
Overview of Castleman disease
pubmed.ncbi.nlm.nih.gov/32106302/
キャッスルマン病(CD)は、病理学的に特徴的な症状を持つ少なくとも4つの疾患群であり、病因、症状、治療法、転帰が多岐にわたる。CDには一中心性CD(UCD)と多中心性CD(MCD)があり、後者は特発性MCD(iMCD)ヒトヘルペスウイルス8(HHV8)関連MCD(HHV8-MCD)多発性神経炎、臓器肥大、内分泌障害、単クローン性形質細胞障害、皮膚変化(POEMS)関連MCD(POEMS-MCD)に分類される。iMCDはさらに、iMCD-thrombocytopenia, ascites, reticulin fibrosis, renal dysfunction, organomegaly (iMCD-TAFRO)、またはiMCD-not otherwise specified (iMCD-NOS)に分類される。診断、分類、病因、治療法の進歩は目覚しいものがある。
1954年にBenjamin CastlemanがUCDを発表して以来、診断、分類、病因、治療法は大きく進歩した。効果的なレトロウイルス療法の登場と、HHV8-MCDにおけるリツキシマブの使用により、HHV8-MCDの治療成績は向上している。抗インターロイキン6療法は、多くのiMCD患者に非常に有効であるが、難治性の症例には追加の治療が必要である。最近の進展の多くは、Castleman Disease Collaborative Network (CDCN)によって調整されたものであり、医師、科学者、患者が継続的に関与することによって、さらなる進展が期待される。また、CDCNのACCELERATE自然史登録(#NCT02817997;
www.CDCN.org/ACCELERATE)への患者の自己登録を促すことでも進展が期待されている。(Blood. 2020;135(16): 1353-1364)
はじめに
キャッスルマン病(CD)は、特徴的な病理組織学的特徴のスペクトルを共有するものの、病因、症状、治療法、転帰が多岐にわたる少なくとも4つの疾患群を表する。CDは、1950年代にBenjamin Castlemanによって、局所的な縦隔リンパ節の腫大として最初に記述された。その特徴は、胚中心が消失したリンパ濾胞の数の増加と、濾胞および濾胞間内皮過形成を含む顕著な毛細血管の増殖である1。1980年代半ばには、CDは、単一のリンパ節またはリンパ節領域が腫大した単中心性CD(UCD)と、複数のリンパ節ステーションが腫大した多中心性CD(MCD)に分けられた5,6。また、1980年代から 1990年代にかけて、PC新生物多神経症、臓器肥大、内分泌障害、単クローン性形質細胞障害、皮膚変化(POEMS)症候群(高槻やCrow-Fukaseとしても知られている)とMCDの併発・重複が指摘され、その後、POEMSの原因となっている単クローン性PCがこれらの症例のMCDの原因となっていることが提唱された。ヒトヘルペスウイルス8(HHV8)は、1990年代にすべてのHIV1および一部のHIV2のMCDの病因として同定された。2010年代に入ると、高井らは、HHV8、2の重症型である特発性MCD(iMCD)を認識した。この患者は、血小板減少症、腹水、網状線維症、腎機能障害、臓器肥大(TAFRO)症候群と呼ばれる臨床検査値の異常と臨床症状が同質的に存在していた9,10。最近、Castleman Disease Collaborative Network(CDCN)は、UCDとMCDの命名法を維持しつつ、MCDを病因別ドライバー(HHV8関連MCD[HHV8-MCD]、POEMS関連MCD[POEMS-MCD]、iMCD)に、iMCD内では表現型、iMCD-TAFRO、iMCD-not otherwise specified(iMCD-NOS)に分類する分類法を提案した(図1)11。
疫学
CDの疫学はあまり研究されていない。MCDでは男性の方が女性よりもわずかに多く発症するが、UCDでは男女差はない。UCDの平均診断年齢はMCDよりも若く(第4世代)MCDよりも若く(第6世代)なっているが、幼児を含む全ての年齢層の患者がいずれかのCDと診断される可能性がある5,12-14。
UCD、POEMS-MCD、iMCDの危険因子は知られていない。免疫不全はHHV8-MCDの主要な危険因子であり、HIVはHHV8-MCDの原因となる最も一般的な慢性免疫不全状態の1つです15。HHV8-MCDのその他の危険因子としては、出身国、21 血縁関係、胸腺腫、慢性ウイルス性肝炎、臓器移植、男性との性交渉などが挙げられる17。
形質細胞診
