
Contents
- 要旨
- 1. 序論
- 2. アルツハイマー病のためのMTDLデザイン戦略におけるターゲットの組み合わせ
- 2.1. デュアルChE/BACE-1阻害剤
- 2.2. デュアルChEとGSK-3β阻害剤
- 2.3. デュアル ChE と MAO インヒビター
- 2.4. ChE 阻害剤と他の酵素系
- 2.5. ChE阻害薬とNMDA受容体拮抗薬またはCa2+チャネル遮断薬
- 2.6. ChE阻害剤とセロトニン受容体モジュレーター(5-HT4および5-HT6)
- 2.7. ChE阻害剤とH3Rアンタゴニストまたはσ1Rアゴニスト
- 2.8. エンドカンナビノイド系のデュアルChE阻害剤と調節剤の効果
- 2.9. アミロイドβ凝集、金属誘起毒性、酸化ストレスに対する複数の効果を有するChE阻害剤
- 2.10. BACE-1阻害剤とMTDLへの組み合わせ
- 2.11. ChE や BACE-1 阻害とは無関係な アルツハイマー病 に対するマルチターゲット効果
- 2.12. アルツハイマー病における合成ペプチドの役割
- 3. 結論
Multitarget Therapeutic Strategies for Alzheimer’s Disease: Review on Emerging Target Combinations
www.ncbi.nlm.nih.gov/pmc/articles/PMC7354643/
オンラインで公開2020年6月30日
要旨
神経変性疾患は、現在、大きな健康問題の一つとなっている。神経変性疾患の原因となるメカニズムの解明が進められているが、何が神経変性現象の引き金となり、何が神経変性現象を進行させるのかについては、まだ完全には解明されていない。しかし、神経変性は、タンパク質の凝集、酸化ストレス、神経炎症などの有害なプロセスの結果として、最終的には神経細胞の機能を失うことになると考えられている。
このような背景から、複数の事象に同時に作用する新規な薬剤、いわゆるマルチターゲット指向性リガンド(MTDL)が広く探索されていた。これらの化合物は、異なるファルマコフォリック成分の組み合わせに由来し、異なる酵素系や受容体系に干渉したり、タンパク質や金属の恒常性を調節することで神経保護効果を発揮したりする能力を持っている。
MTDLは、神経変性や認知機能障害を特徴とする認知症の中で最も一般的な形態と考えられているアルツハイマー病の新しい治療法を発見するための最新の戦略の焦点となっている。本レビューでは、試験管内試験で良好な特性を有する新規薬剤や、認知症の動物モデルで既に生体内試験で評価されている最適化された構造についての詳細な議論を交えながら、アルツハイマー病治療のための最新かつ最も興味深いターゲットの組み合わせを収集することを目的としている。
1. 序論
神経変性は、神経細胞機能の進行性の損失を引き起こし、認知障害、記憶喪失、およびいくつかの形態の運動失調をもたらす病理学的プロセスである。この特徴は、アルツハイマー病 (AD)パーキンソン病(PD)ハンチントン病(HD)筋萎縮性側索硬化症(ALS)などの疾患において極めて重要だ。神経変性疾患は、経済的にも社会的にも大きな脅威となっている。世界保健機関(WHO)によると、高齢者を中心に約5,000万人が認知症に罹患しており、そのうち約60~70%がアルツハイマー病であるとされている[1]。世界的に平均寿命が延びていることから、神経変性を抑制したり、逆行させたりすることのできる新しい神経保護薬の開発が求められている。
生化学的な観点から見ると、神経変性疾患には、タンパク質の誤形成や凝集、神経伝達物質(アセチルコリンやドーパミンなど)のレベルの変化、金属イオンの不均一性[2]、ミトコンドリアの機能不全、酸化ストレス、神経炎症[3]などの病理学的なプロセスが共通している。
例えば、アルツハイマー病では、アミロイドβ線維が凝集して形成されたβアミロイドプラークや、高リン酸化TAUタンパク質(pTAU)からなる神経原線維もつれ(NFT)の沈着が組織学的な異常変化の特徴となっている[4]。PDプロテオパシーは、ルイス小体に蓄積されたα-シヌクレインの誤った凝集体と関連している[5]。ALSでは、組織学的研究により、変異型スーパーオキシドジスムターゼ1(SOD1)TAR DNA結合タンパク質(TDP-43)肉腫(FUS)で融合したもの、および変異型第9染色体オープンリーディングフレーム72(C9ORF72)の非カノニカル翻訳からのリピートジペプチドの凝集体の存在が示されている[6]。
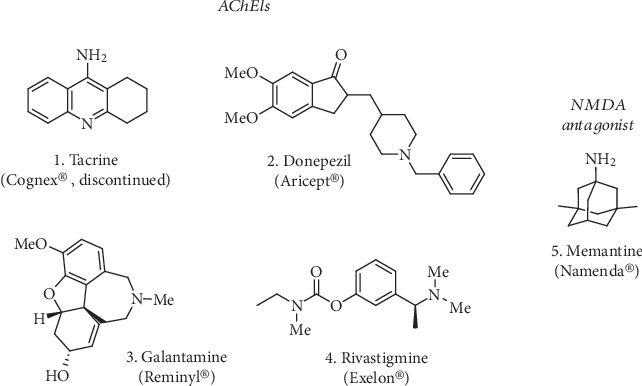
神経変性につながるもう一つの主な病理学的事象は、酸化ストレスである。ヒトの脳は体重の2%しかないにもかかわらず、呼吸器系から運ばれてくる酸素の20%を消費している[7]。このため、脳は酸化ストレスに対してより脆弱になっている。このように、神経細胞の主要な構成要素(脂質、タンパク質、核酸)の酸化は、必ず神経変性を引き起こす [8]。言い換えれば、活性酸素と窒素種(ROSとRNS)の恒常的な蓄積は、神経細胞に不可避的な損傷をもたらす。この酸化ストレスは、ミトコンドリアの機能不全[9,10]、金属イオン(例えば、酸化還元活性Fe2+/Fe3+およびCu+/Cu2+)の恒常性の異常、および凝集しやすいペプチド(例えば、アミロイドβおよびα-シヌクレイン)の沈着を促進する役割[11-13]、および神経炎症[14,15]などの様々な基礎的な要因によって引き起こされる。これらの病因機序が同時に共存し、複数のレベルで互いに影響し合っているという事実については、世界的なコンセンサスがある[16]。その結果、これらの病理学的特徴は、神経細胞の死および神経伝達の機能不全を引き起こし、進行性の認知機能障害および/または運動失調につながる。神経変性疾患の病因におけるそれらの絡み合った役割に基づいて、これらは重要な治療標的となっている。現在、神経変性疾患の治療法として市販されているものは、主に緩和的な治療法であり、患者さんの日常生活を改善するものではない。例えば、現在市販されているアルツハイマー病治療薬は、アセチルコリン(ACh)のレベルを維持し、神経伝達を維持する3種類のアセチルコリンエステラーゼ阻害薬(AChEIs、図1)で構成されている[17];軽度から中等度のアルツハイマー病に承認されているドネペジル、ガランタミン、リバスチグミン(2-4,図1)とともに、中等度から重度のアルツハイマー病に承認されている1種類のNMDA拮抗薬メマンチン(5,図1)がある[18]。タクリン(THA、1,図1)は、アルツハイマー病治療のために販売された最初のAChEIであったが、その肝毒性のために急速に中止された[19]。
図1 AChE阻害薬は、NMDA受容体拮抗薬メマンチン(5)とともにアルツハイマー病(1-4)の治療薬として販売されている
パーキンソン病の治療法としては、主にL-DOPAやカルビドパなどのカテコールアミンの投与によるドーパミン作動性の回復がある(6および7)。図2)またはエルゴット由来のアルカロイド(ブロモクリプチン、アポモルフィン(8および9,図2)カベルゴリン、リスリド、およびペルゴリド)および非エルゴット由来の低分子(プラミペキソール、ロピニロール、およびピリベジル)などのドーパミン作動性受容体アゴニストが挙げられる。L-DOPAの半減期が短いため、カテコール-O-メチルトランスフェラーゼ(COMT)阻害薬(例えば、エンタカポンおよびトルカポン(それぞれ10および11,図2))は、COMT媒介の代謝を遮断するために、しばしばL-DOPAと併用投与され、その結果、より長いドーパミン作動性トーンが維持される。
図2 代表的なドパミン受容体作動薬(6-9)COMT阻害薬(10,11)のPD治療薬

HDに関しては、病状の経過を変える治療法はない。しかし、テトラベナジン(12,図3)のように運動障害を軽減できる薬がある。また、抗精神病薬、抗うつ薬、精神安定剤などが使われることもある。ALSの治療にはリルゾールやエダラボン(13,14,図3)などの緩和薬しかなく、めまいや頭痛などの重篤な副作用に加え、消化器や肝臓の問題もある。
図3 HDに関連した運動障害の治療に用いられる化合物(12)ALSの進行を遅らせるために用いられる化合物(13,14)

神経変性疾患の病因は非常に複雑であり、様々なレベルで多くの生物学的要因が同時進行して神経変性を引き起こす。このことは、神経変性の深遠な原因を標的とした疾患修飾薬の開発のハードルが高いことを意味する。このように、神経変性疾患の多機能性を背景に、「1剤1標的」のドラッグデザイン戦略の失敗から、「設計された複数のリガンド」、「ハイブリッド分子」、「マルチターゲット指向リガンド」(MTDL)と呼ばれる別のドラッグデザイン戦略の有効性が研究されるようになった。この新しい戦略は、少なくとも2つの治療標的と同時に相互作用できる多能性リガンドの開発を中心としている。MTDL のアイデアは、アルツハイマー病 のより効果的な治療法の発見のために大きく追求されており、このポリファーマコロジーの概念に基づく多くの構造が提案されている [20]。最も魅力的な類似体のいくつかは、複数のファーマコフォアの組み合わせは、選択された標的に対する一定の選択性を保持しながら、親化合物の活性を再現すべきであるという分子ハイブリダイゼーションの結果である。これらのハイブリッド構造は、(i)生物学的に活性な部位を空間的に固定するリンカーを使用することによって、(ii)活性部位を融合させることによって、(iii)または単に標的の関与に関与することが知られている機能性を融合させることによって、組み合わせることができる[21]。これらの潜在的な新薬の合理的な設計は、THA [22, 23]、ドネペジル [24, 25]、リバスチグミンなどのよく知られた薬剤や、レスベラトロールやクルクミン[26]などの異なる天然の生理活性誘導体に触発されているが、最近では他にも非常に興味深い構造的な組み合わせや修飾が確認されている。ここでは、異なる生物学的システムと相互作用し、調節することができ、アルツハイマー病の新しい治療法のための潜在的なプロトタイプを代表する、新しく開発されたMTDLの最も最近の、より興味深い例を報告する。
2. アルツハイマー病のためのMTDLデザイン戦略におけるターゲットの組み合わせ
コリン作動性欠損はアルツハイマー病の否定できない原因である。AChは認知プロセスにおいて極めて重要な役割を果たしており、その神経伝達の障害は、アルツハイマー病だけでなく他の加齢に関連した認知症の形態においても、認知と行動のあらゆる側面に影響を及ぼす可能性がある[27]。アセチルコリンエステラーゼ(AChE)は、その加水分解活性の産物としてコリンとアセテートを残して、シナプス裂け目でAChの作用を急速に終了させる。ブチリルコリネテラーゼ(BuChE)もまた、コリン作動性の媒介において重要な役割を果たしている[28]。
コリンエステラーゼ(ChEs)阻害剤は、AChのレベルを増加させ、ニューロンにおけるコリン作動性トーンのアップレギュレーションに寄与し、認知症状を部分的に改善することができる。AChEは、その触媒機能だけでなく、アミロイドβ沈殿、プラーク形成[29]、および炎症に対する効果のために、アルツハイマー病関連症状に対処するための特に魅力的なターゲットである。前述したように、軽度から中等度のアルツハイマー病症状の治療薬として承認されたドネペジルやリバスチグミン(2,4,図1)のように、いくつかの化合物が市場に登場している。
コリン作動性欠損に加えて、細胞外アミロイドβペプチドプラークの存在と高リン酸化pTAUのNFTが他の主な病理学的特徴を表している。したがって、「アミロイドカスケード仮説」は、現在でもアルツハイマー病治療の主要な焦点となっている。アミロイドβは、アミロイド前駆体タンパク質(APP)から、速度制限段階でβセクレターゼβサイトAPP切断酵素1(BACE-1)を含む逐次的な切断によって生成される。長年にわたり、多種多様なBACE-1阻害剤が提案されており、アルツハイマー病におけるこのプロテアーゼの中心的な役割を強調して臨床試験に入っている[30]。
