
Contents
www.ncbi.nlm.nih.gov/pmc/articles/PMC6382748/
Archana Prasad、† Vidhya Bharathi、† Vishwanath Sivalingam、 Amandeep Girdhar、 and Basant K. Patel*.
要旨
TAR DNA結合タンパク質43(TDP-43)は、RNA関連代謝に関与する汎用性の高いRNA/DNA結合タンパク質である。筋萎縮性側索硬化症(ALS)および前頭側頭葉変性症(FTLD)という運動ニューロン疾患の患者の脳および脊髄には、高リン酸化およびユビキチン化されたTDP-43が封入体として存在している。
ALSの大部分(90-95%)は散発性(sALS)であるが、家族性ALSの中では5-10%がTARDBP遺伝子の突然変異によるものであり、残りの90-95%は他の遺伝子の突然変異によるものである。C9ORF72,SOD1,FUS、NEK1などである。
しかし、驚くべきことに、散発性ALS患者の大多数(97%)には、神経細胞内包物に沈着したTDP-43タンパク質が含まれており、このタンパク質がALSの病態に重要な役割を果たしていることが示唆されている。このように、TDP-43の病態の分子機構を解明することは、ALS治療の中心となると考えられている。
本研究では、TDP-43の変異、細胞質内での局在異常、翻訳後修飾の役割について議論する。また、TDP-43の試験管内試験(試験管内試験)でのアミロイド様凝集、生体内試験(生体内試験)での生理的オリゴマー化と病理学的オリゴマー化、液-液相分離(LLPSそしてTDP-43包接体の潜在的なプリオン様伝播性を評価する。最後に、エンドサイトーシス障害やマイトoxicityなどの様々なTDP-43誘発毒性メカニズムを解説するとともに、TDP-43解離とALS治療に向けた新たな戦略についても議論する。
* *
キーワード 筋萎縮性側索硬化症(ALSTDP-43,分裂毒性、液液相分離(LLPSエンドサイトーシス、前頭側頭葉変性(FTLDプリオン、ALS治療薬
序論
TAR DNA結合タンパク質-43(TDP-43)は、1995年にHIV-1転写に関連したリプレッサータンパク質として同定され、ウイルスゲノムのtrans-active response element DNA配列に結合し、ウイルス遺伝子発現の調節に重要な役割を果たしていることが明らかになった(Ou er al)。
また 2001年には、TDP-43は、嚢胞性線維症膜貫通コンダクタンスレギュレーター(CFTR)エクソンのRNAスプライシングにも関与していることが報告された(BuratiおよびBaralle,2001)。TDP-43は、高度に保存され、かつユビキタスに発現するRNA/DNA結合タンパク質であり、このファミリーのメンバーは、1つ以上の高度に保存されたRNA認識モチーフ(RRM)の存在によって達成されるかなりの配列特異性を有するRNAに結合する能力を示す(Sephton et al 2010年 2012年;Geuens et al 2016)。
その後、TDP-43は、ニューロンおよび胚の発生に関与するmRNAを調節することも示されている(Polymenidou et al 2011; Sephton et al 2011; Tollervey et al 2011)。
* *
年に、TDP-43は、筋萎縮性側索硬化症(ALS)および前頭側頭葉変性症(FTLDまたはFTLD-TDP)疾患に罹患している患者の脳内の不溶性およびユビキチン化された介在物の主要成分として同定された(Arai et al 2006;Neuman et al 2006)。
TDP-43の病理学的進展を伴う他の疾患は、原発性側索硬化症および進行性筋萎縮症であり、これらの4つの疾患を合わせてTDP-蛋白質障害として知られている(図1)(Dugger and Dickson、 2017)。ALSおよびFTLD-TDPはともに、いくつかの共通の臨床的、神経病理学的および遺伝的特徴を有する遅発性神経変性疾患であるが、それらは神経系の異なる領域に影響を及ぼす(Neumann et al 2006年;Spires-Jones et al 2017年;Tan et al 2017)。
驚くべきことに、ALS症例の〜97%およびすべてのFTLD症例の〜45%(と呼ばれる:FTLD-TDP)は、TDP-43の凝集を伴う(Ling et al 2013年;Tan et al 2017)。
図1 TDP-43プロテインオパチー
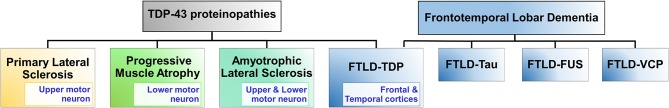
TDP-43プロテインオパチーとは、TDP-43が関与する疾患のことで、筋萎縮性側索硬化症(ALS前頭側頭葉変性症(FTLD-TDP原発性側索硬化症、進行性筋萎縮症などがある
FTLDは、主に認知症を引き起こす脳の前頭側頭葉の障害群である。FTLDの他の形態としては、FTLD-Tau、FTLD-FUS、FTLD-VCPがある。FTLD-Tauは、微小管関連タンパク質であるタウをコードするMAPT遺伝子の変異と関連している
タウのミスフォールディングおよび凝集は、微小管結合機能の喪失、および神経細胞およびグリア内包物の形成につながる(Irwin et al 2015)。FTLD-FUSは、RNA結合タンパク質FUSの変異と関連しており、これは、その核局在の破壊をもたらし、封入体への蓄積を導く(Mackenzie et al 2011)
FTLD-VCPは、バロシン含有タンパク質(VCP)の変異と関連している。FTLD-VCPは、ユビキチンおよびTDP-43陽性の神経細胞の核内包物および細胞質包接体を発現する。FUS、fused in sarcoma、TDP-43,TAR DNA結合タンパク質43,VCP、バロシン含有タンパク質