米国におけるUCDおよびMCDの推定年間発症率は4300~5200人であるが、他の研究ではもっと低い発症率が推定されている22-24。抗レトロウイルス薬併用療法時代に発症率が低下したカポジ肉腫とは異なり、HIV1 HHV8-MCDの認知度は高まっている。HIV1感染者を対象とした大規模な前向きデータベースでは、24名のHHV8-MCDが確認され、全体の発症率は10,000患者年当たり5.3(95%信頼区間、2.4, 6.4)であった25。
図1. CDの分類
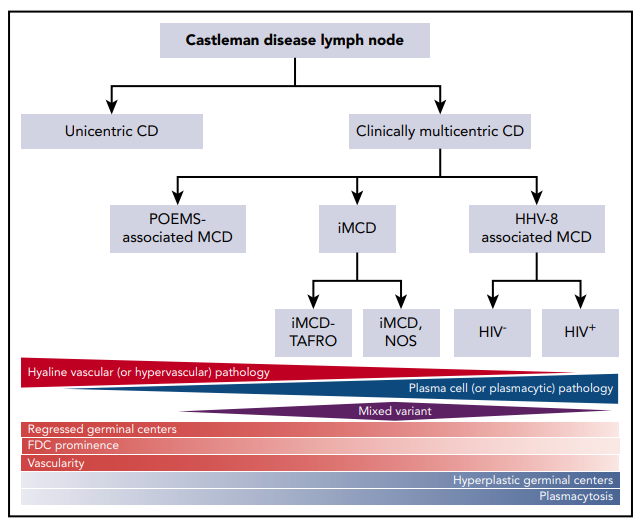
三角形と菱形は、CDのさまざまなサブタイプの中で、これらの病態のそれぞれが発生する相対的な頻度を示す。赤と青の相対的な濃淡は、それぞれの病態タイプに多血性または形質細胞性の病態がどの程度含まれているかを示している。FDC, follicular dendritic cell(濾胞性樹状細胞)。Fajgenbaumら11から引用。
病因
最近の研究では、UCDのリンパ節組織の次世代シークエンスを用いて、約20%の症例で血小板由来成長因子受容体bの体細胞変異が発見された27;この変異はCD452細胞に局在しており、これは間質細胞であると考えられる。In vitroの実験では、この変異が増殖や生存に有利な機能を獲得していることが確認された。
HHV8-MCDでは、制御されていないHHV8感染が病因となっている28。免疫不全の人では、HHV8はリンパ節の形質細胞で複製され、ヒトのIL-6を含む他のサイトカインのカスケードとともに、症状、徴候、リンパ節の病理を引き起こすインターロイキン-6(IL-6; vIL-6)のウイルスホモログを転写する。 15 HHV8は、免疫グロブリンM陽性(IgM1)のナイーブB細胞を、胚中心反応を経ることなく形質細胞に分化させることができるのではないかと推測されている29。ヒトのIL-6の発現は、胚中心の濾胞樹状細胞とPCに局在する30,31。一方、vIL-6はマントルゾーンと濾胞間領域に局在する19,31,32(最近の優れたレビューを参照)33。
HHV-8、2/iMCDの病因および病態は、HHV8-MCDやPOEMS-MCDに比べて理解されていない。IL-6やIL-6受容体の抗体34,35によるiMCDの症状の消失や、マウスにおけるIL-6の過剰発現によるiMCDの表現型の再現などから、一部の患者ではIL-6が重要な病因であることがわかっている36。また、血管内皮増殖因子(VEGF)もiMCD患者では上昇しているが、POEMSに比べて低い37。一部の患者ではIL-6がiMCDの病因の原動力となっているが、iMCDではIL-6が普遍的に上昇しているわけではなく、iMCD患者の約1/2はIL-6の阻害に反応しない。これらの症例には、代替のサイトカインやシグナル伝達経路が関与していると考えられる。最近では、活性化T細胞と哺乳類ラパマイシン標的経路の活性化が重要な役割を果たしていることが確認されており、哺乳類ラパマイシン標的阻害剤の使用が早期に成功している38。
iMCDにおけるIL-6やその他のサイトカインの増加の原因は不明である。iMCDが自己免疫疾患、感染症、またはクローン性疾患であるかどうかに関する推測は数多くあるが、データは限られている39。iMCDがHHV8以外のウイルスの感染によって引き起こされるという仮説は、信憑性が低くなってきている40。25名のCD患者を対象に、RNAハイブリッド、ディープシーケンス、バイオインフォマティクスのヴィロムキャプチャーシーケンスのプラットフォームを用いて、脊椎動物のウイルスを探索した40。新種のウイルスは発見されず、UCDやiMCDと既知のウイルスとの間に明確な関連性は見られなかった。病原体仮説をさらに検証するために、既知の全てのウイルス、バクテリア、真菌、寄生虫のヌクレオチド配列を検出する直交法を用いて、CDサンプルの分析が行われている。最後に、年齢をマッチさせた対照群と比較して悪性腫瘍のリスクが高く、ホジキンリンパ腫や骨髄線維症などの悪性腫瘍と臨床病理学的に重複していることから、iMCDを駆動する腫瘍細胞の仮説は魅力的である41,43。
図2 CDの病理組織像 (A-B) HVの組織病理学的特徴
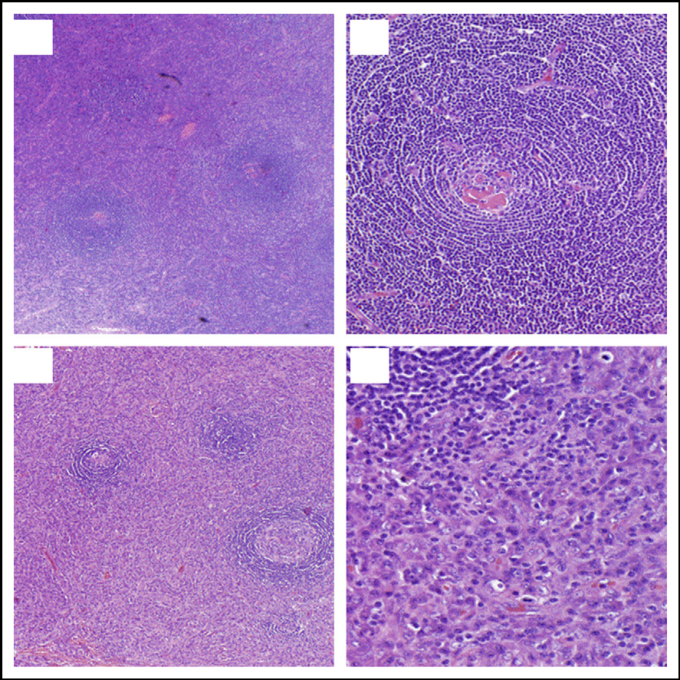
