポトーシスと変性に寄与している可能性がある[55, 60, 116]。
腸内マイクロバイオーム。免疫系と消化管の共通メカニズム
最近のシステマティックレビューおよびメタアナリシス研究によると、COVID-19感染者の約7.8%および5.5%が、それぞれ下痢および吐き気・嘔吐などの消化器症状を経験し、さらに腹痛やGI出血などの他の症状も経験している。これは、そのメカニズムはよくわかっていないが、おそらくACE受容体を介して、GI上皮へのウイルスの浸潤を示唆している[52, 117]。重要なことに、感染した患者の糞便中のウイルスの脱落は、感染後5週間以上で観察されている[53]。さらに、感染を含む様々な遺伝的・環境的要因が腸内細菌叢の構成の鍵を握っており、COVID-19患者ではそのような変化が検出されている[54, 118]。一方、先に述べたように、病原体が末梢神経終末を介して中枢神経系に侵入し、神経変性に寄与している可能性があり、腸内細菌叢の異常がウイルスの拡散や中枢神経系への侵入に大きな役割を果たしている可能性が機械論的に示唆されている。さらに、動物モデルやヒトでの知見に基づき、消化管病変や腸管上皮の透過性の亢進などの消化管の変化は、神経変性疾患に特異的な病態が発症する数十年前に起こり、神経変性疾患の発症に寄与している可能性が示唆されている[119]。
腸内細菌叢の異常がウイルスの中枢神経系への直接的な侵入に寄与している可能性に加え、腸管透過性の亢進、分子模倣、酸化ストレスなどのメカニズムにより免疫系の活性化が亢進し、神経伝達バランスの変化とともに神経変性疾患の発症に寄与している可能性がある[119]。このような事実を考えると、SARS-CoV-2感染は、腸内細菌叢の変化により神経変性疾患を発症させる可能性があると考えられる[119]。しかし、腸内マイクロバイオームの組成の潜在的な変化が永続的なものであるかどうか、また、感染の重症度や消化器症状の有無に依存するかどうかは明らかではない。これに関して、COVID-19の消化器症状に関する最近のシステマティックレビューでは、重症型と非重症型の消化器症状の有病率に有意な差がないことが示されている[52]。
さらに、腸内細菌叢の組成は免疫学的平衡を維持する上で重要な役割を果たしており、COVID-19を含む感染性および炎症性疾患の罹患率の決定因子であることが示唆されている[48, 120]。例えば、重度のCOVID-19感染症や神経変性疾患の共通の危険因子として、高齢になると腸内細菌叢の多様性が低下することが指摘されている[120]。この事実は、腸内細菌叢の多様性がCOVID-19感染と神経変性疾患の発症の共通の危険因子として作用するため、COVID-19感染から生き残った患者では神経変性疾患を発症する確率が高くなることを示唆しているのかもしれない。
腸内マイクロバイオーム、COVID-19感染症、神経変性症との関連を考慮すると、個別化された栄養改善や糞便移植法がCOVID-19感染症の予防的役割を持ち、COVID-19感染症の既往歴のある患者の神経変性症のリスクを軽減するのにも有益であることが明らかになるだろう[120]。
アンジオテンシン変換酵素。神経保護および神経毒性の特徴
アンジオテンシン変換酵素(ACE)は、ACE-1とACE-2から構成され、レニン-アンジオテンシン系(RAS)の重要な構成要素である。ACE-2は多くの細胞型に発現する膜結合酵素であり、アンジオテンシンIIをアンジオテンシン(1-7)などのより小さなタンパク質に切断し、Mas受容体に結合してACE-2/アンジオテンシン(1-7)/Mas軸を形成する役割を担っている[49]。前述したように、ACE-2は、SARS-CoV-2スパイク蛋白質と結合する受容体として作用し、それによって細胞への侵入を可能にする[51]。SARS-CoV-2とACE-2の結合はACE-2枯渇を引き起こす可能性があり、ACE-2/アンジオテンシン(1-7)/Mas軸が保護作用を持つことが知られているように、多臓器傷害をさらに永続化させる可能性がある[50]。