
Old Drugs as New Treatments for Neurodegenerative Diseases
pubmed.ncbi.nlm.nih.gov/29751602/
Received: 2018年4月27日; Accepted: 2018年5月8日;公開:2018年5月11日
概要
世界の一般人口が高齢化していることから、神経変性疾患は増加している。これらの疾患は、多くの場合、完全には解明されていないメカニズムで現れ、記憶、認知、運動を障害する。現在、神経変性疾患は完治する病気ではなく、治療法は症状を抑えるか、病気の進行を止めることしかできない。世界保健機関(WHO)は、運動機能に影響を及ぼす神経変性疾患が、今後20年間で2番目に多い死因になると予測していることから、この種の疾患に対する新しい治療法が急務となっている。新しい治療法は、合成、天然物、そして既存の医薬品という3つの主要なソースから得られる。この最後のソースは、薬剤の再利用として知られており、薬剤の薬物動態および薬力学的プロファイルがすでに確立されているため、最も有利であり、この戦略に投入される投資は、古典的な新薬の開発ほど大きくはない。アルツハイマー病、パーキンソン病、ハンチントン病、多発性硬化症、筋萎縮性側索硬化症など、最も関連性の高い神経変性疾患に対する旧薬の可能性について、いくつかの研究が行われている。
キーワード:神経変性疾患、ドラッグ・リパーパス、アルツハイマー病、パーキンソン病、ハンチントン病、多発性硬化症、筋萎縮性側索硬化症
1. はじめに
薬剤の再利用(Drug Repurposing)は、薬剤の再配置(Drug Repositioning)や薬剤の再ファイリング(Drug Reprofiling)とも呼ばれ、創薬において増加傾向にある。これは、既存の薬剤の新たな治療法を見つけることを意味し、合成や天然物と並んで、新たな治療法につながる低分子リード化合物を見つけるための主要なアプローチの一つとなっている。最近では、特に新しい併用療法や、孤児や顧みられない病気など、臨床的にニーズが満たされていない病気を対象とした、薬剤の再利用への関心が高まってきている。その利点は、再利用される薬剤のプロファイルが既に確立されているため、創薬や最適化、安全性や薬物動態の研究への投資が少なくて済むことである。古い医薬品の再利用は、セレンディピティ、副作用の観察、ターゲット探索、または新規の洞察によって特定され、従来の使用方法と同じ作用機序で作用する場合と、新しい作用機序で作用する場合がある[1-3]。
薬剤の再配置のために最も使用される戦略の1つは,新しい標的での化合物ライブラリのin silicoスクリーニングである[2]。薬剤の再利用の顕著な例として,サリドマイドという薬剤がある。サリドマイドは、妊娠に伴うつわりの治療のために市販の制吐剤として使用されていたが、催奇形性や記憶障害の報告を受け、すぐに使用を中止した。しかし、1998年、FDA(米国食品医薬品局)はサリドマイドを結節性紅斑の皮膚症状の治療薬として承認した。2006,サリドマイドは、その血管新生阻害作用により、骨髄腫の治療薬として承認された[4]。この例は、創薬において薬剤の再利用がいかに重要であるかを証明している。しかし,古い薬が新しい用途に使われる例は,他にも数多く報告されている[5,6]。
神経変性疾患(ND)は、加齢に依存する疾患であり、その病態は非常に異なっており、その原因やメカニズムが理解されていないため、治療法が確立されていないのが現状である。世界的に高齢化が進むにつれ、記憶、認知、運動などに障害をもたらすこれらの疾患の有病率も高くなっている[7,8]。世界保健機関(WHO)は、20年後には主に運動機能に影響を及ぼすNDが、がんを抜いて心血管疾患に次ぐ2番目に多い死因になると予測しており、NDに対する治療の必要性は急務となっている[9,10]。
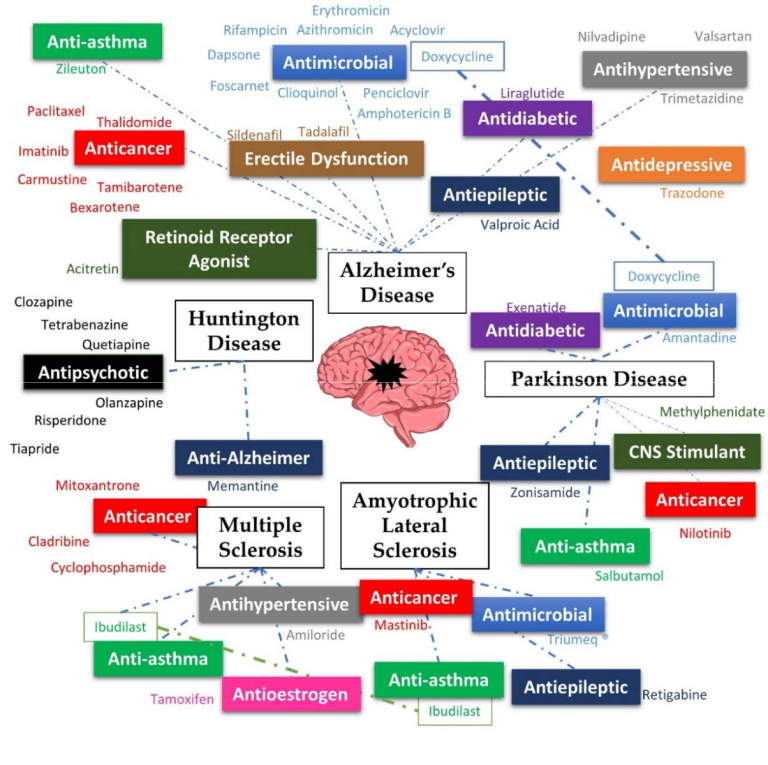
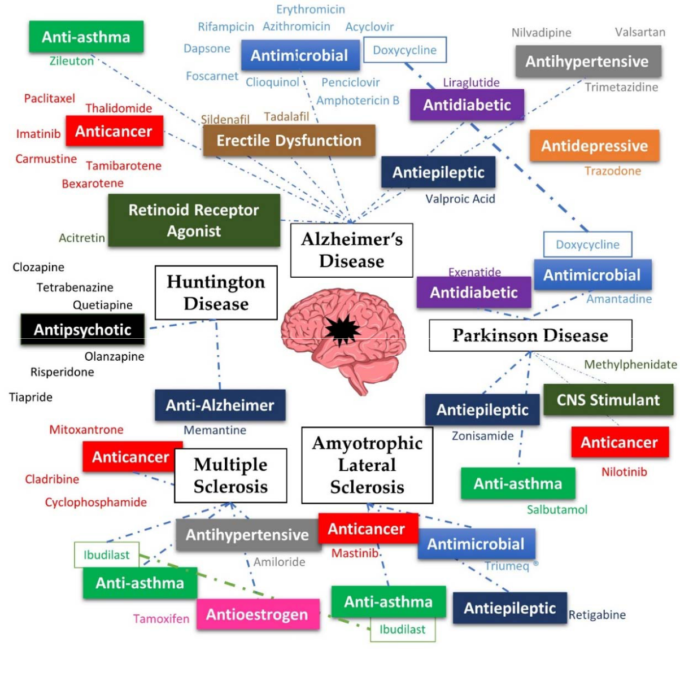
このようにNDの治療に対するニーズが高まっている中で,薬剤の再利用が期待されていることから,既存の薬剤がこれらの疾患に対して試験されていることは理にかなっている。このレビューでは、最も研究されている神経変性疾患であるアルツハイマー病(AD)パーキンソン病(PD)ハンチントン病(HD)多発性硬化症(MS)筋萎縮性側索硬化症(ALS)に対して再利用された薬剤を取り上げる(図1)。
しかし、これらの疾患をまとめて取り上げた包括的なレビューはまだなく、これらの疾患がどのように関連しているのか、より広い視野で理解することができる。
図1. 本レビューで紹介する疾患と再利用医薬品の概要
2. アルツハイマー型認知症
アルツハイマー型認知症は、高齢者の認知症の80%を占める代表的な認知症の一つである。その症状は、記憶力の低下、学習能力の低下、行動や機能の低下が進行することである。ADの原因はまだ完全には解明されていないが、脳内にアミロイド斑が沈着し、最終的には神経細胞やシナプスの損失につながることが関係していると考えられている[14-16]。
現在のところ、ADの治療法は確立されておらず、この病気の範囲内で使用される薬は、主に認知症状などを治療するためのもので、早期に投与することでより良い機能を発揮する[14]。不思議なことに、ADの治療薬として販売されている薬の一つであるガランタミン(1,図2)は、それ自体が再利用されたものである。実際、ガランサス属に含まれるこのアルカロイドは、筋肉のアセチルコリンエステラーゼを阻害することがわかり、筋疾患や末梢神経疾患の治療薬として、また、神経インパルスの伝達を促進するガランタミン(1)の能力を利用して、麻酔後の神経筋遮断の回復薬として注目された。ガランタミン(1)は3級アンモニウム塩基を持つため、血液脳関門を通過しやすく、脳のアセチルコリンエステラーゼを阻害することができ、特に注目されている[17,18]。1980年代、ガランタミンのAD治療効果が研究され始め 2000年には抗アルツハイマー病薬として導入されたが、現在でもアルツハイマー病患者の重篤な症状の出現を遅らせるために最も使用されている薬剤の一つである[18]。
抗がん剤がADの治療に再利用できるかどうかを確認するための研究が行われている。この考えの背景には、がんと神経変性が、ミトコンドリア機能障害、酸化ストレス、細胞代謝の低下、ミスフォールドタンパク質の発生など、シグナル伝達経路を共有しているという事実がある。乳がん生存者が化学療法を受けた場合、対照群と比較して高齢になってからADを発症するリスクが低いことが報告されている[19]。
カルムスチン(2)は、脳腫瘍のアルキル化剤として使用されているニトロスレアである。小さな親油性の非イオン化分子であるため,血液脳関門を通過することが可能である[20]。アミロイドβタンパク質前駆体を過剰に発現させた細胞において、カルムスチン(2)は無毒性量でアミロイドβ産生の強い減少を示した[21]。
皮膚T細胞リンパ腫の治療に用いられているレチノイドX受容体拮抗薬のベキサロテン(3)は,家族性AD変異を過剰発現させたマウスにおいて,神経変性を回復させ,認知機能を改善し,アミロイドβレベルを低下させる能力があることが証明された[22]。
日本で急性前骨髄球性白血病の治療薬として承認されているレチノイン酸受容体アゴニストのタミバロテン(4)は、脳細胞からの炎症性サイトカインおよびケモカインの分泌を減少させ、老化を促進したマウスの行動を改善し、皮質のアセチルコリンを減少させるなど、ADの病態生理に関連する複数の経路に作用することができる[23]。
慢性骨髄性白血病などの治療薬として承認されているチロシンキナーゼ阻害剤であるイマチニブ(5)は、アミロイドβの減少と神経保護という2つのメカニズムにより、ADの治療に有用であることが示唆されている。しかし、イマチニブ(5)は、血液脳関門への浸透性が低く、P-glycoprotein(P-gp)によって容易に排出されてしまうという問題がある[24]。
また、卵巣がん、乳がん、非小細胞肺がんなどで承認されている抗ミトコンドリア薬のパクリタキセル(6)も、ADの治療薬として研究されている。特に、中枢神経系の細胞に多く存在し、微小管を安定化させる機能を持つタウタンパク質の欠損であるタウオパチーの治療に効果があるという。このタンパク質がリン酸化されると、微小管に結合する能力が低下し、その結果、線維化が促進される。パクリタキセル(6)はこのリン酸化を抑制する。しかし、パクリタキセル(6)には、イマチニブ(5)と同様の問題がある:P-gpの基質になりうるし、中枢神経系への浸透性も悪い[25,26]。
また、抗AD作用が期待できる抗がん剤として、サリドマイド(7)がある。サリドマイドは、内皮細胞の増殖、血管新生、血液脳関門の破壊を抑制することが実証されている。また、腫瘍壊死因子-αの阻害により、海馬の神経細胞の減少を抑えることができた[11,27]。