ALSは、細胞質内包物を呈する上位運動ニューロンおよび下位運動ニューロンの両方の進行性変性によって特徴づけられる致死的な神経変性疾患である(RowlandおよびShneider 2001;Kiernan et al 2011)。
上位運動ニューロンの変性は痙縮と興奮性を引き起こし、下位運動ニューロンが死滅すると脱力、筋痙攣、最終的には筋萎縮を引き起こし、それに続いて進行性麻痺が生じる。初期の症状としては、けいれんや筋肉の硬直があり、腕や脚に影響を与える筋力低下につながる。
患者は言語の不規則化、咀嚼や嚥下の困難を呈する(Mitchell and Borasio、 2007; Rothstein、 2009)。最終的には、発症から3~5年以内に呼吸不全や肺炎などの合併症により死亡する。平均発症年齢は約50歳だ(Logroscino er al 2007; Chio er al 2009)。
ALSの有病率は世界で毎年10万人中5人程度である。ALS症例の大部分(〜90〜95%)は原因不明の散発性(sALS)と考えられているが、〜5〜10%の症例は家族性遺伝子変異のメンデルパターン継承を伴い、家族性ALS(fALS)として知られている(Renton et al 2014;Taylor et al 2016)。
* *
TARDBP遺伝子をコードするTDP-43に加えて、他のいくつかの遺伝子の変異もまた、以下のようなALSと関連している。SOD1(スーパーオキシドジスムターゼ1)(Rosen、1993;Kunst et al 1997FUS(Fused in sarcoma)(Kwiatkowski et al 2009;Vance et al 2009C9ORF72(C9ORF72におけるヘキサヌクレオチドリピート拡張)(Dejesus-Hernandez et al 2011;Renton et al 2011ATXN2(Renton et al 2016)などの他のいくつかの遺伝子もまた、ALSと関連している。
2011ATXN2(アタキシン-2)(Elden et al 2010;Ross et al 2011OPTN(オプティニューリン)(Maruyama et al 2010VCP(バロシン含有タンパク質)(Johnson et al 2010;Koppers et al 2012PFN1(プロフィリン1)(Wu et al 2016PFN1(プロフィリン1)(Wu et al 2016PFN1(プロフィリン1)(Wu et al 2016)。
2014bCHCHD10(Coiled-coil-helix-coiled-helix domain containing 10)(Woo et al 2017SETX(Senataxin)(Hirano et al 2011TBK1(TANK-binding kinase 1)(Oakes et al 2017およびKIF5A(Kinesin重鎖アイソフォーム5A)(Nicolas et al 2018)などが挙げられる。これらの遺伝子に変異を持つ対応するタンパク質は、様々なメカニズムでALSの発症に関与している。
* *
FTLDは、神経細胞の核内包および細胞質内包を伴う前頭葉および側頭葉の変性を伴う進行性の神経疾患である(Mackenzie et al 2007年;Dugger and Dickson 2017)。稀に認知症を伴うALSとは異なり、FTLDは65歳未満の個人ではアルツハイマー病に次いで有病率が高く、推定有病率は10万人あたり~15~22人である(Van Langenhove et al 2012年;Onyike and Diehl-Schmid 2013)。
この疾患は、著しい人格と行動の変化、および言語能力の漸進的な障害を特徴とする。顕著なことに、FTLD-TDPのTDP-43包接体もまた、ALSで観察されるように、高リン酸化、ユビキチン化、およびN末端切断されている(Neumann et al 2007a; Hasegawa et al 2008; Igaz et al 2008)。また、TARDBP遺伝子の変異は、FTLD-TDP疾患と同様にALSを引き起こす可能性がある。
TDP-43の構造
TDP-43タンパク質は414アミノ酸を含み、コード化遺伝子TARDBPは染色体番号1に位置している。核局在化シグナル(NLS、aa 82-98)を持つN末端領域(aa 1-1022つのRNA認識モチーフから構成されている。
RRM1(aa 104-176)およびRRM2(aa 192-262核輸出シグナル(NES、aa 239-250プリオン様グルタミン/アスパラギンリッチ(Q/N)ドメイン(aa 345-366)およびグリシンリッチ領域(aa 366-414)を含むC末端領域(aa 274-414)からなる(図2)(Cohen et al 2011; Lukavskyら、図2)。
2011; Lukavsky et al 2013; Kuo et al 2014; Qin et al 2014; Jiang et al 2016; Mompeán et al 2016b)。) TDP-43は、主に核内に局在しているが、いくつかの機能のために細胞質にシャトルすることもある(Ayala et al 2008)。
ALSでは、細胞質のTDP-43濃度の増加があり、細胞質包接形成をもたらす(Neumann er al 2006; Winton er al 2008a)。TDP-43のミトコンドリア局在化は、疎水性アミノ酸の連続的な伸張からなる内部モチーフM1(aa 35〜41M3(aa 146〜150およびM5(aa 294〜300)に依存する(Wang et al 2016)。
その試験管内試験(試験管内試験)での溶解性の悪さと高い凝集性のために、TDP-43の完全な構造は、これまでのところ不明瞭なままであった。しかし、いくつかのグループがいくつかのドメインの高分解能構造を決定し(図2その全体的な構造は現在進化している。
