
Contents
オンラインで公開2020年6月14日
要旨
NRF2は、遺伝子の発現を制御することで作用し、第II相抗酸化応答のマスターレギュレーターであり、神経炎症の制御の鍵を握っている。NRF2の活性は、プロテアソームによるタンパク質分解、転写、転写後など、いくつかのレベルで制御されている。本レビューの目的は、NRF2制御の主なメカニズムと、神経変性疾患の治療標的としての実際の、あるいは潜在的な利用について、簡潔かつ批判的な概観を提供することである。
キーワード
NRF2-ARE経路、神経変性疾患、酸化ストレス、KEAP1,NRF2制御
1. 共通の病態としての神経変性疾患と酸化ストレス
過去数十年の間に、アルツハイマー病、パーキンソン病(PD)ハンチントン病(HD)フリードリヒ失調症(FRDA)筋萎縮性側索硬化症(ALS)多発性硬化症(MS)脳卒中など、加齢に関連した慢性疾患の有病率が増加し、平均寿命が継続的に伸びている[1]。従来の「一病一機構一医療」というアプローチとは対照的に、慢性疾患には多因子性の病因があり、疾患発症に関与する機構についてのより正確な知識が必要とされている[1]。
ヒトの疾患ネットワークの発展は、異なる疾患(NDD)が共通の分子機構を持つ可能性があることを強調している[2]。このように、神経変性疾患は、ミトコンドリアの機能不全、慢性炎症、酸化ストレスの増大に伴う代謝・保護機能のアンバランス、活性酸素種(ROS)の病理学的な形成を伴うホメオスタシスの喪失によって特徴づけられている[1]。
このような背景から、核内因子(赤血球由来2)様2(NRF2)は、複数の細胞保護反応の制御に関与する魅力的な薬剤化可能な標的であると指摘する多くのエビデンスが存在する[1]。本レビューでは、神経変性疾患(NDD)に対する薬剤開発のための新たな戦略を提供するために、NRF2の調節に関与するさまざまなメカニズムを提示する。NRF2の活性制御は、プロテアソーム分解や転写・転写後レベルでの制御を含む複雑なものである[3]。
2. NRF2-ARE経路
1994 年に NRF2 転写因子が発見された後 [4]、抗酸化応答エレメント(ARE)と呼ばれるエンハンサー配列を含む約 250 個の遺伝子の発現制御に関与していることが明らかになった [5]。これらの遺伝子は、プロオキシダント、電気化学物質、炎症性物質からの細胞保護、外来物質の生体内変換反応、ミトコンドリア機能の維持、タンパク質のホメオスタシス、抗酸化代謝に関わるいくつかの酵素をコードしている[6]。
NRF2レベルの維持には、NRF2の合成とプロテアソーム分解の適切なバランスが必要である[7,8]。図1に示すように、生理的条件下では、NRF2は通常、細胞質に存在している[7]。NRF2の主な陰性制御系であるKEAP1(Kelch-like ECH-associated protein 1)は、NRF2を細胞質に維持し、Cullin 3(Cul3)/Rbx1ベースのE3-ユビキチンリガーゼと複合体を生成し、NRF2のユビキチン化とそれに続くプロテアソーム分解を誘導することができる[8]。このように、NRF2はストレスのない条件下では、プロテアソームによって構成的に合成され、迅速に分解される[8]。しかし、酸化ストレスに応答して、KEAP1の「センサー」領域に存在するシステイン残基が酸化され、KEAP1からNRF2を放出する構造変化を引き起こす。あるいは、これらのシステイン残基は、電気泳動剤との反応によって活性化され得る[9]。NRF2がKEAP1から解放されると、それは核内に移動し、そこでsMaf(small masculoaponeurotic fibrosarcoma)タンパク質のような異なるアクチベーターとヘテロ二量体を形成する [1,910]。
図1 正常条件下での核内因子(赤血球由来2)様2(NRF2)の負の調節と病理学的条件下での活性化
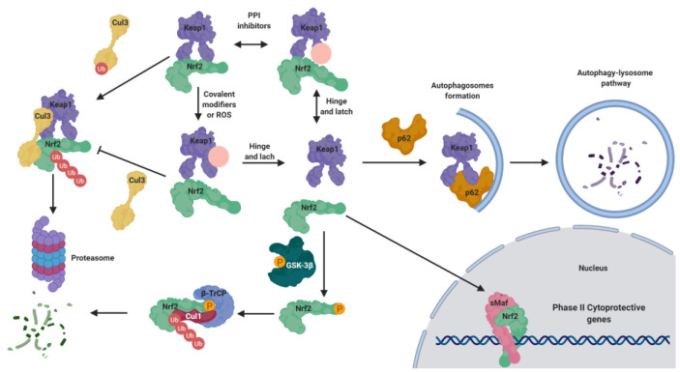
通常の条件下では、NRF2はKelch-like ECH-associated protein 1 (KEAP1)と結合し、細胞質に維持される。KEAP1は、NRF2をユビキチン化するカリン3(CuI3)ユビキチンリガーゼのアダプターとして作用し、プロテアソーム26Sによる分解につながる。病理的条件下では、活性酸素種(ROS)や共有結合修飾剤の存在下で、KEAP1の構造が変化し、細胞質でNRF2が解放され、その後、それは核に移動し、それは抗酸化応答要素(ARE)配列に結合するために小さな男性ホルモン性線維肉腫(sMaf)タンパク質とヘテロ二量体を形成し、フェーズII遺伝子の発現を促進する。このメカニズムは、「ヒンジ&ラッチ」NRF2活性化メカニズムとして知られている。さらに、NRF2は、追加のメカニズムによって緊密に制御されている。それは、オートファジーリソソーム経路によるKEAP1分解を誘導することができるので、p62の作用によって過剰に活性化することができる。逆に、キナーゼ・グリコーゲン合成酵素キナーゼ-3β(GSK-3β)が過剰に活性化されている病理学的条件下では、NRF2を直接リン酸化し、その分解を誘導するためにCul1リガーゼアダプターβ-transducing repeat-containing protein(β-TrCp)との相互作用を促進することができる。
3. NRF2の構造
NRF2は、基本領域ロイシンジッパー(bZIP)転写因子、より具体的にはキャップ”n”カラー(CNC)サブファミリーに属している[11]。Neh1-7として知られる7つのNRF2-ECH相同性ドメインによって形成され、それぞれが特定の機能を持つ(図2)[12]。Neh1ドメインにはCNC-bZIP領域が含まれており、これはNRF2がDNAを認識することを可能にし、sMafタンパク質などのパートナーとのヘテロ二量化を促進する[13]。この領域のアミノ酸配列の保存性の高さは、広範な種の間で見出されており [14]、NRF2の転写活性の重要性を強調している。Neh2は主なNRF2ネガティブレギュレーターであるKEAP1との相互作用に関与している[15]。この相互作用は、このドメインに存在するデグロンモチーフ、すなわちDLG(低親和性)およびETGE(高親和性)を介して行われる[15]。さらに、Neh2領域にも含まれる7つのリジン残基の存在は、NRF2のユビキチン化後のプロテアソーム分解を促進する[9]。C末端のNeh3,Neh4,およびNeh5は、転写機械の様々な構成要素に結合するトランザクティベーションドメインであり、NRF2標的遺伝子の転写を促進する[16,17]。Neh6は、DSGISおよびDSAPGSという2つの保存されたペプチドモチーフを含むセリンに富んだ領域である[18]。グリコーゲン合成酵素キナーゼ-3β(GSK-3β)は、DSGIS配列をリン酸化することができ、それによってβ-transducing repeat-containing protein(β-TrCP)のNeh6への結合効率を高め、NRF2のプロテアソーム分解を促進する[18]。最後に、Neh7はRXRα(レチノイン酸受容体α)との結合に関与し、CBP([CREB(cAMP-応答性エレメント結合タンパク質)結合タンパク質])のNeh4およびNeh5ドメインへの結合を阻害し、ARE配列の転写を阻害する[19]。
図2
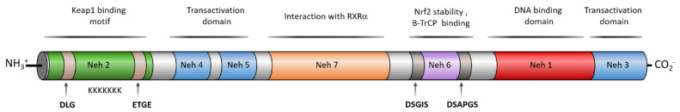
ヒトNRF2のドメイン構造と各ドメインに関連する活性 NRF2は7つのNehドメインで構成されており、それぞれ異なる相互作用パターンと特異的な活性を持っている。Neh1は、sMafタンパク質との二量化に関与するbZip領域を提示し、ARE領域やc-JUN、Sp-1,JDP2(Jun二量化タンパク質2)などの他の転写因子と結合する。また、Neh1はアセチル化感受性ドメインであり、Crm1によって輸出されるNES配列を含み、AMPK(AMP-activated protein kinase)によるリン酸化の標的となる。Neh2は、KEAP1や他のユビキチンリガーゼの結合ドメインを提示する分解用ドメイン(デグロンドメイン)である。KEAP1は、DLGおよびETGEモチーフをそれぞれ異なる親和性で標的とする。Neh2は、ユビキチン化の標的である7つのLys残基、NLS配列、PKCδのリン酸化標的であるセリン残基(S40)からなるαヘリックスを含み、両方のモチーフはNRF2の核内転座に関連している。Neh3ドメインは、別のNLS配列と、核内転座に関連するアセチル化の標的である2つのLys残基を含む。Neh4とNeh5は、CBP、P300,Hrd1,RAC3のNRF2への結合に関与するトランザクティベーションドメインである。Neh6は、GSK-3βとDSAPGSモチーフによってリン酸化された後、DSGISモチーフとの相互作用によってβ-transducing repeat-containing protein (β-TrCP)によって標的とされる第二のデグロンドメインを含む。最後に、Neh7は、RXRαによって標的化されたリプレッサードメインである。
古典的なNRF2標的遺伝子は、いくつかの小器官および細胞内コンパートメントに分布する酵素をコードしている[20]。これらの酵素は、活性酸素を消去し、スーパーオキシドジスムターゼ(SOD)グルタチオンペルオキシダーゼ、カタラーゼ、グルタチオン還元酵素(GR)グルタミン酸システインリガーゼ(GCL)またはNAD(P)H/キノンオキシドレダクターゼ1(NQO1)などの電解質を中和する代謝反応に関与している[20]。
また、NRF2は、非シトクロムP450フェーズIおよびフェーズIIの薬物代謝酵素をコードする遺伝子を含む、薬物の代謝および分配に関与する遺伝子の誘導にも重要な役割を果たしている[21]。
さらに、ヘムオキシゲナーゼ-1(HO-1)を誘導することでヘム基の分解を促進し、誘導された酵素の一部は、細胞内に最も多く存在する抗酸化物質・求電子中和分子であるグルタチオン(GSH)の生合成に不可欠なグルタチオン合成酵素のような抗酸化小分子の生成に関与している[22]。
NRF2はまた、オートファジー[23]、ミトコンドリア機能の維持[24]、炎症性サイトカインの発現抑制[25]などの重要なプロセスに関連するタンパク質を制御している。
4. 神経変性疾患の特徴とNRF2とのクロストーク
4.1. 酸化ストレス