ChEsおよび/またはBACE-1阻害と追加の酵素/受容体系への活性、および金属異常恒常性および酸化ストレスなどの他のアルツハイマー病関連の変化への効果の組み合わせ(図4)は、非常に興味深いMTDLの同定のための道を開き、新しい治療法の発見のための好ましいアプローチを表している。
図4
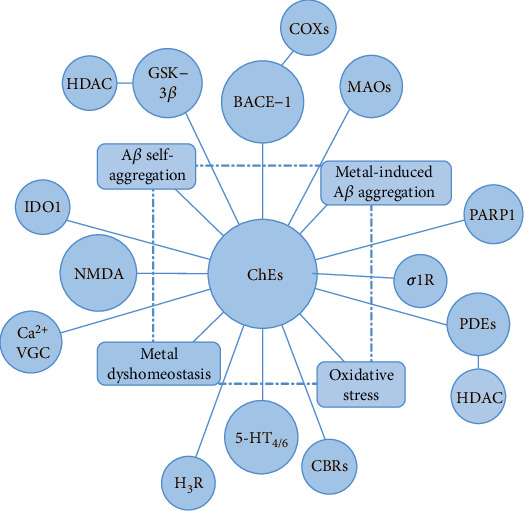
ChEsとBACE-1阻害剤、および/またはアルツハイマー病に関与する他のターゲットに関する最近の興味深い組み合わせを模式的に示している。選択されたシステムへの二重作用に加えて、新たに開発された類縁体のほとんどは、酸化ストレスと金属の恒常性障害とともに、アミロイドβペプチド凝集に影響を与える可能性を持っている。
2.1. デュアルChE/BACE-1阻害剤
上述したように、ChEs と BACE-1 は アルツハイマー病 の重要な標的である。新しいMTDLの発見のための賢明なアプローチは、これら、2つの酵素系を同時に阻害することによって代表されるかもしれない。この目的のために、様々な構造が提示されている。
GabrおよびAbdel-Raziqは最近、AChEおよびBACE-1に対する二重活性のために、ドネペジルの剛体類似体を探索した(15および16,図5)。化合物15[31]は、ドネペジルおよび他のBACE-1阻害剤、例えばAZD3839[32]または単純な2-アミノキノリン環からの特徴の組み合わせであり、酵素活性部位における二座相互作用をAZD化合物と共有している。インデノン部位を分子の残りの部分に連結する二重結合をさらに加えることで、IC50 = 14.7 nMおよび13.1 nMのナノモルのAChEおよびBACE-1阻害剤を得ることができた。AChEの速度論的研究から、AChEの阻害は濃度依存性の混合型であることが明らかになり、BACE-1の活性が改善されたことから、アミノキノリン基が重要な役割を果たしていることが確認された。また、SH-SY5Y神経芽腫細胞の生存率は、50μMまでの濃度では影響を受けなかった。さらに、化合物15は、PAMPA-BBBアッセイで高い伝染性を示し、ラット肝ミクロソームではかなりの代謝安定性を示し、脳への浸透性を持つ可能性を持ってた。
図5
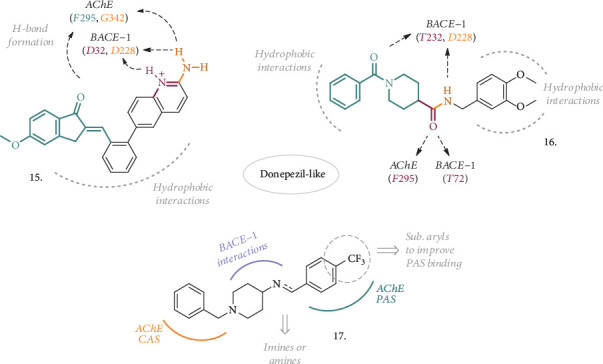
AChEおよびBACE-1阻害剤としてのドネペジルのインスパイアまたはリジッドアナログの構造。主にH結合と疎水性相互作用に代表される2つの酵素残基との主な相互作用が強調されている。
構造16[33]に代表される別の興味深い類似体シリーズは、良好な基の組み合わせを有しており、その結果、アミド部分のおかげでキレート能力を有するデュアルAChE/BACE-1阻害剤が得られる可能性がある。実際、化合物16は2つの標的酵素に対して低ナノモルの阻害剤であった(IC50,AChE = 4.11 nM、BACE – 1 = 18.3 nM)。50μMまでのSH-SY5Y細胞では細胞毒性効果は検出されず、バランスのとれた親油性と高い膜伝染性により、良好な脳への浸透性と代謝安定性を予測することができた。さらに、本化合物はCu2+をキレートすることで、これらの金属イオンの濃度や神経変性の促進に影響を与えることが期待される。
ベンジルピペリジン側でドネペジルに構造的に関連する一般構造17(図5)の化合物は、AChEに対する親化合物の特性を維持した上で、適切に装飾されたアリール基を導入してBACE-1に拡張したMTDLのもう一つの最近の例である[34]。このアリール基が4-CF3置換環で表される場合、アミンおよびイミンの両方がヒト酵素アイソフォームのサブマイクロモルインヒビターをもたらし、さらなる特性評価のために選択された。4-CF3置換環は、PAMPA-BBBアッセイで確認されたように、より高い膜伝染性を付与した。また、アミンをベースとした化合物は、PAS-AChE からヨウ化プロプリジウムを置換するのに重要な効果があり、それゆえに、さらなる進行のためのより興味深いものであった。おそらく、PAS-AChEに結合する能力のために、この化合物の5〜20μM濃度は、自己誘発性のアミロイドβ凝集だけでなく、AChE誘発性のものに対しても抗凝集特性を有していた(それぞれ50%および89%)。AFM試験では,本化合物を用いたインキュベーション後のアミロイドβ凝集体の減少が確認された。SH-SY5Y細胞において80μMまでの濃度では神経毒性効果は観察されず、認知への効果はスコポラミン誘発性健忘症動物モデルで最大10mg/kgの用量で試験された。その結果、本化合物が空間記憶や即時記憶を改善し、認知機能障害に影響を与える可能性が確認された。Ex vivo解析の結果、化合物を投与した動物では、スコポラミン投与群と比較してマロンジアルデヒド(MDA)のレベルが減少し、スーパーオキシドジスムターゼ(SOD)のレベルが増加したことが明らかになり、抗酸化作用が示唆された。また、アミロイドβ1-42誘発ICVラットモデルを用いたモリス水迷路実験で化合物を評価したところ、認知機能および記憶機能の頑健な改善が認められた。
これらのデュアルAChE/BACE-1阻害剤は、アルツハイマー病の病態におけるこれらの酵素の重要性を改めて確認し、それらに対する複合的な作用が、認知機能障害やアミロイドβ関連機能障害に対処するための貴重なアプローチであることを示している。
2.2. デュアルChEとGSK-3β阻害剤
グリコーゲン合成酵素キナーゼ-3β(GSK-3β)は、主に中枢神経系で発現するマルチタスク型のセリン/スレオニンキナーゼである。GSK-3βは、いくつかの細胞プロセスやシグナル伝達経路に関与しており、その制御異常は様々な障害の発生に関与している[35]。また、GSK-3βはpTAUのリン酸化過程にも関与しており[36]、その活性の増加はAPP消去酵素を阻害してアミロイドβ産生と相関しており[37]、神経毒性を引き起こしている。さらに、トランスジェニックマウスにおけるGSK-3βの過剰発現は認知障害の発症に関与することから、GSK-3βはアルツハイマー病病理学的に有効な標的となっている[38]。過去10年間、GSK-3βは集中的に標的とされており、AChEとの併用による阻害は、プラーク沈着とpTAUの高リン酸化に影響を及ぼすアルツハイマー病の多因子性に対処するための十分に統合された効率的なアプローチである。
既知のGSK-3β阻害剤[39]とAChEバインダーとしてのTHA部位の組み合わせから、新規なデュアルGSK-3β/AChEクラス[40]となる可能性のあるチアゾール系化合物が合成された(18,図6)。アミド結合が2-C鎖または3-C鎖で2つのファルマコホリック要素を連結する役割を果たし、THA芳香環上のいくつかの置換も評価された。THAの導入はGSK-3βの阻害力に影響を与えず、新規化合物は後者の酵素とhAChEに対してナノモルの活性を示し、ほとんどすべての化合物はhBuChEに対しても選択的であった。しかし、これらの化合物はSH-SY5Y細胞に対して顕著な抗増殖効果を示し、THA置換基と3C-リンカーを有する類似体のみが進行し、神経芽腫細胞株に対して30μMのIC50を有し、肝細胞においても安全であり、これらの後者の細胞の生存率への影響は同じ濃度では低かった。この化合物はまた、アミロイドβ自己オリゴマー化に対しても中程度の活性を示した。その後、マウス神経芽腫N2a-TAU細胞を用いて、pTAUの過リン酸化に対する効果を評価するために、濃度を上げて試験したところ、このプロセスを有意に阻害した。スコポラミンによって誘導される認知障害の動物モデルでは、化合物を処理したマウスは、モリス水迷路テストで有意に改善された記憶性能を示し、化合物の生体内試験活性を確認した。
図6 二重AChE/GSK-3β阻害剤18および19,および主要な酵素残基との相互作用のためのそれらの相対的なファーマコフォリック要素
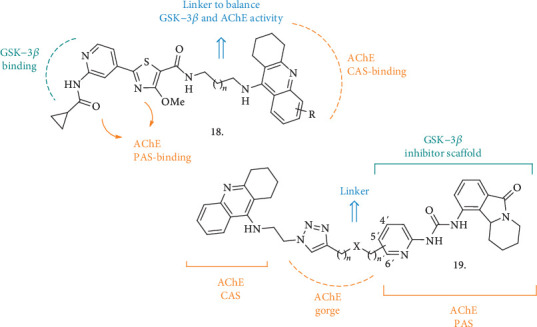
ChEおよびGSK-3βのデュアルインヒビターとしての別の新規な化合物シリーズ(19,図6)が最近報告された[41]。ここでは、THA(ChE阻害剤側)の構造とバルメリン(イソインドロン、GSK-3β阻害剤側)の足場をハイブリダイズした。TcAChEとの複合体である新規MTDLの結晶構造を分子ドッキング研究と組み合わせて解析した結果、1,2,3-トリアゾール基を、両酵素系の間で阻害力を保持または増強するための最良のリンカーとして同定することができた。最終的なハイブリッド化合物とともに、THAおよびイソインドロンをベースとしたフラグメントもまた、酵素阻害能力について試験を行い、各酵素阻害に対する単一部分の寄与についての更なる情報を得ました。その結果,ヒトAChEとGSK-3α/βの両方をナノモルの範囲で阻害することができ,他のキナーゼに対する選択性は高くないものの,トリアゾール環がGSKに対する阻害力を高める上で重要な役割を果たしていることは間違いないと考えられた。興味深いことに、新規化合物はいくつかの細胞株(肝臓HuH7細胞株を含む)において、対応するTHAおよびイソインドロンフラグメントよりも細胞毒性が低かったが、SH-SY5Y細胞において100μMまでの濃度で試験した場合、24時間後に細胞生存率の低下が観察された。新規化合物は、この系を介して良好な伝染性を示し、P-gpとの相互作用を示さなかった。
これらのクラスの化合物によって示されたすべての試験管内試験および生体内試験の生物学的特性は、アルツハイマー病の治療のための非常に興味深い可能性を強調表示する。選択性を向上させ、細胞毒性を低下させる必要があるとしても、これらのハイブリッド構造は、適切かつ特異的に設計されたリンカーによって結合された異なるファーマコフォリック要素が、MTDL治療薬として進歩するに値する貴重な出発点であることを改めて証明している。
2.3. デュアル ChE と MAO インヒビター
マルチターゲットの目的のためのもう一つの組み合わせは、ChEsとモノアミノ酸化酵素(MAO)の二重阻害から生じる。中枢神経系では、MAOはドーパミンやセロトニンなどのいくつかのモノアミン神経伝達物質の作用を停止させ、ヒトの脳内で優勢なアイソフォームであるMAO-Bは、その阻害薬であるラサジリンがPD症状の治療に承認されているなど、すでに神経変性疾患の有効な標的として認められている[42]。MAO-B の発現は アルツハイマー病 患者でも増加しており、MAO-B の活性と細胞内 アミロイドβレベルとの間に相関関係が観察されており、γセクレターゼとの相互作用によるものと考えられている[43]。アルツハイマー病発症におけるMAO-Bの役割は明らかにされていないが、MAO-Bの阻害剤は神経保護効果を示しており、この酵素をアルツハイマー病の魅力的なターゲットにしている[44]。