(A)低倍率(ヘマトキシリン・エオジン染色、原寸320)、(B)高倍率(ヘマトキシリン・エオジン染色、原寸3100)。UCDに最もよく見られるこのリンパ節は、しばしばリンパ節を横断する幅広い線維性バンドを伴う被膜線維化を特徴とする。増加したリンパ濾胞が皮質と髄質に散在し、しばしば0.1個の胚中心が同じマントルゾーンを共有している。マントルゾーンは広く、小さなリンパ細胞の同心円から構成されている(「オニオンスキンパターン」)。胚中心はしばしばB細胞を失い、主に濾胞性樹状細胞で構成されており、顕著なヒアルロン酸沈着を伴う。胚中心内には硬化した血管が入り込み、いわゆる “ロリポップ病変 “を形成しているのが観察される。卵胞樹状細胞は、異形成の特徴を示すことがある。毛包間領域は、ふっくらとした内皮細胞を有する顕著な高い内皮静脈で構成されており、しばしば形質細胞性樹状細胞のクラスターと間質の増殖に囲まれている。毛包間浸潤にはPC、免疫芽細胞、好酸球も含まれるが、PCのシートは見られない。(C-D) PCの病理組織学的特徴。(C)低倍率(ヘマトキシリン・エオジン染色、原寸320倍)(D)高倍率(ヘマトキシリン・エオジン染色、原寸3100倍)。PCの組織病理学を示すリンパ節は、濾胞間領域にPCのシートが存在することで区別される。濾胞間領域には、顕著な高内皮静脈瘤が存在することもある。また、好酸球やマスト細胞の存在も確認できる。卵胞/胚中心の過形成が見られ、マントルゾーンと胚中心の極性が明確に定義され、頻繁な有糸分裂と核破片を伴う組織球が見られる。
モノクローナルPCは、POEMS-MCDの病因と考えられる。POEMS-MCDと古典的POEMS症候群との間で、細胞やサイトカインのプロファイルがどのように異なるのかは正確には分かっていないが、PCの体細胞変異によるVEGFやIL-12の過剰産生が、古典的POEMS症候群のドライバーとして確立されている44。45 驚くべきことに、POEMS患者のPCでは、VEGFAの発現は増加していなかった。一方、Wangらは、POEMS症候群の患者において、骨髄中のCD1381細胞(PC)は、CD1382細胞よりも高いレベルのVEGFメッセンジャーRNAの発現を示した46。
病理組織学
CDの4つのサブタイプを診断するためには、まず、古典的なCDの病理組織学的所見と一致するリンパ節生検標本を切除する必要がある。しかし、HV型、PC型、および混合型の組織学的サブタイプに見られる特徴は、簡単に定義できる3つのグループに当てはまるというよりも、むしろスペクトラムのようになっている。2017,iMCDの診断基準を確立するために、複数の病理学者と専門家の合意に基づいた論文が発表された(図1および図2)11。
専門家パネルは、iMCDの病理組織学的特徴のスペクトルを次のように定義した。すなわち、退行した胚中心と顕著な血管形成を有する患者は、スペクトルの過血管側に属すると考えられ、顕著な形質細胞症を伴う過形成の胚中心は、スペクトルの形質細胞側に属すると考えられ、両方の特徴が重なる患者は、混合した病理組織学的特徴を示すと考えられたのである。これらの病理学的所見は、iMCDのコンセンサス診断基準に組み込まれた(表1)。
iMCDグループの名称をHVからhypervascularに変更した理由の一つは、病理学者が「hyaline vascular」という用語をUCDと関連付けることが多いが、これらの病理学的特徴を持つiMCD患者が存在することを認識していたからである。各変異は幅広い臨床的特徴と関連しているため、病理組織学は患者がCDであるかどうかを判断するために使用されるべきであるが、それだけでは臨床管理を導くことはできない。
HV(またはハイパーバスキュラー)の病理組織学的特徴
最も一般的にUCDに見られるHVの病理組織は、リンパ節を横断する幅広い線維帯を伴う被膜線維化、退行した胚中心を持つリンパ濾胞の数の増加、同じマントルゾーン内の胚中心の数が0.1個であることが多いことが特徴である(図2A-B)3,47-49 ハイパーバスキュラーは、iMCDに見られるこれらの特徴を説明するために使用される用語である。このhyper-vascular pathologyは、iMCD-TAFROの患者にしばしば見られる。
表1. iMCDの診断基準
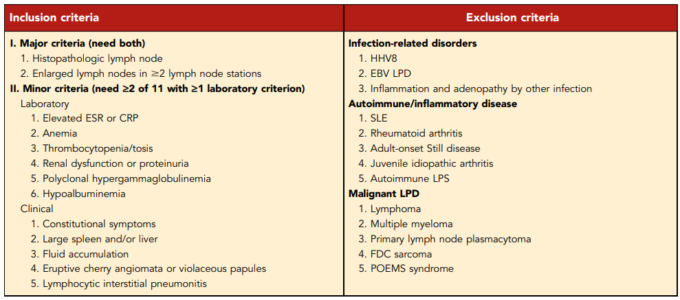
診断に必要ではないが、支持される特徴には以下のものがある。IL-6、可溶性IL-2受容体(sIL-2R)、VEGF、免疫グロブリンA(IgA)、IgE、乳酸脱水素酵素(LDH)、および/またはb2-ミクログロブリン(B2M)の上昇;骨髄の網状線維症(特にTAFRO症候群の患者において)。腫瘍随伴性天疱瘡、閉塞性細気管支炎-組織化肺炎、自己免疫性細胞減少症、多発性神経炎(POEMSと診断されない場合)、糸球体腎症、炎症性筋線維芽細胞性腫瘍。Fajgenbaumらより転載11
PC(または形質細胞)の組織病理学
3,50,51 PCの病理組織は、HHV8-MCD、iMCD、POEMS-MCDで最もよく見られるが、UCDではほとんど見られないことから、PCの病理組織を示すリンパ節は区別される。