図2 TDP-43の構造的特徴
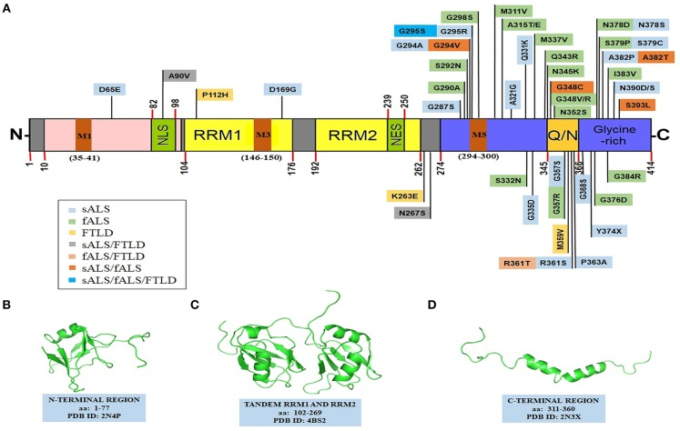
(A) TDP-43のドメイン構造は、ALSとFTLD関連の変異を示している。TDP-43は、NTDドメイン、2つのRRMドメイン、核輸出シグナル(NES核局在化シグナル(NLSプリオン様無秩序C末端ドメイン(グルタミン/アスパラギンリッチ領域(Q/N)とグリシンリッチ領域ミトコンドリア局在化モチーフ(M1-35-41; M3-146-150; M5-294-300)から構成されている
TDP-43のアミノ酸置換を生じる散発的変異と家族性遺伝子変異を分類した。いくつかのTDP-43変異は、ALSとFTLD、およびsALSとfALSの間で重複している(Baumer et al 2009; Xiong et al 2010; Fujita et al 2011; Janssens et al 2011; Budini et al 2012; Chiang et al 2012; Cruts et al 2012; Lattante et al 2013; Moreno et al 2015)
以下のPDB構造。(B) N末端領域(PDB id-2N4P(C) タンデムRRM1およびRRM2セグメント(PDB id-4BS2(D) C末端領域(aa: 311-360)(PDB id-2N3X)。(B-D)の構造は、John Wiley and Sons (Mompeán er al 2016b)、Springer Nature (Lukavsky er al 2013)、およびSpringer Nature (Jiang er al 2016)からそれぞれ許可を得て翻案されたものである
2016, creative commons attribution 4.0 license)。fALS、家族性筋萎縮性側索硬化症;NES、核輸出シグナル;NLS、核局在シグナル;NTD、N末端ドメイン;Q/N、グルタミン/アスパラギン;RRM、RNA認識モチーフ;sALS、散発性筋萎縮性側索硬化症;TDP-43,TAR DNA結合タンパク質43
N末端ドメイン(NTD
蓄積されたエビデンスは、TDP-43がネイティブに二量体であるか、少なくとも通常の生理学的条件下ではモノマーと二量体の平衡状態で存在することを示唆している(Shiina er al 2010; Zhang Y. J. er al 2013)。
TDP-43の二量体化は、明らかにN末端残基の相互作用を介して行われており、いくつかの報告では、TDP-43のN末端ドメイン(NTD)の二量体化がRNAスプライシングのような生理機能に必要であることが示唆されているが、他の報告では、TDP-43のN末端ドメイン(NTD)の二量体化がRNAスプライシングのような生理機能に必要であることが示唆されている。
また、他の研究者は、NTDの二量化が、実際には、その凝集に関与している可能性があると主張している(Shiina er al)。 注目すべきことに、TDP-43のN末端領域は、β1-β2-α1-β3-β4-β5-β6配列の1つのαヘリックスおよび6つのβ鎖からなるユビキチン様の折り畳みを示す(図2)(Qin et al 2014; Mompeán et al 2016b)。
TDP-43分子のホモ二量化は、RRM2ドメインが外側に延びている間に、2つのNTDのヘッドツーヘッド相互作用によって起こる(Wang Y. T. er al)。 実際、Zhangらは、NTDの最初の10残基が機能的ホモダイマーの形成に決定的に重要であり、完全長TDP-43の凝集にも関与していることを報告している(Zhang Y. J. et al 2013)。
注目すべきことに、N末端領域は、その核酸結合特性を調節する濃度依存的な方法で自己オリゴマー化を促進することができる(Chang et al 2012)。最近では、単一分子蛍光法を用いて、NTDが可逆的なオリゴマー化を経て、本質的に乱れたC末端領域が凝集する傾向を高めるという証拠も提供されている(Tsoi et al 2017)。
* *
対照的に、NTDを介したTDP-43の二量体化は、パートナータンパク質および標的RNAとの相互作用を可能にし、それによって凝集を防止する可能性があることも論じられている。実際、二量体化されたTDP-43のNTDは、そのプレmRNAスプライシング活性を高め、溶解性を改善し、細胞質のTDP-43介在物の形成から保護することが示されている(Jiang er al 2017)。
最近、TDP-43 NTDの1〜80残基の2.1Å分解能構造により、凝集を起こしやすいC末端領域を空間的に分離する動的ソレノイド様構造の存在が明らかになり、おそらく病的凝集を減少させていると考えられる(Afroz er al 2017)。
NTDにおける核局在化シグナル(NLS)配列の欠失または変異は、細胞質再局在化およびTDP-43の凝集を誘導する(Winton et al 2008a; Barmada et al 2010)。実際、核局在化シグナル(NLS)に存在するALS関連A90V変異は、内因性のTDP-43を不溶性の細胞質凝集体に隔離することができる(Winton et al 2008b)。
RNA認識モチーフ(RRM)
RNA結合タンパク質(RBP)は、真核生物において最も豊富なタンパク質ドメインの一つである高度に保存されたRNA認識モチーフ(RRM)を含む(RomanoおよびBuratti 2013;Gerstberger et al 2014;Marchese et al 2016;ConlonおよびManley 2017)。