Sangらは、一連のカルコン-O-カルバミン酸誘導体(20,図7)を報告しており、ChEsおよびMAO-A/MAO-B阻害剤として機能し、抗酸化活性、抗アミロイドβ42凝集および金属キレート特性、およびH2O2誘発PC12細胞傷害に対する神経保護効果を有している[45]。新規化合物は、カルコン[46]の興味深い生物学的活性と、リバスチグミンのよく知られたAChEおよびBuChE阻害活性を組み合わせるように設計されている。カルコンのカルボニル基に隣接する水酸基の付加は、金属キレート剤としての可能性を告げる。L1=MeおよびL2=EtとR=HまたはN(Me)Etを置換基とした組み合わせが、期待される特性の点で最良の結果を与えた。この化合物は、選択的なBuChEおよびMAO-B阻害剤であり、両方の酵素に対してμMの範囲で活性であり、50%以上の値でアミロイドβ42の自己誘導性凝集を阻害することができた(最も活性の高いものでは63.蛍光による酸素ラジカル吸収能(ORAC-FL)法による強力な抗酸化活性が観察され、水酸基(R=H)を有する化合物はカルバメート類似体よりも強力であった。また、同じ水酸基を持つ化合物は、Cu2+やAl3+を選択的にキレートする金属キレート剤であった。したがって、Cu2+誘導アミロイドβ42凝集に対するその能力が評価され、参照としてのクルクミンよりも高い高い阻害値を表示した。2つの化合物は、MTTアッセイを使用して、H2O2誘発PC12細胞傷害に対する神経保護の可能性を評価するためにさらに進んで、それらはヒドロキシルラジカルを捕捉する能力と相関して細胞生存率を増加させることが示された。また、PAMPA-BBBアッセイでは人工膜を伝染し、最終的にはスコポラミン誘発性認知障害アッセイで生体内で試験された。ヒドロキシル誘導体は、最高用量(23.4mg/kg)では神経毒性を示したが、マウスの短期作業記憶の改善に有効であった。
図7 新規なChE/MAO-Bデュアルインヒビターと相対的なファーマコフォリック要素

新しい類縁体はまた、金属をキレートし、酸化ストレスから保護する能力を付与する構造的な要素に恵まれている。
Xuらはまた、ChEおよびMAO阻害剤としてのプロパルギルアミン修飾足場(21,図7)の素晴らしい例を提示している[47]。より詳細には、彼らは、イミダゾールで置換されたピリミジニルチオ尿素部位(AChE阻害剤ファーマコフォア)を、異なるリンカーによってスペーシングされたセレギリンのプロパルギルアミン基(MAO-B阻害剤ファーマコフォア)と組み合わせた。すべての化合物は、BuChEに対して無視できる活性を有するサブミクロンモルのAChE阻害剤をもたらし、R=HまたはMe置換は、特に単一の炭素原子リンカー(n = 1)と結合した場合、効率的な阻害のために最も適切であった。これらの心強い予備的な結果に続いて、MAO阻害試験を行ったところ、上記の化合物はマイクロモルの範囲で酵素を効率的に阻害し、R=Me化合物はMAO-Bに選択的であることが明らかになった。また、ORAC-FLアッセイにおいても良好な抗酸化活性を示し、チオ尿素フラグメントが金属キレート部分として働き、Cu2+を選択的にキレートし、Cu(II)関連レドックスにより生成する活性酸素を抑制した。この化合物はアミロイドβの自己会合には影響を与えなかったが、予想通りCuを介在するアミロイドβの会合を効率的に阻害することができた。それはラットの初代皮質ニューロンに安全に許容された最大30μMの濃度で、100μMで軽度の神経毒性を示し、それは、細胞の生存率を増加させ、Cu誘発アミロイドβ毒性損傷からニューロン細胞を保護することができた。PAMPAアッセイはBBBを横断する良好な可能性を示し、マウスのスコポラミン誘発性認知障害における生体内試験効果を評価した。HCl塩を30 mg/kgで経口投与したところ、スコポラミン投与群と比較して、学習・記憶障害を改善し、脱出潜時が短くなり、ミスの頻度が減少した。
これらのデータは、これらのデュアルChE/MAO阻害薬の有望なプロファイルを示したものであり、他のシリーズの化合物と合わせて、神経変性疾患に関連した認知症の動物モデルでの追加解析など、さらなる開発と解析の価値がある。
2.4. ChE 阻害剤と他の酵素系
ChEs とインドールアミン 2,3-ジオキシゲナーゼ 1 (IDO1) の同時阻害は、アルツハイマー病 における有益な効果に恵まれた別のターゲットの組み合わせをもたらした。IDOは細胞内の細胞質ヘム含有酵素であり、キヌレニン経路(KP)でトリプトファン(Trp)のN-ホルミルキヌレニンへの分解を制御し、第一段階の速度制御因子として作用する[48]。KPはいくつかの神経変性疾患では不均衡であり、その結果、Trpの異化は3-ヒドロキシクヌレニンなどの神経毒性代謝物を引き起こす。IDO1はこの経路に必須であり、その活性化はアルツハイマー病のアミロイドβ関連炎症と関連しており[49]、神経変性疾患治療に関する様々な研究の焦点となっている[50-51]。Lu らは、BuChE と IDO1 の二重活性を有する新規構造を同定した[52]。BuChE の選択的活性は、アルツハイマー病 の進行期には Ch の加水分解において AChE の欠乏に代わるものとして注目されており、より重要な標的となっている。一般構造22(図8)を有する化合物は、既にIDO1阻害剤[53]としての活性を示し、参照化合物として生体内試験で試験された抗真菌薬ミコナゾールをベースにしたプロトタイプである。3-OMeまたは4-OMe置換類似体は、AChEに対する活性が最も弱く、BuChEおよびIDO1に対する最高の阻害IC50値(3-OMeではそれぞれ8.3および2.8μM、4-OMeでは16.5および1.0μM)を示し、ミコナゾールよりも高い活性を示した。空間記憶に対する効果をスコポラミン誘発性障害マウスで評価したところ,THA投与群よりも良好な成績を示した。さらに、急性肝障害は観察されなかった。これらすべての特徴は、これらの構造体が革新的なアルツハイマー病関連の標的に対して興味深い活性を発揮する可能性を強調している。
図8 デュアルBuChE/IDO1,ChE/PARP-1,およびChE/PDE阻害剤の構造

構造22がミコナゾールに着想を得ている一方で、MTDL 23の合成においては、オラパリブが参照構造として取り上げられ、一方で、ドネペジルおよびリバスチグミンは、既知のPDE9A阻害剤のピラゾロピリミジノン構造と相まって、一般的な構造24および25を有する化合物の同定につながった。
MTDLの検索における別の興味深いアプローチは、デュアルChEおよびポリ(ADP-リボース)ポリメラーゼ-1(PARP-1)阻害剤を発表したGaoらによるものである[54]。PARP-1阻害剤は、その抗がん活性のために広範囲に研究されてきたが[55]、神経変性疾患、特にアルツハイマー病およびパーキンソン病に注目した場合には、潜在的な治療薬としての役割を果たす可能性がある[56]。PARP-1の役割はまだ完全には解明されていないが、その阻害剤の神経保護効果についてのエビデンスが出てきている。PARP-1 阻害と ChEs に対する阻害を融合させるために、既知の PARP-1 阻害剤オラパリブの構造を選択し、シクロプロパンの代わりに置換アリール ビニル基を導入して修飾した(23,図 8)。この組み合わせは、3-芳香族-α,β-不飽和カルボニル部位の形成につながったので、PARP-1とChEに対する二重の活性が保証されるはずである。試験管内試験の結果、これらの類縁体はいずれも親化合物オラパリブよりも強力なPARP-1阻害剤ではなかったが、低マイクロモルのPARP-1阻害剤であることが明らかになった。しかし、それらはAChEおよびBuChEに対して中等度のマイクロモル活性を示し、後者の酵素のより強力な阻害剤であった。また,Arを3-または4-ニトロ置換フェニル環とした場合,BuChEに対して最も高い阻害力を示し,IC50はそれぞれ9.2,5.9μMであり,基準となるNeostigmineよりもさらに高い阻害力を示した。上述の解析により、これらの類縁体の二重の活性が確認され、生体内試験での解析には十分な効力が得られないかもしれないが、分子ドッキング研究により、すでに活性を向上させる方法が発見されている。このように、これらのファルマコホリック要素の組み合わせにより、ChEとPARP-1の二重阻害につながるMTDLの別の興味深いクラスが生まれるかもしれない。
ホスホジエステラーゼは、最近、アルツハイマー病患者の認知機能を改善する可能性のために関心を集めている。これらの酵素は、cAMPとcGMPレベルの調節に関与することができる[57]と、それらの阻害は、記憶と学習プロセスの結果的な改善と、海馬と大脳皮質の2つのセカンドメッセンジャーのレベルを増加させるための貴重なツールを提供することができる。cGMPに対して加水分解活性を発揮するホスホジエステラーゼ9(PDE9)は、現在、アルツハイマー病を含む中枢神経系疾患の潜在的なターゲットとして研究されており[58]、最近開発された阻害剤PF-04447943[59]は、アルツハイマー病治療を対象とした第II相臨床試験(NCT00930059)で試験されている。
デュアルPDE9A/AChE阻害剤の効果を評価するために、Huら[60]は、ドネペジルのベンジルピペリジン部位と、報告されているPDE9A阻害剤[61]のピラゾロピリミジノン構造(24,図8)との組み合わせを利用した。2つのファーマコフォアのリンカーとして、異なるアミドまたは(環状)アミン鎖が検討されたが、4員のエーテルまたは炭素のテザーで最良の結果が得られ、その結果、PDE9AおよびAChEに対してサブマイクロモルの阻害活性を有する化合物が得られた。連鎖の種類と長さは、PDE9Aの阻害において極めて重要な役割を果たし、AChEの混合型の阻害にも関与し、酵素の触媒アニオン性部位(CAS)と周辺アニオン性部位(PAS)の両方への結合を可能にした。10〜20μMの間の濃度では、SH-SY5Y細胞の生存率に影響は観察されず、限定的な神経毒性の可能性を示した。すべての化合物は、PAMPAアッセイで試験され、その結果は、それらがBBBを横断することも可能であることを示した。中等度の代謝安定性にもかかわらず、急性毒性が評価され、有害な有害事象の欠如に続いて、化合物は、認知および学習障害のスコポラミン誘発マウスモデルで生体内試験で試験された。これらの類似体は、モリス水迷路試験において空間記憶と認知を有意に改善することができ、したがって、それらはまた、アミロイドβ25-35のICV注射によって生成された空間学習と記憶障害のマウスモデルで検討された。その結果、これらの化合物の中で最も性能の高い化合物で処理することによって誘導された欠損の部分的な改善が見られた。
同様のアプローチに従って、ピラゾロピリミジノン骨格とリバスチグミンとのハイブリダイゼーションは、有望なMTDLの別のシリーズ(25,図8)の同定につながった[62]。リバスチグミンのカルバメート官能性を置換するために様々な基が使用され、2つのファーマコフォリック要素間の連結を作成するために異なる鎖が採用された場合でも、一般的な構造25を有する化合物は、このシリーズの中で最も有望で効果的な類似体として結果として得られた。興味深いことに,これらの薬剤は,IC50値が0.96~18.8μMの範囲で,BuChEの選択的阻害剤として作用し,また,ナノモルの範囲で強力なPDE9A阻害剤であった。また、PDEスーパーファミリーに対する選択性についての研究も少し報告されており、PDE9A酵素に対する十分な選択性が確認された。それにもかかわらず、カルバメート化合物は、抗酸化剤として活性がなく、この機能性を水酸基で置換しただけで抗酸化力が回復した。さらに、試験した化合物のいくつかは、100μMまでの濃度ではSH-SY5Y細胞において細胞毒性を示さず、50μMの濃度ではアミロイドβの自己凝集をある程度抑制することができた。
これらのデータはいずれも、ピラゾロピリミジノンと他のChE阻害剤のファーマコフォアとの組み合わせが、アルツハイマー病治療のための新規候補を開発するための最適な戦略であることを指摘している。
2.5. ChE阻害薬とNMDA受容体拮抗薬またはCa2+チャネル遮断薬