HHV8-MCDの症例では、他のMCDタイプでは見られない微妙な変化が見られる。歴史的に、形質細胞病理学は、これらの症例を説明するために使用されてきた。52 反応性卵胞のいくつかは、大きな免疫芽細胞や形質細胞を含むマントルゾーンが不鮮明である。これらの細胞は、通常、HHV8潜伏核抗原に陽性で、しばしばvIL-6にも陽性である。29,52 これらの形質芽細胞は、小さなクラスター(微小リンパ腫)またはコンフルエントなシート(顕性リンパ腫)を形成することがあり、分子遺伝学的研究により、微小リンパ腫はポリクローナルまたはモノクローナルのいずれかである可能性がある。
混合組織病理学
HVとPCの両方の特徴を持つリンパ節は、混合組織病理学と考えられ、UCDやiMCDで観察されることがある。最も一般的には、これらのリンパ節は、広範囲に退縮した胚中心とシート状の形質細胞症を示する。
臨床所見と診断
これらの古典的な病理組織学的特徴が観察されたら、これらの病理組織学的特徴は他の疾患でも見られるため、正式な診断を下す前に追加の研究が必要である。最初のリンパ節生検がCDと一致せず、臨床的な疑いが強い場合は、追加のリンパ節生検が必要となることがある。PET-CTスキャンを行った場合は、診断用サンプルを得るためだけでなく、リンパ腫を除外するためにも、標準化された取り込み値(SUV)が最も高い部位の生検が推奨される。CDがUCD、HHV8-MCD、POEMS-MCD、iMCDのいずれであるかに関わらず、最大SUV(SUVmax)の中央値は通常;3~8であり、これより高い値はリンパ腫を示唆する17。
患者は、全身状態の確認、身体検査、全血球計算、赤血球沈降速度(ESR)C反応性タンパク質(CRP)直接抗グロブリン検査(DAT)肝機能検査、クレアチニン、免疫固定法による血清タンパク質電気泳動、HIV血清検査、尿検査、胸部・腹部・骨盤のCTスキャン(またはPET/CT)を受ける必要がある。肺症状がある場合は、肺機能検査を検討する必要がある。画像診断でリンパ節が1つだけ腫れている場合はUCD、2つ以上腫れている場合はMCDと診断される。
全てのCD患者は、CDCNのACCELERATE自然史登録(#NCT02817997, …www.CDCN.org/ACCELERATE)に自己登録するように勧められ、研究のために血液サンプルを提供する機会について知らされるべ� サンプル)。
UCD
無症状の患者が UCD を診断するには、胸部、腹部、骨盤の画像診断で単一のリンパ節またはリンパ節の領域が確認され、CD の組織学的特徴があれば十分である。B症状、発疹、呼吸困難、末梢神経障害がある場合は、ベースラインを明らかにし、パラネオプラスチック天疱瘡(PNP)閉塞性細気管支炎、POEMS症候群を除外するためにさらなる評価が必要である。
UCD患者は通常、圧迫症状を呈するか、またはリンパ節が偶発的に発見される(表2)17,53。UCD 患者の 70%~90%は HV の病理組織を有している。UCDは、縦隔、頸部、腹腔・骨盤腔に最も多く発生するが、どのリンパ節にも発生する可能性がある。重篤な合併症として、PNP、多発性神経障害、肺合併症、自己免疫性溶血性貧血などが起こる可能性がある53。
表2 CDの臨床的特徴
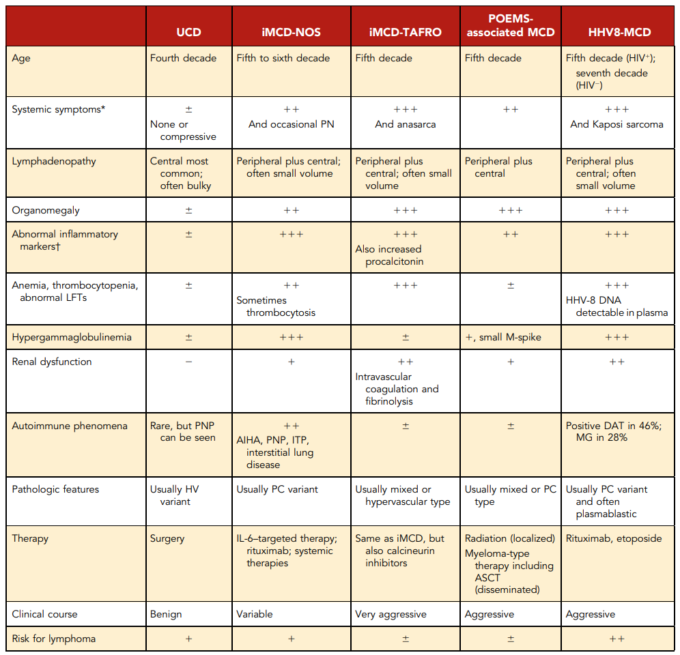
データは、Weisenburgerら、6 Iwakiら、10 Chronowskiら、13 Oksenhendlerら、17 Frizzeraら、54 Oksenhendlerら、56 Menkeら、57 Fujimotoら、64 and Nishimuraら、65から収集した。
AIHA(自己免疫性溶血性貧血)、ASCT(自家幹細胞移植)、DAT(直接抗グロブリン検査)、ITP(免疫性血小板減少性紫斑病)、LFT(肝機能検査)、MG(単クローン性ガンマグロブリン血症)、PN(末梢神経障害)、PNP(腫瘍随伴性天疱瘡)。
*発熱、発汗、体重減少、倦怠感、胸水、自己免疫疾患、呼吸器症状など。
†ESR、CRP、コリンエステラーゼ、フェリチンの上昇、アルブミンの低下。
MCD
MCD 患者は,0.1 カ所のリンパ節にリンパ節腫脹を呈し、広範囲の臨床的および検査的異常を示す。
再発や寛解の可能性がある(表2)。6,13,54-58 MCDのリンパ節腫脹は、どのリンパ節局にも発生する可能性があり、しばしば血管性であり、フルオロデオキシグルコースPET検査でも確認される43。