抗菌薬もまた、ADとその症状の治療に適しているかどうかが研究されている。マクロライド系抗生物質であるアジスロマイシン(8)とエリスロマイシン(9)は、いずれもアミロイド前駆体タンパク質を阻害し、その結果、脳内のアミロイドβレベルを低下させることがわかっている。また、テトラサイクリン系抗生物質は、アミロイドβの形成を抑制し、トリプシン消化に対する抵抗性を高め、形成された前駆体の分解を促進することが証明されている。また、酸化ストレスも減少させており、多様な作用機序が示唆されている[28]。
ドキシサイクリン(10)は、この点において、単独およびリファンピシン(11)との併用で可能性を示している[29-31]。リファンピシン(11)は,マイコバクテリウム感染症に最も頻繁に処方されており,アミロイドβの生成を抑え,クリアランスを増加させることにより,用量依存的にアミロイドβを減少させる効果があるとされている[32]。
ダプソン(12)はハンセン病の治療に用いられる抗生物質で,1990年代には,ダプソン(12)を投与されたハンセン病患者で認知症の発症率が低下したことが注目された。ダプソン(12)が老人斑を減少させることができるかどうかについては、相反するデータがあり[33,34]、この事象がアミロイド沈着に対する保護因子であるという仮説が立てられた。この仮説は、ハンセン病と結核の患者において、薬物治療を受けた患者の割合に違いがあるにもかかわらず、ADの類似した事例を示した研究によって、さらに裏付けられた[35,36]。
抗ウイルス剤のアシクロビル(13)、ペンシクロビル(14)、フォスカルネット(15)は、AD細胞モデルにおいて、リン酸化されたタウタンパク質とアミロイドβを減少させることに成功しており、ADの治療に適していることを意味していると言える[37]。
抗真菌薬であるアンフォテリシンB(16)は、アミロイドβの形成を遅らせることが示されている[38]。しかし,より最近の研究では,同じ結果が得られておらず[39],アンフォテリシンB(16)に関連する毒性は,ADの治療に適した候補とは言えないだろう。クリオキノール(17)は、抗真菌薬および抗寄生虫薬で、トランスジェニックマウスにおいて、良好な忍容性をもって、脳内のアミロイドβプラークの減少を引き起こすことが示されている[40]。
抗てんかん薬のバルプロ酸(18)は,トランスジェニックマウスにおいて,アミロイドβプラークの形成を抑制し,記憶障害の改善を示したことから,ADの神経保護剤として示唆されている.その作用機序は複雑であるが,炎症性サイトカインの産生を抑制し,ミクログリアによるアミロイドβの貪食を促進することによるものと考えられている[41-44]。
バルサルタン(19)は、アンジオテンシン受容体拮抗薬で、降圧剤として使用されている。このクラスの薬剤をADに使用する根拠は、ADの発症および進行の主要な環境的原因の1つである慢性的な有害ストレスが、AT1およびAT2受容体サブタイプで作用する脳内アンジオテンシンIIの上昇を引き起こすことが可能であるという事実から来ている。さらに、アンジオテンシンIIの上昇は、アミロイド生成との関連性が示唆されており、AT1を遮断するアンジオテンシン受容体拮抗薬の使用は、認知処理能力の低下を遅らせるのに有用であると考えられている。バルサルタン(19)は、この作用機序とは別に、炎症、血管収縮、ミトコンドリア機能障害を抑制し、アセチルコリンの放出を促進する[45,46]。バルサルタン(19)を試験管内試験および生体内試験で投与することにより、アミロイドβの減少が報告されており、このエビデンスは認知症の軽減を示唆している。さらに、この薬剤は脳への浸透性が高いが、この薬剤をADの治療に取り入れるにはさらなる研究が必要である[47-49]。
カルシウム拮抗薬は、高血圧や狭心症の治療に用いられる薬剤である。ニルバジピン(20)などのジヒドロピリジン系のカルシウム拮抗薬は,試験管内試験でのアミロイドβの産生,オリゴマー化,蓄積を抑制し,細胞生存率を向上させ,神経毒性を低下させるとともに,血液脳関門への浸透性が高く,血管拡張作用により脳血流を増加させることができる[50-52]。
トリメタジジン(21)は、ピペラジン系の抗虚血薬である。その作用機序は、一酸化窒素の産生増加、細胞のアポトーシス抑制、抗酸化剤として内皮機能を高めることなど多岐にわたる[53]。血液脳関門を通過できるだけでなく、その抗酸化作用によりフリーラジカルの生成を抑えることができる。また、健康な神経と損傷した神経の軸索再生と効果的な髄鞘形成を改善することができる[54]。
2型糖尿病がADの危険因子として同定されていることから、抗糖尿病薬もADに再利用されている。アルツハイマー病患者の脳では、インスリンシグナルの脱感作が報告されている。インスリンはまた、神経幹細胞の活性化や細胞の成長・修復を誘導することができ、インスリンを投与することで、神経保護やリン酸化タウタンパク質のレベルの調節、さらには記憶や認知の改善が認められている[55-57]。このことから、インスリン分泌を促進する化合物は、ADにも有用であると考えられる。インスリン分泌を促進するグルカゴン様ペプチド1アナログ(GLP-1)は、アミロイドβの減少、神経細胞の機能低下や細胞死、さらにはタウリン酸化など、ADに関連する多くの経路にも作用する可能性がある[58,59]。リラグルチド(22)は、これらの基準を満たし、脳への浸透性が確立されており、脳内で生理的効果を示し、学習能力を向上させ、アミロイドβの形成や脳内炎症を抑制する[60,61]。
グレリン(23)は,消化管で産生されるペプチドホルモンで,食欲を調節するとともに,中枢神経系の神経ペプチドとしても機能する。 グレリン(23)およびその脱アシル化体は、アポトーシスを抑制し、アミロイドβによる炎症性インターロイキンの増加を緩和し、神経保護作用を示すことが明らかにされている。脳内の免疫防御の第一線であるミクログリアがアミロイドβプラークと相互作用すると、炎症反応が始まるという仮説がある。NDでは、ミクログリアが過剰に活性化され、神経細胞の機能障害と死を招く。成長ホルモン刺激剤として用いられる合成ヘキサペプチドであるヘキサレリン(24)とその誘導体EP80317(25)は,成体ラットの海馬前駆細胞の増殖を促進し,成長因子の除去による壊死やアポトーシスからこれらの細胞を保護することができた[62-64]。
レチノイド受容体活性化剤は,にきびや乾癬などの皮膚疾患の治療に用いられる。レチノイン酸自体は、神経の機能と修復に不可欠であり、この化合物のシグナル伝達の減少は、ADの一因となる可能性がある[65-67]。アシトレチン(25)を用いた研究では、アミロイド消去酵素のアップレギュレーション、および抗酸化物質の調節が示されている[68]。
ジレトン(26)は、5-リポオキシゲナーゼを阻害することで作用する抗喘息薬であるが、ADの改善効果があると仮定された。これは、アルツハイマー病患者では5-リポオキシゲナーゼの活性が高いことを発見したためで、この範囲では有望なターゲットとなる。実際,ジレイトン(26)をマウスに投与した研究では,アミロイドβの沈着が減少した[69]。
勃起不全治療薬であるシルデナフィル(27)とタダラフィル(28)は,cGMPの制御因子であるホスホジエステラーゼ-5の阻害剤であり,ADとの関連が指摘されている。シルデナフィル(27)は神経炎症を抑制することに成功し,加齢マウスモデルにおいてアミロイドβを低下させることができた[70]。タダラフィル(28)も認知機能の向上と神経保護作用を示し、27よりも効果的にホスホジエステラーゼ-5を阻害するのに十分な高濃度で血液脳関門を通過した[71]。
最近では,抗うつ薬であるトラゾドン(29)が,細胞内のタンパク質合成の制御に有害な役割を果たし,アルツハイマー病患者では過剰に活性化されているunfolded protein responseのPERK/eIF2α-P分岐を介したシグナル伝達を抑制する可能性を示した。トラゾドン(29)は、試験管内試験および生体内試験において、eIF2α-Pの翻訳機能の低下を回復させることに成功した。さらに、神経保護、記憶の回復を示し、神経変性を予防した[72]。
3. パーキンソン病
パーキンソン病は、ADに次いで2番目に多い病気で、世界中の人々が罹患している。パーキンソン病は、その運動症状で最もよく知られており、主に黒質内のドーパミン作動性ニューロンの消失から生じると考えられているが、他の神経伝達系も影響を受けるようである[73]。現在、PDには、認知障害、睡眠障害、うつ病などの非運動関連症状があることが知られている。PDには治療法がないとはいえ、現在利用可能な治療法は効果的で、ドーパミン置換や脳深部刺激からなる病気を管理している[74]。
ADと同様に、抗パーキンソン薬の中にも、それ自体が再利用された薬剤がある:アマンタジン(30,図3)。 実際、アマンタジン(30)は、最初はインフルエンザの治療薬として開発されたが、その後、弱いグルタミン酸受容体拮抗薬として、ドーパミンを増加させ、その再取り込みを阻害することで、PDの治療に向けられた[12]。