ChEsの同時阻害とN-メチル-D-アスパラギン酸受容体(NMDAR)への拮抗効果は、新規MTDLの同定に向けた最も有望な戦略の一つであることは間違いない。NMDAイオントロピー性受容体は興奮性神経伝達物質であるグルタミン酸によって活性化され、Ca2+を含む様々な正イオンに対して伝染性を持ち、そのためシナプス可塑性や長期的な変化だけでなく、興奮毒性プロセスにも寄与している[63]。細胞内Ca2+濃度が病的なレベルに達すると、シナプス機能の低下や神経細胞死が生じ、認知機能の低下が進行する。近年、NMDARの活性化はアルツハイマー病関連のシナプス機能障害と関連しており[64]、NMDAR非競合アンタゴニストであるメマンチンは中等度から重度のアルツハイマー病の対症療法として承認されている[65]。NMDARアンタゴニストとChEsに対する阻害活性の組み合わせは、すでにアルツハイマー病の動物モデルで証明されているように、アルツハイマー病の症状に対して有益なまたは相乗的な効果を有する可能性がある[66, 67]。さらに、ドネペジルとメマンチンの合剤(ナムザリック®として知られている)は、現在、アルツハイマー病に伴う中等度から重度の認知症の治療に使用されている[68]。
この仮説に沿って、ベンゾホモアダマンタン-クロロタクリンハイブリッドの新しいシリーズが、新規のChE阻害剤およびNMDARの脳内浸透性アンタゴニストとして提案されている。THAベースのAChEおよびBuChE阻害剤およびNMDA受容体のアンタゴニストとしての多環式アミンの開発における彼らの以前の知識に続いて、著者らは、ベンゾホモアダマンタンモチーフを6-クロロタクリンに連結するために、長さと連結位置を変えて異なるリンカーを利用した(26および27,図9)[69]。特に、2つの連結位置は、ベンゾホモアダマンタンコアのベンゼン環上の橋頭アミノ基(26)または同じ系上の付加的なアミノ基(27)によって表された。これにより、新規なMTDLが同定された。これは、CHEを阻害し、NMDA受容体に拮抗するだけでなく、2つの別々のファーマコフォリック要素について既に報告されているように、BACE-1活性およびアミロイドβ42およびpTAU凝集を阻害することも可能である可能性がある。
図9

ここでは、ChE阻害剤やNMDARアンタゴニストとして作用する新規なTHA-アダマンタンアミン26,27を表現している。また、一般構造28のタクリピリミジン類は、ここでは、電圧依存性Ca2+チャネルを遮断することにより、Ca2+の流入や細胞内濃度に影響を与える薬剤として表されている。
いずれのシリーズにおいても、新規化合物は親類似体の主要標的に対する活性を保持しており、それぞれサブナノモルおよびサブマイクロモルの範囲でAChEおよびBuChE阻害剤であり、マイクロモルの範囲でNMDA受容体に結合していた。それにもかかわらず、2つの部位の関連性が当初期待されていたように、他のタンパク質やアルツハイマー病に関連する標的に対しては活性がなかった。しかし、いくつかの薬剤の効力が参照化合物と比較して増加していることは、抗アルツハイマー病薬の創薬分野でより詳細な評価を行うための正当な理由を表している。
新しいMTDLの発見に向けたもう一つの注目すべき進歩は、ChE阻害活性とカルシウムチャネル遮断能力を融合させて、電位差チャネル(VGC)を介したCa2+の侵入を制限し、神経細胞の損傷を防止する構造を作る可能性を考慮に入れたことである[70]。この目的のために,一連のタクリピリミジン類が提案されている(28, 図 9)[71]。3-Br置換アナログは、3-メトキシフェニル誘導体も良好なμM活性を示したが、IC50 = 0.037μMで最も強力かつ選択的なhAChE阻害剤として得られた。BuChEに対する選択性は一般的に高く,4-(ハロ)置換された化合物はこの酵素に対してわずかにより強力であった。4-Cl誘導体は逆の傾向を示し、BuChEに対してより強い活性を示したが、3-OMe置換化合物は2つの酵素に対して同等の活性を示した。さらに解析を進めた結果、非競合的なタイプの阻害が認められ、分子ドッキング研究により、酵素のCASとPASにおけるこれらの化合物の重要な相互作用が明らかになり、ハロゲン化合物がChEに対する活性と選択性の重要な置換として指摘された。また、SH-SY5Y細胞を用いて、K+脱分極によるCa2+流入量を測定することで、Ca2+チャネル遮断作用を調べた。すべてのタクリピリミジン系化合物はCa2+流入を阻害し,最も有望で強力なAChE阻害剤は参考文献のノモジピンと同様の阻害活性を示した。これらの化合物は100μMまでのHepG2細胞に対して比較的安全であったが,いくつかの誘導体は高濃度でTHAよりも高い肝毒性を示した。自己誘発性アミロイドβ凝集に対する有意な効果は予想通りなかったが、予測されたアルツハイマー病MET特性は、この候補の生体内試験での評価に有利な可能性を示した。このシリーズの中で最も優れたタクリピリミジンは、選択された標的に対してバランスのとれた活性を示し、優れたアルツハイマー病ME特性とTHAよりも低い毒性を有しており、アルツハイマー病におけるMTDLとしてのさらなる研究開発に値する魅力的な構造であると考えられた。
2.6. ChE阻害剤とセロトニン受容体モジュレーター(5-HT4および5-HT6)
セロトニンとその受容体(5-HTR)は、過去数十年の間に、特に記憶と学習に関連する脳領域に特異的に分布しており、認知においてこのシステムが果たす役割のために注目されていた[72]。したがって、特定の5-HTRサブタイプの調節は、アルツハイマー病との闘いにおける主要な治療戦略となり得る[73]。5-HT4Rと5-HT6Rの選択的リガンドが最新の研究の主な焦点となっており、5-HT4Rアゴニストと5-HT6Rアンタゴニスト(またはこれらの有効性の組み合わせ)の効果が評価されている[74]。
5-HT4R は G タンパク質共役型受容体ファミリーに属し、末梢領域と中枢神経系に局在し、黒質実質、線条体、海馬に高密度に分布している。ここでは、海馬のシナプス応答性と可塑性の調節因子として作用し、情報の貯蔵と認知において中心的な役割を果たしている[75]。したがって、5-HT4Rアゴニストは、記憶と行動パフォーマンスへの効果だけでなく、5-HT4Rがコリン作動系とACh放出に影響を与え、APP活性とアミロイドβ産生に関連しているため、アルツハイマー病において治療的に有用である可能性を持っている[76]。5-HT6RはGタンパク質共役型受容体でもあり、その発現は中枢神経系に限定されている。それらの薬理学に関する情報はまだ限られているが、認知機能、記憶、学習を司る脳領域(海馬や大脳皮質など)に5-HT6Rが存在することから、5-HT6Rはアルツハイマー病の別の興味深い標的となっている[77]。これらの受容体のアンタゴニストは、承認されたAChE阻害薬との併用により増強されるプロコリン作用との相関もあり、アルツハイマー病に関連した機能障害の一部に対処するための魅力的な治療薬となっている[78]。
抗ChE活性と5-HT4/5-HT6受容体の調節を融合させる目的で、さまざまな構造の組み合わせが検討されてきた。最近、Donecopride(30,図10)を発見した同じグループは、AChEを阻害し、5-HT4Rを活性化し、5-HT6Rをブロックすることができる新規なクラスのMTDLを開発することができた。Donecopride [79]は、5-HT4RアゴニストRS67333(29,図10)に触発された部分的な5-HT4Rアゴニスト活性を持つAChE阻害剤であり、NMRIマウスでは生体内試験での認知作用と抗アムネス作用を示し、C57BL/6マウスではsAPPα放出を促進した[80]。Donecoprideのベンジルアナログのマイナーな構造修飾により、R=3-Me置換(フマル酸塩として)で非常に有望な試験管内試験トリプル効果を有するいくつかの誘導体が得られた(31,図10)[81] 、化合物は、Ki(5 – HT4R)=210 nMおよびKi(5 – HT6R)=219 nMおよびAChE上のIC50=33. 7 nMであり、h5-HT4Rに対する部分的なアゴニスト(Donecoprideと同様)として作用し、h5-HT6Rに対する逆アゴニストとして作用する。さらに、NMRIマウスを用いた生体内試験評価では,0.3 mg/kgの用量で抗アムネス作用を示し、100 mg/kg以上の濃度では有害な作用は認められなかったことから、アルツハイマー病治療薬として有望なMTDLの候補となった。
図10

5-HT4RアゴニストRS67333(29)と、ChE阻害剤および/またはセロトニン5-HT4および5-HT6受容体モジュレーターとして作用する構造的に関連したMTDL(31および32)に触発されて、Donecopride(30)が発見された。一般構造33は、その活性は5-HTRからσ1Rにシフトしたが、RS67333とドネコプライドの足場のさらなる修飾として追加された。
Donecoprideのさらなる構造的修飾は、マルチターゲットプロファイルを有する他の興味深い化合物を発見することができた。そのようなデュアル5-HT4R部分アゴニストと5-HT6Rアンタゴニスト、AChEには活性がなく(32,図10)[82] アルツハイマー病におけるセロトニン作動性の役割を研究するのに有用な、またはAChEのデュアル阻害剤とσ1受容体(σ1R)のモジュレータ(33,図10)アルツハイマー病関連機能障害に対処するためのより最近の組み合わせとして、次の段落で議論の対象となる。
Marcinkowskaらによって報告された別の一連の化合物は、リンカーとして異なる長さの炭素テーターを使用して、N-ベンジルインドール-ピペラジン骨格をフタルイミド[83]またはTHA-moieties[84]と融合させた(34および35,図11)。インドール-ピペラジン部分は5-HT6Rに対する潜在的な拮抗作用をもたらしたが[85]、インドール単独では抗酸化作用があり、フタルイミド基またはTHA基はChEsの阻害に寄与する可能性があり、このように多面的な活性を持つハイブリッド化合物を作成することができる。フタルイミドサブシリーズ(34)では、新規化合物は5-HT6Rに対する親和性を示し、Kiは21~252nMであり、結合力とフタルイミドとのリンカーの長さとの間に明確な相関が見られた。また、これらの誘導体の拮抗活性は、細胞機能研究により確認された。さらに、これらの誘導体はBuChEのマイクロモルの阻害剤であり、AChEに対しては活性がないか、またはわずかに活性があり、再び短いリンカーは長いものよりも優れた性能を示した。抗酸化活性は、すべての化合物が10μMから始まる抗酸化力を示したFRAPアッセイによって決定され、それらのいくつかは、リファレンスアスコルビン酸よりも優れた活性を持っていた。
図11 最近のChE阻害剤/5-HT6Rアンタゴニストの一般構造

それぞれの構造について、二重活性のファーマコフォリック要素が強調されている。これらの薬剤の中には、複合活性とともに、抗酸化活性やアミロイドβ凝集に対する効果を有するものもある。
THA部位(35)がフタルイミドの代わりに存在する場合、R=H化合物はAChEおよび同程度にはBuChEに対してより高いナノモル活性を示し、非選択的で非競合的なChE阻害剤となったが、アルツハイマー病治療のための更なる開発に値する。5-HT6Rに対するアンタゴニスト活性も保存されており、5-C原子鎖は72 nMの最高のKi値を与えた。アミロイドβの自己凝集に対するチオフラビン-T(ThT)アッセイは、参照として使用されるレスベラトロールと同等またはそれ以上の阻害力(92%以上)を明らかにし、PAMPA-BBB予測は、膜を越えて拡散するために有利な可能性を決定した。
5-HT6Rアンタゴニスト[86]として知られており、同じリンカーによって間隔をあけて配置されたトルアミノフラグメントとTHA部位の別の組み合わせ(36,図11)は、上記のシステム上で同様の活性を示した[77]。この場合、AChEおよびBuChE上のKi値は、以前の構造と比較してさらに高く(最良のアナログについてはそれぞれ10nMおよび22nMに達する)依然として5-HT6Rに対する強力な結合および拮抗作用を示した。アミロイドβの自己凝集に対する効果と膜伝染性が良好であることに加え、ヒト肝ミクロソームにおけるこれらの類似体の試験管内試験での代謝安定性は、既知のTHA関連の肝毒性代謝物のいずれも同定されず、120分であることが強調された。