これらのタンパク質は、mRNA処理、RNA輸出およびRNA安定性のようないくつかのRNA代謝プロセスに関与している。TDP-43のようないくつかのRBPは、神経変性疾患にも関与しており、したがって、原因因子としてのRNA代謝の障害を示唆している(Maris et al 2005; Lunde et al 2007; Clery et al 2008)。
TDP-43は、15個のアミノ酸で区切られた2つのRRMドメイン(RRM1およびRRM2)を含む(Kuo et al 2009,2014;Lukavsky et al 2013)。これらのRRMドメインは、β1-α1-β2-β3-α2-β4-β5パターンで配列された5つのβ鎖と2つのα鎖からなる(Lukavsky et al 2013;Sun and Chakrabartty 2017)。
TDP-43 RRMドメインの両方は、RNA/DNA分子の短いUG/TGに富んだ配列に向かってより高い特異性を有するコグナートRNA/DNA分子との結合に関与している(Lukavsky et al 2013;Kuo et al 2014)。
RRMMにおけるいくつかの変異は、RNA認識を有意に妨げない一方で、RNA結合能力を破壊することが示されている(Lukavsky et al 2013)。注目すべきことに、2つのALSに関連したミスセンス変異もまた、この領域において同定されている:P112Hおよびカスパーゼ切断感受性D169G(Burati 2015;Moreno et al 2015;Change et al 2016)。
提案的には、RRM2ドメインはまた、TDP-43タンパク質の二量化に寄与する可能性がある(Kuo er al 2009)。二本鎖DNA(dsDNA)には結合せず、一本鎖DNA(ssDNA)または一本鎖RNA(ssRNA)に結合することは、TDP-43の溶解性を高め、予想されることに加えて、その凝集を防ぐことが示されている(Huang et al 2013; Sun and Chakrabartty 2017)。重要なことに、TDP-43は、数千のmRNA転写物の3′未翻訳領域(UTR)に積極的に結合し、さらにはそれ自身のmRNAにも自己調節機構として結合して、それ自身の細胞内濃度を制御し、おそらくはその溶解性も制御する(Ayala et al 2011)。
C末端ドメイン(CTD
TDP-43 (aa 277-414) の C 末端領域は高度に乱れており、グリシンに富む領域と、無荷電極性アミノ酸であるグルタミンとアスパラギン(Q/N)に富むセグメントから構成されている(図 2)。この異常な構成は、Sup35,Rnq1,Cyc8などのいくつかの酵母タンパク質のプリオン様ドメインに類似している(Patel er al)。
Patel et al 2009; King et al 2012; LiebmanおよびChernoff 2012)。プリオン原性ドメインを含む酵母タンパク質は、無秩序な構造から、時には適応的な生理的応答として、自己模倣的なクロスβシートリッチなアミロイド様の構造に切り替わることがある(Liebman and Chernoff、 2012)。
驚くべきことに、潜在的なプリオン様ドメインを有する約240個のヒトタンパク質のうち、約70個のタンパク質は、RRRMモチーフを含むRNA/DNA結合タンパク質であり、そのうちのいくつかは、TDP-43,FUS、hnRNP、TATA-ボックス結合タンパク質関連因子15(TAF15およびEWS RNA結合タンパク質1(EWRS1)等を含み、様々な神経変性疾患の病態に関与している(March et al 2016;HarrisonおよびShourter 2017)。
TDP-43のC末端領域は、TDP-43の病理学的挙動に特別に関連しているように思われる。第一に、プリオン様ドメインと同様に、それは本質的に無秩序であり、凝集しやすい(Santamaria et al 2017)。第二に、それは、ALSに関連するTARDBP変異およびリン酸化部位の大部分を有している。
第三に、カスパーゼの異常な活性によってTDP-43から産生される25〜35kDaのサイズの特定のC末端フラグメントは、非常に細胞毒性が高く、ALSに罹患した脳から同定された封入体中に見られる顕著な種である(Zhangh et al 2007,2009)。
TDP-43のC末端領域はまた、短く、高度に動的で不安定ならせん-ターン-らせん領域(aa 311-360)を含む(Jiang et al 2013,2016)。この領域からのペプチドは、試験管内試験(試験管内試験)で効率的にアミロイド様フィブリルを形成することができ、このフィブリルは、可溶性TDP-43を発現する細胞に対してプリオン様感染性播種能力を示すことができる(Chen et al 2010; Guo et al 2011; Jiang et al 2013)。
興味深いことに、TDP-43のC末端領域は、液-液相分離(LLPS)を経て、ダイナミックなタンパク質飛沫を形成することができる。これらの飛沫の中で、C末端残基は、ストレス顆粒の形成に重要であると思われる軽度の一過性の相互作用を示す(Conicella er al 2016)。
突然変異、持続的なストレス条件、または老化は、これらの飛沫が液体-固体相分離(LSPS)を経て、それによって不可逆的な病理学的凝集体を形成する原因となることが提案されている(Patel et al 2015)。
TDP-43の生理機能
TDP-43-RNA相互作用
TDP-43は多面的な機能を有し、転写、翻訳、mRNA輸送、mRNA安定化、マイクロRNA(miRNA)およびロングノンコーディングRNA(lncRNA)処理などのRNA代謝のいくつかのステップに関与している。Ling et al 2013; Coyne et al 2017)(図3)。