ニロチニブ(31)は、チロシンキナーゼAbl阻害剤で、慢性骨髄性白血病の治療に用いられている。神経変性では、α-シヌクレインの発現量が増加し、その結果、蓄積されることでAblが活性化されることが観察されている。ニロチニブ(31)はAblのリン酸化を阻害するため、α-シヌクレインの分解を増加させる[75,76]。
以前,抗ADの候補として取り上げた抗生物質ドキシサイクリン(10,図2)は,抗PD作用についても研究されている。実際、ドキシサイクリン(10)の濃度を変えることで、抗菌作用と抗炎症作用を選択することができる。抗生物質として使用される量よりも低用量であれば、細菌の感受性は変わらず、抗炎症作用を示すことが研究で明らかになっており、これが神経保護作用につながっている。神経保護に寄与する他のメカニズムとして、ドキシサイクリン(10)の抗酸化作用と、α-シヌクレインオリゴマーの初期種を無毒性で播種しない種にリモデリングする能力が挙げられる。さらに、ドキシサイクリン(10)はα-シヌクレインのオリゴマー種にのみ結合することが報告されており、生理的なモノマー種は保存されている[77]。
ゾニサミド(32)はスルホンアミド系の抗てんかん薬で、作用機序が混在しているため、異なる疾患への使用に適している。これらの作用機序には,ナトリウムおよびカルシウムチャネルの遮断,GABAA受容体の調節,炭酸脱水酵素の阻害,グルタミン酸放出の抑制などがある。ラットを用いた研究では、治療量を使用した場合、線条体のドーパミンが増加することが示された。一方、高用量では、細胞内ドーパミンの減少が認められた。PDに関しては,運動症状と非運動症状の両方に良好な効果を示しているが,その作用機序はまだ明らかになっていない[78]。ゾニサミド(32)もモノアミンオキシダーゼB阻害剤である。この酵素は,主にアストロサイトに存在し,神経細胞やグリア細胞におけるドーパミンの分解に関与し,最終的にはフリーラジカルの生成につながり,PDの発症に決定的な役割を果たしうるものである。その阻害により、シナプス間隙のドーパミンレベルが安定し、ドーパミンの効果が高まる[79,80]。モノアミンオキシダーゼB阻害剤のもう一つの例は、抗パーキンソン薬のセレギリン(33)で、線条体損傷後のアストロサイトの活性化を高める[81,82]。
メチルフェニデート(34)は,中枢神経系の興奮剤で,線条体と前頭前野において,シナプス前のドーパミントランスポーターとノルアドレナリントランスポーターを遮断し,ドーパミンとノルアドレナリンの再取り込みを阻害することで作用する。本剤は、注意欠陥多動性障害の治療に用いられている。本剤を用いた複数の研究により、34はPDの歩行障害に加え、非運動症状の軽減にも有効であることが示されている[83]。
エクセナチド(35)は、前述のリラグルチドと同様に、2型糖尿病の治療に用いられるグルカゴン様ペプチド-1である。PDの治療薬としても研究されており、疾患の進行を遅らせたり予防したりする神経保護作用や神経可塑性の変化が認められている。GLP-1は血液脳関門を通過することができ、GLP-1受容体の活性化を介して神経保護作用を発揮する[84-86]。エクセナチド(35)についても、ADの治療において肯定的な報告がなされている[87]。前述のリラグルチド(22)は、現在、第2相臨床試験が行われており 2019年には成果が期待されている(clinicaltrials.gov, ID: NCT02953665)。
抗喘息薬、すなわちβ2アドレナリン受容体アゴニストは、抗PD活性について研究されている。最近の研究では、β2アドレナリン受容体がαシヌクレイン遺伝子SNCAの制御に関係していることが分かってきた。より具体的には、β2アドレナリン受容体の活性化が神経保護作用を示すことが示された。試験された薬剤の中では、3種類の抗喘息薬が最も有望であり、中でもサルブタモール(36)は血液脳関門を通過することができ、現在治療薬として承認されている。実施された研究では、3つの薬剤すべてがSNCA-mRNAとα-シヌクレインの存在量を減少させることができた[88]。
4. ハンチントン病
ハンチントン病は常染色体優性遺伝の疾患で、先進国では最も一般的な単遺伝性の神経疾患である。ハンチントン病は、常染色体優性遺伝の疾患であり、先進国で最も多い単発の神経疾患である。本疾患は、多機能タンパク質であるハンチンチンの突然変異を起源とする遺伝子変異によって発症し、その毒性によって神経細胞の死や機能障害が引き起こされる。成人になってから発症し、数年で死に至るまで症状が進行していく。この病気の治療法は知られていないため、唯一の選択肢は症状の管理である[89,90]。
テトラベナジン(コレアジン)(37,図4)は、レセルピン様の抗精神病作用を持つ単純な化合物を設計することを目的とした研究の一環として最初に開発され、シナプス前ニューロンのモノアミン取り込みの高安全性可逆的阻害剤として、また、シナプス後ニューロンのD2ドーパミンの弱い遮断剤として作用した。この化合物を用いた抗精神病薬の研究は不明確であったため、この薬剤はHDのような不随意の過運動性異常を示す疾患に再利用された。さらに、テトラベナジン(37)は、運動異常症状を引き起こすことが記録されていないため、ドパミン受容体遮断薬よりもHDに使用する方が安全である[91]。
このことを踏まえて、ドパミン拮抗作用を持つ他の薬剤がHDの治療のために試験されている。これは、D2受容体拮抗薬であるティアプリド(38)が抗精神病薬として使用されているケースである。しかし、ヨーロッパではハンチントン舞踏病の治療にセレギリン(33)が頻繁に選択されている[92]。
クロザピン(39)は統合失調症の治療に用いられる神経弛緩薬である。本薬は、ドーパミンD1およびD4受容体に対して高いアフィニティを示し、ドーパミンD2受容体に対しては低い拮抗作用を示する。錐体外路系の副作用が少ないことから、舞踏病の対症療法薬としての可能性が示唆されたが、臨床試験では相反する結果が示された[93]。
もう一つの抗精神病薬であるオランザピン(40)も、HDの運動・行動症状の治療に広く処方されている。この薬剤は,セロトニン受容体への親和性が高く,ドーパミンD2受容体には拮抗する。また、安全性と忍容性が高く、イライラ、睡眠障害、体重減少のほか、舞踏病が見られる場合に推奨することができる[94,95]。
統合失調症や双極性障害の治療に用いられている抗精神病薬のリスペリドン(41)は、D2受容体拮抗薬とセロトニン作動薬として作用するため、HDの舞踏病の治療にも用いることができる。運動機能の低下や精神症状の安定化など、ベネフィットのある効果を示した[96]。
非定型抗精神病薬のクエチアピン(42)は、セロトニンとドーパミンの受容体に高い親和性を示する。HD症状の治療にクエチアピンを使用した例は多くないが、記載されている数少ない例では、特に精神症状を伴う場合の舞踏病の治療に有用であることが強調されている[97]。
メマンチン(43)は、ADの治療に使用されるアダマンタン誘導体である。メマンチンは、非競合的なN-methyl-D-aspartate(NMDA)阻害剤である。NMDA受容体が過剰に刺激されると、細胞内にカルシウムが大量に流入し、最終的には細胞死に至る。メマンチン(43)は、このカルシウムの神経細胞への流入を防ぎ、脳の細胞死を防ぐことができる。メマンチン(43)は,HDの治療薬として研究され,グルタミン酸を介した興奮毒性に対する神経細胞の脆弱性を減少させることができることが注目された[98,99]。
5. 多発性硬化症
MSは、中枢神経系の自己免疫疾患である。ミエリンと軸索が程度の差こそあれ破壊される、慢性の炎症性疾患である。その経過は予測不可能であり、最初は可逆的な神経学的障害を特徴とするが、時間の経過とともに進行する。MSの治療法はないが、症状や病気の進行を抑えるための治療法がすでに承認されている[100,101]。
抗がん剤の多くは、MSとその症状の治療に再利用されている。これは、アントラセンジオン系の合成化合物であるミトキサントロン(44,図5)の場合で、乳がん、前立腺がん、急性白血病、リンパ腫の治療に使用される広範囲の抗腫瘍剤として確立されている[102]。ミトキサントロン(44)は,その免疫抑制作用により,抗原に対する中枢神経系T細胞およびB細胞の不規則な反応,マクロファージを介したミエリン損傷,および軸索損傷を伴うMSの治療薬としても承認されている。ミトキサントロン(44)は、T細胞の活性化を抑制し、T細胞およびB細胞の増殖を止め、抗体産生を低下させ、マクロファージを不活性化することができる。また、ミトキサントン(44)は高い忍容性を示した[103]。