MCDのその他の特徴としては、自己免疫性、血球貪食性、炎症性、または特発性の細胞減少、肝脾腫、第二級アミロイドーシスや膜増殖性糸球体腎炎を含む多くの腎障害、末梢神経障害などが挙げられる。肺炎、拘束性肺疾患、リンパ性間質性肺炎、閉塞性細気管支炎などの肺の異常、発疹、色素沈着、桜色血管腫症、PNP、カポジ肉腫などの皮膚の異常。 43 リンパ性間質性肺炎と閉塞性細気管支炎は、アジア人に多く見られる可能性がある。
診断時には、iMCDとHHV8-MCDを区別するために、HIVの状態とリンパ節のHHV8の状態を評価することが不可欠である。同様に、免疫固定法を用いた血清・尿蛋白電気泳動や神経学的評価など、POEMS症候群との併発の可能性を評価することも重要だ。
HHV8-MCD
リンパ節組織のLANA-1によるHHV8の陽性反応、および/または循環中のHHV8のポリメラーゼ連鎖反応により、多中心性リンパ節腫脹とCDの組織病理学的特徴を有する患者のHHV8-MCDの診断が確立される59。1つのシリーズでは、HHV8-MCD症例の約2分の1に血球貪食症候群が認められた21。これらの患者の4分の3は、血液や神経の障害のために集中治療を必要とした。
HIV1とHIV2のHHV8-MCD患者の間には,いくつかの臨床的な違いが報告されている.HIV1のHHV8-MCD患者は、年齢が若く(42歳対65歳)白人で男性が多く、発熱、脾臓肥大、血球貪食症候群を有する傾向があり、単クローン性ガンマグロブリン血症やDAT陽性の可能性は低かった17。
iMCD
iMCDは臨床病理学的にはHHV8-MCDと似ているが、HHV8は認められない。さらに、関節炎、皮膚症状、腎疾患、全身性エリテマトーデス様症状は、HHV8-MCDよりもiMCDに多く見られる。iMCDの患者は、表1に示すように、幅広い臨床症状と検査異常を示する。診断基準としては、iMCDの病理組織学的に一致するリンパ節、少なくとも2カ所以上のリンパ節の腫大、少なくとも2つのマイナーな基準があり、そのうち少なくとも1つは検査基準となっている。他の疾患は、臨床病歴、病理学的評価、および必要に応じて追加の検査によって除外する必要がある。
症状や視力が全く異なるため、iMCDをiMCD-TAFROとiMCD-NOSにさらに分類することが推奨される。最も重篤な患者に対しては、iMCD-TAFROの臨床的サブタイプを考慮すべきである。
iMCD-TAFRO
iMCD-TAFROは、血小板減少、腹水、網膜線維症、腎機能障害、臓器肥大を伴うiMCDの積極的な臨床サブタイプを記述している60。iMCD-TAFRO患者は、UCDで記述されているHVの病理組織学的特徴に類似した血管拡張リンパ節を有し、iMCD-NOSとは異なるサイトカインスペクトルを示する61,62。iMCD-NOS患者は血小板数が多く、高ガンマグロブリン血症を示すのに対し、iMCD-TAFRO患者は血小板数が減少し、ガンマグロブリンは正常または軽度の上昇にとどまる17。
表3 iMCD-TAFROの診断基準

Iwaki et al10 から許可を得て引用している。
CTはコンピュータ断層撮影、ECは内皮細胞、GCは胚中心、LNはリンパ節。
iMCD-TAFROが疑われる患者では、血管内凝固および線溶の検査を行うとともに、網状赤血球線維症および巨大核細胞過形成を評価するために骨髄生検を行う必要がある。iMCD-TAFROの診断基準案を表310に示する。
iMCD-NOS
iMCD-NOSは、iMCD-TAFROの診断基準を満たさないiMCD患者を記述する。iMCD-TAFROと比較して、iMCD-NOS症例は、より積極的でない臨床経過、コルチコステロイドへのより高い反応性、血小板症、より少ない頻度のアナザーカ、低いアルカリホスファターゼ、およびg-グロブリンレベルの増加を示す傾向がある10,63-65。
POEMS-MCD
時折、HHV-8,2 MCDの患者が同時にPOEMS症候群と診断されることがある。我々はこのような併発をPOEMS-MCDと定義し、POEMS症候群を引き起こしている病理学的なPCが同時にMCDを引き起こしているのではないかと考えている。古典的なPOEMS症候群は、骨硬化性骨髄腫との関連性が最も高い稀なパラ新生物症候群である。末梢神経障害、臓器肥大(肝脾腫)内分泌障害、単クローン性ガンマグロブリン血症(通常はL型軽鎖)皮膚変化など、いくつかの特徴が頭文字をとって表現されている。66 POEMS-MCD の診断に必要な POEMS の診断には、多発性神経炎、モノクローナル・ガンモパ ー、および硬化性骨病変、高 VEGF、CD と一致するリンパ節のうち少なくとも 1 つが必要である。興味深いことに、骨硬化性骨病変のないPOEMS-MCD患者は、骨病変のある患者よりもはるかに悪い経過をたどる58。