これらの化合物は、認知障害の動物モデルでの生体内試験が行われていない場合でも、試験管内試験での分析により、アルツハイマー病において有用なMTDLとなる可能性が高いことが明らかになった。
2.7. ChE阻害剤とH3Rアンタゴニストまたはσ1Rアゴニスト
ChE阻害薬と前述の受容体ファミリーとのより古典的なカップリングの他に、他の魅力的な組み合わせが最近探究されている。これまでのところ、ヒスタミンH3受容体(H3R)は、認知障害の治療のための多くの研究の焦点となっている[87]。H3Rは主に大脳皮質、海馬、尾状体、および被膜に発現しており[88]、それらの活性化は様々な神経伝達物質(AChEを含む)の放出に影響を与え、したがって、アルツハイマー病のような脳障害に影響を与えている。このため、H3Rのアンタゴニストは、アルツハイマー病に関連した認知機能障害におけるその潜在的な役割について研究されていた[89]。上述したように、σ1Rもまた、神経変性疾患の文脈でその場所を見つけた[90]。これらの受容体は主に小胞体に位置しており、ここでは通常、生存促進および抗アポトーシス作用を発揮するが、脂質、タンパク質、およびイオンの輸送に影響を与える他の小器官にも存在することがある[91]。この多機能タンパク質の機能や発現の変化は、アルツハイマー病 [92] や HD を含む様々な疾患と関連している。ここに、H3Rアンタゴニストまたはσ1Rアゴニスト活性と組み合わせたChE阻害剤のいくつかの例が提案されている理由がある。
Wangらは、異なる鎖でイソフラボンコアの7位に連結されたアミノ基またはTHA基を有するいくつかの新規なイソフラボン誘導体を、既知のH3Rアンタゴニストに触発されたいくつかのジアミノ置換類似体とともに提示した(37および38,図12)[93]。既知のAChE阻害活性を有するイソフラボン系化合物のH3Rアンタゴニストプロファイルの予備的な分析は、SAR研究のための適切なリンカーおよびアミンの同定を導くものであった。モノアミノ置換誘導体(37)は選択された標的に対して良好な活性を示したが、第2のアミノ官能基を有する類似体(38)は、より強力な低マイクロモルのH3Rアンタゴニストとして結果を得た。また、これらの化合物は、低またはサブマイクロモルレベルでAChEとBuChEの二重の阻害作用を示し、最も優れた阻害剤のIC50値はそれぞれ0.08μMと2.9μMであった。また、これらの化合物はORAC-FL試験においても抗酸化活性を示し、LPS刺激を受けたBV-2ミクログリア細胞において抗炎症作用を示し、細胞生存率に影響を与えることなくIL-6およびTNF-αの産生を抑制した。さらに、SH-SY5Y細胞では100μMまでの濃度では細胞毒性を示さず、SH-SY5Y-APPsw細胞株(ヒトAPPのスウェーデン変異体を過剰発現)では、銅によるアミロイドβ凝集体毒性を有意に抑制し、細胞の生存率を高めた。急性毒性をマウスで評価したところ,1000 mg/kgまでの忍容性を示し,良好なPKパラメータと薬物様特性から,著者らは本化合物を認知障害の動物モデルで試験することになった。マウスでは、スコポラミンによるステップダウンの潜伏が用量依存的に有意に延長され(10~30 mg/kg)脳内コリン作動性が増加し、認知障害が改善された。これらの結果は、これらの化合物の絶妙なマルチターゲットプロファイルを強調しており、生体内試験での解析は、アルツハイマー病治療への興味深い可能性を示唆している。
図12 H3Rアンタゴニスト(37および38)またはσ1Rアゴニスト(33)活性を有するデュアルChE阻害剤の発見のための新規な組み合わせの一般的な構造

Lalutらは、先に述べたDonecopride(30,図10)[79]の構造から、AChE阻害活性とσ1R親和性を有する新規リガンド[94]の研究に焦点を当てました。Lalut らは、AChE 阻害活性と σ1R 親和性を有する新規リガンドの研究に注目した[94]。σ1Rに対する結合親和性とともに、インドール足場はAChEの末梢アニオンサイト(PAS)との相互作用を増加させていた。
すべての化合物は、hAChEを阻害し、モルモット(gp)の5-HT4Rに結合する能力について評価した。これらの化合物は、5-HT4Rに対する親和性が全体的に低下し、N-ベンジルピペリジンが最も弱い化合物であった。これらはすべてAChEを強力に阻害し、ピペリジンのN-ベンジル基がこの活性を増強し、非競合的な阻害剤として作用し、PASと活性部位のサブアニオン部位との相互作用の可能性を示唆していた。これらの相互作用は、その後、選択された化合物の酵素との結晶構造のX線解析により確認された。σ1R親和性については,本シリーズの化合物のほとんどがこの受容体の非常に強力なリガンドであり,その活性にはインドール環の存在が重要であることがわかった。その中でも特に優れた化合物(IC50(AChE)=28.8 nM、Ki(5 – HT4R)=37.5 nM、Ki(σ1R)=5.1 nM)は、ジゾシルピン誘発性健忘症モデルの試験管内試験でも評価され、その試験管内試験での強力なσ1R親和性と相関した受動的回避試験での保護効果を示した。これらすべての理由から、この新しい化合物のシリーズは、アルツハイマー病の治療に有用であることが大きな可能性を持っており、さらなる調査に値する。
2.8. エンドカンナビノイド系のデュアルChE阻害剤と調節剤の効果
MTDLの探索におけるもう一つの革新的なアプローチは、ChEsやBACE-1に作用し、この活性をエンドカンナビノイド系(ECS)の調節と結びつける可能性を想定している。ECSは、エンドカンナビノイド(ECB)とその細胞標的であるGタンパク質共役型カンナビノイド受容体(type-1およびtype-2 CBR)ECBの生合成および代謝を担うトランスポーターや酵素などの内因性脂質シグナリング分子から構成されている。N-アラキドノイルエタノールアミン(アナンダミド(AEA))および2-アラキドノイルグリセロール(2-AG)は、ECBシグナル伝達分子の2つのメンバーであり、CBRを活性化して、痛み、炎症、および体温調節を含む幅広い反応およびプロセスを調節する [95]。これらのシグナル伝達脂質の作用は、細胞内での再取り込みとそれに続く加水分解によって、多くの酵素によって作動し、急速に終了する。後者の中でも、脂肪酸アミドヒドロラーゼ(FAAH)は、もともとAEAの加水分解を担当する酵素として同定された[96]一方、モノアシルグリセロールリパーゼ(MGL)は、2-AGレベルの調節において極めて重要な役割を果たしている[97]。ECBシグナル伝達は、いくつかの神経変性疾患で変化していることがわかっている。例えば、AEAの低下はアミロイドβの低下と逆の傾向を示すことが指摘されている[98]。また、神経炎症時に免疫系やミクログリアの活性化に関わるCB2Rは神経斑領域で選択的に発現しており、この受容体がアルツハイマー病に伴う炎症に関与している可能性が示唆されている[99]。これらの知見は、ECSの調節がアルツハイマー病に大きな影響を与える可能性を示唆している。
ECSは、CBRの直接的な刺激によって、またはECB異化酵素の阻害によって、ECBのレベルを増加させることによって調節することができる[100-104]。この後者の目的のために、Montanariらは最近、FAAH、AChE、およびBuChEに対する阻害活性を有するいくつかの化合物(ここでは39(図13)で表される)を提案している[105]。彼らは、39のトリアゾールリンカーをより柔軟なN-メチルアルキル鎖で置換したいくつかの構造を評価したが、そのうちのいくつかは、AChEとBuChEの両方を同程度に阻害し、FAAH酵素に対して良好な活性を保持することができた。しかし、よりバランスのとれたプロファイルが得られた39は、rFAAH、hAChEおよびhBuChEに対するIC50がそれぞれ922,42.7および27.9 nMであり、BuChEに対して良好な選択性を示していた。50μMまでのSH-SY5Y細胞に対する細胞毒性評価では急性毒性は認められず、生体内試験での評価は行われていないが、これらの化合物はアルツハイマー病治療のためにさらに検討する価値があると考えられる。
図13

ChEs(およびBACE-1)に作用し、CBRとの直接的な相互作用を介してECSを調節することができる新規化合物の構造(40および41)またはAEA代謝酵素FAAHの阻害を介してECBのトーンを増加させることによってECSを調節することができる。
CBRの直接刺激を介してECSを調節することができるアルツハイマー病治療用MTDLを発見する目的で、Nuñez-Borqueらは、BACE-1および/またはBuChE阻害剤としても作用する2つのCBRアゴニストを同定した[106]。CBRのKi値は報告されていないが、機能実験で40と41のアゴニストプロファイルが確認された(図13)40は10μMでBACE-1を60%阻害したが、化合物41は同濃度で38%阻害し、BuChEに対するIC50は2.5 nMであった。ラット初代皮質神経細胞培養において、両化合物はアミロイドβ誘導細胞死を効率的に減衰させ、細胞生存率を増加させたが、化合物40のみがアルツハイマー病の動物モデル、すなわちTgAPPトランスジェニックマウスにおいて性能を向上させることができた。さらに、本化合物はアルツハイマー病リンパ芽球の異常な特徴を回復させることができ、この疾患の全身症状と考えられている非神経細胞周期の変化に影響を与えた。これは、血清刺激による細胞増殖の亢進を抑制するか、血清欠乏による細胞死への抵抗性が高い状態でアポトーシスに感作することで達成された。これらの MTDL の背後にある効果とメカニズムを完全に理解するためには、より詳細な研究が必要であるが、彼らは異なる観点から アルツハイマー病 に対処するための素晴らしい方法を表している。
2.9. アミロイドβ凝集、金属誘起毒性、酸化ストレスに対する複数の効果を有するChE阻害剤
他の酵素活性と結合していない場合でも、(i)アミロイドβ凝集に対するそれらの可能性のある作用、(iii)予め形成されたアミロイドβフィブリルに対するそれらの解離効果、および(iiii)金属キレート特性を考慮すると、ChE阻害剤はマルチターゲットプロファイルを有する可能性があり、金属の恒常性障害および酸化プロセスに影響を与える。この組み合わせにより、神経保護作用を有するMTDLとなる可能性のある化合物が得られることが多かった。
Patelらは、神経保護および抗酸化作用を有するChE阻害剤の探索において、同じインドール部位に基づくメラトニンがフリーラジカル消去能を有し、アミロイドβ誘導毒性に対する神経保護作用を有するという観察から、インドール環を利用して新規な多活性構造を構築することを再考した[107]。彼らは、この環を、いくつかの薬剤にも共通する特徴である 1,2,4-トリアジン足場と融合させ、チオおよびアミノ結合したアリール/ベンジル/アミノアルキル側鎖(42で代表される、図14)の効果を探った。すべての化合物は、AChEおよびBuChEに対してマイクロモルからサブマイクロモルの活性を示した。最も活性な類似体のいくつか(IC50<5μM)は、10から 20μMの範囲の濃度で1,1-ジフェニル-2-ピクリルヒドラジル(DPPH)アッセイにおいて抗酸化活性を示し、アスコルビン酸と比較して中等度から良好なフリーラジカル消去活性(54.9-64.3%)を示した。SH-SY5Y細胞株(80μMまで)およびH2O2-およびアミロイドβ誘導毒性試験では細胞毒性は観察されなかった;化合物42は濃度依存的な方法で毒性障害に対して実質的な保護を示した。マルチターゲット活性に必要な本質的な相互作用と特徴を確立するための詳細な計算解析を行い、トリアジノインドールコアの重要な役割と鎖に塩基性中心を導入することによってもたらされる増強を確認したほか、本化合物はアルツハイマー病の動物モデルにおける認知機能改善効果についても試験を行った。また、アミロイドβ誘発性ADモデルでは、アミロイドβ1-42により低下した自発的交替が同量で有意に逆転した。スコポラミン投与群と化合物投与群の神経化学的解析から、アミロイドβ1-42が酸化ストレス誘発性認知症の管理だけでなく、参照記憶や作業記憶の障害を逆転させる能力を有することが確認された。これらの解析結果は、アルツハイマー病METの顕著なin silicoでの特性と相まって、このアナログがアルツハイマー病関連疾患の治療薬として有用である可能性を強調している。