活性酸素の濃度が中程度であれば、セカンドメッセンジャーとして生理的な役割を果たすが[26]、活性酸素濃度の不均衡は酸化ストレスを引き起こし、いくつかの病理学的プロセスの発症と関連している。酸化ストレスは神経変性の主要な特徴の一つであり、活性酸素の過剰産生と抗酸化防御機能の低下の組み合わせから生じる。このアンバランスは分子およびオルガネラの損傷を引き起こし、重症化すると神経細胞死に至る。O2の酸化的代謝から生じる活性酸素種は、リノール酸やアラキドン酸などの不飽和脂肪酸と反応する [27]。結果として生じる反応性の高いペルオキシラジカルは、近くの脂肪酸との連鎖反応を開始する[28]。活性酸素促進鎖反応から生じるこれらの分子のいくつかは、アルツハイマー病およびパーキンソン病における4-ヒドロキシ-2-ノネナール(HNE)[28,29]、アルツハイマー病におけるアクロレインおよびF2-イソプロスタンス[30]、パーキンソン病におけるマロンジアルデヒドなどの特定の神経変性疾患に特徴的である。また、パーキンソン病患者の脳内では8-ヒドロキシグアニンと8-ヒドロキシ-2-デオキシグアノシンのレベルが上昇していることが示されているように、DNAは酸化的損傷を受けやすくなっている[31]。アクロレインやHNEは,マイケル付加を介してシステイン,リジン,ヒスチジン残基と架橋することで毒性を誘導する[32]。これらの付加体の形成は、酵素や受容体を損傷させ、DNA変異を誘発し、様々な生化学的経路の誤作動を引き起こす[32]。さらに、活性酸素はタンパク質の構造変化を誘導し、それによってタンパク質のフォールディングや凝集のミスフォールディングを引き起こす [33]。また、これらのタンパク質の構造変化は、電圧依存性カルシウムチャネル(VDCC)やN-メチル-D-アスパラギン酸(NMDA)受容体などの複合体の機能不全を引き起こし、細胞内カルシウム過負荷や興奮毒性の原因となる[33]。さらに、酸化ストレスは酸化還元感受性経路を活性化し、持続的なM1ミクログリアの活性化につながる[34]。
細胞は、第I相(チトクロームP450s)および第II相(解毒および抗酸化タンパク質)酵素の転写を誘導することにより、細胞の酸化還元恒常性を維持するための多くの解毒機構を発達させてきた[34]。以上のように、NRF2は細胞の酸化還元恒常性のマスターレギュレーターであり、この経路の活性化は酸化的ストレスまたは求電子的ストレスに対する有効なメカニズムである[35]。
4.2. 神経炎症
4.2.1. NRF2と神経炎症。一般的な側面
炎症は、腫瘍や感染症に対する防御において重要な役割を果たしており、組織の修復に不可欠である;しかしながら、慢性的な炎症は有害であることがあり、神経変性過程における共通の特徴である[36,37]。
神経炎症のプロセスは、主に、2つの異なる活性化経路によって、ミクログリア細胞が安静状態M0から活性化状態(M1またはM2)に切り替わることによって媒介される[38]。炎症性のM1表現型は、IL-1β、IL-6,腫瘍壊死因子α(TNF-α)などの炎症性サイトカインを放出し、活性酸素の形成を増加させ、組織損傷を引き起こす [38]。代替的な活性化経路は、組織再生過程に関与し、抗炎症性サイトカイン(IL-4,IL-10,およびIL-13)の産生によって特徴づけられる神経保護的なM2状態を促進する[38]。M1/M2表現型間の不均衡は、神経保護因子または神経変性因子としてのミクログリアの複雑な役割を理解するために広く研究されている[39]。
NRF2の活性化は、ミクログリアおよびアストロサイトにおけるTNF-α、IL-1β(インターロイキン1β)IL-6,および一酸化窒素合成酵素(iNOS)の発現などのいくつかの炎症性サイトカインの量を減少させることにより、抗炎症性の特性を発揮する[10,39]。アルツハイマー病(APP/PSEN1)マウスモデルの海馬と皮質におけるNRF2経路の誘導は、ミクログリア症、アストログリア症、およびプロ炎症性サイトカインTNF-αとIL-17Aの分泌を減少させることが示されている[40]。最も重要なことは、ADマウスの長期記憶の低下が有意に減衰したことである[40]。さらに、いくつかのヒトの研究では、NRF2の活性化によって達成されたアミロイドβペプチド誘発毒性に対する細胞保護特性と、ミクログリアの状態をM1からM2の表現型にシフトさせることによる抗炎症効果との関連が示されている[41]。NRF2活性化は、ALS(筋萎縮性側索硬化症)マウスモデルにおいて、神経炎症の減衰、神経細胞の生存、および活性化されたミクログリアの劇症的効果の抑制に有用であることが示されている[42]。同様に、薬理学的に刺激されたNRF2活性化は、神経炎症の主要な細胞メディエーターであるマウスのミクログリアとアストロサイト、およびHD患者の血液単球における炎症反応を抑制した[43]。NRF2はまた、以下に詳述するように、主要な炎症反応経路のうち2つの経路を阻害する。
4.2.2. Nrf2とToll-like Receptorシグナル伝達
Toll様受容体(TLR)は、核内因子κB(NF-κB)などの転写因子の活性化を介して炎症性免疫応答を開始し、炎症性分子の合成に関与している(図3)。TLRシグナル伝達に関与するいくつかのタイプの免疫細胞が脳内に存在し、アルツハイマー病、PD、およびMSなどの自己免疫過程を含む様々な神経変性疾患における病理学的神経炎症反応の発現に重要な役割を果たしている[44]。
図3 Toll様受容体(TLR)と核内因子(赤血球由来2)様2(NRF2)経路間のクロストーク
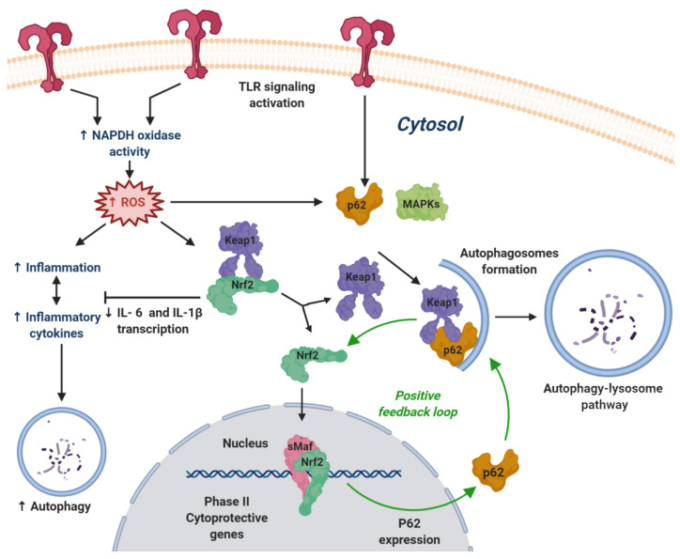
TLRのシグナル伝達開始は、NOX (NADPHオキシダーゼ)酵素の活性化を誘導して大量の活性酸素を生成し、それがKEAP1に存在するセンサーCys残基と反応してNRF2の遊離と核内移動を誘導し、第II相抗酸化反応を活性化させる。NRF2制御遺伝子は、プロ炎症性サイトカインの産生を減少させ、フリーラジカルの形成を減少させ、プロ炎症反応を停止させるのに役立つ。TLRの活性化および活性酸素の産生は、p62の発現およびマイトジェン活性化プロテインキナーゼ(MAPK)の活性を増加させる。p62タンパク質はKEAP1を標的とし、オートファジーを介してその分解を誘導し、NRF2-ARE経路の活性化を促進する。ターンでは、NRF2は、p62を含むオートファジーフラックスに関与する様々なタンパク質の発現を増加させ、正のフィードバックループを生成し、TLR活性化によって誘導されたプロ炎症性シグナル伝達を解決するために抗酸化フェーズII応答を強化する。
TLRシグナル伝達とNRF2経路の間のクロストークの存在はよく知られており、両方の経路の協調した作用は、抗炎症応答の微調整された制御を可能にする。図3に要約されるように、TLRシグナル伝達とNRF2経路の間の接続は、主にNADPH酸化酵素に関連した活性酸素の生成、またはp62またはマイトジェン活性化プロテインキナーゼ(MAPK)キナーゼの誘導から生じる。全体的なプロセスは、プロ炎症性サイトカインの転写を阻害し、抗炎症性分子のNRF2介在性発現を阻害することにより、オートファジーを介したサイトカインの調節を介して炎症を調節する[45]。
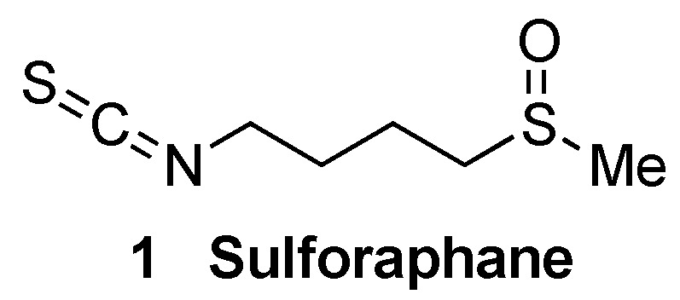
NRF2を誘導するいくつかの化合物がTLR活性を調節し、全体的に改善された抗炎症プロファイルにつながることは興味深いことである。スルフォラファン(化合物 1,図 4)は良い例で、TLR3 媒介の NF-κB シグナルを阻害し、KEAP1 に共有結合することで NRF2 を誘導することができる(セクション 5.1.2 でさらに議論される)[46]。ポリフェノールは、このようなマルチターゲット挙動のもう一つの例である[47]。
図4 TLR3 介在性 NF-κB シグナルを阻害し、KEAP1 に共有結合して NRF2 を誘導するマルチターゲット化合物スルフォラファンの構造
4.2.3. NRF2 と NFκB
核内因子-κB ファミリーは、TLR シグナルの下流で活性化され、TNF-α やいくつかのインターロイキンなどの炎症性サイトカインの発現によって炎症反応を誘導するいくつかのタンパク質から構成されている。NRF2との関連は非常に複雑であり、その主な特徴は図5に要約されている[48]。
図5 核内因子-κB(NF-κB)と NRF2 パスウェイ間のクロストーク
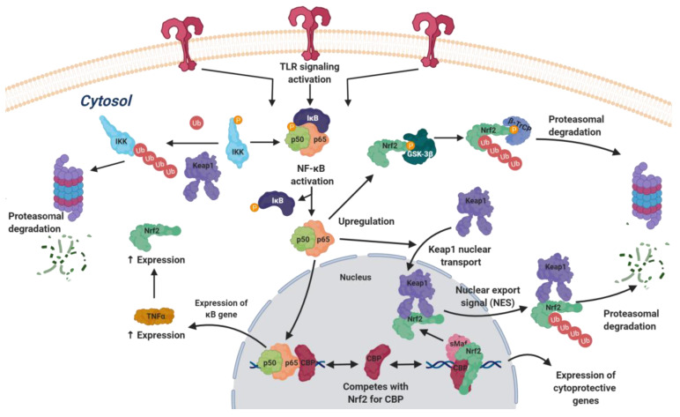
TLR 活性化に伴い、NF-κB は IKK (IκB キナーゼ) 複合体によって促進されるリン酸化によって、その天然リプレッサーである IκB から解放される。解放された NF-κB 二量体は、CREB 結合タンパク質(CBP)と結合して NRF2 と競合し、腫瘍壊死因子α(TNF-α)シトカインを含む炎症性遺伝子の発現を開始する。NF-κB は、そのモノマーである p65 を介して NRF2 を負に制御し、p65 は KEAP1 の核内への取り込みを促進し、NRF2 と結合し、第 II 相関連遺伝子の発現を終了させる。KEAP1-NRF2複合体は細胞質へと輸送され、Cul3ユビキチンリガーゼと結合してNRF2のプロテアソーム分解を誘導する。さらに、GSK-3βはNRF2を直接リン酸化し、β-TrCPとの相互作用によりNRF2の分解をコードしている。また、β-TrCPはIκB(inhibitor of nuclear factor kappa B)の分解を誘導し、NF-κBの活性を高め、NRF2の活性化を制限する。一方、KEAP1はプロテアソーム分解を介してIKKの分解を誘導することでIκBを安定化させる。このように、NF-κB と NRF2 の間のクロストークを閉じ、炎症と抗酸化反応の間に微調整されたループを形成する。
NF-κB は、いくつかのメカニズムによって NRF2 経路に作用する p65 を介して NRF2 駆動の遺伝子発現に負の影響を与える [48]。一方、p65 は、ヒストンのアセチル化によって作用する転写共活性化因子である CREB 結合タンパク質(CBP)を NRF2 と競合する [49];一方で、p65 は KEAP1 の核への輸送を促進し、そこで NRF2 と結合し、核輸出シグナル(NES)を介して核から除去する [49]。一方、p65はTNF-αレベルの上昇を介してNRF2遺伝子の発現を増加させることができる。
4.3. プロテオスタシス
特定のタンパク質のフォールディングの誤りと、それに続く毒性のある形での凝集は、神経変性疾患の最も一般的な特徴である。これらの異常なタンパク質は、細胞ストレス、突然変異、またはタンパク質の生産ミスから生じる [50]。タンパク質静態系は、タンパク質の合成、適切な折り畳み、分解、およびクリアランスを制御しているが、加齢に伴い、誤って折り畳まれたタンパク質の蓄積により、タンパク質静態系が崩壊し、ユビキチン化された包接体(IB)の量が増加する [51]。疾患によって、パーキンソン病ではα-シヌクレイン(α-シヌクレイン)アルツハイマー病ではβ-アミロイド(アミロイドβ)プラークと高リン酸化タウ神経原線維タングル(NFT)HDではハンチン(Htt)ALSではスーパーオキシドジスムターゼ1(SOD1)TAR DNA結合タンパク質43(TDP-43)海綿状脳症ではスクレイピープリオンタンパク質(PrPSc)など、異なるタンパク質の凝集体が見られる[52]。誤って折り畳まれたタンパク質沈着物の形成と適応免疫との間には強い関連性が見出されている [53]。