一方、神経変性におけるACE-2/アンジオテンシン(1-7)/Mas軸の保護的役割は、酸化ストレス、神経炎症、アポトーシスの増強に関与し、それによって神経変性に寄与することが示されているACE-1とアンジオテンシンIIとは対照的に、神経変性におけるACE-2/アンジオテンシン(1-7)/Mas軸の同様の保護的役割が取り上げられている[121]。ACE-2およびアンジオテンシン(1-7)活性の低レベル、およびACE-1活性およびアンジオテンシンⅡの高レベルは、対照群と比較してアミロイドβおよびリン酸化タウの高レベルと関連しており[68]、また、動物のADモデルにおける同様の証拠[69]も検出されている。
神経細胞の終末におけるACE-2とその下流軸の役割とは別に、遺伝子発現研究により、ACE-2遺伝子は、L-3,4-ジヒドロキシフェニルアラニン(L-DOPA)のドーパミンへの変換に関与するドーパミン脱炭酸酵素との最も有意な共発現を示すことが明らかになった。このことは、SARS-CoV-2感染によって誘導されるACE-2のダウンレギュレーションは、パーキンソン病の病態生理に関与するドーパミン合成経路の変化と関連している可能性を示唆している[122]。さらに、ACE-2遺伝子多型は、COVID-19感染および多臓器不全などの合併症に対する感受性に影響を及ぼすことが示されているが、正確な関連性は明らかにされていない[123, 124]、一方で、特定のACE-2遺伝子多型はPD[125]およびアルツハイマー病[70]のリスクを潜在的に増加させる可能性がある。
したがって、アンジオテンシンIIレベルを低下させるACE阻害薬は、神経変性疾患の進行を予防することが期待されている[121, 126]。その後、再び問題となるのは、ACE-2レベルのこのような変化が持続するかどうかである。さらに、ACE阻害剤の投与により、重症COVID-19感染症患者における臓器損傷の永続と神経変性疾患への進行の可能性を食い止めることができるかどうかは、依然として疑問である。
視床下部-下垂体軸:神経内分泌機構
ウイルス感染症の急性期における免疫系の過剰活性化とサイトカイン産生は、視床下部-下垂体-副腎皮質(HPA)軸を刺激すると推測されている[68, 69]。この現象は、サイトカインストームの劇症的な影響を防ぐために、炎症性細胞の活性化と過剰な量のサイトカインの産生をダウンレギュレートするグルココルチコイドの分泌量の増加につながる[122]。ウイルス感染に対する生理的反応としてのグルココルチコイド産生の増加に加えて、COVID-19を有する重症患者における外因性グルココルチコイドの投与は、これらの患者におけるグルココルチコイドの増加レベルをさらに増加させる可能性がある。重要なことに、グルココルチコイドの長期投与は、無数のメカニズムを介して神経変性プロセスを促進することが知られている。グルココルチコイドの障害は、ミトコンドリアの機能障害を引き起こし、アポトーシスを促進し、重要な細胞骨格タンパク質であるタウの過リン酸化を活性化し、それによってシナプスの喪失および神経伝達の障害に寄与することがある[127]。これと一致するように、de novo パーキンソン病患者では、HPA軸の異常が一般的な所見として観察されている[128]。
一方、SARS-CoVは副腎皮質刺激ホルモン(ACTH)と構造的に類似したアミノ酸配列を発現し、抗体を形成することで宿主の免疫応答が内因性ACTHを破壊することができる[129]。さらに、SARS-CoVに感染すると、肺や心臓などの様々な組織でACE-2の発現が低下することが示唆されている[130, 131]。一方、視床下部のコルチコトロピン放出ホルモン(CRH)産生細胞におけるACE-2の過剰発現は、HPA軸の活性化の低下と関連している[132]。SARS-CoV-2感染がこれら視床下部ニューロンにおけるACE-2発現を低下させるかどうかは明らかではないが[133]、もしそうであれば、このメカニズムはさらにHPA軸の活性化につながると考えられる[132]。