アルキル化剤のシクロホスファミド(45)は、さまざまな固形腫瘍の治療に使用されており、白血病、リンパ腫、乳がんなどの治療に承認されている。シクロホスファミドは、ナイトロジェンマスタードに関連しており、DNAに結合して有糸分裂と細胞複製を阻害し、主に急速に分裂する細胞を標的とする。MSでの使用は、シクロホスファミド(45)が免疫抑制および免疫調節の役割を果たすことに由来する。具体的には、T細胞とB細胞に作用し、細胞媒介性免疫と液性免疫を抑制する。また、シクロホスファミド(45)は、脳や血液中のプロ炎症性Tヘルパー1サイトカインであるインターフェロン-γやインターロイキン-12の分泌を減少させ、抗炎症性サイトカインの分泌を増加させることが明らかになっている。また、Tリンパ球を低炎症性の表現型に変化させる。シクロホスファミド(45)は、血液脳関門を伝染し、中枢神経系でのバイオアベイラビリティが高く、免疫調節と免疫抑制を行い、病気の安定化と進行防止を図る[104]。
代謝拮抗薬であるクラドリビン(46)は、ヘアリーセル白血病やその他の血液がんの治療に使用されている。クラドリビンはデオキシアデノシン類似体で、活性化するためには細胞内でリン酸化されて三リン酸になる必要があり、これにより細胞死がもたらされる。この薬はMSの治療に再利用されていたが 2013年に却下された。最近では 2017年に、クラドリビン(46)が欧州医薬品庁からこの病気の治療薬として販売を許可された。その作用機序は、循環しているBリンパ球とTリンパ球の減少に関連している。さらに、インターフェロン-αを産生する骨髄性樹状細胞の誘導や、インターロイキン-1βのシナプス作用の阻害などのメカニズムも示唆されており、クラドリビン(46)は神経保護作用を示す可能性があると結論づけられている[105,106]。
アミロライド(47)は,高血圧症や心不全・肝疾患によるむくみの治療に用いられる利尿薬である。アミロライドは、MSの神経保護作用について研究されている。アミロライドは、MS病変部の軸索やオリゴデンドロサイトに過剰に発現している神経のプロトンゲート型酸感知イオンチャネル1(ASIC1)を遮断することで、神経保護作用や骨髄保護作用を発揮する。さらに、アミロライドの保護効果が炎症の下流で起こるという事実は、炎症の発症時にも有効であるという利点をもたらしている[107]。
イブジラスト(48)は、気管支喘息や脳血管障害の治療薬として一部の国で承認されている。イブジラストは、抗炎症作用で知られるホスホジエステラーゼを阻害することで作用するが、MSに関係するロイコトリエンや一酸化窒素の合成機構を阻害することもできる。脳では、イブジラスト(48)は、ミクログリアやアストロサイトからの腫瘍壊死因子の放出を抑制し、神経細胞の変性を抑制する。さらに、アストロサイトをアポトーシスから守り、オリゴデンドロサイトのアポトーシスと脱髄を抑制することができるため、MSに有用である。研究では、高用量で脳の萎縮率を減少させながら、その安全性と忍容性が示されている[108]。
6. 筋萎縮性側索硬化症
筋萎縮性側索硬化症(ALS)は、随意筋を制御する上下の運動ニューロンが死滅する疾患である。これにより、筋肉が徐々に弱くなり、サイズが小さくなる筋萎縮が起こる。症状としては、筋肉の硬直や痙攣、呼吸・嚥下・会話の障害などがある。ALSの原因は、ほとんどの場合、原因不明であり、約10%は遺伝性であるとされている[109]。
現在、ALSの治療に使用できる薬剤が検討されているが、一度現れた症状を元に戻すことはできないものの、病気の進行を遅らせることができる薬剤は、リルゾールとエダラボンの2種類のみである[110,111]。
マシチニブ(49,図6)は、犬のがん治療に使われるチロシンキナーゼ阻害剤である。ALSで増殖する異常なグリア細胞は、チロシンキナーゼ阻害剤に感受性があるのではないかという点に着目して、ALSへの使用が検討されている。マスチニブ(49)は、適切なラットモデルにおいて、グリア細胞の活性化を阻害し、生存率を高めることが証明されている[112]。
先に述べたイブジラスト(48,図5)も、神経保護作用があることから、ALSの治療法として研究されている[13]。
ALS患者の血清中の逆転写酵素濃度がHIV感染者と同等であること、ALS患者の脳にヒト内因性レトロウイルスの発現が認められたことから、抗HIV治療薬として使用されている抗レトロウイルス剤「トリュメック®」がALS治療薬として研究されている。このことからも、抗HIV薬がALSに有効であることがわかる。Triumeq®は、インテグラーゼ阻害剤であるドルテグラビル(50)抗レトロウイルス剤であるアバカビル(51)とラミブジン(52)の配合剤で、ALS患者に対する安全性と忍容性が確認されている[13]。
レチガビン(53)は,てんかん治療薬として承認されている薬剤で,電位依存性カリウムチャネルに結合してM電流を増加させ,膜の過分極をもたらす作用がある。レチガビン(53)は、運動ニューロンの生存期間を延長し、興奮性を低下させることができる。この病気では、ニューロンが通常よりも過剰に興奮し、最終的には細胞死に至ると考えられているため、ALSの治療に有利である。 この薬はALSの治療薬として現在も臨床試験中である[13]。
タモキシフェン(54)は抗エストロゲン薬で,乳がんの化学療法や化学予防のために承認されている。タモキシフェン(54)を投与した乳がん患者の神経学的改善と病状の安定化が観察されたことから、この薬剤のALS治療への再利用が偶然にも始まった。タモキシフェンの神経保護作用はこれまでにも報告されており、ALS患者の脊髄で過剰に発現しているプロテインキナーゼCの阻害に関係していると考えられている。さらに、タモキシフェン(54)は、オートファジーを調節することで、ALSに見られるプロテオパシーを調節することができることがわかった[113-115]。
本章で紹介した化合物はすべて、近年注目されているALSの治療薬として臨床試験が行われている。
7. 再利用の失敗
医薬品の再利用には多くの成功例があるが、多くの再利用の試みが失敗していることも事実である。計算機レベルや試験管内試験のアッセイでは有望に見えても、生体内試験では活性がなく、新たな活性を求めて研究を断念してしまうことがある。これは、ロシアでアレルギー性鼻炎の治療薬として承認された抗ヒスタミン薬であるラトレピルジンのケースで、ADやHDへの再利用が試みられた。作用機序が明確に確立されていないにもかかわらず、チャネルや神経伝達物質の活性を調節し、アミロイドβの毒性をブロックすることができることなどが報告されていた[116,117]。実際には、第2相試験ではプラセボに比べてアルツハイマー病患者の改善が見られたものの、第3相試験では疾患の進行に有意な変化を検出できなかった[118]。HDの第3相試験でも同じことが起こった[119]。
高コレステロール血症の治療薬であるシンバスタチンとアトルバスタチンもまた、ADへの再利用が試みられている。この背景には、ADと心血管疾患がしばしば重なり合っているという重大な観察結果があった。スタチン系薬剤は、アミロイドβの濃度を低下させ、神経保護作用を高めることがわかってた。 しかし、どちらもADの治療には役立たないことが証明された[120,121]。
抗うつ剤として使用される選択的セロトニン再取り込み阻害剤も、AD治療への使用を評価するために研究されてきた。ノルトリプチリンとパロキセチンは、当初、認知機能の改善を示したが、さらなる評価により、これらの薬剤が気分障害を治療した後でも、認知行動の改善は見られなかったという結論に達した[122]。
抗菌薬のセフトリアキソンは、ALSの治療のための第2相試験では有望と思われたが、第3相試験では臨床的な有効性を示すことができなかった[123]。
クラドリビンでさえ、承認される前にMSの再利用された治療法として最初は却下された[105]。これらの例から、NDの新しい治療法の発見において薬剤の再利用が励みになることが示されているにもかかわらず、その承認に至るまでの道のりには問題があり、再利用の試みを放棄してしまうことが非常に多いことが推察される。
8. 結論
上述のデータを分析した結果、医薬品の再利用は、元々治療薬として使用されていたものとは別の疾患を治療するための候補の興味深い供給源となり得ることが明らかになった。また、安全性と忍容性がすでに確立されているため、薬剤の再利用にかかるコストは、新薬の設計や最適化にかかるコストよりも大幅に低く、臨床試験の費用対効果が高く、必要なサンプル数も少なくて済み、臨床開発の全体的なコストを削減できるという魅力的な選択肢でもある。また、ハイスループットスクリーニングや計算化学などの技術が発達したことで、古い薬でも、簡単かつ迅速に新しいターゲットのテストができるようになり、これも利点となっている。
ここで紹介する神経変性疾患は不治の病であり、患者の生活の質に大きく影響する。薬剤の再利用の例では、主に症状の管理や病気の進行を止めることにメリットがあるとされている。残念ながら、神経変性のメカニズムを完全に元に戻すことができる薬剤はまだ見つかっていない。しかし、この総説で紹介したいくつかの薬剤は、すでに第2相および第3相の臨床試験が行われている。つまり、数年後には、神経変性疾患に対する武器は、現在よりもはるかに多くなると考えられる。また、第7章で紹介したように、臨床試験を経たものの、有効性を証明できなかったり、高い毒性を示したりした化合物もある。しかし、これらの薬剤を見捨てるべきではない。むしろ、分子修飾やリガンドベースのデザインによって新しい化合物を合成するためのモデルとなる出発点と考えるべきである。精神疾患の背後にある神経科学の完全な理解に向けた継続的な進歩と相まって、NDのための薬の再利用は将来的にさらに有望なものとなるであろう。