NRF2とタンパク質分解経路との間には興味深いクロストークが存在する [54]。NRF2が介在するこれらのシステムの構成要素の転写 [23,55] は、ミスフォールドされたタンパク質の分解を促進する一方で、NRF2はそのプロテアソーム分解により、またオートファジーを介してもダウンレギュレートされる [54]。いくつかの試験管内試験および生体内試験モデルでは、NRF2を介して薬理学的にタンパク質クリアランスを活性化し、オートファジーを介してリン酸化された不溶性のタウの分解を増加させることで、保護的な役割を果たすことが証明されている[56, 57]。さらに、NRF2 誘導剤であるスルフォラファン(1,図 4)は、NRF2 誘導を介して mHtt(変異型ハンチンチン蛋白質)のプロテアソーム分解を亢進させた[23]。最後に、5.1.2節で詳しく述べるMS治療薬として承認されているフマル酸ジメチルの神経保護効果は、パーキンソン病のα-シヌクレイン障害モデルにおけるオートファジー活性化に一部起因していることが示されている[58]。
5. 神経変性疾患治療への有用なアプローチとしてのNRF2活性化
NRF2欠損はいくつかの神経変性疾患で発見されている;例えば、アルツハイマー病患者の海馬ニューロンでは核内NRF2の劇的な減少が見られ[59]、NRF2ノックアウトマウスなどのパーキンソン病の動物モデルではドーパミン作動性ニューロンの特異的な欠損が見られ[60]、ALS患者の死後の研究では運動野のKEAP1 mRNAの増加が見られ[61]、NRF2活性の低下につながることが示されている。他の研究では、タウやアミロイド障害を受けたニューロンではNRF2とその標的タンパク質であるシークエストソーム1(SQSTM1/p62)のレベルが上昇しており、おそらくオートファジーを介してこれらの有害タンパク質を放出するためのクリアランスメカニズムであると考えられることが示されている[23,62]。これらの結果と一致して、HO-1,NQO1,GCLM、およびSQSTM1レベルのレベルは、アルツハイマー病およびパーキンソン病脳で増加している[62,63]。この文脈では、NRF2活性化がアルツハイマー病 [64]、PD [65]、およびALS [42]動物モデルにおいて生存期間を延長することが実証されていることは驚くべきことではない。
神経変性モデルにおけるNRF2の発現または活性の低レベルとNRF2誘導による神経変性過程の減少は、NRF2-ARE経路が神経変性疾患sの有望な標的であることを示している[35,66]。さらに、NRF2 は酸化ストレス、神経炎症、プロテオスタシス異常など、神経変性過程に関与する複数の病態を制御しており、NRF2 を標的とすることで、複数の病態を同時に制御することが可能となる[23,25,35,54]。
5.1. KEAP1依存性の制御
NRF2 の制御は、その安定性や核内転位を制御するいくつかの細胞因子によって行われているが、その中でも特に重要なのは、先に述べた KEAP1 タンパク質である[9]。
5.1.1. 酸化ストレスセンサーとしての KEAP1
KEAP1は、625個のアミノ酸残基を持つZnメタロタンパク質であり、5つのドメインで形成された分岐ステム二量体構造を有する[67]。N末端領域(NTR)KEAP1のホモ二量化とCul3結合に関与するBTB/POZ(Bric-a-brac, tramtrac, broad-comple/proxvirus zinc fingers)ドメイン、BTBとDGRドメイン間のリンカーとして働く介在領域(IVR)NRF2の制御とアクチン相互作用に重要な二重グリシンリピート(DGR)ドメイン、C末端ドメイン(CTR)である[67](図6)。
図6
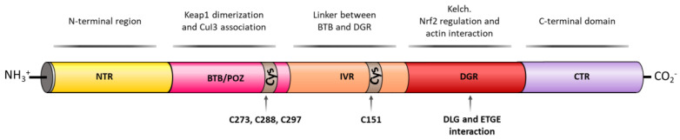
KEAP1の構造ドメインと関連する活性 KEAP1はN末端領域(NTR)、KEAP1のホモ二量化を可能にするTramtrack and Bric-á-Brac (BTB)ドメインを含み、Cul3 E3リガーゼとの結合に関与している。BTBドメインはまた、3つの重要なCys残基(Cys273,Cys288,Cys297)を含み、フリーラジカルや電気泳動剤と反応してKEAP1の構造変化を誘導し、細胞質内でNRF2を解放する。次のドメインは、重要なCys残基であるCys151を含む介在領域(IVR)であり、この残基はフリーラジカルや電気泳動剤と反応することができ、また、KEAP1の細胞質内での位置を調節するNESモチーフを含んでいる。ダブルグリシンリピート(DGR)ドメインは、NRF2,p62,および関連するE/STGEタンパク質の結合部位を含む6つのケルヒリピートを含む。最後に、KEAP1は、C末端ドメイン(CTR)を含む。
KEAP1はシステインを多く含むタンパク質であり、酸化還元活性であり、局所環境に対して反応性である[68]。KEAP1のシステイン残基の大部分は塩基性アミノ酸に囲まれており、それらのpKa値を下げることで反応性を高めている[69]。したがって、KEAP1の27個のシステイン残基は、タンパク質が求電子的または酸化的ストレスを感知する主要な部位であると提案されている[70]。KEAP1の配列全体に沿ってシステインが分布しているにもかかわらず、BTB/POZドメイン[71]のC273,C288,およびC297残基とIVRドメイン[72]のC151残基は、より反応性が高く、電着剤、活性酸素種(ROS)およびCd2+、As3+、およびSe4+などの金属の酸化還元センサーとして機能していることが証明されている[73]。スルフヒドリル基の化学修飾は、NRF2のユビキチン化を減少させ、KEAP1-NRF2の解離を引き起こし、NRF2の核内移動を促進する [74]。
ヒンジとラッチ」モデル [75] は、KEAP1に依存した制御のための最も広く受け入れられているメカニズムである。このモデルでは、Neh2ドメイン内のNRF2の2つの異なる結合部位が、KEAP1のDGRドメイン内の1つの重複部位と異なる親和性で相互作用し、ETGEモチーフはDLGよりも高い親和性を示す[76]。ホメオスタティック条件下では、ETGEモチーフのβヘアピン(高親和性)は、最初のステップでKEAP1サブユニットの1つと相互作用し、これはオープンコンフォメーションと呼ばれている[76,77]。このオープン構造は、NRF2が空間内で比較的自由に動くことができる「ヒンジ」を表している。第二段階では、DLGモチーフのβヘアピン(低親和性)は、いわゆるクローズドコンフォメーションでKEAP1の第二のプロトマーと同様に相互作用し、「ラッチ」を表す[76,77]。この閉じた構造は、NRF2の移動能力を制限し、NRF2のNeh2領域に存在するLysに富んだαヘリックスの最適な配置を可能にする。
酸化ストレス条件下では、NRF2誘導因子はKEAP1に存在するCys残基を酸化し、構造変化を促進する [77]。KEAP1のこの構造変化は、標的となるリジンの空間的配置の不適切な変化を引き起こし、その結果、NRF2はもはやユビキチン化も分解もされなくなる [78]。このことは、KEAP1の飽和をもたらし、その結果、新たに合成されたNRF2は、抑制を回避して直接核に蓄積し、標的遺伝子の発現を促進することができる[79]。
5.1.2. KEAP1 共有結合修飾因子
NRF2は酸化ストレスの増加によって生理的に活性化されるが、化学物質によって外因性に誘導されることもある[76]。既知のNRF2誘導剤の大部分は、酸化またはアルキル化によって、KEAP1のチオールリッチドメイン[80,81]に存在するシステイン残基を共有結合的に修飾する電着剤である。これらの親電性付加体は、2つの異なる方法でKEAP1を阻害することができる。すなわち、(1) KEAP1のNRF2との結合を阻害する構造変化を促進することによって、あるいは(2) KEAP1とCul3/Rbx1との相互作用を阻害し、NRF2のユビキチン化を阻害し、その隔離とそれに続く新たに合成されたNRF2の安定化を促進することによってである[82,83]。
過去数年の間に、このメカニズムを介してNRF2経路を活性化する可能性のある多くの天然および合成化合物が記載されており、これらを以下に要約する。
天然および半合成のNRF2活性化剤とその類似体
カフェイン酸(2,図 7)はコーヒーに含まれるポリフェノールであり、その構造にマイケルアクセプターを持つ。その求核性部位(カテコール部位)はクリアランスを提供するが、NRF2 の誘導には直接関与していないことが明らかにされている[84]。さらなる研究では、カフェ酸はまた、NRF2を活性化するためにKEAP1の発現を減少させることができ、HO-1(ヘムオキシゲナーゼ1)およびNQO1(NAD(P)H:キノンオキシドレダクターゼ1)の発現の増加につながることが示されている[84]。
図7 天然のNRF2活性化剤とその類似体の化学構造
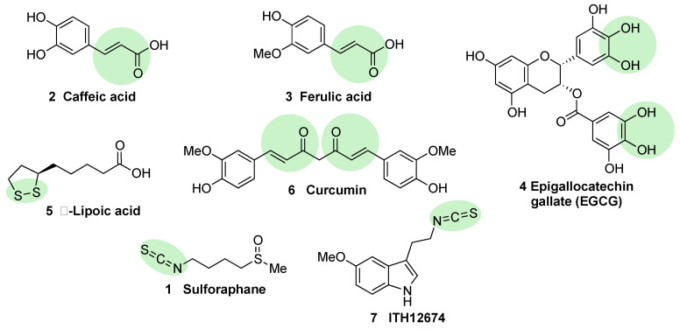
カフェイン酸(2)、フェルラ酸(3)、クルクミン(6)は、二重結合がカルボニル基と共役して強力な求電剤となる古典的なマイケルアクセプターと考えられている。エピガロカテキンガレート(4)は、酸化後にキノン誘導体を生成することができるポリフェノール化合物であり、対応するキノンは、強力な求電性特性を発揮する。リポ酸(5)は、KEAP1のCys残基と反応してリポイルシステニルジスルフィドを形成し、その構造を変化させることができる求電子性化合物と考えられている。最後に、スルフォラファン(1)とそのハイブリッドメラトニン誘導体であるITH12674(7)のイソチオチアン酸部位が、これらの化合物の強力な求電子性特性の原因となっている。各化合物の親電子性部位またはプロモイエットは緑色で強調表示されている。
フェルラ酸(3,図7)は野菜に多く含まれており、カフェイン酸との主な構造的な違いはベンゼン環上にメトキシ基が1つあることである。フェルラ酸は、ヒト神経芽腫細胞株SH-SY5Yにおいて、NRF2経路を制御し、トリメチルスズ(TMT)誘発の神経細胞障害を抑制することが証明されている[84]。さらに、活性酸素消去による酸化ストレスに拮抗し、非相同末端結合DNA修復プロセスを活性化することができる[84]。
緑茶に含まれるカテキンであるエピガロカテキンガレート(EGCG)(4,図7)[85]は、そのカテコール部位のオルトキノンへの事前の自己酸化を介した求電子的な混乱を介してNRF2経路をアップレギュレートする[86]だけでなく、p38-MAPKおよび細胞外シグナル調節キナーゼ(ERK)1/2シグナル伝達経路の活性化を介して[87]。EGCGは、MS、PD、および外傷性脳損傷のさまざまなモデルにおいて、生体内試験および試験管内試験で複数の神経保護効果を示しており、そのすべてがNRF2誘導の増加、抗酸化活性、および炎症性反応の減少に関連している[88,89,90]。