ゲノムワイドRNA免疫沈降法(CLIP-seq)を用いて、6,000以上のmRNAターゲットがTDP-43と関連することが同定され、これはトランスクリプトーム全体の約30%に相当することになる(Polymenidou et al 2011; Tollervey et al 2011; Xiao et al 2011)。
これまでの従来のRNA免疫沈降法でも、特定のRNA標的が明らかになっている(Buratti and Baralle、 2001; Sephton er al 2011)。TDP-43は、RNAのUGに富んだ配列に高い特異性で結合するが、細胞質に局在する場合には、ほとんどがmRNA/pre-mRNAの3′UTRに結合する(Colombrita er al 2012)。このことは、mRNAの安定性、成熟および輸送の維持におけるTDP-43の幅広い役割を示唆している(Tollervey er al 2011; Colombrita er al 2012)。
図3 TDP-43の機能
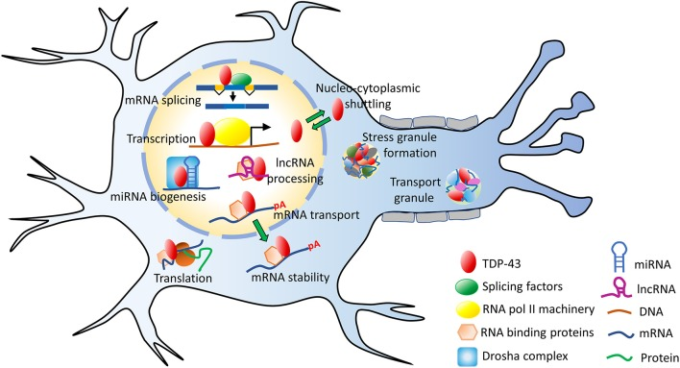
TDP-43 は、転写、スプライシング、RNA の安定性維持、miRNA や lncRNA の処理など、核内でいくつかの mRNA 関連のプロセスを実行する。TDP-43は、主に核内のタンパク質であるが、核と細胞質の間をシャトルする役割も持っている。細胞質ではストレス顆粒形成、リボ核タンパク(RNP)輸送顆粒形成、翻訳などに関与している
mRNAの転写とスプライシング
TDP-43はサイレントヘテロクロマチンの領域には存在しないが、転写やスプライシングの部位に局在している(Casafont er al 2009)。TDP-43は、嚢胞性線維症膜貫通コンダクタンスレギュレーター(CFTRTARDBP、FUS、SNCA(α-シヌクレインHTT(ハンチンチンAPP(アミロイド前駆体タンパク質)などの重要な遺伝子の転写物のスプライシングパターンを制御している(Buratti and Baralle、 2001; Pol. (Buratti and Baralle、 2001; Polymenidouら、 2011, 2012)。実際、TDP-43の核欠乏は、mRNAスプライシング異常をもたらす(Arnold et al 2013; Highley et al 2014; Yang et al 2014)。
同様に、TDP-43の過剰過剰は、結合パートナータンパク質の供給が限られているために、機能不全複合体を形成し得る。実際、TDP-43の過剰発現によって引き起こされる不均衡は、神経細胞に有害である(Cannon et al 2012;HeyburnおよびMoussa 2016;Lu et al 2016)。
TDP-43の核枯渇はまた、運動ニューロンにおけるスプライシングイベントの広範な調節障害を誘発することが見出された(Highley et al 2014)。TDP-43における2つのALS関連変異、Q331KおよびM337Vもまた、トランスジェニックマウスモデルにおいてmRNAスプライシングプロセスを変化させることが示されている(Polymenidou et al 2011年 2012年;Lagier-Tourenne et al 2012年;Arnold et al 2013)。
mRNAの成熟と安定性
TDP-43は、mRNA転写物と結合することにより、自身のmRNAを含むいくつかのmRNAの安定性を調節する(Strong et al 2007; Volkening et al 2009; Ayala et al 2011; Colombrita et al 2012; Costessi et al 2014)。
TDP-43は、これらのmRNAの調節性3′UTR配列と相互作用し、ヒト低分子量ニューロフィラメントmRNAについて観察されたように正に、または血管内皮増殖因子およびプログラニューリンmRNA転写物について観察されたように負に、半減期に影響を与える(Strong er al 2007; Volkening er al 2009; Ayala er al 2011; Colombrita er al 2012; Costessi er al 2014)。
mRNA輸送
TDP-43はRNA分子と結合してリボ核タンパク質(RNP)顆粒を生成し、mRNAを遠方へ輸送する。軸索細胞では、RNP顆粒は微小管の助けを借りて輸送される(Alami er al 2014)。実際、ALSに関連したTDP-43変異株は、RNP顆粒の輸送を損なうことが明らかになった(Wang et al 2008;Alami et al 2014)。
mRNAの翻訳
プロテオミクスにより、TDP-43のグローバルなタンパク質相互作用プロファイルが明らかになり、スプライシングや翻訳などのRNA代謝に関与するいくつかのパートナータンパク質も同定された。これらの相互作用のうちのいくつかは、ALSに関連した突然変異であるA315TとM337Vの影響を受けませんであった(Freibaum er al 2010; Kim er al 2010)。
最近のショウジョウバエの研究では、TDP-43が神経筋接合部におけるFutsch(Map1bのオルソログ)mRNAの局在化と翻訳を制御していることが報告されている(Coyne er al)。 TDP-43はまた、翻訳機械系に関与する他のタンパク質、例えば、リボソームタンパク質、活性化Cキナーゼ1の受容体(RACK1)との複合体を形成し得る(Russo et al 2017)。
ある研究では、細胞質TDP-43の増加は、神経芽腫細胞においてグローバルなタンパク質合成の抑制を引き起こし、これはRACK1の過剰発現によって救済され得る(Russo et al 2017)。TDP-43はまた、ストレス顆粒への翻訳因子の隔離を介して、いくつかのmRNAの翻訳を変化させることができる(Aulas and Vande Velde、 2015)。