資金調達
本研究は、科学技術・高等教育省(PIDDAC)の科学技術財団(FCT/MCTES)による国家資金と、欧州地域開発基金(ERDF)のCOMPET-Programa Operacional Factores de Competitividade(POFC)プログラム(戦略的資金 UID/Multi/04423/2013)による支援を受けている。欧州地域開発基金(ERDF)を通じて、ポルトガル2020パートナーシップ協定の下、北ポルトガル地域運営プログラム(NORTE 2020)の支援を受け、PT2020プログラムの枠組みおよびプロジェクトINNOVMAR-Innovation and Sustainability in the Management and Exploitation of Marine Resources(参照番号:NORTE-01-0145-FEDER-000035,研究ラインNOVELMAR内)の支援を受けている。
利害の衝突
著者は利益相反のないことを宣言している。
参考文献
1. Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [CrossRef] [PubMed]
2. Doan, T.L.; Pollastri, M.; Walters, M.A.; Georg, G.I. Chapter 23—The future of drug repositioning: Old drugs,new opportunities. In Annual Reports in Medicinal Chemistry; Macor, J.E., Ed.; Academic Press: Waltham, MA, USA, 2011; Volume 46, pp. 385–401.
3. Fava, M. The promise and challenges of drug repurposing in psychiatry. World Psychiatry 2018, 17, 28–29. [CrossRef] [PubMed]
4. Richardson, P.; Hideshima, T.; Anderson, K. Thalidomide: Emerging role in cancer medicine. Annu. Rev. Med. 2002, 53, 629–657. [CrossRef] [PubMed]
5. Sleire, L.; Forde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.O. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [CrossRef] [PubMed]
6. Baker, N.C.; Ekins, S.; Williams, A.J.; Tropsha, A. A bibliometric review of drug repurposing. Drug Discov. Today 2018, 23, 661–672. [CrossRef] [PubMed]
7. Agnati, L.F.; Zoli, M.; Biagini, G.; Fuxe, K. Neuronal plasticity and ageing processes in the frame of the ‘red queen theory’. Acta Physiol. Scand. 1992, 145, 301–309. [CrossRef] [PubMed]
8. Agnati, L.F.; Benfenati, F.; Solfrini, V.; Biagini, G.; Fuxe, K.; Guidolin, D.; Carani, C.; Zini, I. Brain aging and neuronal plasticity. Ann. N. Y. Acad. Sci. 1992, 673, 180–186. [CrossRef] [PubMed]
9. Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Models Mech. 2017, 10, 499–502. [CrossRef] [PubMed]
10. Gammon, K. Neurodegenerative disease: Brain windfall. Nature 2014, 515, 299–300. [CrossRef] [PubMed]
11. Appleby, B.S.; Nacopoulos, D.; Milano, N.; Zhong, K.; Cummings, J.L. A review: Treatment of alzheimer’s disease discovered in repurposed agents. Dement. Geriatr. Cogn. Disord. 2013, 35, 1–22. [CrossRef] [PubMed]
12. Lee, H.-M.; Kim, Y. Drug repurposing is a new opportunity for developing drugs against neuropsychiatric disorders. Schizophr. Res. Treat. 2016, 2016, 6378137. [CrossRef] [PubMed]
13. Martinez, A.; Palomo Ruiz, M.D.; Perez, D.I.; Gil, C. Drugs in clinical development for the treatment of amyotrophic lateral sclerosis. Expert Opin. Investig. Drugs 2017, 26, 403–414. [CrossRef] [PubMed]
14. Kumar, A.; Singh, A. A review on alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [CrossRef] [PubMed]
15. Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [CrossRef]
16. Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of alzheimer disease—Insights from amyloid-beta metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [CrossRef] [PubMed]
17. Mucke, H.A.M. The case of galantamine: Repurposing and late blooming of a cholinergic drug. Future Sci. OA 2015, 1, FSO73. [CrossRef] [PubMed]
18. Heinrich, M. Chapter 4—Galanthamine from galanthus and other amaryllidaceae—Chemistry and biology based on traditional use. In The Alkaloids: Chemistry and Biology; Cordell, G.A., Ed.; Academic Press: Waltham, MA, USA, 2010; Volume 68, pp. 157–165.
19. Monacelli, F.; Cea, M.; Borghi, R.; Odetti, P.; Nencioni, A. Do cancer drugs counteract neurodegeneration? Repurposing for alzheimer’s disease. J. Alzheimers Dis. 2017, 55, 1295–1306. [CrossRef] [PubMed]
20. Blakeley, J.; Grossman, S.A. Chapter 17—Chemotherapy with cytotoxic and cytostatic agents in brain cancer. In Handbook of Clinical Neurology; Aminoff, M.J., Boller, F., Swaab, D.F., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 104, pp. 229–254.