αリポ酸(5,図7)は、ニンジン、ビーツ、ブロッコリー、ほうれん草など、多くの植物に含まれている。アルファリポ酸が NRF2 を誘導する正確なメカニズムは現在のところ知られていないが、その構造から、アルファリポ酸は KEAP1 とリポイルシステインイルジスルフィドを形成し、NRF2 の分解を防ぐことができる[91,92]。この可能性のあるメカニズムに加えて、アルファリポ酸は、代替経路を介してNRF2を活性化することができるキナーゼの一つであるプロテインキナーゼC(PKC)を活性化することも示されている[93]。アルファリポ酸の神経保護効果に関する研究がいくつか行われているが、その多くでは、NRF2の活性化がこれらの効果に関与しているかどうかは調査されていない。マウスMSモデルでは、αリポ酸は炎症を抑制した[94]。また、PDモデルでは、活性酸素の減少、ATPレベルの回復、ドーパミン作動性ニューロンの保存、ミトコンドリア形成のアップレギュレーションなどの効果があった[95]。
クルクミン(6,図7)は、ウコン酸に存在する主なクルクミノイドであり、強力な抗酸化および抗炎症特性を持っている[96,97]。クルクミンは KEAP1 の Cys-151 を求電子的に修飾することで NRF2 を活性化し、活性酸素消去活性も修飾する [98]。一方、クルクミンはKEAP1の発現を抑制することでNRF2を活性化する。クルクミンは、脳内出血や外傷性脳損傷モデルにおいても神経保護作用を示した [25,100]。また、クルクミン投与によりミクログリア細胞のNF-kB活性化を抑制し、炎症性遺伝子の発現を低下させることが確認されている[101]。
スルフォラファン(1,図7)は、ブロッコリー、芽キャベツ、カリフラワー、キャベツなどのアブラナ科植物に含まれる有機硫黄化合物グルコラファニンを酵素で切断することで生成されるイソチオシアネートである[1]。スルフォラファンの放出に必要な触媒反応は、腸内細菌叢に存在するβ-チオグルコシダーゼによって駆動される[102]。スルフォラファンはKEAP1のCys151残基と直接相互作用し、それによってNRF2の活性化を促進する[103,104]。スルフォラファンは、純粋な形でもスプラウト抽出物としても、非常に優れた安全性プロファイルを示しており、慢性疾患に対する30以上の臨床試験が実施されている[105,106]。スルフォラファンは血液脳関門(BBB)を通過する能力を持ち、神経細胞のアミロイドβ1-42ペプチドに対する神経保護能力を示している[107]。In vivoでは、スルフォラファンはマウスのADモデルにおいて認知機能障害を緩和することが示されている。PDモデルでは、スルフォラファンはパーキンソン病の毒素である6-ヒドロキシドパミンからドーパミン作動性ニューロンを保護した[109]。さらに、スルフォラファンはリン酸化されたタウのレベルを低下させ、Beclin1とLC3-IIを増加させることができ、NRF2の活性化はオートファジーを介したタウの分解を促進するとの結論に至った[56]。それにもかかわらず、スルフォラファンは親水性媒体中での安定性が低い油性物質であり、その薬物動態学的特性を改善する必要がある[1]。この点、スルフォラファン-β-シクロデキストリン複合体であるスルフォラデックス(SFX-01)は優れたバイオアベイラビリティーを示し、くも膜下出血や転移性乳癌の治療薬として臨床試験が行われている[1]。最後に、スルフォラファンと天然の抗酸化物質メラトニン生成物質であるITH12674(7,図6)とのハイブリダイズが行われており、脳虚血の治療薬として二重の薬物-薬物作用機序を持つように設計された化合物である[110]。
抗酸化性炎症調節剤(AIM)として記載されているバルドキソロン(RTA401,CDDO、8a、図8)由来の半合成シアノエノントリテルペノイドは、マイケルアクセプター部位を示し、KEAP1のCys151と相互作用する最も強力な既知の求電子的NRF2活性化剤の一つである[111]。プルーフ・オブ・コンセプト研究では、これらのトリテルペノイドの変性疾患への使用が強く支持されている[111]。例えば、CDDO-エチルアミド(RTA405,8b、図8)およびCDDO-トリフルオロエチルアミド(RTA404,8c、図8)は、PDモデルにおいて毒性レベルの有意な低下を誘導した[65]。CDDO-メチルエステル(CDDO-Me、RTA402,8d、図8)は、糖尿病性腎症の治療のために臨床試験に到達した最初のCDDOであったが、その後、NRF2誘導とは関係のない心血管系の安全性の問題のために第III相(BEACON試験)で取り下げられた[112]。安全性の向上を目指して、CDDO-ジフルオロプロピオンアミド(RTA408,オマベロキソン、8e、図8)が合成され、現在、FRDA、眼炎症、眼手術後の疼痛の治療薬として第II相試験が行われている[113]。
図8 いくつかの半合成シアノエノントリテルペノイド(8)とニトロ脂肪酸(9)の化学構造
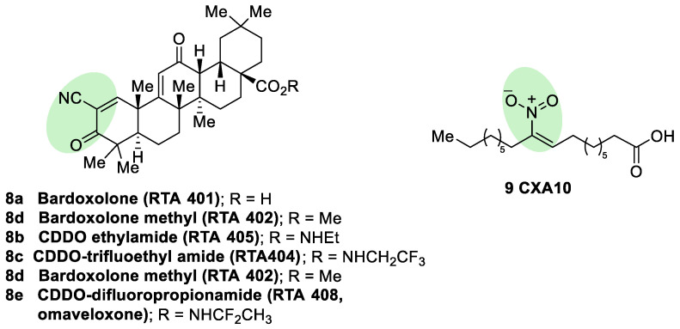
これらの化合物は、カルボニル基、シアノ基、またはニトロ基と共役したα,β-不飽和の存在により、求電子的なNRF2誘導剤と考えられている。これらの求電子性部位は緑色で強調表示されている。
ニトロ脂肪酸(NO2-FA)は、代謝性および炎症性疾患の前臨床動物モデルにおける抗炎症および抗線維化活性を有する内因性シグナル伝達メディエーターである[114]。ニトロアルケン構造は、そのβ炭素に求電子性を付与し、システインなどの求核剤とのマイケル付加体の形成を促進する。NO2-FAはKEAP1のCys-273およびCys-288と反応し、NRF2を活性化することが証明されている[115]。この反応の可逆性[116]により安定な付加体が形成されず、毒性の原因となる可能性がある。さらに、化合物9(10-NO2-OA、CXA10,図8)は、第I相安全性試験で活性用量でヒトで安全であることが証明されており[117]、現在、焦点性細分化糸球体硬化症の治療のために試験が行われている。
合成NRF2活性化剤
フマル酸エステルは、製薬業界から最も注目されている合成NRF2活性化剤のグループである[20]。フマル酸ジメチル(DMF)(10,図9)は、このファミリーの中で最も臨床的に成功しているメンバーである。乾癬の治療薬として1994年に最初に承認されたが[118]、多発性硬化症マウスモデルでの有効性のため 2013年にBiogen社が再発寛解型MSの治療薬としてTecfideraの名称で再利用し[119]、ここ数年で最も成功した新薬の1つとなっている[1]。チオール反応性マイケルアクセプター構造により、DMFは主にKEAP1のシステイン修飾を介してNRF2を活性化し[120]、ホスファチジルイノシトール-3-キナーゼ(PI3K)およびERK1/2経路を介してNRF2のリン酸化に影響を与えることも証明されている[121]。
図9 フマル酸エステル誘導体の化学構造
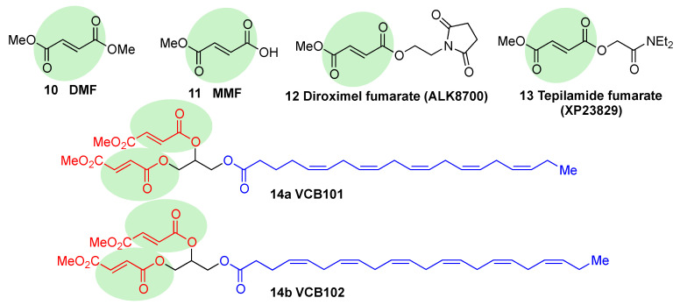
それらの求電子性部位は緑色で強調表示されている。
DMFは、ADモデルにおいて広範囲の神経変性状態を保護することが示されており[122]、脳卒中マウスモデルにおいて虚血後の海馬傷害を予防し、BBBの完全性を保護することが示されている[123]。さらに、α-Synおよびアミロイドβ毒性からの保護およびタウの高リン酸化の減少も観察された[58,124,125]。
DMFは、ほとんどが腸内エステラーゼによってフマル酸モノメチル(MMF、11,図9)に変換される。さらに、MMFはKEAP1のCys151と反応してNRF2を活性化することが示されている[126]。いくつかのバイオ製薬会社は、MMFのバイオアベイラビリティを改善し、DMFに関連する副作用を軽減するために、MMFの徐放性形態を開発している[111]。アルカーネス社は、副作用を軽減したMMFプロドラッグであるALK8700(フマル酸ジロキシメル、12,図9)を開発し、現在、MSを対象とした第III相試験を実施中である[127]。Tepilamide fumarate(XP23829,13,図9)は、XenoPort社[20]によって開発された別のMMFプロドラッグである。この化合物は、DMFと比較して溶解性および透過性が良好であり、また、有効性が改善され、消化管副作用が減少したことが示されており、現在、尋常性乾癬の治療のための第II相臨床試験が実施されている[20]。MMFとドコサヘキサエン酸の複合体(CAT4001)は、カタバシス社によって開発されている。動物モデルでは、FRDAやALSなどの神経変性疾患の治療に有望な活性を示している[20]。V ClinBio社は、同様のデザインで、エイコサペンテン酸(VCB101,14a、図9)またはドコサヘキセン酸(VCB102,14b、図9)のいずれかに連結された2分子のMMFを含む、中央のグリセロールユニットを中心に構築されたコンジュゲートをMSや乾癬の治療薬として開発した[20]。
OT551(15,図10)は、Othera Pharmaceuticalsによって開発されたN,N-二置換ヒドロキシルアミンである[1,128]。活性代謝物TEMPOL(4-ヒドロキシTEMPO、4-ヒドロキシ-2,2,6,6,6-テトラメチルピペリジン-1-オキシル)が、ラジカル消去活性に加えて、急性炎症におけるNF-κB[129]の活性化の低下やKEAP1を標的としたKEAP1-NRF2経路の活性化を間接的に、また、KEAP1を標的とした複合的なメカニズムにより、酸化ストレスや疾患に関連した炎症を抑制するプロドラッグです[130]。局所用に設計され、前臨床試験において網膜色素上皮および光受容体を酸化的損傷および炎症から保護した。また、加齢黄斑変性症を対象とした第II相臨床試験でも有効性が示されている[20]。
図10
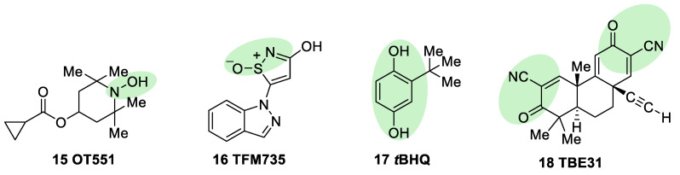
OT551,TFM735,tBHQ、およびTBE31の化学構造。それらの求電子性部位またはプロモイエットは緑色で強調表示されている。
ハイスループットなスクリーニング研究では、TFM735(16,図10)がCys151依存性のメカニズムを介してNRF2の活性化剤として同定された[131]。この化合物は現在、持田製薬がMSの治療薬として前臨床開発を行っている[20]。
tBHQ、tert-ブチルヒドロキノン(17,図10)は、KEAP1/NRF2複合体を破壊することができる親電子性キノンの代謝前駆体である。食品保存料として広く使用されており、その抗酸化特性のため、神経保護目的でも使用されている。tBHQによる処理は酸化ストレスを減少させ、NT2N細胞株における神経毒性やアミロイドβ形成を抑制した[132]。さらに、外傷性脳損傷後の二次損傷の減少や機能回復の改善にもつながった[133]。
完全合成のアセチレン系三環式ビス(シアノエノン)であるTBE-31(18,図10)は、最も強力な既知のNRF2誘導剤の一つである[91]。TBE-31 は主に癌モデルで研究されており、強い抗酸化活性と抗炎症活性を示している。さらに、TBE-31を投与すると、FRDAモデルでは線維芽細胞や小脳顆粒ニューロンのミトコンドリア機能が改善され、酸化ストレスから保護されることが示されている[134]。