末梢神経障害は、CD患者の約10%に見られると報告されており、UCDよりもMCDに多く見られる43,57,70。しかしながら、末梢神経障害を有するHHV8,2 MCD患者のすべてがPOEMS症候群の診断基準を満たすわけではなく、POEMS-MCDであるとされている。症状的にも客観的にも末梢神経障害の程度と重症度は、末梢神経障害はあるがPOEMSを併発していないCD患者では少なく、次いでPOEMS-MCDとなり、MCDを併発していない古典的なPOEMSでは最悪となる71。
HHV8,2 MCD の診断を下す際には、神経障害を併発している場合には、CT や CT/PET スキャンの骨画像を慎重に検討し、硬化性骨病変を探す必要がある。血清蛋白質電気泳動と尿蛋白質電気泳動でM蛋白質を検索する必要がある。広範な内分泌検査(甲状腺、副腎、下垂体、性腺軸)も実施する必要がある。骨髄生検では、クローン性PC、巨核球の増殖と異型化を調べることも重要だ。また、肺機能検査や神経学的評価も行うべきである。
鑑別診断
CDの様々なサブタイプの鑑別診断は、CDと一致するリンパ節生検を行った場合でも、広範囲に及ぶ。しかし、UCD、HHV8-MCD、POEMS-MCDはiMCDに比べて鑑別範囲が狭く、除外すべき疾患も少ない。リンパ腫以外の疾患では、UCDのようにCDに類似した組織病理学的特徴を持つ孤高のリンパ節腫脹を呈するものはほとんどない。HHV8-MCDではHHV8,POEMS-MCDではM-proteinという診断用バイオマーカーが陽性であることが診断の助けとなる。臨床的には、全身性エリテマトーデス(SLE)、関節リウマチ、自己免疫性リンパ増殖症候群(ALPS)などの複数の自己免疫疾患や、急性感染症、悪性腫瘍などがiMCDの鑑別診断に含まれる。iMCDは非常に不均一であるため、iMCD-TAFROとiMCD-NOSでは鑑別診断が異なる。iMCD-TAFROはSLE、骨髄線維症、急性HIV、血球貪食性リンパ組織球症との鑑別が難しく、iMCD-NOSは自己免疫性リンパ増殖症候群、IgG4関連疾患、ローザイ・ドルフマンとの鑑別が難しいとされている。
病理学的には、HVの病理組織学的特徴は、胸腺腫、1 血管免疫芽球性T細胞リンパ腫のような退行性胚中心を有するリンパ増殖症、およびHIV関連リンパ節症の進行期との重なりを示する51,54,70。
PCの病理組織学的特徴は、感染症、自己免疫疾患、原発性または後天性の免疫不全、悪性腫瘍など、他の多くの疾患で見られることがある。
二次悪性腫瘍
CDでは二次悪性腫瘍は珍しくない。UCD患者は、濾胞性樹状細胞肉腫43やホジキンおよび非ホジキンリンパ腫を発症するリスクが高いようである17,58。HHV8-MCDのHIV感染者は、MCDを発症していないHIV感染者と比較して、リンパ腫の発症頻度が15倍になると推定されている72。また、HIV2のHHV8-MCD患者は、悪性腫瘍、特にリンパ腫(;15%)54,57,73-75を発症し、カポジ肉腫は50%もの症例で発症する17,54。フランスの共同研究では、HIV1型HHV8-MCD患者において、リツキシマブ投与前の時代にはリンパ腫の発生率は1000人年に69.6人であったが、リツキシマブを用いた治療法の導入後は1000人年に4.2人に減少したと報告している76。
治療法
UCD
UCDの治療法は、病理学的に関わらず、可能な限り外科的に切除するという単純なものである。外科的に完全に切除すれば、ほぼ一様に治癒し、すべての症状と検査異常は正常に戻る。PNP が存在する場合、多くの場合 77,78 は、常にではないが、79 は 1 年以内に改善する。非アミロイドーシスに関連した腎疾患も、外科的切除後 12 ヶ月以内に治癒することが報告されている。81,82 UCD の完全切除後に閉塞性細気管支炎が回復しなかったという報告は、早期の介入(すなわち、線維化前)によって肺の変化が回復したかどうかが不明であるため、解釈が難しい。
手術が不可能な場合は、放射線照射、塞栓術、またはリツキシマブやシルトキシマブ/トシリズマブ(急性炎症状態の証拠がある場合)を用いたネオアジュバント療法を検討すべきである43,53,84,85。
発表された278例のUCDのシステマティックレビューでは、249例が外科的切除のみ、16例が免疫抑制療法のみ、13例が外科的切除と免疫抑制療法の併用であった85。UCD患者71名を対象とした最近のシリーズでは、発症時に切除可能なUCDリンパ節を有していたのは54%に過ぎなかった53。残りの切除不能なUCD患者33人のうち、19人がネオアジュバント療法(例えば、ステロイド、アルキル化剤、リツキシマブ、トシリズマブ、塞栓術)を受け、7人が切除に至った。完全奏効が4例、部分奏効が14例であった。8人の患者が放射線治療を受けた。4人が完全奏効、4人が部分奏効を示した。合計すると、11人の切除不能なUCD患者は、治療を受けずにアクティブサーベイランスで長期的に安定していた。これは興味深い観察結果であるが、二次的な悪性腫瘍や、PNPや閉塞性細気管支炎のような関連する傍腫瘍性疾患の進行に注意しなければならない。