21. Hayes, C.D.; Dey, D.; Palavicini, J.P.; Wang, H.; Patkar, K.A.; Minond, D.; Nefzi, A.; Lakshmana, M.K. Striking reduction of amyloid plaque burden in an alzheimer’s mouse model after chronic administration of carmustine. BMC Med. 2013, 11, 81. [CrossRef] [PubMed]
22. Tousi, B. The emerging role of bexarotene in the treatment of alzheimer’s disease: Current evidence. Neuropsychiatr. Dis. Treat. 2015, 11, 311–315. [CrossRef] [PubMed]
23. Fukasawa, H.; Nakagomi, M.; Yamagata, N.; Katsuki, H.; Kawahara, K.; Kitaoka, K.; Miki, T.; Shudo, K. Tamibarotene: A candidate retinoid drug for alzheimer’s disease. Biol. Pharm. Bull. 2012, 35, 1206–1212. [CrossRef] [PubMed]
24. Netzer, W.J.; Dou, F.; Cai, D.; Veach, D.; Jean, S.; Li, Y.; Bornmann, W.G.; Clarkson, B.; Xu, H.; Greengard, P. Gleevec inhibits -amyloid production but not notch cleavage. Proc. Natl. Acad. Sci. USA 2003, 100, 12444–12449. [CrossRef] [PubMed]
25. Brunden, K.R.; Yao, Y.; Potuzak, J.S.; Ferrer, N.I.; Ballatore, C.; James, M.J.; Hogan, A.M.; Trojanowski, J.Q.; Smith, A.B., III; Lee, V.M. The characterization of microtubule-stabilizing drugs as possible therapeutic agents for alzheimer’s disease and related tauopathies. Pharmacol. Res. 2011, 63, 341–351. [CrossRef] [PubMed]
26. Zhang, B.; Maiti, A.; Shively, S.; Lakhani, F.; McDonald-Jones, G.; Bruce, J.; Lee, E.B.; Xie, S.X.; Joyce, S.; Li, C.; et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc. Natl. Acad. Sci. USA 2005, 102, 227–231. [CrossRef] [PubMed]
27. Ryu, J.K.; McLarnon, J.G. Thalidomide inhibition of perturbed vasculature and glial-derived tumor necrosis factor-alpha in an animal model of inflamed alzheimer’s disease brain. Neurobiol. Dis. 2008, 29, 254–266. [CrossRef] [PubMed]
28. Diomede, L.; Cassata, G.; Fiordaliso, F.; Salio, M.; Ami, D.; Natalello, A.; Doglia, S.M.; De Luigi, A.; Salmona, M. Tetracycline and its analogues protect caenorhabditis elegans from beta amyloid-induced toxicity by targeting oligomers. Neurobiol. Dis. 2010, 40, 424–431. [CrossRef] [PubMed]
29. Costa, R.; Speretta, E.; Crowther, D.C.; Cardoso, I. Testing the therapeutic potential of doxycycline in a drosophila melanogaster model of alzheimer disease. J. Biol. Chem. 2011, 286, 41647–41655. [CrossRef] [PubMed]
30. Loeb, M.B.; Molloy, D.W.; Smieja, M.; Standish, T.; Goldsmith, C.H.; Mahony, J.; Smith, S.; Borrie, M.; Decoteau, E.; Davidson, W.; et al. A randomized, controlled trial of doxycycline and rifampin for patients with alzheimer’s disease. J. Am. Geriatr. Soc. 2004, 52, 381–387. [CrossRef] [PubMed]
31. Molloy, D.W.; Standish, T.I.; Zhou, Q.; Guyatt, G. A multicenter, blinded, randomized, factorial controlled trial of doxycycline and rifampin for treatment of alzheimer’s disease: The darad trial. Int. J. Geriatr. Psychiatry 2013, 28, 463–470. [CrossRef] [PubMed]
32. Tomiyama, T.; Shoji, A.; Kataoka, K.; Suwa, Y.; Asano, S.; Kaneko, H.; Endo, N. Inhibition of amyloid beta protein aggregation and neurotoxicity by rifampicin. Its possible function as a hydroxyl radical scavenger. J. Biol. Chem. 1996, 271, 6839–6844. [CrossRef] [PubMed]
33. Kimura, T.; Goto, M. Existence of senile plaques in the brains of elderly leprosy patients. Lancet 1993, 342, 1364. [CrossRef]
34. Chui, D.H.; Tabira, T.; Izumi, S.; Koya, G.; Ogata, J. Decreased beta-amyloid and increased abnormal tau deposition in the brain of aged patients with leprosy. Am. J. Pathol. 1994, 145, 771–775. [PubMed]
35. Goto, M.; Kimura, T.; Hagio, S.; Ueda, K.; Kitajima, S.; Tokunaga, H.; Sato, E. Neuropathological analysis of dementia in a japanese leprosarium. Dementia 1995, 6, 157–161. [CrossRef] [PubMed]
36. Endoh, M.; Kunishita, T.; Tabira, T. No effect of anti-leprosy drugs in the prevention of alzheimer’s disease and beta-amyloid neurotoxicity. J. Neurol. Sci. 1999, 165, 28–30. [CrossRef]
37. Wozniak, M.A.; Itzhaki, R.F. Antiviral agents in alzheimer’s disease: Hope for the future? Ther. Adv. Neurol. Disord. 2010, 3, 141–152. [CrossRef] [PubMed]
38. Hartsel, S.C.; Weiland, T.R. Amphotericin b binds to amyloid fibrils and delays their formation: A therapeutic mechanism? Biochemistry 2003, 42, 6228–6233. [CrossRef] [PubMed]
39. Smith, N.W.; Annunziata, O.; Dzyuba, S.V. Amphotericin b interactions with soluble oligomers of amyloid abeta1-42 peptide. Bioorg. Med. Chem. 2009, 17, 2366–2370. [CrossRef] [PubMed]
40. Grossi, C.; Francese, S.; Casini, A.; Rosi, M.C.; Luccarini, I.; Fiorentini, A.; Gabbiani, C.; Messori, L.; Moneti, G.; Casamenti, F. Clioquinol decreases amyloid-beta burden and reduces working memory impairment in a transgenic mouse model of alzheimer’s disease. J. Alzheimers Dis. 2009, 17, 423–440. [CrossRef] [PubMed]
41. Mark, R.J.; Ashford, J.W.; Goodman, Y.; Mattson, M.P. Anticonvulsants attenuate amyloid beta-peptide neurotoxicity, Ca2+ deregulation, and cytoskeletal pathology. Neurobiol. Aging 1995, 16, 187–198. [CrossRef]
42. Smith, A.M.; Gibbons, H.M.; Dragunow, M. Valproic acid enhances microglial phagocytosis of amyloid-beta(1–42). Neuroscience 2010, 169, 505–515. [CrossRef] [PubMed]
43. Qing, H.; He, G.; Ly, P.T.; Fox, C.J.; Staufenbiel, M.; Cai, F.; Zhang, Z.; Wei, S.; Sun, X.; Chen, C.H.; et al. Valproic acid inhibits abeta production, neuritic plaque formation, and behavioral deficits in alzheimer’s disease mouse models. J. Exp. Med. 2008, 205, 2781–2789. [CrossRef] [PubMed]
44. Tariot, P.N.; Schneider, L.S.; Cummings, J.; Thomas, R.G.; Raman, R.; Jakimovich, L.J.; Loy, R.; Bartocci, B.; Fleisher, A.; Ismail, M.S.; et al. Chronic divalproex sodium to attenuate agitation and clinical progression of alzheimer disease. Arch. Gen. Psychiatry 2011, 68, 853–861. [CrossRef] [PubMed]
45. Culman, J.; Blume, A.; Gohlke, P.; Unger, T. The renin-angiotensin system in the brain: Possible therapeutic implications for at(1)-receptor blockers. J. Hum. Hypertens. 2002, 16 (Suppl. 3), S64–S70. [CrossRef] [PubMed]
46. Wright, J.W.; Harding, J.W. Brain renin-angiotensin—A new look at an old system. Prog. Neurobiol. 2011, 95, 49–67. [CrossRef] [PubMed]
47. Wang, J.; Ho, L.; Chen, L.; Zhao, Z.; Zhao, W.; Qian, X.; Humala, N.; Seror, I.; Bartholomew, S.; Rosendorff, C.; et al. Valsartan lowers brain -amyloid protein levels and improves spatial learning in a mouse model of alzheimer disease. J. Clin. Investig. 2007, 117, 3393–3402. [CrossRef] [PubMed]
48. Li, N.C.; Lee, A.; Whitmer, R.A.; Kivipelto, M.; Lawler, E.; Kazis, L.E.; Wolozin, B. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: Prospective cohort analysis. BMJ 2010, 340, b5465. [CrossRef] [PubMed]
49. Danielyan, L.; Klein, R.; Hanson, L.R.; Buadze, M.; Schwab, M.; Gleiter, C.H.; Frey, W.H. Protective effects of intranasal losartan in the app/ps1 transgenic mouse model of alzheimer disease. Rejuvenation Res. 2010, 13, 195–201. [CrossRef] [PubMed]
50. Hanyu, H.; Hirao, K.; Shimizu, S.; Iwamoto, T.; Koizumi, K.; Abe, K. Favourable effects of nilvadipine on cognitive function and regional cerebral blood flow on spect in hypertensive patients with mild cognitive impairment. Nucl. Med. Commun. 2007, 28, 281–287. [CrossRef] [PubMed]
51. Zhao, W.; Wang, J.; Ho, L.; Ono, K.; Teplow, D.B.; Pasinetti, G.M. Identification of antihypertensive drugs which inhibit amyloid-beta protein oligomerization. J. Alzheimers Dis. 2009, 16, 49–57. [CrossRef] [PubMed]
52. Bachmeier, C.; Beaulieu-Abdelahad, D.; Mullan, M.; Paris, D. Selective dihydropyiridine compounds facilitate the clearance of beta-amyloid across the blood-brain barrier. Eur. J. Pharmacol. 2011, 659, 124–129. [CrossRef] [PubMed]
53. Zou, H.; Zhu, X.X.; Ding, Y.H.; Jin, Q.Y.; Qian, L.Y.; Huang, D.S.; Cen, X.J. Trimetazidine in conditions other than coronary disease, old drug, new tricks? Int. J. Cardiol. 2017, 234, 1–6. [CrossRef] [PubMed]
54. Hassanzadeh, G.; Hosseini, A.; Pasbakhsh, P.; Akbari, M.; Ghaffarpour, M.; Takzare, N.; Zahmatkesh, M. Trimetazidine prevents oxidative changes induced in a rat model of sporadic type of alzheimer’s disease. Acta Med. Iran. 2015, 53, 17–24. [PubMed]
55. Carro, E.; Torres-Aleman, I. The role of insulin and insulin-like growth factor i in the molecular and cellular mechanisms underlying the pathology of alzheimer’s disease. Eur. J. Pharmacol. 2004, 490, 127–133. [CrossRef] [PubMed]
56. Watson, G.S.; Craft, S. Modulation of memory by insulin and glucose: Neuropsychological observations in alzheimer’s disease. Eur. J. Pharmacol. 2004, 490, 97–113. [CrossRef] [PubMed]
57. Zhao, W.Q.; Chen, H.; Quon, M.J.; Alkon, D.L. Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 2004, 490, 71–81. [CrossRef] [PubMed]
58. Perry, T.; Lahiri, D.K.; Sambamurti, K.; Chen, D.; Mattson, M.P.; Egan, J.M.; Greig, N.H. Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (abeta) levels and protects hippocampal neurons from death induced by abeta and iron. J. Neurosci. Res. 2003, 72, 603–612. [CrossRef] [PubMed]