5.1.3. KEAP1 タンパク質-タンパク質相互作用阻害剤
KEAP1は、NRF2の分解を促進することで、NRF2の主な調節因子となっている[8]。KEAP1に含まれるKelchドメインによるNRF2の認識は、創薬標的としてのタンパク質-タンパク質相互作用の古典的な例であり、一般的には標的表面が大きく、阻害剤との相互作用のためのポケットがないため、創薬の課題として残されている。しかし、KEAP1の場合、標的表面の大きさは約300~1000Å2であり、低分子を安定して認識できる残基を持つよく定義されたポケットを有している[135]。これらのトポロジカルな特徴は、KEAP1の3次元構造の利用可能性と相まって、KEAP1を認識し、他のタンパク質との結合を破壊することができる低分子の開発を可能にする[136]。
NRF2のNeh2ドメインに含まれるETGEモチーフは、KEAP1ケルチドメイン結合ポケットと相互作用するためにβヘアピン構造を採用しており、これは人工的に5つのサブポケットに細分化することができる(図11a)[136]。P1およびP2サブポケットは、複数の正に荷電したアルギニン残基(Arg415,Arg483,およびArg485)の存在によって特徴づけられ、これらのサイトが基質の負に荷電したアミノ酸残基を認識することを可能にしている[137]。特に、NRF2のETGEモチーフのGlu79およびGlu82のカルボキシル側鎖(図11b)は、P1およびP2サブポケットとの相互作用によって結合を促進する。中央のP3サブポケットは、ETGEモチーフを安定化する小さな極性残基および非極性残基によって形成される。外側のP4およびP5サブポケットは、脂肪族鎖およびチロシンに富んだ残基によって形成され、疎水性相互作用と水素結合を介して基質認識を向上させる[136,137]。
図11
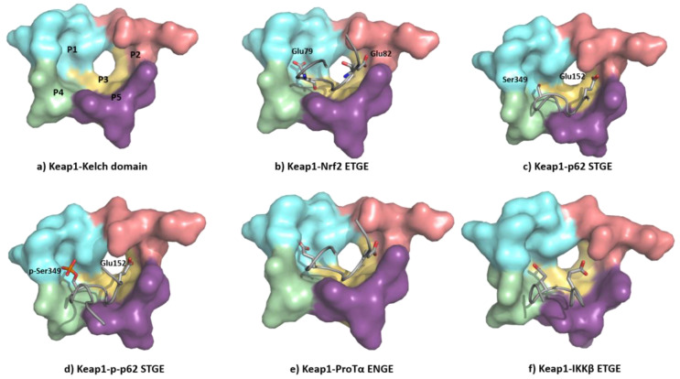
(a) KEAP1のKelchドメインの構造(PDB: 5WTV)結合ポケットの強調表示 P1: 青(残基415,461,462,478,483,508)P2: 赤(363,380,381,414)P3: 黄(364,509,556,571,602,603)P4. 緑(334,572,577);P5:紫(525,530,555);(b)KEAP1に結合したNRF2のETGEモチーフの構造詳細(PDB:5WTV);(c)p62のSTGEモチーフの構造詳細(PDB:3ADE)。KEAP1と相互作用する領域(KIR)にあるSTGEモチーフは、NRF2のETGE様モチーフと類似しており、両者とも似たようなβターン構造をとっている。しかし、ETGEのGlu79はKEAP1 P1サブポケットに含まれるArg415と相互作用するが、STGEの対応するSer349は短すぎて結合親和性が低い;(d)Ser349-リン酸化p62 STGEモチーフの構造詳細(PDB:3WDZ リン酸化されたSer439はP1サブポケットに深く挿入され、負の電荷はArg415とArg483との静電的相互作用を介して親和性を高める;(e) Protα ENGEモチーフの構造詳細(PDB 2Z32);(f) 分子動力学法によって得られたIKKβ ETGEモチーフの構造詳細[149]。Thr残基(DEETGE、NRF2)がAsn残基(NEENGE、ProTα)で置換されることにより、βターン構造が不安定になり、ENGEモチーフとKEAP1の間の結合は、NRF2のETGEモチーフと比較して、より強力ではない。
KEAP1のKelchドメインは、NRF2と同様の方法でETGE様配列を含むいくつかの追加のタンパク質と相互作用することができる[138]。それらのうちのいくつか、例えばSQSTM1/p62およびプロチモシンα(ProTα)はまた、調節またはNRF2-ARE経路に関連しているか、または神経炎症におけるIKK-β(核因子κBキナーゼサブユニットβの阻害剤)のような神経変性の他のホールマークとのクロストークに暗示されている[138]。
KEAP1ケルチドメインは、KEAP1相互作用領域(KIR)を介してp62を認識し、これがKEAP1-NRF2系を混乱させ、NRF2-ARE経路とp62との間のクロストークを導く[139](図11c)。このSer349のmTORC1依存的なリン酸化は、側鎖を伸長させ、それに負の電荷を付加し、KEAP1による認識を改善する(図11d)[140]。p62タンパク質は選択的オートファジーのためのカーゴ受容体であり、KEAP1との強い相互作用により、初期のオートファゴソームにp62タンパク質を隔離し、オートファジー-リソソソーム経路を介した分解を促進することができる[141]。KEAP1-NRF2相互作用のp62媒介による破壊とKEAP1のp62依存性分解の両方が、NRF2の安定性を高め、神経保護につながる[142]。KEAP1を結合することができる別のETGE様基質は、そのENGEモチーフがKEAP1ケルチドメインとの相互作用を媒介するProTαである(図11e)。KEAP1-ProTα複合体の結晶構造は、NRF2 ETGEモチーフと同様の相互作用ネットワークを示している[143]。ProTαは、虚血再灌流傷害に対する神経保護プロファイルを示し、これは、大量に存在する場合には、NRF2-KEAP1複合体を破壊する能力によって説明することができる[144,145]。
KEAP1はまた、リン酸化およびいくつかの下流ステップの後、プロ炎症性NF-κB経路の制御において中心的な役割を果たすIKKβと相互作用する[146]。KEAP1とIKKβの間の認識は、後者の分解を促進し、IKKβのリン酸化を防ぎ、神経炎症を減少させる[147]。NRF2-AREとNF-ĸB経路の相互作用は、神経変性疾患や脳血管疾患において特に興味深いようであり、KEAP1とNRF2およびIKKβとの相互作用によって部分的に説明することができる[147]。KEAP1-IKKβ複合体の結晶構造は得られていないが[148]、タンパク質-タンパク質ドッキングと分子動力学解析によって研究されており、IKKβのETGEモチーフはβターン構造を採用しており[148]、NRF2のETGEモチーフと同様の方法で基質結合ポケットを占有していることが示されている(図11f) [149]。
KEAP1基質との構造的類似性に基づく小型ペプチドは、KEAP1阻害剤として作用するように設計された分子の初期の例であった[150]。しかし、これらの分子には、BBBを横断することができないこと、および安定性が低いことなど、いくつかの制限がある。したがって、KEAP1結合阻害剤としての低分子の開発は、ペプチドに関連するバイオアベイラビリティと安定性の問題を克服するための主要なアプローチであり続けている[151]。30万以上の化合物のライブラリーのHTSにより、テトラヒドロイソキノリン19(図12)がファーストインクラスの低分子KEAP1-NRF2 PPI阻害剤として開発された[151]。この足場に関する更なる研究努力により、新たな構造-活性関係が明らかになった[152]。また、ナフタレンスルホンアミド誘導体20a(図12)も、蛍光異方性アッセイでKEAP1-NRF2阻害剤として同定された[153]。構造決定により、化合物20aとKEAP1のケルチドメインのP3-P5サブポケットとの相互作用が確認された。この知見に基づき、2つのカルボキシル基が複数の水素結合を形成し、P1とP2に存在する塩基性Arg残基と静電的に相互作用し、KEAP1-NRF2界面との親和性を向上させる化合物20b(図12)を設計した[153]。開発過程では、ナフタレンコアをイソキノリンコアに置き換えて 20c を得た(図 12)ため、溶解性、代謝安定性が向上し、ナフタレン足場の固有毒性が低減した[154]。最近では、両方のカルボン酸のバイオイソステリック置換の研究により、ジテトラゾールアナログ20d(図12)が得られた。別の研究では、オキサジアゾール-ウレアベースの化合物21a-c(NK-252,図12)は、カルボキシル基とウレア-オキサジアゾール構造が水素結合を介して相互作用し、フェニル基がそれぞれP1,P2およびP3サブポケットとのπスタッキングによって相互作用することが示されている[156]。
図12 KEAP1タンパク質-タンパク質相互作用阻害剤19-28の化学構造
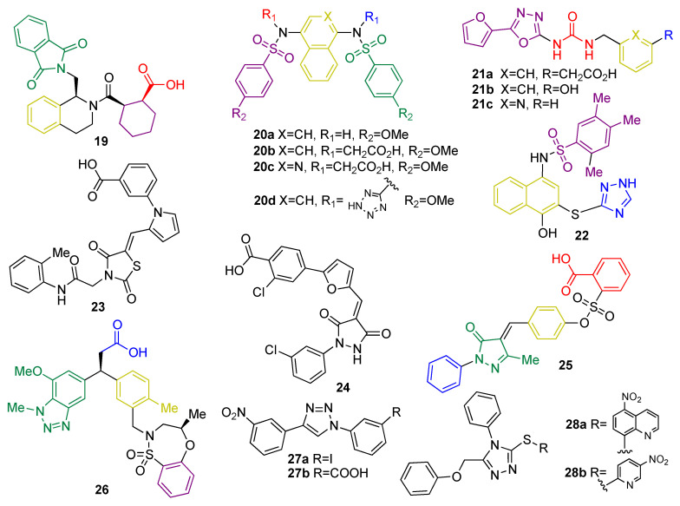
化合物19,20,21,22,22,25および28は、KEAP1との結晶化および/またはそれらの相互作用パターンが分子モデリングおよび分子動力学を用いて提案されている。KEAP1との相互作用パターンは、各ポケットと相互作用する各分子のモチーフに異なる色を与えることでコード化されている。
30万個の化合物ライブラリーを仮想的にスクリーニングしたところ、KEAP1-NRF2界面と相互作用する化合物22,23,24を含むいくつかのヒット構造が得られた[157]。20に構造的に関連する化合物25は、KEAP1-NRF2相互作用を阻害することで、PD動物モデルの神経保護剤として非常に興味深いプロフィールを示している[158]。フラグメントベースの創薬は、新規のフェニルプロパン酸ベースのKEAP1-NRF2 PPI阻害剤である26の開発に成功した[159]。結晶学的研究により、オリジナルのフラグメントと26の結合ポケット内での重複が明らかになった[159]。この化合物は、nMの範囲で細胞ベースのアッセイで効力を示し、生体内でNRF2経路を活性化することが示された[99]。同様に、1,4-ジフェニル-1,2,3-トリアゾール27a-bのようなより単純な5員複素環は、NRF2のGlu残基を模倣するようにシリコで設計され、KEAP1-NRF2認識プロセスを妨害し、生細胞におけるNRF2標的遺伝子の発現を誘導することが示された[160]。1-フェニル-1,3,4-トリアゾール足場を含む異性体化合物も特許文献に記載されている[161]。化合物28aおよび28b(図12)は、中程度の抑制力でKEAP1-NRF2相互作用を阻害し、NRF2下流の標的遺伝子の発現を誘導し、試験管内試験のPDモデルにおける神経保護を導く[161]。特に、化合物28は、HDモデルを用いた生体内試験試験で中棘神経細胞の神経変性を抑制することが明らかになったことから、抗神経変性剤として特に有望であると考えられている[162]。
KEAP1-NRF2 PPI阻害に関する研究は、薬用化学の分野で急速にホットな話題となっており、急速に発展している。この文脈では、KEAP1-NRF2複合体における認識過程に関連する結晶構造および広範な構造解析の利用可能性は、現代的な創薬戦略によるいくつかのリード化合物の開発および新しい化学源の探索に貢献してきた[152,157,161]。しかしながら、これらの分子はしばしば極性を有し、比較的大きな分子量を有し、限定的なBBB浸透性を有するため、神経変性疾患における薬物力学的および薬物動態学的プロファイルの貧弱さにつながっている[136]。したがって、高い潜在的活性と改善された物理化学的特性を有するPPI阻害剤の設計は、KEAP1共有結合阻害剤と比較して、より優れた安全性プロファイルを有する新世代の薬剤を開発するために極めて重要である。