ウイルス感染の急性期に記載されたHPA軸の変化にもかかわらず、SARS-CoVからの回復後3ヵ月後には、ウイルス感染前やウイルス感染中のHPA軸の異常を伴わないHPA軸の低活性化を示す患者のサブセットが存在した[134]。この現象は、トランスフォーミング成長因子β(TGF-β)やTNF-αなどのサイトカインに起因する可能性があり、これらのサイトカインは特定の状況下でHPA軸の活性化をダウンレギュレートすることが示唆されており、重度のSARS-CoV-2感染に関与していることが示されている[135, 136]。しかし、COVID-19感染によるHPA軸機能への影響がSARS-CoVと類似しているかどうか、TGF-βがそのような影響を媒介しているかどうか、そしてHPA軸に対するこの感染の長期的な影響があるとすればどのようなものであるかを決定するためには、さらなるデータが必要である。
低酸素と血栓症 脳への全身的または局所的な傷害
COVID-19の最も重篤な臨床症状と考えられている急性呼吸窮迫症候群(ARDS)は、特に長期の機械的換気を必要とする場合 [137] 、生存者では退院後数年経っても、その後の認知機能障害および実行機能障害の発生率が高い [138]。ARDS患者における認知症および/または進行性神経変性の発症リスクは、既往の神経学的損傷、認知症、アミロイドβ病理またはせん妄、およびそれに伴う低灌流または低酸素血症の存在、サイトカインレベル、およびBBBの完全性の欠如などの複数の因子に依存する[138]。実際、ARDSに関連した低酸素血症、全身性炎症/敗血症、およびBBB透過性の亢進は、脳損傷に寄与することが知られている主なメカニズムである。これと関連して、COVID-19患者を対象とした最近の死後調査では、脳組織内でのSARS-CoV-2感染は最小限であったにもかかわらず、大多数の患者で低酸素障害と一致する神経病理学的変化が示されている[5]。また、最近では、中等度のSARS-CoV-2感染者でも、グリア線維性酸性タンパク質(GFAP)とニューロフィラメント軽鎖タンパク質(NFL)のレベルが上昇し、それぞれアストロサイト反応と軸索損傷の指標となることが明らかにされている[139]。それにもかかわらず、海馬の損傷、特にCA1およびCA2における損傷は、低酸素血症よりもむしろ高レベルのサイトカインに起因することが明らかにされている[140]。実際、末梢性サイトカインと敗血症のレベルのアップレギュレーションは、BBBの上皮細胞の機能の変化とアミロイドβペプチドの蓄積とアルツハイマー病病理学[141]に続く傾向に関連している。先に述べたように、敗血症はまた、脳内の神経炎症過程を誘発し、グルコース代謝と神経伝達を変化させ、酸化ストレスなどの全身性の障害に対する脳組織の脆弱性を増加させる可能性がある[142]。同様に、敗血症性ショックは、特にハイパーカプニアと関連している場合には、脳血行の自己調節機構を障害し[143]、低酸素血症をさらに助長する可能性がある。ARDSとそれに伴う血行動態の変化とは別に、中枢神経系の局所血管閉塞は、全身の炎症により末端臓器に微小血栓症が形成される傾向のある敗血症誘発性凝固障害によって引き起こされる可能性があり、重篤なCOVID-19患者では顕著であり、脳循環に障害をもたらし、低酸素-虚血変化を引き起こす [144, 145]。さらに、SARS-CoV-2はACE-2と結合することでACE-2枯渇を引き起こす可能性があり、ACE1/アンジオテンシンIIへと平衡状態を移行させることで、その後の血管収縮はさらに灌流低下に寄与する可能性がある[144]。前節で述べたように、ACE-2はアンジオテンシンIIを切断してアンジオテンシン(1-7)に変換するが、これは血管拡張を誘導するとともに、Mas受容体に結合することで抗炎症作用を発揮し、神経保護因子として認識されている[146]。重要なことは、アルツハイマー型認知症をはじめとする神経変性疾患は、慢性的な脳低灌流や血管イベントによって引き起こされる可能性があることが明らかになってきたことである[46]。さらに、血栓症や内皮機能障害は、抗酸化活性の低下と関連しており、低酸素とともに酸化ストレスや炎症性サイトカインの産生を引き起こし、神経変性の主な原因となっている[47]。酸化ストレスは、神経炎症およびその他のメカニズムを介して、パーキンソン病におけるα-シヌクレイン凝集[147]、アルツハイマー病におけるβ-アミロイドペプチド蓄積[62]を増強する可能性がある。