ストレス顆粒の形成
真核生物の細胞は、様々な細胞傷害から細胞を保護するいくつかのメカニズムを開発していた。ストレス顆粒(SG)の形成は、酸化ストレス、熱ショック、ウイルス感染、化学物質への曝露などのストレスに曝されると速やかに起こる。アンダーソンとKedersha 2009; AulasとVande Velde 2015)。
SGは通常、RNA結合タンパク質、翻訳的に停止したmRNA、および停止した前導入複合体のための安全な「貯蔵および選別ステーション」である。SGの形成は可逆的なプロセスであり、SGはストレスが終わった後に溶解する(Anderson and Kedersha、 2008)。
神経細胞はストレスに対して非常に脆弱であり、欠陥のあるストレス応答は、ALSやFTLDに罹患した脳で見られるように、SGの病的包接体への変換を促進する可能性がある(Wojcik et al 2006; Van Damme et al 2008; Colombrita et al 2009; DormannおよびHaass 2011)。
TDP-43はストレス顆粒に組み立てることが可能であり、細胞障害に対する保護的役割を示す(Colombrita et al 2009;AulasおよびVande Velde 2015)。実際、TDP-43は、SGの組み立ておよび維持の両方に関与し、また、主要なSG核形成タンパク質であるrasGAP SH3ドメイン結合タンパク質1(G3BP)およびT細胞制限細胞内抗原-1(TIA-1)の発現を調節する(McDonald et al 2011)。
ALSに関連した変異は、ストレス顆粒のダイナミクスに影響を与え得る。ソルビトール誘導浸透圧ストレス下では、G348C変異株TDP-43は、徐々に大きなストレス顆粒に局在することが判明した(Dewey et al 2011)。
反対に、TDP-43のR361S変異株は、ストレス顆粒の集合を混乱させることが示された(McDonald er al)。 ストレス顆粒ダイナミクスに対する他のいくつかのALS関連変異の異常な効果については、本レビューの「TDP-43変異の役割」のセクションでさらに議論されている。
miRNAとlncRNAの処理
また、TDP-43は、マイクロRNA(miRNA)などのノンコーディングRNAの生合成や処理を促進する(川原・稗田里 2012)。最近の研究では、TDP-43とDroshaおよびDicer複合体との相互作用が確認されている(Ling er al 2010; Kawahara and Mieda-Sato、 2012)。
TDP-43は核内のDrosha複合体と結合し、プライマリーmiRNAに直接結合して、前駆体miRNAのサブセット(pre-miRNA)の産生を促進する(Kawahara and Mieda-Sato、 2012)。ヒト胚性腎293細胞(HEK293)では、細胞質のTDP-43がダイサー複合体と相互作用してpre-miRNAの処理を促進することがわかっている(川原・三枝里 2012)。
実際、TDP-43のダウンレギュレーションは、培養HeLa細胞、げっ歯類ニューロン、人工多能性幹細胞(iPSC)由来のヒトニューロンにおいて、いくつかのmiRNAの発現変化をもたらする(Buratti et al 2010; Zhang Z. et al 2013)。
ゲノムワイドな研究では、タンパク質をコードしないが、様々なメカニズムで遺伝子発現を制御している200ヌクレオチド以上の転写産物であるいくつかのロングノンコーディングRNA(lncRNA例えば、nuclear enriched abundant transcript 1(NEAT1)やmetastasis associated in lung adenocarcinoma transcript 1(MALAT1)がTDP-43と結合していることが明らかになった。興味深いことに、NEAT1およびMALAT1は、FTLD-TDPにおいても高いレベルで見出される(Tollervey et al 2011)。
TDP-43 タンパク質-タンパク質相互作用
グローバルなインタラクトーム研究により、TDP-43が多様な生理機能に関与するタンパク質と相互作用することが明らかになった(Freibaum et al 2010)。最近の研究では、Blokhuisらは、免疫沈降、プルダウンアッセイ、質量分析法を用いて、神経細胞内のALS関連タンパク質の結合パートナーを同定するためのインタラクトーム解析を行った。
TDP-43のインタラクトームからは、RNA処理、遺伝子発現、RNAスプライシング、遺伝子発現および翻訳の転写後制御に関与する多くのDNAおよびRNA結合タンパク質が検出された(Blokhuis et al 2016)。TDP-43は、いくつかのタンパク質との直接的な物理的相互作用、またはRNA依存的な相互作用を有しており、主要な相互作用のいくつかを表1に概説した。
表1 TDP-43タンパク質の他のタンパク質との鍵となる相互作用
| プロテイン | 備考 | 参考文献 |
|---|---|---|
| RNA結合蛋白質 | ||
| エフエス | TDP-43はFUSのごく一部と相互作用している。TDP-43のALS変異はFUSとの相互作用を増強する。この相互作用を阻害すると、ヒストン脱アセチル化酵素6(HDAC6)mRNAの発現が減少することが観察された
|
Kim et al2010; Ling et al2010; Kabashi et al2011 |
| hnRNPA1およびhnRNPA2/B1 | hnRNPはTDP-43のC末端領域と相互作用し、mRNAのスプライシングやTDP-43のフィードバック自動制御を制御している
|