5.1.4. KEAP1発現のエピジェネティック制御
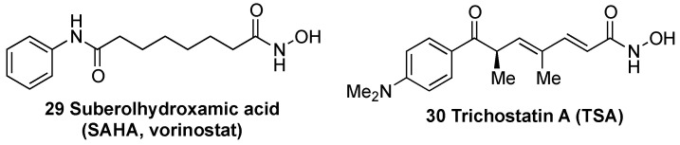
ヒストン脱アセチル化酵素(HDAC)は、KEAP1プロモーター領域に関連するヒストンのアセチル基を除去し、KEAP1転写の増加を誘導する[163]。その結果、この作用を打ち消すHDAC阻害剤は、HD [163]、ALS [164]、脳卒中の動物モデル [165] などのいくつかの神経変性疾患において、酸化還元バランスを改善し、神経細胞の変性を減衰させる。一過性脳虚血のモデルでは、ボルリノスタットとしても知られるスベロールヒドロキサム酸(SAHA)(29,図13)を投与すると、梗塞容積が30~40%減少した[166]。トリコスタチンA(TSA)(30,図12)は、KEAP1発現の阻害剤として同定され、NRF2活性の改善と脳虚血に対する保護につながる[167]。
図13 ヒストン脱アセチル化酵素(HDAC)阻害剤であるサブロールヒドロキサム酸(SAHA)(29)およびトリコスタチンA(TSA)(30)の化学構造。
マイクロRNA(miRNAまたはmiR)は、標的mRNAの3′非翻訳領域(UTR)に結合することができる長さ18〜25ヌクレオチドの小さなノンコーディングRNAである[168]。miRと標的mRNAが結合した後、両者の配列の相補性に応じて、標的mRNAが分解されるか、またはその転写が抑制される[168]。SH-SY5Y細胞株では、miR-7はKEAP1の3′-UTRとの相互作用後、KEAP1の発現を低下させている[169]。KEAP1の発現のこのダウンレギュレーションは、NRF2の安定化をもたらし、いくつかの細胞保護タンパク質の発現を増加させる。miR-7の効果は、miR-7の過剰発現が酸化ストレスから神経細胞を保護するPDモデルにおいて特に興味深いようである[169]。また、黒質や線条体などのパーキンソン病の影響を受けやすい脳領域では、大脳皮質や小脳などの非影響領域と比較してmiR-7のレベルが高いことも関連している[170]。さらに、PDマウスモデルで観察されたmiR-7の発現ダウンレギュレーションは、パーキンソン病の発症に関与していることを示唆している[170]。
これらの前例に基づき、エピジェネティクスやmiRによるKEAP1の発現制御は、NRF2-ARE経路を介して神経保護を改善するための有効な戦略を提供することができる。
5.2. KEAP1に依存しない制御
上述したように、NRF2の活性化は主にKEAP1によって制御されており、これはカノニカル経路として知られているメカニズムである。しかし、多くの非KEAP1関連のプロセスがNRF2の調節に寄与しており、本節ではそれらをまとめている。
5.2.1. GSK-3βによる NRF2 制御
GSK-3βは、グリコーゲン代謝、細胞増殖、アポトーシスなどの細胞内機能に関与するSer/Thrキナーゼである[171]。神経変性疾患sに関しては、GSK-3βの活性がアルツハイマー病で上昇することが報告されており、タウの高リン酸化に重要な役割を果たしていると考えられている[172]。そのため、GSK-3βはアルツハイマー病に対する薬剤開発において最も重要なターゲットの一つとなっている。2006年には、Rojoらは、このキナーゼがNRF2-ARE応答を調節する能力を報告している[173]。GSK-3βはFynをリン酸化し、その活性を高める; そして、FynはTyr568でNRF2をリン酸化し、その核外化[175]と分解をもたらす。
さらに、別の研究では、上述のように、GSK-3βがNRF2のNeh6でDSGIS [17] デグロンモチーフをリン酸化することが実証されている[176]。この翻訳後修飾は、β-TrCPのNRF2に対する親和性を高め、そのプロテアソーム分解を誘導することを示している[17]。さらに、GSK-3βの活性はいくつかのキナーゼによって制御されており、PI3K/Akt経路はSer9のリン酸化によってGSK-3βをダウンレギュレートし、不活性化を誘導している[177]。
表1にまとめたように、NRF2のGSK-3β調節機構は、この重要なキナーゼの潜在的なターゲットとしての関心を新たにしており、特にアルツハイマー病に重点を置いた神経変性疾患の標的となっている。
表1 NRF2誘導能を有するGSK-3β阻害薬
| 化合物 | 疾患 | 臨床試験 | 参照 |
|---|---|---|---|
ピオグリタゾン |
II型糖尿病 | 承認済み | |
| いくつかの病気 | 300件の臨床試験 | – | |
チデグルシブ |
広告 | フェーズII | NCT01350362 |
| 先天性筋緊張性ジストロフィー | フェーズII / III | NCT03692312 | |
| 自閉症スペクトラム障害 | フェーズII | NCT02586935 | |
| 進行性核上性麻痺 | – | NCT01049399 | |
TDZD-8 |
広告 | 前臨床評価 | |
SB216763 |
広告 | 前臨床評価 | |
YQ138 |
脳虚血 | 前臨床評価 | |
オバクノン |
いくつかの病気に向けて位置づけられた天然物 | 前臨床評価 | |
物体名は、biomolecules-10-00904-i006.jpg 複数の疾患に対応した天然物 前臨床評価
GSK-3β阻害薬は、アルツハイマー病、軽度認知障害、自閉症スペクトラム障害、癌を含む様々な疾患を対象とした臨床試験が行われている。当初、双極性障害の治療に用いられるGSK-3β阻害薬であるLiClやHO-1発現を増加させる化合物TDZD-8[180]では、GSK-3β阻害を介したNRF2活性化が実証されていた。最近では、LiClによるNRF2活性化が試験管内試験と生体内試験の異なるモデルで報告されており、後者のGSK-3βによる制御が確認されている[181]。TDZD-8によるNRF2活性化は、腎虚血/再灌流傷害の生体内試験モデルでも実証されている。TDZD-8はNRF2活性化を介して酸化ストレスと細胞のアポトーシスを減少させた[182]。
臨床的に進んだ例としては、チアジアゾリジノン誘導体であるチデグルシブがあり、これはアルツハイマー病治療のための非ATP競合GSK-3β阻害薬として開発され、第II相臨床試験(ARGO試験)[183]に達したが、臨床的な有用性は得られなかった。研究者らは、非線形な用量反応の負の効果の可能性を示唆し、初期のアルツハイマー病症例におけるさらなる用量設定試験を提案している。チアジアゾリジノン誘導体であるチデグルシブとピオグリタゾンは、NRF2誘導および第II相反応活性化を介したMPPT毒性に対する神経保護作用を示した。このモデルでは、GSK-3β IC50(5 nM)に比べて高濃度(5 μM)で神経保護効果が認められたが、抗酸化応答の活性化はGSK-3β阻害を介していることが著者らによって示された[184]。
GSK-3β阻害剤のNRF2誘導能の別の例として、化合物SB216763がある。SB216763は、GSK-3βの病理学的活性化と酸化ストレスを介して、初代ポッドサイトにおいてドキソルビシンによって誘導される毒性に対して興味深い細胞保護効果を示した[185]。SB216763はNRF2誘導と第II相抗酸化応答活性化を介してポッドサイトを保護し、この応答はGSK-3β阻害能と直接関連していた[185]。最後に、GSK-3β阻害を介したNRF2誘導は、YQ138 [186]、オバキュノン [187]、コリラギン [188]などの他の多くのGSK-3β阻害薬で実証されているが、後者の場合、NRF2の作用機序は、化合物の構造的特徴とマルチターゲット活性により、KEAP1または他の活性化経路との直接的な相互作用にも関連している可能性がある。
5.2.2. KEAP1に依存しないNRF2安定性の制御
KEAP1以外にも、NRF2の安定性を制御するメカニズムが存在する。最も重要なものは、PI3K/AKT経路に接続されたβ-transducin repeat containing protein(β-TrCP)とHrd1である(図14)。
図14 KEAP1に依存しないNRF2の負の調節
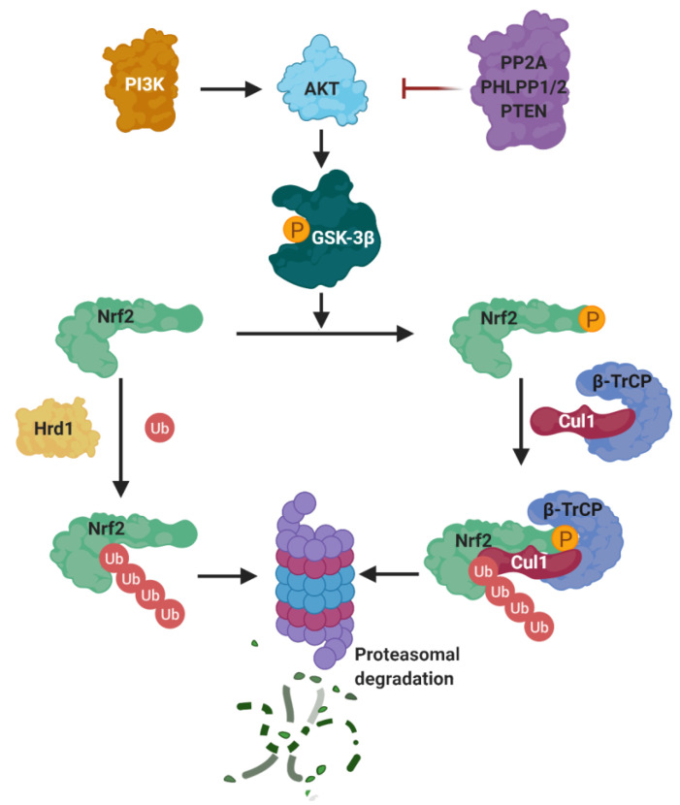
後者のタンパク質は、ストレス条件下でのみ第二相応答の活性化を確実にするために、その主要なリプレッサーKEAP1とは独立して、異なるシステムによってタイトに制御されている。左から、異なる経路の活性化は、右から、順番に、プロテインホスファターゼ2A(PP2A)PHLPP1/2(PHドメインおよびロイシンリッチリピートプロテインホスファターゼ1および2)または腫瘍抑制ホスファターゼおよびテンシンホモログ(PTEN)によって阻害されたリン酸化によってAKTを活性化するPI3Kの活性化につながる可能性がある。活性化された後、AKTはGSK-3βをSer9でリン酸化し、その活性を阻害することで、このキナーゼによるNRF2の負の制御を減少させている。GSK-3βは、いくつかのメカニズムによってNRF2の負の調節因子として作用することが知られている。NRF2は、このキナーゼによってSer335およびS338でリン酸化され、β-TrCPによって認識されたホスホデグロンドメインDSGISを形成し、これがCul1/Rbx1 NRF2のユビキチン化およびプロテアソーム分解を導く。また、NRF2は、NRF2を標的にしてプロテアソーム分解を行うユビキチンリガーゼであるNeh4トランザクティベーションドメインにおいて、Hrd1との相互作用により第3のユビキチン化分解系に制御されている。
(a) PI3K/Akt。PI3K/Akt経路は、代謝、増殖、増殖、細胞生存、遺伝子転写を制御する。Aktは、ホスファチジルイノシトール-3-キナーゼ(PI3K)によって触媒されるホスファチジルイノシトール-(3,4,5)-三リン酸(PIP3)を産生するいくつかのシグナルによって活性化されたSer/Thrキナーゼである[189]。PIP3はAktとその活性化キナーゼであるPDK1のアンカーとして機能し、Thr308でのリン酸化によりAktを活性化する。リン酸化されたAktは、Ser473でリン酸化されることにより、mTORC2によってさらに活性化される[189]。Aktの負の調節は、プロテインホスファターゼ2A(PP2A)PHLPP1/2,および腫瘍抑制ホスファターゼおよびテンシンホモログ(PTEN)によって発揮される[189]。