図14

アミロイドβ凝集/解離、金属誘発性毒性、および酸化ストレスなどのアルツハイマー病の古典的な特徴に作用して、ChE阻害と神経保護効果を組み合わせた最近の化合物のいくつかが提案されている。
漢方薬のよく知られた五環式インドロピリドキナゾリノンアルカロイドであるRutaecarpineのコア構造とスルホンアミド部位の組み合わせは、Wuらによって有望なMTDLとして提案されている(43,図14)[108]。これらの類縁体はいずれも BuChE に対して良好から中等度の濃度依存性を持ち、μM の範囲で親化合物よりも高い活性を示したが、THAほど効率的ではなかった。また、これらの類縁体は、AChEに対してほとんど無効な効果を持つ、上記の酵素の選択的な阻害剤であることが判明した。100 μMの濃度では、アスコルビン酸とドネペジルを参照抗酸化物質として使用したDPPHフリーラジカルスカベンジャーアッセイでは、軽度のスカベンジャー活性の下に、有意な効果があった。また、H2O2で処理したSH-SY5YとPC12細胞では、細胞内の活性酸素の発生に対して堅牢な効果があり、化合物の処理は、ほぼブランク群に活性酸素レベルを復元し、神経抗酸化の可能性を強調している。同じモデルおよび細胞株を用いて、表題化合物の濃度を増加させたインキュベーション後の細胞生存率を測定することにより、神経毒性を評価した。いずれの場合も、生存率は用量依存的に増加した。シリーズの最高性能のアナログとアミロイドβ42の共沸は、ペプチドの自己組織化プロセスとの干渉をもたらし、TEM分析は、100μMの濃度でドネペジルのそれに匹敵するか、またはそれ以上の抗凝集効果を確認した。また,化合物中のカルボニル基とスルホンアミド基の存在がキレート作用を付与している可能性があり,特異な波長での紫外線吸収率の違いから,Cu2+を1:1のストイキオメトリーでキレートする能力があることが示唆された。ルタエカルピンの足場に新たに導入された特徴は、興味深い活性の組み合わせにつながり、このコアとその類似体は、アルツハイマー病に対するMTDLとしてのさらなる開発のためのスポットライトの下に置かれている。
別の最近の研究では、THAモチーフを(ポリ)フェノールまたはメトキシ置換環と結合させるために異なるテザーを使用する可能性を評価し、アルツハイマー病治療に有用な可能性のある新規MTDLを生成した(一般構造44および45,図14)[109]。より詳細には、新しい類似体は、古典的なTHA由来の副作用を克服し、神経細胞の酸化還元状態、アミロイドプラークの沈着、神経保護につながるなどの他の重要な治療標的へのアクセスを提供するように設計された。分子ドッキング解析によって導かれたリンケージ戦略は、炭素原子の数を増やすことでテザーの長さを変化させながら、イミノ、アミノ(44)エーテル(45)結合を形成することを想定している。9原子のエーテル型鎖とジメトキシ置換環を有するシリーズ44の最高のパフォーマンスのアナログは、非常に強力な(サブナノモル範囲)と選択的なBuChE阻害剤として動作し、THAと比較して85倍の活性の増加と、アミロイドβの自己凝集を妨害するために良い能力を示した5μMまでの濃度での神経毒性を欠いている。その神経保護特性は、それが10μMまでの濃度で神経保護を示した血清とK +の脱離によって神経細胞の損傷を誘導し、一次ラットのニューロンで評価された。これらすべての活性は、低い肝毒性と生理的条件下での良好な安定性と相まって、このリード化合物は、さらなる分析とアルツハイマー病治療薬のリストでの進行に値する有望なファーマコフォアの組み合わせとして指摘されている。
2.10. BACE-1阻害剤とMTDLへの組み合わせ
前節で既に述べたように、BACE-1はアルツハイマー病治療のもう一つの鍵となる酵素であり、ChE/BACE-1阻害剤の重要性と効果が報告されている(第2.1節)。MTDLの探索においては、神経保護作用と抗炎症作用を有する新規BACE-1阻害剤の探索から、他の興味深い組み合わせが生まれる可能性がある。
クマリン、アシルグアニジン、環状グアニジンにヒントを得て、さまざまな化合物がBACE1阻害剤として同定されており、抗酸化作用や金属キレート作用を有している(46-48,図15)。それらのいくつかは、フェニルイミノ-2H-クロメンコアとアミノメチレン-1,2,3-トリアゾール環の組み合わせに由来する(46)が、3-ヒドラジニル-1,2,4-トリアジン構造の使用は、化合物47および48の同定につながった。後者はまた、金属のキレート化における1,2,4,4-トリアジンおよび1,2,3-トリアゾール部位の寄与を評価することを促した。一般構造46[110]の化合物は、BACE-1酵素の中等度の阻害剤であり、最も活性の高い化合物はフタルイミドペンダントを有し、IC50=2.2μMであった。これらの化合物は神経保護剤としての可能性を示し、アミロイドβ25-35で処理したPC12細胞の生存率を増加させ、細胞毒性効果を示さなかった。また、4-ブロモフェニル置換されたアナログは、金属と3:2の錯体を形成し、Fe2+をキレートする能力を有していた。
図15 金属キレート特性とラジカル消去能を有するBACE-1阻害剤

一連のトリアジン系化合物[111]では、ジ-(チオフェン-2-イル)置換と異なるアリールヒドラゾン部位を有する興味深い類似体のセットが報告されている(47,図15)。以前に報告された他の環状グアニジンMTDLに触発されて、著者らはチオフェン環を利用して化合物の親油性特性を調節し、BACE-1活性部位内での相互作用を増加させ、一方でヒドラゾンの機能性に連結されたアリールペンダントを変化させながら、抗酸化およびラジカル消去の可能性を探索した。これらの化合物はBACE-1に対して良好な阻害活性を有しており、アリールペンダント周辺のSARを良好に評価した結果、2-インドール置換アナログが最も強力な阻害剤となり、IC50 = 0.69μMとなった。また、ヒドロキシルフェニル置換化合物は、特にDPPHアッセイで示された高い消去能(IC50 = 7 μM、ケルセチンのIC50値が4.6 μMであるのに比べて)に注目していた。上述の2-インドール置換アナログは、最大10μMの濃度でPC12神経細胞において非細胞毒性を示し、異なる化学量論でZn2+, Fe2+/3+, Cu2+をキレートする能力を示し、金属キレート活性を試験するために選択された。
トリアジンコアへの追加の修飾として、一般構造48(図15)を有する化合物が報告された[112]。ここでは、アリールフェノキシメチル-1,2,3-トリアゾール部位の導入により、金属キレート作用および抗酸化作用を示す更なる可能性が追加された。これらのハイブリッドでは、置換フェニル基が2つのチオフェン環を置換し、ヒドラゾン官能性に結合したペンダントアリールが1,2,3-トリアゾール基にもO-結合していた。これらの化合物をBACE-1を阻害する能力について試験したところ、Ar基がプロピルイソインドリンフラグメントである場合、18μMのIC50(30μMで67.09%阻害)に対応する最高の効力が得られた。これらのツール化合物をDPPHおよびMTTアッセイで評価したところ、アミロイドβ25-35で処理したPC12細胞において、抗酸化剤としての軽度の活性しか示さず、中等度の神経保護活性を示した。しかし、ペンダント型の4-ニトロベンジル基を含む化合物は、より高い抗酸化作用を示し、1:1のストイキオメトリーでFe2+とZn2+をキレートすることができた。
これらの興味深い一連の化合物は、異なるファルマコフォリック要素を組み合わせており、さらなる最適化が必要であるが、アルツハイマー病の最も一般的なホールマークのいくつかに対処することができるMTDLとして確かに興味を持っている。
炎症プロセスを阻害することができるBACE-1阻害剤は、Sagarらがアルツハイマー病用の新規MTDLの合成に着手するために使用したチアゾリル-チアジアジン足場から生まれた(49,図16)[113]。彼らは、Verubecestat [114]およびBACE-1阻害剤として作用する他のチアジン-1,1-ジオキシド足場に触発され、急性炎症状態の軽減に有用なチアゾール系化合物とともに、COX活性を阻害することができる可能性がある。
図16 COX相互作用に関連した抗炎症活性を有するBACE-1阻害剤

プロトタイプ化合物49(CF3置換基を有する)は、IC50が9μMであり、シリーズの中で最も強力なBACE-1阻害剤であり、カラギーナン誘発性急性炎症の動物モデルでも評価された。浮腫抑制効果はジクロフェナクと同等の高い割合(70%)が認められたが、ホルマリン誘発性慢性炎症に対する効果はセレコキシブを参考薬とした場合と比較してわずかに劣った(58%)。記憶増強効果は、AlCl3誘発ADラットモデルで評価したところ、高架プラス迷路やY迷路などの行動テストにおいて有意かつ頑健な改善が認められた。また、AlCl3を投与した動物では、脂質過酸化と酸化ストレスのマーカーであるMDAレベルの有意な上昇が認められた。注目すべきことに、処理したラットではMDAレベルが減少し、SOD活性が増加したことから、本化合物の抗酸化能力が示唆された。血液学的パラメータには有害な影響は観察されず、健康な海馬領域は、処理によって誘導される神経細胞の変性からの保護の最も説得力のある証拠であった。さらに、動物の胃腸では出血性の損傷や病変は報告されておらず、消化管の安全性が高いことが示された。この化合物は、BACE-1阻害作用と抗炎症活性を組み合わせて、アルツハイマー病に対する効率的な薬剤として機能する可能性があることを示す貴重な例を表している。
BACE-1阻害剤として作用し、炎症を調節するのに有用なキノキサリンベースの分子の別の良い例は、同じ著者によって報告されている(50,図16)[115]。これらの新規化合物は、これらの酵素の残基とH結合を確立し得るチアゾール環の導入のおかげで、BACE-1とBACE-2を介して相互作用し、COX-1およびCOX-2酵素に対する結合親和性を増加させるように合理的に設計された。すべての類似体は、マイクロモルの範囲でBACE-1を阻害し、特に置換アミノ官能性(R2=H)を有する類似体を阻害した。急性および慢性抗炎症作用をカラギーナンおよびホルマリン誘発ラット前足浮腫試験で評価した。50 mg/kgの用量では,COX-1およびCOX-2の試験管内試験阻害は認められなかったが,2つの動物モデルにおいて浮腫を69%および55%まで抑制することが示された。必要とされる活性プロファイルを有していることを確認し、Y迷路、条件付き回避反応、高架プラス迷路などの行動試験を実施した結果、いずれの試験においても有意な浮腫抑制効果が認められた。いずれの試験においても、AlCl3注射による健忘効果の減少、条件付回避反応の有意な減少、空間作業記憶の改善などの有意な効果が認められた。また、ラット脳内の脂質ペルオキシダーゼ(LPO)およびSOD測定により、抗酸化作用とフリーラジカル消去作用が確認され、対照と比較してMDA値が低下した。また、COX阻害剤としての可能性については、消化管での安全性を確認したところ、胃部では損傷はなかったが、病変は最小限であった。BACE-1 阻害と抗炎症活性を組み合わせることで、アルツハイマー病 に対する有益な効果を促進することができ、生体内で観察された抗酸化および抗アミロイド電位は、これらの構造を興味深い MTDL にした。