APOEと脂質代謝。共有された遺伝的および代謝的危険因子
せん妄および意識障害は一般的な臨床所見であり、特にCOVID-19の重症症例では[75]、既往の認知症は死亡率を増加させるだけでなく、COVID-19感染の重症化リスクを約3倍に増加させることが認められている[63]。さらに、COVID-19で報告されている症状の大部分が遺伝性であることが決定されており、特にせん妄はホモ接合性双生児を対象とした研究によると50%の遺伝性があると報告されている[61]。興味深いことに、ApoE e4-e4遺伝子型は認知症およびせん妄のリスクの有意な増加と関連しており[64]、ApoE e4ホモ接合体は、既存の認知症や他の併存疾患とは無関係に、COVID-19が陽性である可能性が高く、より重篤な疾患を経験する可能性が高いことが示されている[66, 148]。アルツハイマー病とCOVID-19のリスクを増加させる同じ遺伝的素因は、COVID-19を生き残った患者のアルツハイマー病型認知症発症リスクの高さを少なくとも部分的に説明する交絡因子を構成しているかもしれない。
メカニズム的には、高レベルの血中コレステロールがApoE受容体に結合することで、ACE-2受容体を介したSARS-CoV-2の細胞への侵入が改善されることが示されている[149]。一方、ApoE e4遺伝子型はマクロファージの炎症促進状態を好み、神経細胞膜およびミエリン鞘の維持のための必須脂肪酸の送達効率が低く、また、誤ったタンパク質の蓄積を促進し、樹状突起棘の形成およびシナプス可塑性を混乱させる[150]。
中国で行われたSARS-CoV生存患者を対象としたある研究では、感染から回復してから 12年後、患者は代謝変化、特にホスファチジルイノシトールやリゾホスファチジルイノシトールのレベルの上昇、高インスリン血症、グルコース代謝障害、心血管系異常を伴う高脂血症などの脂質代謝障害を発症したことが示されている[151]。このような代謝変化は、メチルプレドニゾロンの高用量パルスとの関連が示唆され、肺の初期障害の重症度も示唆された[151]。また、ウイルスの細胞への結合は、2型糖尿病(2型糖尿病)の既往歴のない50%の患者において、SARS-CoV感染中に急性2型糖尿病を引き起こしたが、感染解消後も糖尿病を維持したのはわずか5%であり、血糖値は兄弟姉妹と比較して有意な差はなかった[67]。DMのような恒久的な代謝変化は、膵臓の損傷や敗血症の結果とそれに伴う代謝変化のために起こる可能性がある退院後の合併症を伴う重症患者で特に観察された[65]。さらに、自己免疫機構やウイルス感染後の膵島細胞の損傷による1型糖尿病(1型糖尿病)は、これらの患者で発生する可能性のある現象である[67]。さらに、ARDSや低酸素症後の長期的な認知機能障害[138]、うつ病や心的外傷後ストレス障害(PTSD)[152]は、エピソード記憶力や作業記憶力の低下、コントロールできない摂食行動と関連しており、体重増加につながる可能性がある[153]。SARS-CoV-2感染後のこのような代謝変化の発生はまだ確立されていないが、予想されるべきである。重要なことは、肥満、メタボリックシンドローム、脂質代謝異常、糖尿病は神経変性疾患の発症確率を高めることが知られていることである[154]。