PI3K/Akt経路は、Cuadradoら[190]によって記載されているように、KEAP1非依存的なモードでNrf2誘導に寄与する。PI3K阻害剤LY294002を使用することで、カルノゾルが媒介するNrf2誘導が大幅に阻害されることが観察された。これはシアノエノントリテルペノイドファミリーに属する強力なNrf2誘導剤であり、PTENの活性部位内のCys124と共有結合的に反応してその活性を阻害するため、PI3K/Akt経路を活性化する[191]。同様に、4-ヒドロキシノネナール(Nrf2誘導剤)はPTENのCys71を修飾する[192]。PTENのCys71とCys124残基は酸化還元センサーとして作用し、酸化されるとジスルフィド橋を形成し、PTENのPIP3 3-リン酸化酵素活性を不活性化し、Aktの活性を増加させる[193]。したがって、PTENは細胞内の酸化還元センサーとして作用するNrf2のネガティブリプレッサーと考えられている;一度不活性化されると、Nrf2はPI3K/Akt経路の活性化を介して活性化される。
PI3Kの活性化は、Akt活性を増加させることでNrf2の誘導につながり[173,194]、その結果、Ser9[195]とSer21[196]でのリン酸化によりGSK-3βの活性を阻害することが示された。さらに、GSK-3βは、PDK1[199]を介してPI3Kによって制御されているPKC[197]、P70S6K、およびP90RSK[198]によっても阻害され得る。
b)β-TrCP。PI3K/AKT経路は、GSK-3βの阻害を介してNRF2を活性化し、NRF2をリン酸化[190]し、β-TrCPによる認識を可能にし、これによりNRF2はユビキチン化とそれに続くプロテアソーム分解のためにマークされる[200]。β-TrCPはWD40サブファミリーのF-Box含有タンパク質であり、Skp1-Cul1-F-box(SCF)ユビキチンリガーゼSCFβ-TrCP内で基質アダプター/受容体として機能し[201]、NRF2以外にもIκB、β-カテニン、CDC25,APC[70]などのユビキチン化にも関与している。β-TrCPは、リン酸化された破壊モチーフに直接結合することで基質を認識し、例えば、343DSGIS347や382DSAPGS387の配列でリン酸化されたNRF2に結合している[202]。興味深いことに、この配列はGSK-3β標的モチーフを模倣しており、したがって、GSK-3β標的タンパク質の多くは、分解のためにβ-TrCPによって処理されることが知られている[203]。
選択的なGSK-3β阻害剤またはGSK-3βを標的としたsiRNAを用いることで、NRF2の安定化と核内局在化が最初に観察された。β-TrCPを標的としたsiRNAを用いても同様の結果が得られ、両タンパク質によるNRF2の制御が実証された。NRF2のNeh6ドメインには、DSGISとDSAPGSという2つのモチーフがあり、それらがリン酸化されるとSCFβ-TrCPによって認識される[204]。このように、GSK-3β/β-TrCPシステムは、リン酸化後にβ-TrCP/Cul1in1/Rbx1 E3リガーゼ複合体を介してNRF2のユビキチン化を誘導しており、受容体シグナル伝達により活性化されるKEAP1に依存しない機構であると考えられる[205]。この調節機構を考えると、KEAP1とβ-TrCPによる「二重調節」という仮説がCuadradoらによって提案されている[77]。したがって、β-TrCPは、代謝適応に大きな影響を与えるNr2によって制御されるタンパク質の数が多いことを考慮すると、細胞シグナル伝達のためのNRF2の微調整因子であると考えられる[78]。
(c) DJ-1。神経変性疾患sとの別の関連では、パーキンソン病に関与するタンパク質であるDJ-1とNrf2との間の直接的な関連が記述されている。パーキンソン病におけるDJ-1の不活性化は、酸化ストレスに対するニューロンの感受性の高さと関連している[206]。説得力のある証拠は、DJ-1がPTENのネガティブレギュレーターとして作用することを示している[207]。したがって、DJ-1のダウンレギュレーションは、PTENの活性の増加により、Nrf2の活性を低下させる。さらに、DJ-1は、酸化ストレスセンサーとして作用し得るいくつかのROS感受性Cys残基を提示する。最後に、DJ-1はPTENのCys残基に対して硝酸性酸化物伝達タンパク質として作用し、その活性を阻害することが提案されている[208]。
5.2.3. NRF2に関連するその他のARE転写調節因子
a)AHR。アリール炭化水素受容体(AHR)は、多くの外来物質(例えば、多芳香族炭化水素、ダイオキシン)および内因性化合物の作用を媒介するbHLH/PASファミリーのリガンド活性化転写因子である[209]。これは神経系に広く分布しており、特に神経細胞の機能を制御している[209]。Miaoらは、NRF2の転写がAHRによって直接制御されていることを示しており[210]、実際、それらはそのプロモーター領域にAREとXRE応答要素の両方を示している。さらに、それらはいくつかの共通の抗酸化標的遺伝子を共有している[211]。
b)P53。ゲノムの守護者として知られるP53タンパク質は、DNA損傷によって活性化され、細胞周期停止、DNA修復、老化、アポトーシスの経路を調節する転写因子である。実際、p53はすべての神経変性疾患に共通する神経細胞死のプロセスに寄与している可能性が提案されている[212]。NRF2とp53経路の間にはいくつかの関連性がある [213]。まず、p53の活性化はp21の発現を誘導し、これはKEAP1と競合してNRF2に結合し、NRF2の分解を防ぎ、そのレベルを上昇させる[214]。さらに、NRF2は、20Sプロテアソームによるp53の分解を妨げるNQO1の発現を誘導する[215]。
(c) エピジェネティックな調節。KEAP1の場合と同様に、NRF2の転写は、CpG島におけるNRF2プロモーターのメチル化、H3ヒストンメチル化、およびH4ヒストンアセチル化を介してエピジェネティックな調節の対象となる[216]。脳の発達中にDNAメチルトランスフェラーゼを阻害すると、βアミロイド前駆体タンパク質などのアルツハイマー病関連遺伝子のプロモーターのメチル化を介して、老化した脳の酸化的DNA損傷に対する感受性が増加することを示すいくつかの証拠がある[217]。興味深いことに、5-アザシチジン(31,図15)によるDNA脱メチル化は、アルツハイマー病細胞モデルにおいてNRF2発現をアップレギュレートすることが示された。218] 同じ文脈で、マウス神経芽腫細胞におけるスルフォラファン(1,図15)による神経保護のメカニズムの一つは、NRF2発現のアップレギュレートであり、NRF2プロモーター遺伝子のメチル化レベルを低下させることでNRF2核内転座を促進することが証明されている[219]。
図15
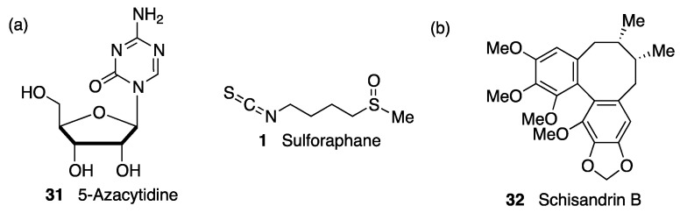
(a) NRF2発現のエピジェネティックなアップレギュレーションにより神経保護作用を示した2つの化合物。(b) Schisandrin Bは、マイクロRNA miR-34aによるNRF2活性の抑制を阻害することにより、パーキンソン病(PD)細胞モデルにおいて神経保護作用を示した。
NRF2活性は、標的mRNAのUTR内の相補的なシード配列と相互作用してタンパク質産生を抑制するマイクロRNAによっても制御され得る。いくつかのmiRはNRF2の直接的な調節に関与しており、酸化ストレスに関連する神経疾患に関連している[168]。そのため、miR-27a、miR-142-5p、miR-144,miR-153はin silico解析によりNRF2抑制因子として同定された。これらのmiRの過剰発現は、NRF2のmRNAを標的とすることでNRF2の活性を抑制し、制御されない酸化還元恒常性をもたらすことが、神経細胞SH-SY5Y細胞での研究で明らかになった[220]。したがって、中国の伝統的な薬草であるSchisandra chinensisに含まれるジベンゾシクロオクタジエンリグナンであるschisandrin B(32,図15)は、miR-34aの作用の阻害剤として作用し、6-OHDA PDモデルにおいて神経保護効果を示している[221]。脳梗塞後の病態には多くのmiRが関与しており、その中でもmiR93はNfr2発現のネガティブレギュレーターであり、虚血時の酸化還元バランスの調節に大きく寄与していることが明らかになっている[222]。一方、miR-424はNRF2をアップレギュレートしており、その過剰発現はマウスの局所脳虚血・再灌流において抗酸化作用を示し、NRF2のノックダウンやSOD阻害剤による治療によって抑制される[223]。
5.2.4. NRF2 の翻訳後リン酸化を制御する様々な経路
定型的な経路を超えて、細胞内の第II相抗酸化応答の厳密な制御は、異なる翻訳後機構によってNRF2を制御する多くの経路を網羅している[224,225]。これまでに議論されたGSK-3β [173]、PI3K/Akt [190]、MAPK [226,227]、β-TrCP [190]、PKC [228,229]などの異なる陽性および陰性のNRF2調節タンパク質が記載されている。このように非定型的なメカニズムを介してNRF2を細かく制御することで、特定の標的に向けた薬剤を設計する機会が開かれる。
(a) MAPKs。これらの酵素は、細胞生存[226,230]、アポトーシス[228,231]、および遺伝子発現[227,230]を含む重要な生物学的プロセスを調節するセリン/スレオニンキナーゼである。ERKによって構成される3つの主要なMAPK経路がある:c-jun N末端キナーゼ(JNK)およびPro残基に近いSer残基またはThr残基をリン酸化するp38キナーゼです[227]。異なるMAPK経路によるNRF2の制御は、選択的阻害剤の使用やトランスフェクション研究によって広く説明されており、ERKシグナルがNRF2活性を増加させ[232]、P38がNRF2シグナルを減少させることが実証されている[233]。Sunらは、Ser215,Ser408,Ser558,Thr559およびS577を含むNRF2の多くのリン酸化部位が、このキナーゼファミリーのメンバーによって転写後に修飾されることを実証した[234]。
(b) P38。P38 MAPKファミリーは、特定の分布と基質を持ついくつかのアイソフォーム(P38α、β、γ、δ)から構成されている。一旦リン酸化によって活性化されると、それらは、炎症、細胞分裂、および分化またはアポトーシスを含むプロセスを制御するために、様々なタンパク質や転写因子をリン酸化する[235]。P38キナーゼは主に神経変性疾患の病理学的特徴である酸化ストレスによって活性化される[236]。P38は酸化ストレスを介してアミロイドβペプチドや高リン酸化タウによって活性化されることから、アルツハイマー病と関連している[237]。この系統では、アルツハイマー病脳では初期段階でP38経路が過剰に活性化されていることが記述されている[238]。一般的なコンセンサスは、NRF2タンパク質をリン酸化してその核蓄積を減少させることができる活性なp38アイソフォームを記述したKeumらによって示されるように、P38 MAPKによるNRF2のネガティブな調節を指摘している[239]。P38 MAPKはNRF2を3つのSer残基(Ser215,Ser408,およびSer577)でリン酸化し、KEAP1との相互作用を改善し、したがってその分解を促進する[9]。逆に、p38の活性化は、いくつかのキセノバイオティクスの投与後にNRF2-ARE経路を活性化させた[240,241]が、これはGSK-3βに対するP38の阻害活性と相関する観察である[239]。おそらく、NRF2に対するP38の活性は、恒常性平衡の小さな変化に依存しており、最終的な応答は刺激、タイミング、細胞タイプに依存していると考えられる。