2.11. ChE や BACE-1 阻害とは無関係な アルツハイマー病 に対するマルチターゲット効果
酸化ストレスとともにアミロイドβ媒介毒性と自己および金属誘発性の凝集を標的とし、共通のアルツハイマー病関連系のいずれに対しても酵素活性を持たないという少し変わったアプローチは、神経保護的なMTDLの発見に大きなチャンスを提供するかもしれない。
ヒストン脱アセチル化酵素(HDACs)は、アルツハイマー病脳におけるその役割に注目が集まっている。HDACsは、ヒストンと非ヒストン蛋白質の両方のリジン残基からアセチル基の除去を触媒する酵素のクラスである[116]。既存の18のアイソフォームは、脱アセチル化プロセスを達成するために、亜鉛依存性またはNAD+依存性のいずれかのメカニズムを使用することができる。クラスI(アイソフォーム1,2,3,および8)IIa(アイソフォーム4,5,7,および9)IIb(アイソフォーム6および10)およびIV(アイソフォーム11)は亜鉛依存性のメタロアミダーゼであり、一方、クラスIIIのHDAC(サーチュイン)はNAD依存性の酵素である。選択されたアイソフォームのいくつかの阻害剤は、有望な抗がん剤としてすでに試験に成功している[117-118]。それにもかかわらず、HDACの阻害はまた、神経保護を提供し、シナプス可塑性だけでなく、学習や記憶を強化することができ、したがって、アルツハイマー病治療のための貴重なアプローチを表している[119]。特に、HDAC2とHDAC3は記憶関連遺伝子の制御に重要な役割を果たしており[120]、一方でHDAC6の活性を減衰させることでpTAUとアミロイドβクリアランスが向上する[121-122]。さらに、HDAC2とHDAC6はアルツハイマー病患者の大脳皮質と海馬で過剰発現しているようである[123]。
HDACsと他のアルツハイマー病関連タンパク質に対する効果を組み合わせる目的で、Cuadrado-Tejedorらは、HDAC6とPDE5の併用阻害剤、すなわち化合物51(CM-414,図17)の効果を探っている[124]。以前、これら、2つの酵素に作用する2つの異なる薬剤(VorinostatとTadalafil)のカクテルは、ADマウスの認知障害を緩和し、海馬ニューロンの密度低下を逆転させることで、生体内試験で肯定的な効果をもたらした[125]。この場合、単一の化合物が、中等度のクラスIのHDAC活性とHDAC6およびPDE5の強力な阻害という二重の活性を担っている。化合物51はピラゾロピリミジノンであり、既知のHDACおよびPDE5阻害剤から着想を得たものであり、必須のファーマコフォリック機能を有するいくつかの重要な要素を有している。また、良好なアルツハイマー病ME特性を有し、毒性と心血管系の安全性の観点から安全性の高いプロファイルを有している。CM-414のHDAC1-3,HDAC6,PDE5に対するIC50はそれぞれ310,490,322,91,60nMであり、ヒドロキサム酸部位がHDAC活性を担っている。HDAC/PDE5阻害の相乗効果は、10 nMでのWTニューロンにおけるヒストン3リジン9(AcH3K9)アセチル化の増加をもたらし、これは64 nMからのSH-SY5Yに対する同様の効果と相関している。Tg2576ニューロンでは、100 nMで同様の効果が観察され、ここではhAPP処理とpTAUへの効果も評価され、アミロイドβ42前駆体とpTAUレベルの低下が強調された。海馬スライス(200 nM)でプレインキュベートすると、化合物51は、シナプス増強とAPP/PS1 ADマウスのシナプス可塑性障害を救済した。PKパラメータ、毒性、試験管内試験PAMPAアッセイでのBBB浸透性を評価した後、本アナログをTg2576マウスで試験し、許容可能な脳内濃度と半減期を有する最適なものとして40 mg/kgの用量を選択した。2週間の投与後、化合物51は恐怖条件付け(FC)試験でWTマウスと同様の凍結反応を示し、記憶障害を救済することができ、3週間の投与後にMorris水迷路試験を行ったところ、空間記憶に肯定的な効果を示した。これらの活性をさらに説明するために、著者らは、可溶性アミロイドβ42とpTAUのレベルが治療マウスでは減少し(WTマウスでは減少しなかった)同時にGSK-3βの不活性型が増加することを発見した。さらに、51は先端CA1樹状突起の棘密度を増加させ、シナプス可塑性のマーカーをアップレギュレートし、Tg2576のダウンレギュレートされた遺伝子のいくつかの修復を誘導した。また、4週間の治療により、海馬領域における遺伝子発現および関連するシナプス伝達の濃縮がもたらされ、これらの変化はエピジェネティックな作用モードによって引き起こされた。全体として、化合物51は、アルツハイマー病関連機能障害を治療するための新規かつ有望な薬剤として、新規HDACおよびPDE5阻害剤の発見における最適な出発点を表している。
図17 ChEsやBACE-1酵素に対する作用とは無関係な新規MTDLの一般的な構造

同様の程度に、De Simoneらは最近、化合物52(図17)に代表される一連のHDAC/GSK-3β阻害剤を提案している[126]。既に述べたように、GSK-3βは、pTAUのリン酸化を介してアルツハイマー病の発症メカニズムにおいて中心的な役割を果たしており、後者とHDACとの間の密接な関係が既に出現している。例えば、HDAC1によって誘導される神経毒性はすでにGSK-3β活性と関連しており[127]、一方、GSK-3βによるHDAC6のリン酸化の亢進は、このHDACの活性とpTAUのリン酸化の亢進と関連している[128]。化合物52は、HDACと相互作用する部分が再びヒドロキサム酸によって表され、フタルイミド様の足場がGSK-3βのATPサイトのためのバインダーとして機能しているファーマコフォリック要素の組み合わせである。このアナログは、低マイクロモル濃度範囲(それぞれ12.78,3.19および2.69μM)でHDAC1,HDAC6およびGSK-3βを阻害することができる。SH-SY5Y細胞を用いて、リジン9および14におけるチューブリンおよびヒストンH3のアセチル化のレベル、およびpTAUのリン酸化のレベルを分析することにより、試験管内試験で本化合物の効果を決定した。処理した細胞はα-チューブリンの過剰アセチル化を示したが、AcH3K9やK14には効果が観察されず、HDAC6を介して優先的に作用することが強調された。HDAC阻害剤は抗がん剤として使用されているという事実にもかかわらず、この分子は100μMまでこの細胞株で安全であり、また、それはまた、効率的にH2O2誘発酸化ストレスを対比することができ、p53発現のレベルにも効果があった。さらに、化合物52は、GAP43,N-myc、MAP-2などの認識された神経新生マーカーの発現誘導によって確認されるように、神経新生を促進することができ、ミクログリアに対して免疫調節活性を有し、神経毒性から神経保護表現型へのシフトをもたらした。また、50μMからはゼブラフィッシュの発生にも明らかな効果が認められ、GSK-3βの阻害と相関していた。さらなる研究が必要であるが、化合物52のプロファイルは、その低分子量と高い溶解度と相まって、革新的なアルツハイマー病修飾薬の開発のための有望なヒット化合物となっている。
Kaurらは、アミロイドβ凝集、金属誘発性アミロイドβ凝集、金属代謝異常、および酸化ストレスを含む4つの主要なアルツハイマー病ホールマークに対処することができる2つのトリアゾール系化合物シリーズ(53および54,図17)を同定した[129, 130]。これらの類似体は、疎水性部分、すなわち、アミロイドβペプチドとの接触と抗凝集効果を担う置換フェニル環と、銅媒介のアミロイドβ42凝集を調節し、酸化ストレスを減少させることができるジトリアゾール部位を含む金属キレート部分の組み合わせから生まれた。
一般構造53を有するトリアゾール含有化合物の中で、R=o-CF3置換アナログは、シリーズの中で最も優れた性能を示し、ThT蛍光結合アッセイにおいて自己媒介アミロイドβ42凝集に対する最も強力な阻害活性を示し、IC50は8.065μMであった(参照クルクミンよりもさらに優れている)。また、有意に減少したアミロイドβ42フィブリルの形成は、TEMアッセイによって確認された。さらに、予備形成されたフィブリルを本剤の40μM溶液で24時間同時計回りに同調させると、TEMアッセイにおいてアミロイドβフィブリルの量を減少させる能力があり、予備形成されたアミロイドβ42フィブリルに対する破壊的な効果が強調された。また、本化合物は、UV-Vis分光法で評価されるように、金属キレート能を示し、その結果、40μMの濃度でCu2+誘導アミロイドβ42凝集を阻害し、Cu2+誘導アミロイドβ42フィブリルの解離を促進した。さらに、多官能性配位子は、Cu-アスコルビン酸レドックス系の銅酸化還元サイクルを阻害することで、活性酸素の発生に影響を与えた。この化合物は細胞毒性がなく、50μMまでの濃度ではSH-SY5Y細胞の生存率に影響を与えなかった。注目すべきは、同じ神経細胞株においても、アミロイドβ42凝集体によって誘導される毒性を阻害することができたことである。以上のことから、本化合物は、試験管内試験/生体内試験でのさらなる研究に値する神経保護作用を有する有望な候補化合物であると考えられる。
置換されたフェニル基(疎水性部分)にN-連結された1,2,3-トリアゾールコア(金属キレート部分)を構築するためにアセトアミドマロン酸ジエチルを使用することにより、アミロイドβ凝集体の疎水性ポケットと相互作用することを意図した一般構造54を有する化合物が得られた。2-ヨードフェニル基は、TEM分析によっても確認されたように、IC50=4.6μMで78%のアミロイドβ42自己凝集を阻害し、ThTアッセイにおいて最高の性能を発揮した。さらに、このアナログは、20μMの濃度で事前に形成されたフィブリルを解離することができた。その金属キレート特性を試験したところ、Cu2+を2:1の化学量論で錯形成させることができ、これにより金属を酸化還元不活性状態に維持し、活性酸素の産生を防止することができた。また、Cuを介したアミロイドβ凝集への影響を評価したところ、Cu単独ではアミロイドβ42フィブリルの形成が増加したのに対し、選択された化合物で処理することにより、アミロイドβ42フィブリルの形成が急激に減少した。金属の存在によって沈殿が誘導された予備形成されたフィブリルに対しても同様の破壊的効果が観察された。化合物はSH-SY5Yニューロン細胞の生存率には影響を与えなかったが、アミロイドβ誘発毒性障害の後、50μMで最大78%まで生存率を増加させることができた。したがって、これらの構造は、アルツハイマー病の主要な特徴に作用し、神経保護効果を有する新規MTDLの発見に実質的に貢献できることを示す証拠がたくさんある。