MCD
ほとんどの治療データは、症例報告やシリーズから得られたもので、その多くは歴史的にMCDのサブタイプを特定しておらず、HHV8-MCD、POEMS-MCD、iMCD-NOS、iMCD-TAFROのケースが混在している。幸いなことに、これらの区別がなされている最近のシリーズがいくつかある10,17,41,63,86
また、転帰や予後に関するデータは限られている。Oksenhendlerらは253人のCD患者の素晴らしいレビューの中で、フランスの彼の施設における20年間のCD患者の全生存期間(OS)について報告している17。iMCDの推定OSは非常に良好であったが、27名のうちiMCD-TAFROを発症したのは2名のみであった。抗IL-6療法が承認される前の古いケースシリーズでは、HIV2(推定HHV8,2)MCDの5年OSは65%であった58。
図3 特発性多中心性CDの治療に関するコンセンサス・ガイダンス
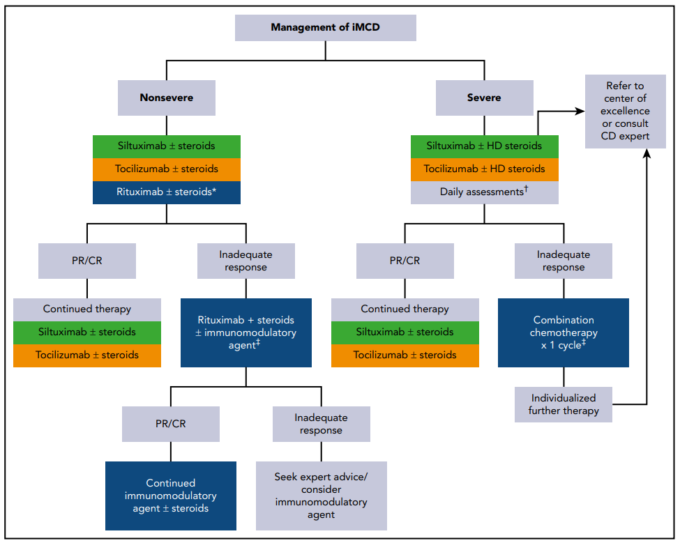
84 iMCD 患者は表 4 に従って疾患の重症度に応じて層別化されるべきである。*症状が軽度の患者には、リツキシマブの限定経過を選択することができる。†最初の1ヶ月間は、抗IL-6療法を毎週加速して投与するとともに、患者の状態を毎日評価することが推奨される。臓器障害が悪化した場合は、併用化学療法の開始を検討する必要がある(本文参照)。4.治療法の例を表5に示す。緑はカテゴリー1のエビデンス:高レベルのエビデンスに基づいており、その介入が適切であるという一様なコンセンサスが得られている。金色はカテゴリー2Aのエビデンス:低レベルのエビデンスに基づくもので、介入が適切であるという一様なコンセンサスが得られている。青はカテゴリー2Bのエビデンス:低レベルのエビデンスに基づいており、介入が適切であるというコンセンサスが得られている。CR、完全奏功、HDステロイド、高用量ステロイド、mAb、モノクローナル抗体、PR、部分奏功。
HHV8-MCD
87 HHV8 に感染した形質細胞は CD20 をあまり発現していないことが多いのであるが、複数のケースシリーズと 3 つの 非盲検試験でリツキシマブの適用が成功している。88,89 このようなアプローチにより、95%の患者が臨床的寛解を達成し、5年OSは92%、5年無再発生存率は82%であった。すべての患者は、再発時にリツキシマブベースの治療を受けることができた。リツキシマブベースのアプローチは、HHV8 関連リンパ腫のリスクも低減している。88,89 リツキシマブとリポソーム・ドキソルビシンを 3 週間ごとに併用することで、カポジ肉腫の増悪を減衰させることができるようである。1 件のパイロット試験では、バルガンシクロビルとジドブジンの高用量併用により、90%に近い臨床的奏効率が得られたが、無増悪生存期間はわずか 6 ヶ月であった91。
POEMS-MCD
17 POEMS-MCD POEMS 症候群と MCD を併発し、標準的な骨髄腫治療を受けた骨硬化性病変と末梢神経障害症状が優勢な患者には、できれば自家 幹細胞移植(ASCT)を伴う大量化学療法を行うことが望まれる。ASCT の候補とならない患者に対しては、メルファラン、シクロホスファミ ッド、レナリドミド、サリドマイド、ボルテゾミブ、カーフィルゾミブ、ダラツムマブなどの他の骨髄 腫治療を検討することができるが、これらの推奨事項のほとんどが小規模なケースシリーズから得られた ものであることを理解している。
骨病変のない POEMS-MCD 患者については、データがさらに少なくなっている。IL-6が高値の場合、iMCDの概要と同様にsiltuximabとrituximabを考慮することができるが、PCを中心とした免疫調整療法への移行も考慮すべきである。
表4. iMCDの重症度スコアリング
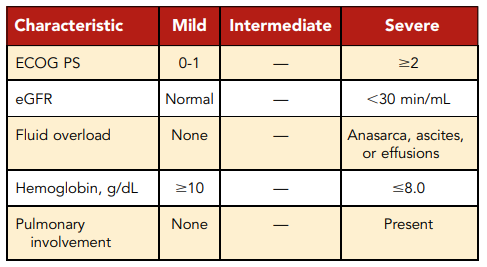
van Rhee et al.84 から引用している。
重度のiMCDは、5つの重度基準のうち2つを満たす必要がある。軽度のiMCDは、5つの軽度基準のすべてを満たすことが必要である。軽度iMCDは、軽度の基準を5つすべて満たすことが必要である。中間症例は、軽度および重度の基準を満たさない。軽度および 治療アルゴリズムの目的上、軽度および中間症例は非重度とみなされる。