c)JNK。c-jun-NH2末端キナーゼ(JNK)経路は、主にストレス刺激によって活性化され、MAPKシグナル伝達経路の重要なメンバーであり[242]、発生、細胞増殖、炎症、およびアポトーシスにおいて重要な役割を果たしている[243]。JNKは、3つの異なるアイソフォーム(JNK1,JNK2,JNK3)を含むスレオニンプロテインキナーゼのファミリーであり、後者は中枢神経系で特異的に発現しており[244]、神経変性疾患sの重要な標的と考えられている。JNK3は、アルツハイマー病 [245]、PD [246]、ALS、脳卒中 [247]などの神経変性疾患で活性化されていることが分かっている。JNKの持続的な活性化は、細胞の損傷と死を促進する病理学的状態と広く関連しており、また、NRF2シグナル伝達の低下とも相関している。最近では、アセトアミノフェンによる肝障害の予防に関連して、JNKとNRF2の直接的なクロストークが明らかにされている。活性化されたJNK(p-JNK)は、NRF2のNeh1ドメインと直接相互作用することで、NRF2のターンオーバーを増加させた。この相互作用により、NRF2のNeh6デグロンドメインのSer残基がリン酸化される。このように、p-JNKはNRF2のDSGISモチーフのSer335をリン酸化し、KEAP1に依存しない機構を介してNRF2の分解を成文化することが実証された[249]。
(d) ERKs。細胞外シグナル制御キナーゼ(ERK)は、多数の生物学的プロセスに関与する主要なMAPKサブファミリーである。ERKの活性化は細胞保護、特に神経保護に強く関連しており、このキナーゼ経路は神経変性疾患に対する潜在的なターゲットにリンクしている[250]。ERK経路によるNRF2の制御は多くの異なる細胞タイプで報告されており、いくつかのNRF2誘導剤の作用機序に関与している。ERK活性化は、HepG2細胞におけるピロリジンジチオカルバメート[252]、カフェイン酸[253]、スルフレチン[254]、ガリン酸[254]、ルテオリン[255]、およびブチル化ヒドロキシアニソール[256]などの他のいくつかのNRF2誘導剤に応答して、NRF2-AREシグナル伝達に必要であることが実証された;RAW 264のlico A. 7細胞(26576227)またはPC12細胞におけるフェニルエタノイド配糖体。いくつかの報告では、活性化された ERK1/2 が NRF2 を Ser40 で直接リン酸化し、その核内安定化をコード化して NRF2-ARE パスウェイの活性化を増加させることによって、NRF2 と ERK1/2 の間の直接的なクロストークの可能性が示唆されている [257]。この仮説は、ウエスタンブロットで測定したp-Ser40-NRF2タンパク質レベルの上昇によって支持されている。しかし、ERK1/2とNRF2の直接的な相互作用は完全に証明されておらず、PKC、JNK、またはAktのような他の経路が寄与している可能性がある[257]。
(e) PKC。このタンパク質は、アポトーシス、生存、および分化を含む多くの細胞シグナル応答に関連するセリン/スレオニンキナーゼのファミリーの一部である。PKCの活性化は伝統的に受容体依存性と考えられているが、活性酸素依存性の活性化機構も存在する[258]。ホルボル酸エステル(PKC活性化剤)がNRF2関連タンパク質の発現を誘導することが発見されたことから、PKCによるNRF2-phase II応答の制御が仮説として導かれた[228]。PKCは他のキナーゼ[229]とともに、NRF2Neh2ドメインでSer40をリン酸化し、KEAP1-NRF2相互作用を阻害することが証明された[259]。Ser40のリン酸化はその核内移動に必要であるが、核内蓄積を促進するものではない[260]。PKCによる第二のNRF2調節機構は、不活性化部位でGSK-3βをリン酸化するいくつかのPKCアイソフォーム(α、β1,γ>β2;εではない)の能力に関連している[261]。神経変性疾患に注目すると、アルツハイマー病脳ではPKCタンパク質のレベルと活性が低下していることが明らかになった[262]。PKCによるNRF2のポジティブな調節および他の潜在的に有益な特性を考慮すると、いくつかのPKC活性化剤がアルツハイマー病治療のための潜在的な薬剤として提案されている[263]。
f)CK2。カゼインキナーゼⅡ(CK2)は、300 以上の基質に作用する Ser/Thr キナーゼであり、シグナル伝達、遺伝子転写、複製過程、生存経路などを含む多くの異なるプロセスの制御に関与している。CK2によって制御されるプロセスの中で、CK2は細胞ストレスに対するいくつかの重要な調節因子を制御している[264]。CK2はNRF2の制御にも関連しており、その配列はCK2が標的とする少なくとも13の潜在的なリン酸化位置を示している[265]。NRF2の欠失解析を用いて、Neh4およびNeh5ドメインがCK2の標的領域として同定され、このキナーゼがいくつかの残基をリン酸化し、NRF2の核蓄積および経路活性化を誘導していることがわかった[265]。
g)Fynキナーゼ。Srcファミリーの一員であるFynキナーゼは、NRF2のネガティブな制御に密接に関連している。Fynキナーゼは、NRF2をTyr568で直接リン酸化し、その核輸出をコード化する[266]。Tyr568のリン酸化は、NRF2の核内排泄に関連するタンパク質であるexportin1によるNRF2の認識を促進する[266]。
6. 結論
NRF2は、酸化還元電位の恒常性維持や神経炎症の制御に重要な役割を果たしており、その活性を高めることは、神経変性疾患の治療への興味深いアプローチである。NRF2の制御は、タンパク質の安定性、転写・後転写レベルでの制御など、いくつかのレベルで行われている。これらの過程に関与するタンパク質や酵素の多くは、図16に示すように、神経変性疾患の治療のための創薬標的となり得るものである。NRF2制御の調節は、まだ黎明期にある分野であり、未踏の潜在的な創薬標的という点でチャンスに満ちており、このレビューがこの魅力的な分野の革新的な研究を刺激することを期待している。
図16 NRF2の主要な調節機構の概要
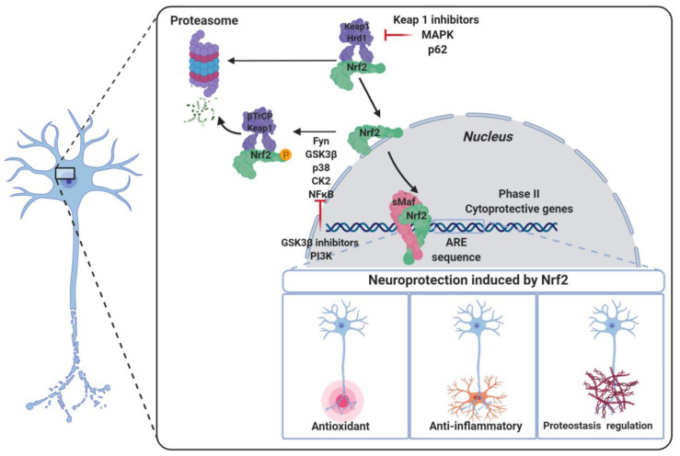
KEAP1は、低レベルでの発現を維持するためにそのプロテアソーム分解をコードするNRF2の主要な負の調節タンパク質を表す。KEAP1とNRF2の相互作用は、タンパク質-タンパク質阻害剤、MAPK、p62の活性によってブロックすることができる。Hrd1はまた、NRF2のプロテアソーム分解を誘導し、その調節を助ける。活性化されると、NRF2は、抗酸化、抗炎症、およびプロテオスタシス関連遺伝子の発現を誘導し、神経細胞の生存を促進する。病的条件下では、いくつかのシグナルが異なるキナーゼや因子の活性化を誘導し、NRF2の分解を促進するネガティブな制御を行う。この複雑な制御システムが病理学的条件によって制御不能になると、NRF2-ARE経路の障害は病気の進行と相関している。
略語
Aβ:βアミロイド;AD.アルツハイマー病;AHR:アリール炭化水素受容体、AIP:AHR相互作用タンパク質;ALS:筋萎縮性側索硬化症;AMPK:AMP活性化プロテインキナーゼ;ARE:抗酸化応答エレメント;ARNT:アリール炭化水素受容体核トランスロケーター;α-syn:α-シヌクレイン;BBB:血液脳関門;bHLH/PAS:塩基性らせん-ループ-らせん-PER-ARNT-SIM;BTB/POZ.ブリック・ア・ブラック、トラムトラック、ブロード・コンプレックス/プロキシウイルス・ジンクフィンガー;β-TrCP:β-transducing repeat-containing protein Bzip:basic-region leucine zipper;CBP:[CREB(cAMP-response-element-binding protein)-binding protein];CDK:サイクリン依存性キナーゼ;CK2:カゼインキナーゼII;CNC:cap ”n” collar;CTR:C末端ドメイン;Cull3.カリン3;DGR:ダグルグリシンリピート;DMF:フマル酸ジメチル;EGCG:ガレートエピガロカテキン;ERK:細胞外シグナル制御プロテインキナーゼ;FRDA.フリードリッヒ失調症;GCL:グルタミン酸システインリガーゼ;GCLc:グルタミン酸システインリガーゼ;GI:胃腸;GR:グルタチオン還元酵素;GSH:グルタチオン;GSK-3β.グリコーゲン合成酵素キナーゼ-3β;HD:ハンチントン病、HNE:4-ヒドロキシ-2-ノネナール;HO-1:ヘムオキシゲナーゼ1;Hrd1.ERAD関連E3ユビキチンリガーゼ;HSP90:熱ショックタンパク質90;Htt:ハンチントン、IBs:ユビキチン化包接体;IκB:核内因子κBの阻害剤;IL-1β:インターロイキン1β;IKK.IκBキナーゼ;iNOS:誘導性一酸化窒素合成酵素;IVR:介在領域;JDP2:jun二量化タンパク質2;JUNK:c-jun N末端キナーゼ;KEAP1:Kelch様ECH関連タンパク質1;MiRNAs.マイクロRNA;MMF:フマル酸モノメチル;MS:多発性硬化症;神経変性疾患S:神経変性疾患;NF-κB:核因子-κB;NO2-Fas.ニトロ脂肪酸;NQO1:NAD(P)H/キノン酸化還元酵素1;NOX:NADPH酸化酵素;NRF2:核内因子(赤血球由来2)様2;NTR:N末端領域;PD:パーキンソン病;PHLPP1/2:PHドメインおよびロイシンリッチリピートタンパク質ホスファターゼ1,2;PI3K.PI3K:ホスファチジルイノシトール-3-キナーゼ;PIP3:ホスファチジルイノシトール-(3,4,5)-三リン酸;PKC:プロテインキナーゼC;PP2A:プロテインホスファターゼ;PrTa:プロチモシンa;PrPSc:スクレイププリオンタンパク質;PTEN:ホスファチジルイノシトール-3,4,5-三リン酸-3-ホスファターゼ;ROS.活性酸素種;RXRα:レチノイン酸受容体α;SAHA:サブエロールヒドロキサム酸;SCF:Skp1-Cul1-F-box、SMaf:小男性器線維肉腫、SOD:スーパーオキシドジスムターゼ、tBHQ:tert-ブチルヒドロキノン;TDP-43.TAR DNA結合タンパク質43,TLRs.Toll様受容体;TMT:トリメチルスズ;TNF-α:腫瘍壊死因子α;TSA:トリコスタチンA;UTR:3′-翻訳されていない領域;VDCC:電圧依存性カルシウムチャネル、XRE:xenobiotiotic Respongelement.