また、2型糖尿病患者の血糖値の上昇は、インターフェロン調節因子5(IRF5)活性のアップレギュレーションを介して、フィブリル形成やアルツハイマー病病理を悪化させる可能性がある[35]。一方、高血糖値に反応してIRF-5が増加すると、COVID-19感染症への感受性が促進される[35]。さらに、肥満やメタボリックシンドロームがCOVID-19感染症への罹患率を高めることが最近明らかになっている[155]。このように、COVID-19感染後の代謝変化率が有意に増加しないと仮定しても、既存の代謝異常を有する患者はCOVID-19の発症リスクが高く、より重症化することが認められており[156]、また、COVID-19を生き残った場合には、同じ集団が神経変性疾患を発症するリスクが高いことを考慮することができる[154]。
結論
この記事全体で議論し、表1,図1および図2にまとめたように、SARS-CoV-2の直接または間接的な中枢神経系への侵入、および末梢白血球の脳への移動および浸潤は、サイトカインの産生を誘導し、ミクログリアを活性化し、それらのM1表現型への分極を誘導し、それによって神経変性過程の伝播に寄与する。また、ウイルスの複製は、神経細胞のタンパク質合成機構をハイジャックし、アンフォールドタンパク応答を促進し、プロテオスタシスを阻害することで、誤ったタンパク質蓄積を誘発する可能性がある。一方、敗血症やサイトカイン産生の亢進は、HPA軸やコルチゾール分泌を亢進させ、タウの高リン酸化をさらに促進させる。SARS-CoV-2の過剰な占有によるACE-2枯渇とそれに伴うACE-2/アンジオテンシン(1-7)/Mas軸活性の低下は、神経保護メカニズムを減衰させ、神経変性を促進すると考えられる。さらに、ARDSや敗血症による高凝固は、脳を低酸素障害に陥れ、酸化ストレスの原因となり、神経変性過程を促進することが知られている。また、APOE e4-e4遺伝子型などの遺伝的変異や、糖尿病や高脂血症などの代謝性危険因子など、COVID-19感染症や神経変性疾患発症の危険因子が共通していることから、回復した患者の神経変性疾患発症リスクが高いことが示唆されている。
図1 SARS-CoV-2 は中枢神経系への侵入を介して神経変性に寄与する
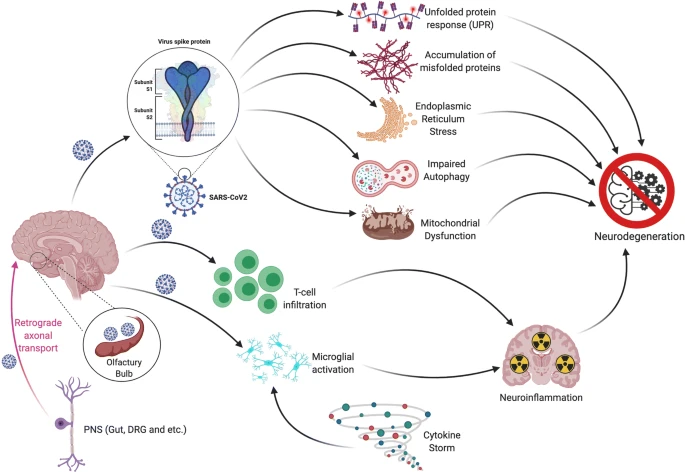
SARS-CoV-2は嗅球に直接侵入し、末梢神経末端からの逆行性軸索輸送を介して脳に侵入するか、あるいは血行性経路を介して脳に到達する。ウイルスの侵入は、敗血症による末梢性サイトカインの過剰産生とともに中枢性サイトカインの産生を亢進させ、ミクログリアの活性化を促進し、T細胞の浸潤とともに神経炎症を引き起こし、神経変性の一因となることが明らかになった。さらに、SARS-CoVスパイクタンパクは小胞体(ER)アンフォールドタンパク応答を促進し、ミトコンドリアのオートファジーとプロテオスタシスを障害し、ミスフォールドタンパク蓄積とアポトーシスを引き起こすことが示されている。
図2 SARS-CoV-2の全身作用を介した神経変性への寄与
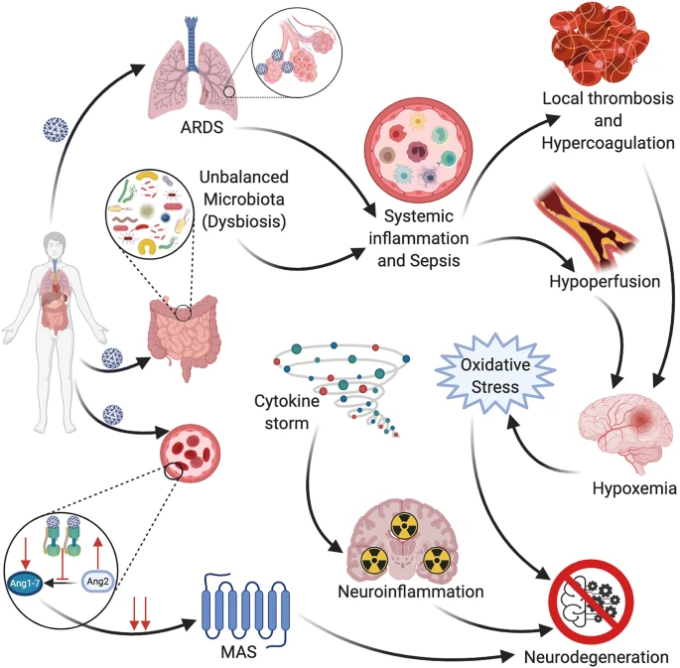
アンジオテンシン変換酵素2(ACE-2)をウイルスが占有すると、神経保護作用を発揮するACE-2/アンジオテンシン(1-7)/Mas軸の活性が低下する。
急性呼吸窮迫症候群(ARDS)に伴う低酸素血症は、敗血症に伴う血液凝固の亢進を伴い、脳血管に局所血栓症を形成する傾向があるため、低灌流を引き起こし、酸化ストレスを悪化させ、神経変性を促進する。
このようなメカニズムが持続するかどうか、また、疾患の重症度が変化する患者における神経変性のプロセスをどの程度加速させることができるかどうかは明らかではない。また、特に重症化した患者や、神経変性のリスクを高める併存疾患を有する患者では死亡率が高く、他の併存疾患による早死にもつながることから、神経変性疾患の全体的な有病率・発生率は今後数年の間に上昇しない可能性がある。全体として、現時点では、これらの関連性に関する研究は仮説に基づいたものであり、この点でのエビデンスを得るためには、プロスペクティブなコホート研究を計画する必要がある。また、神経変性疾患の発症リスクが高まることは必然ではなく、例えば、ビタミンDの補給による神経変性機序の修飾が示唆されている[157]。同様に、ACE阻害剤を投与してACEやRAS系を標的とすることは、神経変性を食い止めるための有望な方法であると考えられる。