-、軽度または重度の基準を満たさない;ECOG PS、Eastern Coopera
iMCD
van Rhee らによるコンセンサスガイダンスでは、iMCD に対する治療法の推奨事項が概説されている(図 3)84。iMCD 患者の治療法を選択する際には、疾患の重症度(表 4)を考慮する必要がある。
重症度に関わらず、すべての患者に対して、アルゴリズムは、抗IL-6指向性治療から始まる。抗IL-6抗体であるSiltuximabは、米国食品医薬品局(FDA)がiMCDの治療法として承認している唯一の薬剤である93。79名の患者を対象とした登録試験では、Siltuximab投与群の34%が症状および腫瘍の持続的な改善を示したが、プラセボ投与群では全くなかった。
IL-6 値が高い患者の奏効率が高い傾向にあったが、低値または正常値の iMCD 患者の中には siltuximab に反応した患者もいたが、高値の患者の中には反応しなかった患者もいた。94 試験では、免疫グロブリン、CRP、フィブリノゲンが高く、ヘモグロビンが低い患者が siltuximab に最も反応しやすかった。日本でiMCDに承認された抗IL-6レセプター抗体であるtocilizumabの単群非盲検試験のデータは、治療を中止すると再燃することを示唆している96。IL-6 濃度は、患者が抗 IL-6 療法を行っている間は解釈できず、Siltuximab の最終投与後 18~24 ヶ月は奏効のモニタリングに使用できない。さらに、抗IL-6抗体単剤療法を受けている患者では、化学療法に比べてリンパ節の反応が遅れることが多い。
最も信頼性の高い測定方法は、ヘモグロビン、ESR、CRP、アルブミン、および臨床症状である84。
IL-6 ブロッキングに反応しない非重症の iMCD 患者に対しては、多くの治療法を試すことができ、あるものを別のものより推奨することは困難である。しかし、一般的には、進行性の臓器障害を伴う重篤な疾患でない限り、細胞毒性化学療法は避けるべきである。38,41,58,63,84,97,98 患者の約半数は、コルチコステロイドで一時的に改善するが、再発が起こり、長期的に大量のコルチコステロ イドを投与すると重大な病的状態に陥る。リツキシマブは、一部の患者に再反応を引き起こすことができ41 、軽度のiMCDの症状アトロジーを持つ患者の第一選択薬として、また抗IL-6剤が効かない場合の第二選択薬として検討されている。過去に行われた解析では、Siltuximab を投与した患者の無増悪生存期間は、リツキシマブ/リツキシマブをベースとした治療法または細胞毒性のある化学療法と比較して優れてた63。ただし、これらの解析では疾患の重症度を補正していない。サリドマイドは、iMCD患者にも使用され、一定の成果を上げている。新たにiMCDと診断された25名の患者を対象に、サリドマイド、シクロホスファミド、プレドニゾンを経口投与した第2相試験では、48%の患者が主要評価項目である24週間以上の持続的な腫瘍および症状の改善を達成した。ASCT を用いた大量化学療法はまれに報告されているが、成功率は様々で、POEMS 症候群の患者に見られるものよりも劣っているようである99。
表5.治療法
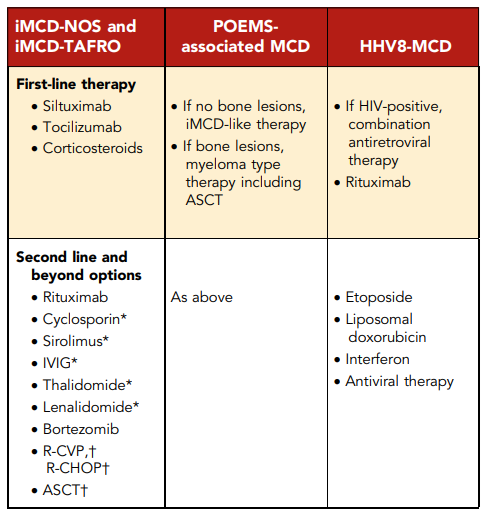
IVIG、IV免疫グロブリン、R-CHOP、リツキシマブ、シクロホスファミド、ドキソルビシン、ビンクリスチン、プレドニゾン、R-CVP、リツキシマブ、シクロホスファミド、ビンクリスチン、プレドニゾン
*免疫調整療法。
†併用化学療法。
iMCD-TAFROの基準を満たす場合も満たさない場合もある重症患者に対しては、抗IL-6療法が第一選択となるが、最初の1カ月間は週1回の投与とし、高用量コルチコステロイド(メチルプレドニゾロン500mg/日)の同時投与が最初に必要となる。84 リンパ腫、骨髄腫、血球貪食性リンパ組織球症などで検討される細胞傷害性レジメンは、集中治療室にいる患者にも使用されている。これらの患者のサイトカイン/ケモカインストームを解消するためには、このような集中的なレジメンが必要なのかもしれない。これらの積極的なレジメンの全体的な奏効率は75%程度であるが、再発はよくあることである。維持療法はその場その場で行われる。
iMCD-TAFROは比較的新しく認識されたサブタイプであるため、これらの患者に対する最良の治療法に関する情報はさらに少ない。現在、ペンシルバニア大学とアーカンソー大学で、抗IL-6療法に抵抗性を示すiMCD患者を対象に、sirolimusを評価する臨床試験が行われている(NCT03933904)。