59. Perry, T.; Lahiri, D.K.; Chen, D.; Zhou, J.; Shaw, K.T.; Egan, J.M.; Greig, N.H. A novel neurotrophic property of glucagon-like peptide 1: A promoter of nerve growth factor-mediated differentiation in pc12 cells. J. Pharmacol. Exp. Ther. 2002, 300, 958–966. [CrossRef] [PubMed]
60. McClean, P.L.; Parthsarathy, V.; Faivre, E.; Holscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of alzheimer’s disease. J. Neurosci. 2011, 31, 6587–6594. [CrossRef] [PubMed]
61. Xiong, H.; Zheng, C.; Wang, J.; Song, J.; Zhao, G.; Shen, H.; Deng, Y. The neuroprotection of liraglutide on alzheimer-like learning and memory impairment by modulating the hyperphosphorylation of tau and neurofilament proteins and insulin signaling pathways in mice. J. Alzheimers Dis. 2013, 37, 623–635. [PubMed]
62. Wagner, J.; Vulinovic, F.; Grunewald, A.; Unger, M.M.; Moller, J.C.; Klein, C.; Michel, P.P.; Ries, V.; Oertel, W.H.; Alvarez-Fischer, D. Acylated and unacylated ghrelin confer neuroprotection to mesencephalic neurons. Neuroscience 2017, 365, 137–145. [CrossRef] [PubMed]
63. Lucchi, C.; Curia, G.; Vinet, J.; Gualtieri, F.; Bresciani, E.; Locatelli, V.; Torsello, A.; Biagini, G. Protective but not anticonvulsant effects of ghrelin and jmv-1843 in the pilocarpine model of status epilepticus. PLoS ONE 2013, 8, e72716. [CrossRef] [PubMed]
64. Bulgarelli, I.; Tamiazzo, L.; Bresciani, E.; Rapetti, D.; Caporali, S.; Lattuada, D.; Locatelli, V.; Torsello, A. Desacyl-ghrelin and synthetic gh-secretagogues modulate the production of inflammatory cytokines in mouse microglia cells stimulated by beta-amyloid fibrils. J. Neurosci. Res. 2009, 87, 2718–2727. [CrossRef] [PubMed]
65. Ding, Y.; Qiao, A.; Wang, Z.; Goodwin, J.S.; Lee, E.-S.; Block, M.L.; Allsbrook, M.; McDonald, M.P.; Fan, G.-H. Retinoic acid attenuates -amyloid deposition and rescues memory deficits in an alzheimer’s disease transgenic mouse model. J. Neurosci. 2008, 28, 11622–11634. [CrossRef] [PubMed]
66. Jarvis, C.I.; Goncalves, M.B.; Clarke, E.; Dogruel, M.; Kalindjian, S.B.; Thomas, S.A.; Maden, M.; Corcoran, J.P. Retinoic acid receptor-alpha signalling antagonizes both intracellular and extracellular amyloid-beta production and prevents neuronal cell death caused by amyloid-beta. Eur. J. Neurosci. 2010, 32, 1246–1255. [CrossRef] [PubMed]
67. Shudo, K.; Fukasawa, H.; Nakagomi, M.; Yamagata, N. Towards retinoid therapy for alzheimer’s disease. Curr. Alzheimer Res. 2009, 6, 302–311. [CrossRef] [PubMed]
68. Tippmann, F.; Hundt, J.; Schneider, A.; Endres, K.; Fahrenholz, F. Up-regulation of the alpha-secretase adam10 by retinoic acid receptors and acitretin. FASEB J. 2009, 23, 1643–1654. [CrossRef] [PubMed]
69. Di Meco, A.; Lauretti, E.; Vagnozzi, A.N.; Praticò, D. Zileuton restores memory impairments and reverses amyloid and tau pathology in aged ad mice. Neurobiol. Aging 2014, 35, 2458–2464. [CrossRef] [PubMed]
70. Zhang, J.; Guo, J.; Zhao, X.; Chen, Z.; Wang, G.; Liu, A.; Wang, Q.; Zhou, W.; Xu, Y.; Wang, C. Phosphodiesterase-5 inhibitor sildenafil prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in app/ps1 transgenic mice. Behav. Brain Res. 2013, 250, 230–237. [CrossRef] [PubMed]
71. Garcia-Barroso, C.; Ricobaraza, A.; Pascual-Lucas, M.; Unceta, N.; Rico, A.J.; Goicolea, M.A.; Salles, J.; Lanciego, J.L.; Oyarzabal, J.; Franco, R.; et al. Tadalafil crosses the blood-brain barrier and reverses cognitive dysfunction in a mouse model of ad. Neuropharmacology 2013, 64, 114–123. [CrossRef] [PubMed]
72. Halliday, M.; Radford, H.; Zents, K.A.M.; Molloy, C.; Moreno, J.A.; Verity, N.C.; Smith, E.; Ortori, C.A.; Barrett, D.A.; Bushell, M.; et al. Repurposed drugs targeting eif2 -p-mediated translational repression prevent neurodegeneration in mice. Brain 2017, 140, 1768–1783. [CrossRef] [PubMed]
73. Antony, P.M.; Diederich, N.J.; Kruger, R.; Balling, R. The hallmarks of parkinson’s disease. FEBS J. 2013, 280, 5981–5993. [CrossRef] [PubMed]
74. Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [CrossRef] [PubMed]
75. Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A.; et al. Nilotinib effects in parkinson’s disease and dementia with lewy bodies. J. Parkinsons Dis. 2016, 6, 503–517. [CrossRef] [PubMed]
76. Hebron, M.L.; Lonskaya, I.; Moussa, C.E.H. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of -synuclein in parkinson’s disease models. Hum. Mol. Genet. 2013, 22, 3315–3328. [CrossRef] [PubMed]
77. González-Lizárraga, F.; Socías, S.B.; Ávila, C.L.; Torres-Bugeau, C.M.; Barbosa, L.R.S.; Binolfi, A.; Sepúlveda-Díaz, J.E.; Del-Bel, E.; Fernandez, C.O.; Papy-Garcia, D.; et al. Repurposing doxycycline for synucleinopathies: Remodelling of -synuclein oligomers towards non-toxic parallel beta-sheet structured species. Sci. Rep. 2017, 7, 41755. [CrossRef] [PubMed]
78. Bermejo, P.E.; Anciones, B. A review of the use of zonisamide in parkinson’s disease. Ther. Adv. Neurol. Disord. 2009, 2, 313–317. [CrossRef] [PubMed]
79. Fox, S.H.; Katzenschlager, R.; Lim, S.Y.; Barton, B.; de Bie, R.M.A.; Seppi, K.; Coelho, M.; Sampaio, C. International parkinson and movement disorder society evidence-based medicine review: Update on treatments for the motor symptoms of parkinson’s disease. Mov. Disord. 2018. [CrossRef] [PubMed]
80. Riederer, P.; Muller, T. Monoamine oxidase-b inhibitors in the treatment of parkinson’s disease: Clinical-pharmacological aspects. J. Neural. Transm. 2018, 1–7. [CrossRef] [PubMed]
81. Biagini, G.; Zoli, M.; Fuxe, K.; Agnati, L.F. L-deprenyl increases gfap immunoreactivity selectively in activated astrocytes in rat brain. Neuroreport 1993, 4, 955–958. [CrossRef] [PubMed]
82. Biagini, G.; Frasoldati, A.; Fuxe, K.; Agnati, L.F. The concept of astrocyte-kinetic drug in the treatment of neurodegenerative diseases: Evidence for l-deprenyl-induced activation of reactive astrocytes. Neurochem. Int. 1994, 25, 17–22. [CrossRef]
83. Devos, D.; Moreau, C.; Delval, A.; Dujardin, K.; Defebvre, L.; Bordet, R. Methylphenidate: A treatment for parkinson’s disease? CNS Drugs 2013, 27, 1–14. [CrossRef] [PubMed]
84. Jankovic, J. Exenatide—A drug for diabetes and parkinson disease? Nat. Rev. Neurol. 2017, 13, 643–644. [CrossRef] [PubMed]
85. Athauda, D.; Wyse, R.; Brundin, P.; Foltynie, T. Is exenatide a treatment for parkinson’s disease? J. Parkinsons Dis. 2017, 7, 451–458. [CrossRef] [PubMed]
86. Aviles-Olmos, I.; Dickson, J.; Kefalopoulou, Z.; Djamshidian, A.; Ell, P.; Soderlund, T.; Whitton, P.; Wyse, R.; Isaacs, T.; Lees, A.; et al. Exenatide and the treatment of patients with parkinson’s disease. J. Clin. Investig. 2013, 123, 2730–2736. [CrossRef] [PubMed]
87. Bomba, M.; Ciavardelli, D.; Silvestri, E.; Canzoniero, L.M.; Lattanzio, R.; Chiappini, P.; Piantelli, M.; Di Ilio, C.; Consoli, A.; Sensi, S.L. Exenatide promotes cognitive enhancement and positive brain metabolic changes in ps1-ki mice but has no effects in 3xtg-ad animals. Cell Death Dis. 2013, 4, e612. [CrossRef] [PubMed]
88. Mittal, S.; Bjørnevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. B2-adrenoreceptor is a regulator of the -synuclein gene driving risk of parkinson’s disease. Science 2017, 357, 891–898. [CrossRef] [PubMed]
89. Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [CrossRef] [PubMed]
90. Roos, R.A. Huntington’s disease: A clinical review. Orphanet J. Rare Dis. 2010, 5, 40. [CrossRef] [PubMed]
91. Paleacu, D. Tetrabenazine in the treatment of huntington’s disease. Neuropsychiatr. Dis. Treat. 2007, 3, 545–551. [PubMed]
92. Roos, R.A.; Buruma, O.J.; Bruyn, G.W.; Kemp, B.; van der Velde, E.A. Tiapride in the treatment of huntington’s chorea. Acta Neurol. Scand. 1982, 65, 45–50. [CrossRef] [PubMed]
93. Bonuccelli, U.; Ceravolo, R.; Maremmani, C.; Nuti, A.; Rossi, G.; Muratorio, A. Clozapine in huntington’s chorea. Neurology 1994, 44, 821–823. [CrossRef] [PubMed]
94. Paleacu, D.; Anca, M.; Giladi, N. Olanzapine in huntington’s disease. Acta Neurol. Scand. 2002, 105, 441–444. [CrossRef] [PubMed]
95. Coppen, E.M.; Roos, R.A.C. Current pharmacological approaches to reduce chorea in huntington’s disease. Drugs 2017, 77, 29–46. [CrossRef] [PubMed]
96. Duff, K.; Beglinger, L.J.; O’Rourke, M.E.; Nopoulos, P.; Paulson, H.L.; Paulson, J.S. Risperidone and the treatment of psychiatric, motor, and cognitive symptoms in huntington’s disease. Ann. Clin. Psychiatry 2008, 20, 1–3. [CrossRef] [PubMed]
97. Alpay, M.; Koroshetz, W.J. Quetiapine in the treatment of behavioral disturbances in patients with huntington’s disease. Psychosomatics 2006, 47, 70–72. [CrossRef] [PubMed]
98. Beister, A.; Kraus, P.; Kuhn, W.; Dose, M.; Weindl, A.; Gerlach, M. The n-methyl-d-aspartate antagonist memantine retards progression of huntington’s disease. J. Neural Transm. Suppl. 2004, 68, 117–122.