Burattiら、2005; D’ambrogioら、2009; Romanoら、2014; Blokhuisら、2016 |
| ティア1 | TIA1はストレス顆粒(SG)の形成に関与し、SGにおいてTDP-43と直接物理的またはRNA依存的に結合していることが知られている。ALSで同定されたTIA1変異は、その相分離傾向を増大させ、SGの正常な分解を妨げ、TDP-43タンパク質を含む非ダイナミックSGの蓄積を促進させる
|
Liu-Yesucevitzら、2010; McDonaldら、2011; Mackenzieら、2017 |
| RBM45 | RBM45はALSやFTLDの患者では封入体に集積している
RBM45はTDP-43の細胞質内凝集体と共局在化する。ALSにおけるRBM45の変異はまだ報告されていない。RBM45の変異は、TDP-43を動員する細胞質凝集体を形成する傾向があり、ミトコンドリア機能に障害をもたらすとされている。
|
コリンズら、2012年;リーY.ら、2015年;益子ら、2016年 |
| アタキシン-2 | アタキシン-2におけるポリグルタミンの増加は、ALSの遺伝的危険因子である
アタキシン-2のグルタミン22個は正常であるが、27-33QはALSのリスクとなり、34Q以上が存在すると脊髄小脳失調症2型(SCA2)に関与するとされている。アタキシン-2とTDP-43はRNA依存的に物理的に相互作用する。ALSで同定されたアタキシン-2のポリグルタミンの拡張は、その安定性を高め、TDP-43の切断とリン酸化を増加させる。
|
Eldenら、2010; Rossら、2011; HartとGitler,2012; 二瓶ら、2012; Kimら、2014 |
| Matrin3 | 共免疫沈降実験により、Matrin3とTDP-43がRNA依存的に相互作用することが明らかにされた。Matrin3のS85C変異はTDP-43との相互作用を増強する
|
Johnsonら、2014b; Gallego-Iradiら、2015; Boehringerら、2017 |
| 免疫反応 | ||
| p62とp65 (NFκB) | TDP-43はNFκBと相互作用し、ALS患者のグリア細胞や神経細胞においてNFκBの共活性化因子として働き、炎症性サイトカインや神経毒性メディエーターの産生を誘導している
|
Swarup et al2011 |
| 熱ショック応答とプロテオスタシス | ||
| Hsp40とHsp70 | Hsp40/Hsp70コ・シャペロン/シャペロンシステムはTDP-43のC-末端領域と相互作用し、熱ショックによるTDP-43の凝集を抑制していることがわかった
熱ショックタンパク質DNAJB2はHsp70と会合し、TDP-43を可溶状態に保つことでそのクリアランスを制御していることが明らかになった。酵母のHsp40ホモログであるSiS1の過剰発現は、酵母モデルにおいてTDP-43の毒性を低下させる。
|
Udan-Johnsら、2014年;Chen H. J.ら、2016年;Parkら、2017年 |
| DNAJB1およびDNAJB6 | DNAJB1 (Hsp40 protein, mammalian SiS1 homolog) の過剰発現は、げっ歯類の初代皮質ニューロンにおいてTDP-43が介在する毒性を減少させることが明らかになった
DNAJB6を過剰発現させると、熱ショックによるTDP-43の核内凝集体の形成が抑制されることがわかった。DNAJB6はTDP-43のC-末端ドメインと相互作用し、TDP-43の凝集を調節し、また他のRNA結合パートナーとの相互作用にも影響を及ぼしていることがわかった。
|
Udan-Johnsら、2014年;Parkら、2017年 |
| ピーディーアイ | シャペロンPDIは変異型TDP-43と相互作用し、脊髄神経細胞で共局在化する。また、PDIはTDP-43の異常なシステイン架橋の防止にも関与している可能性がある
|
ウォーカーら、2013年 |
| パーキン | E3ユビキチンリガーゼであるParkinはTDP-43をユビキチン化し、HDAC6と多タンパク質複合体を形成してTDP-43の細胞質包有体への隔離を誘導している
|
ヘブロンら、2013; ウェンチャンら、2014 |
| ユビキリン1,ユビキリン2 | ユビキリンタンパク質の変異は、プロテアソームやオートファジーの経路の異常に関与していることが知られている。ユビキリン2はTDP-43と高い親和性で結合し、神経細胞内にポリユビキチン化封入体の蓄積を誘導する
|
キムら、2009;ハンソンら、2010;カッセルとライツ,2013;オオサカら、2016 |
| オプティニューリン | オプチニューリン変異は失明や緑内障の原因となる。最近、ALSや散発性封入体筋炎において、オプチニューリンがTDP-43と関連していることが明らかにされた
|
山下ら、2013年;李C.ら、2015年 |
| 酸化ストレス応答 | ||
| SOD1 | ALSに関連するSOD1変異株はTDP-43と相互作用してデタージェント不溶性画分になる。SOD1変異株とTDP-43はニューロフィラメントmRNAの安定性を協調的に調節している
|
Volkening et al2009; Higashi et al2010 |
| CHCHD10 | CHCHD10 は、ミトコンドリア膜間部のクリステ接合部に存在し、ミトコンドリア構造と酸化的リン酸化を制御するミトコンドリアタンパク質である。TDP-43はCHCHD10と相互作用して核局在を誘導し、CHCHD10の機能不全はTDP-43の細胞質への集積を促進することが知られている
|
ジョンソンら、2014a; ウーら、2017 |