アミロイドβ誘発毒性と金属キレート化/抗酸化特性を多面的に結合させたPDE阻害剤の好例は、Huら(55および56,図17)[131]の研究から得られたものである。彼らは、クロロキンの金属イオンキレート骨格と、既にアルツハイマー病の前臨床モデルで試験されている既知のPDE4阻害剤であるロリプラムとロフルミラストの鍵となる結合部位フラグメントを融合させたハイブリッド化合物を提案した[132](NCT01433666およびNCT02051335も参照)。その結果、ジアルコキシ置換アリールにアミド結合を介して連結された一連の新規(ハロ)ヒドロキシキノリンは、PDE4DのμM阻害剤となった。これらの類似体は、参照化合物とは異なり、ORAC試験において良好な抗酸化活性を示し(クリオキノールと同等)Cu2+, Zn2+, Fe2+/3+などの金属をキレートし、Cu2+の場合には2:1の配位子と金属の化学量論でキレートする能力を示した。PAMPAテストは、これらの類似体が脳に浸透し、BBBを横断する可能性を評価し、したがって、著者はさらなる分析を行うことを促した。Cu2+-アスコルビン酸系では、化合物とのコインキュベート後に有意な活性酸素の産生は観察されず、金属キレートによるCuの酸化還元を阻害していた。同様の効果がSH-SY5Y細胞でも観察され、細胞毒性効果の欠如に加えて、BuOOHによる細胞内酸化ストレスに対する濃度依存的な保護効果が認められた。また、金属誘起アミロイドβ凝集に対する影響を評価したところ、ThT及びTEM解析の結果、表題化合物はCu誘起凝集に影響を与えていることが確認された。また 2000 mg/kgまで安全であり、肝ミクロソーム中で比較的安定であることから、いくつかの化合物はアミロイドβ25-35誘発性認知機能障害マウスを用いたMorris水迷路試験においても生体内での試験を行った。その結果、対照的なPDE阻害剤と比較して、海馬ニューロンの保護効果に加えて、認知空間記憶や行動性能が向上することが示された。これらの構造は、新しいクラスの抗アルツハイマー病薬の開発のための有望な候補であることは明らかである。
すでに議論したように、コリンエステラーゼ阻害活性とMAO阻害活性の組み合わせは、パーキンソン病やアルツハイマー病のような神経変性疾患の治療に魅力的なアプローチとなるかもしれない。同様に、MAO阻害剤とヒスタミン受容体H3Rアンタゴニストの組み合わせは、興味深いMTDLの開発への道を開くかもしれない[133]。この程度までに、3-ピペリジノプロピルオキシ部位とラサギリン(MAO阻害剤)の組み合わせから生じる2つの化合物がStarkらによって提案されている(57,図17)が、唯一の構造的な違いはアミノインダン骨格上のアンカーポイントによって表される。2つの類似体はH3Rに対して低いナノモルの親和性を示し、5-linkedのものだけがナノモルの範囲でMAO-Bに対して阻害活性を有しており、このように線状構造と分岐構造への選好性を示している。また、この化合物は、H1RおよびH4R、ドーパミンD2およびD3受容体サブタイプよりもH3Rを優先していた。さらに、いずれの構造も神経芽細胞腫細胞において低い細胞毒性を示し、薬物様の特性を有する最適な候補であった。化合物の不斉性や2つのエナンチオマーの分離の可能性についても、それ以上の評価は行われていない。しかし、これらの類縁体は、神経変性疾患治療に有用な MTDL の設計のためのリード構造となる可能性がある。
アルツハイマー病 誘導プロセスを標的とした薬剤の探索において、Ca2+ VGC に作用し、ホスファターゼ 2A (PP2A) の阻害を防止する新しいクラスの化合物が報告されている (58, 図 17) [134]。すでに観察されているように、変化したVGCの開口部に起因するCa2+過負荷は、いくつかの神経変性疾患で一般的である[70]が、この酵素がpTAUのリン酸化に役割を果たしているため、PP2Aのダウンレギュレーションもまた、アルツハイマー病の進行にリンクされている[135]。Gonzalesらは、神経変性疾患の潜在的な治療薬としてすでに評価されている天然アルカロイドグラミンの構造に触発されて[136]、インドールコアを使用してPP2A阻害およびCa2+過負荷を防止することができる新しい一連のN-ベンジル置換化合物を作成するために使用した。これらの類似体を高K+濃度にさらされたSH-SY5Y細胞にプレインキュベートすると、細胞内Ca2+の増加が抑制され、細胞の脱分極およびVGC開通に対する遮断効果が証明され、1.8から4.8μMの範囲のIC50値を示した。また、ラット大脳皮質胚性ニューロンのNMDA受容体に対しても軽度の拮抗効果が報告されているが、これはPP2Aとの間接的な相互作用に起因するものと考えられる。実際、PP2AはNMDA受容体と安定な複合体を形成し[137]、受容体の脱リン酸化と脱感作を引き起こし、Ca2+の流入を低下させる。この仮説を実証するために、オカダ酸(既知のPP2A阻害剤)で処理したSH-SY5Y細胞におけるPP2A活性を、PP2A機能不全を研究し、酵素活性の回復に対する新規化合物の効果を評価するための一般的なADモデルとして評価した。PP2A活性の低下は、ほとんどの化合物を0.1μMの濃度で事前および同時投与することで防止され、PP2A活性化薬として作用することが確認された。一方、オカダイン酸は細胞生存率に有害な影響を与え、これらの薬剤で処理するとSH-SY5Y細胞の生存率が70%まで上昇し、神経保護を誘導するのに必要な濃度の30倍の濃度ではそれ自体は無毒であることが確認された。これらの結果から、この薬理学的組み合わせがアルツハイマー病関連の認知症に対処するための貴重なツールとなり得ることが確認された。
いくつかのキノリン-インドール誘導体を用いた非常に詳細な研究では、アミロイドβ関連の毒性や酸化ストレスに対する神経保護効果の結果として、これらの化合物が成体マウス海馬の細胞増殖を促進する能力があることが指摘されている[138]。調査に値する類似体は1つ以上あると思われるが、最適化された化合物59(図17)は、クリオキノール構造と置換されたインドールフラグメントの組み合わせから生まれた、シリーズの中で最も有望な候補として同定された。ORAC-FL試験により、この化合物はクリオキノールと比較して高い抗酸化効力(ORAC値=5.0)を有し、PAMPAアッセイにより、受動的拡散によるBBBペネトラントである可能性が確認された。アミロイドβ1-42自己誘導性凝集のThTアッセイでは、親化合物および参照化合物よりも高い阻害活性を示し、予備形成されたフィブリルの解離に有意な効果を示した。この誘導体は、Cu2+, Zn2+, Fe2+, Fe3+をキレートし、Cu2+/化合物複合体を1:2の化学量論で生成することができた。このキレート化ポテンシャルに続いて、金属誘起アミロイドβ凝集に対する効果も評価したところ、Cu2+関連のアミロイドβ凝集とCu2+で促進された予備形成されたフィブリルの解離を実質的な割合で阻害することが示された。化合物は、最大50μMの濃度でPC12細胞株に神経毒性を持っていなかったが、むしろ、おそらくMAPK依存性のメカニズムを介して、細胞数を増加させる能力を明らかにした。また、SH-SY5Y刺激細胞における活性酸素の産生を減少させるという明確な効果もあった。塩酸塩製剤は、肝臓ミクロソームにおいてかなりの代謝安定性を示し(T1/2 = 116分)成体C57BL/6マウス(2000mg/kgまで)では急性毒性も有意な変化も観察されなかった。また、同じ動物モデルでは、塩酸塩をICV注射した後、顆粒下帯の神経幹細胞のリザーバーから、他の海馬領域の神経前駆体から海馬細胞の増殖を誘導する効果があり、マウスの脳の免疫組織化学的分析で確認された。二重トランスジェニック APP/PS1 マウス(アルツハイマー病 のモデル)を使用して、モリス水迷路テストで認知と記憶の強化効果を評価するために、化合物との治療は、記憶障害と認知機能障害の有意な改善を示した。毎日30mg/kgの用量は安全で忍容性が高く、慢性的な経口治療はまた、学習および記憶に対する肯定的な効果と相関して、アミロイドβプラーク沈着の顕著な減少をもたらした。結論として、このアナログは、最適な試験管内試験特性と実証された生体内試験活性を持つMTDLの良い例であり、それはアルツハイマー病治療のためにさらに追求される魅力的なエージェントになる。
2.12. アルツハイマー病における合成ペプチドの役割
MTDLとしての低分子の使用は、アルツハイマー病の新しい治療法の発見に向けた最も一般的なアプローチであるが、アルツハイマー病に対する新規薬剤としての合成ペプチドの役割について少し言葉を費やすことは興味深いかもしれない。アミロイドβオリゴマー、プロトフィブリル、プレフィブリル凝集体は、脳損傷につながる有害な障害において重要な役割を果たしている。ALS、先天性疾患、またはHDなどの他の神経変性疾患は、誤って折り畳まれたタンパク質の凝集によって特徴づけられる。合成ペプチドは、内因性ペプチドと相互作用したり、タンパク質の凝集を妨害したりすることで、このプロセスを調節する可能性を提供する。過去数十年の間に、ペプチドをベースとした薬剤の使用は、様々な病態を治療するための貴重なツールとして浮上してきており[139-141]、疾患修飾ツールとしてのペプチドの効果に関心が高まってきている。また、ペプチドは神経変性の早期診断や発見のためのプローブや診断ツールとしても機能しうる[142]。ペプチドをベースとした治療の潜在的なターゲットは、認知障害(例えば、インスリンの神経保護効果)またはタンパク質-タンパク質類似の相互作用を介したアミロイドβの形成および凝集の調節である可能性がある。これらのペプチドは、天然または改変されたアミロイドβ誘導体のいずれかであり得、アミロイドβの構造および凝集体に干渉することができ、および/または非天然アミノ酸を有するものであり得、これらはアミロイドβ凝集体を阻害し、軽度の認知障害を減衰させる能力を既に証明している[143]。それにもかかわらず、これらの薬剤は脳内に浸透しなければならないため、BBBを越えて標的に到達する能力を評価しなければならない。興味深いことに、いくつかのオリゴペプチドはすでに認知症の動物モデルで有意な効果を示しており[144]、さらに絞り込んだものが臨床研究に進んでいる[143]。これらのペプチドは完全にMTDLの定義には当てはまらないかもしれないが、合成ペプチドはアルツハイマー病機能障害に関連する複数のターゲットにヒットしたり、調節したりする可能性を持っており、その汎用性のおかげで、疾患修飾剤としても機能し、新しいアルツハイマー病治療法の探索に新たな展望と機会を与えてくれている。
3. 結論
アルツハイマー病の多面的で複雑な性質は、マルチターゲット治療戦略の開発に向けた途方もない研究努力を引き起こしていた。これらの治療法は主に、ミトコンドリア機能障害、脳内アミロイド凝集体の沈着、酸化ストレス、脳内グルコース代謝の変化など、アルツハイマー病の進行に関与する複数の因子を標的に開発されている。今回のレビューでは、アルツハイマー病の病因に直接関連する複数の標的と相互作用するように合理的に設計された低分子の開発に焦点を当てた。さらに、リード化合物の構造修飾により、金属キレート作用や抗酸化作用、抗炎症作用など、アルツハイマー病の潜在的な治療薬の同定に有益な特性を持つことが証明されている重要な特徴を導入することができた。アルツハイマー病の進行に関与する酵素の二重阻害剤の設計は、MTDLの開発に関与する最も一般的な戦略の一つである。ChEsの阻害剤は、主に分子ハイブリダイゼーションを介してデュアル酵素阻害剤の設計に広く使用されていた。この分野の研究の大部分は、ChE阻害活性が報告されているTHAおよびドネペジルなどのアルツハイマー病薬の足場を使用することに焦点を当てていた。GSK-3β、MAO、PARP-1,およびPDEに対する阻害活性が知られている足場とのハイブリダイゼーションにより、複数のMTDLの例が得られている。同様に、5-HT6Rアンタゴニストだけでなく、NMDA受容体アンタゴニストとのハイブリダイゼーションは、MTDLの開発のための有望なアプローチとして発展してきた。BACE-1を中心としたMTDLは、近年、研究者の間で注目を集めている。BACE-1阻害作用と抗炎症作用、抗酸化作用を併せ持つMTDLが数多く報告されている。
しかし、これらのMTDLの薬物動態の悪さが、これらの化合物の更なる臨床応用の大きな障壁となっている。したがって、リード化合物の経口バイオアベイラビリティ、代謝クリアランス、および中枢神経系への浸透性を最適化することに焦点を当てた取り組みは、より多くのMTDLを臨床試験に進める上で非常に重要である。さらに、アルツハイマー病の異なる病理学的カスケードを標的として設計されたMTDLは、アルツハイマー病進行の単一経路に対して設計されたMTDLと比較して、多因子性アルツハイマー病に対して効率的である可能性が高いと考えられる。