99. Anitha, M.; Nandhu, M.S.; Anju, T.R.; Jes, P.; Paulose, C.S. Targeting glutamate mediated excitotoxicity in huntington’s disease: Neural progenitors and partial glutamate antagonist–memantine. Med. Hypotheses 2011, 76, 138–140. [CrossRef] [PubMed]
100. Goldenberg, M.M. Multiple sclerosis review. P&T 2012, 37, 175–184.
101. Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [CrossRef] [PubMed]
102. Hrynchak, I.; Sousa, E.; Pinto, M.; Costa, V.M. The importance of drug metabolites synthesis: The case-study of cardiotoxic anticancer drugs. Drug Metab. Rev. 2017, 49, 158–196. [CrossRef] [PubMed]
103. Hartung, H.-P.; Gonsette, R.; Konig, N.; Kwiecinski, H.; Guseo, A.; Morrissey, S.P.; Krapf, H.; Zwingers, T. Mitoxantrone in progressive multiple sclerosis: A placebo-controlled, double-blind, randomised, multicentre trial. Lancet 2002, 360, 2018–2025. [CrossRef]
104. Awad, A.; Stuve, O. Cyclophosphamide in multiple sclerosis: Scientific rationale, history and novel treatment paradigms. Ther. Adv. Neurol. Disord. 2009, 2, 50–61. [CrossRef] [PubMed]
105. Leist, T.P.; Weissert, R. Cladribine: Mode of action and implications for treatment of multiple sclerosis. Clin. Neuropharmacol. 2011, 34, 28–35. [CrossRef] [PubMed]
106. Holmoy, T.; Torkildsen, O.; Myhr, K.M. An update on cladribine for relapsing-remitting multiple sclerosis. Expert Opin. Pharmacother. 2017, 18, 1627–1635. [CrossRef] [PubMed]
107. Arun, T.; Tomassini, V.; Sbardella, E.; de Ruiter, M.B.; Matthews, L.; Leite, M.I.; Gelineau-Morel, R.; Cavey, A.; Vergo, S.; Craner, M.; et al. Targeting asic1 in primary progressive multiple sclerosis: Evidence of neuroprotection with amiloride. Brain 2013, 136, 106–115. [CrossRef] [PubMed]
108. Barkhof, F.; Hulst, H.E.; Drulovic, J.; Uitdehaag, B.M.; Matsuda, K.; Landin, R. Ibudilast in relapsing-remitting multiple sclerosis: A neuroprotectant? Neurology 2010, 74, 1033–1040. [CrossRef] [PubMed]
109. Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [CrossRef] [PubMed]
110. Zoccolella, S.; Beghi, E.; Palagano, G.; Fraddosio, A.; Guerra, V.; Samarelli, V.; Lepore, V.; Simone, I.L.; Lamberti, P.; Serlenga, L.; et al. Riluzole and amyotrophic lateral sclerosis survival: A population-based study in southern italy. Eur. J. Neurol. 2007, 14, 262–268. [CrossRef] [PubMed]
111. Sawada, H. Clinical efficacy of edaravone for the treatment of amyotrophic lateral sclerosis. Expert Opin. Pharmacother. 2017, 18, 735–738. [CrossRef] [PubMed]
112. Trias, E.; Ibarburu, S.; Barreto-Núñez, R.; Babdor, J.; Maciel, T.T.; Guillo, M.; Gros, L.; Dubreuil, P.; Díaz-Amarilla, P.; Cassina, P.; et al. Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J. Neuroinflamm. 2016, 13, 177. [CrossRef] [PubMed]
113. Goodman, A. Tamoxifen, a cancer therapy, explored for als. Neurol. Today 2005, 5, 22–26. [CrossRef]
114. Hu, J.H.; Zhang, H.; Wagey, R.; Krieger, C.; Pelech, S.L. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J. Neurochem. 2003, 85, 432–442. [CrossRef] [PubMed]
115. Wang, I.F.; Guo, B.S.; Liu, Y.C.; Wu, C.C.; Yang, C.H.; Tsai, K.J.; Shen, C.K. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the tar DNA-binding protein 43. Proc. Natl. Acad. Sci. USA 2012, 109, 15024–15029. [CrossRef] [PubMed]
116. Bezprozvanny, I. The rise and fall of dimebon. Drug News Perspect. 2010, 23, 518–523. [PubMed]
117. Bharadwaj, P.R.; Bates, K.A.; Porter, T.; Teimouri, E.; Perry, G.; Steele, J.W.; Gandy, S.; Groth, D.; Martins, R.N.; Verdile, G. Latrepirdine: Molecular mechanisms underlying potential therapeutic roles in alzheimer’s and other neurodegenerative diseases. Transl. Psychiatry 2013, 3, e332. [CrossRef] [PubMed]
118. Doody, R.S.; Gavrilova, S.I.; Sano, M.; Thomas, R.G.; Aisen, P.S.; Bachurin, S.O.; Seely, L.; Hung, D. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate alzheimer’s disease: A randomised, double-blind, placebo-controlled study. Lancet 2008, 372, 207–215. [CrossRef]
119. Cano-Cuenca, N.; Solis-Garcia del Pozo, J.E.; Jordan, J. Evidence for the efficacy of latrepirdine (dimebon) treatment for improvement of cognitive function: A meta-analysis. J. Alzheimers Dis. 2014, 38, 155–164. [PubMed]
120. Sano, M.; Bell, K.L.; Galasko, D.; Galvin, J.E.; Thomas, R.G.; van Dyck, C.H.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of simvastatin to treat alzheimer disease. Neurology 2011, 77, 556–563. [CrossRef] [PubMed]
121. Sparks, D.L.; Sabbagh, M.N.; Connor, D.J.; Lopez, J.; Launer, L.J.; Petanceska, S.; Browne, P.; Wassar, D.; Johnson-Traver, S.; Lochhead, J.; et al. Atorvastatin therapy lowers circulating cholesterol but not free radical activity in advance of identifiable clinical benefit in the treatment of mild-to-moderate ad. Curr. Alzheimer Res. 2005, 2, 343–353. [CrossRef] [PubMed]
122. Nebes, R.D.; Pollock, B.G.; Houck, P.R.; Butters, M.A.; Mulsant, B.H.; Zmuda, M.D.; Reynolds, C.F., 3rd. Persistence of cognitive impairment in geriatric patients following antidepressant treatment: A randomized, double-blind clinical trial with nortriptyline and paroxetine. J. Psychiatr. Res. 2003, 37, 99–108. [CrossRef]
123. Cudkowicz, M.E.; Titus, S.; Kearney, M.; Yu, H.; Sherman, A.; Schoenfeld, D.; Hayden, D.; Shui, A.; Brooks, B.; Conwit, R.; et al. Efficacy and safety of ceftriaxone for amyotrophic lateral sclerosis: Results of a multi-stage, randomised, double-blind, placebo-controlled, phase 3 study. Lancet 2014, 13, 1083–1091. [CrossRef]