CHCHCHD10,コイルドコイルヘリックスコイルドコイルヘリックスドメイン含有タンパク質10;DNAJB1,DnaJホモログサブファミリーBメンバー1;DNAJB6,DnaJホモログサブファミリーBメンバー6;FUS、fused in sarcoma;HDAC6,ヒストン脱アセチラーゼ6;hnRNP A1およびA2/B、異種核リボヌクレオタンパク質A1およびA2/B
Hsp、熱ショックタンパク質;NFκB、活性化B細胞の核因子κ-光鎖エンハンサー;PDI、タンパク質ジスルフィドイソメラーゼ;RBM45,RNA結合モチーフタンパク質45;SCA2,脊髄小脳失調症2型;SOD1,スーパーオキシドジスムターゼ1;TIA1,T細胞制限細胞内抗原-1。
TDP-43 ALSの病理学
TDP-43プロテインオパチーの病理学的特徴には、核から細胞質への局在のずれ、ユビキチン化および高リン酸化TDP-43の包接体への沈着、毒性のあるC末端TDP-43フラグメントの形成につながるタンパク質の切断、およびタンパク質の凝集が含まれる。散発的または家族性の突然変異は、これらの有害な影響を悪化させ、早期に疾患の発症を引き起こす可能性がある。このセクションでは、これらの疾患メカニズムを詳細にレビューする。
TDP-43変異の役割
TARDBP遺伝子における多数の変異が、ALSおよびFTLDと関連していることが確認されている(Sreedharan et al 2008年;Buratti 2015)(図2)。これらの変異がTDP-43タンパク質に及ぼす影響としては、凝集傾向の増加、細胞質の誤局在化の亢進、タンパク質の安定性の変化、プロテアーゼに対する抵抗性または他のタンパク質との結合相互作用の改変などが挙げられる。
TDP-43変異の役割については、以前にも他の場所で包括的にレビューされている(Pesiridis er al 2009; Lattante er al 2013; Buratti、 2015)。これらの突然変異の地理的有病率に関する詳細な情報を提供する専用のオンラインデータベースも利用可能である(Pinto et al 2011;Cruts et al 2012;Abel et al 2013)。
ALSに関連した突然変異のほとんどは、TDP-43のC末端のグリシンに富んだ領域をコードするTARDBP遺伝子のエクソン6に存在する。最もよく発生するミスセンス変異はA382TおよびM337Vであり、最もよく研究されている変異のいくつかはA315T、Q331K、M337V、D169G、G294A/V、およびQ343Rなどであり、これらの変異についてもいくつかのALS疾患モデルが確立されている(Buratti、 2015)。
A90VやN267Sを含むTDP-43変異は、散発性ALSとFTLDの両方の症例で観察されているが、fALSとFTLDの症例ではR361Tが報告されている。また、G294V、 G348C、 A328T、 S393Lなどの変異は、散発性ALSと家族性ALSの両方で認められている。
興味深いことに、TDP-43変異G295Sは、sALS、fALSおよびFTLDを含む様々な病態を包含する(Baumer et al 2009; Xiong et al 2010; Fujita et al 2011; Janssens et al 2011; Budini et al 2012; Chiang et al 2012; Cruts et al 2012; Lattante et al 2013; Moreno et al 2015)。
興味あることに、ミトコンドリア局在化内部モチーフM5におけるG298S変異を含む、fALS関連リン酸化を受けやすいTDP-43変異株は、ミトコンドリアへの輸入を増加させることが見出された(Wang et al 2016)。
* *
TDP-43のC末端領域における変異は、その内在性凝集性を増強する(Johnson et al 2009)。Q331K、M337V、Q343R、N345K、R361S、およびN390DのようなALS関連変異を有する組換え発現TDP-43タンパク質は、試験管内試験(試験管内試験)での凝集を増加させ、また酵母細胞において細胞毒性を促進することが見出された(Johnson et al 2009)。
ALSに関連する変異を含むTDP-43のアミロイド原性コア領域(aa 286-366)に由来するペプチドもまた、効率的にアミロイド様フィブリルを形成することが見出された(Chen et al 2010; Guo et al 2011; Sun et al 2011; Zhu et al 2014)(表2)。
興味深いことに、Zhuらは、A315E変異を有するTDP-43ペプチドの凝集体は、アミロイド-β1-40ペプチドの凝集体をクロスシーディングすることさえ可能であることを報告している(Zhu et al 2014)。また、Guoらは、TDP-43 A315Tが試験管内試験(試験管内試験)でアミロイドフィブリルを形成し、培養神経細胞に添加すると神経細胞死を引き起こすことを示している(Guo et al 2011)。
G294V、A315T、M337V、A382T、およびG376DのようなTDP-43における特定の変異もまた、TDP-43の細胞質の誤局在化を増強することが見出されている(Barmada et al 2010; Mutihac et al 2015; Mitsuzawa et al 2018)。
表2 TDP-43タンパク質またはそのペプチドのアミロイド様凝集・オリゴマー化の観察
| TDP-43タンパク質またはそのペプチド | 使用ツール | 観察結果 | 参考文献 |
|---|---|---|---|
| アミロイド様凝集体 | |||
| FLAT TDP-43 (1-414) | ThT、CR、TEM | アミロイド様フィブリル | Johnson et al2009 |
| FL TDP-43, FL TDP-43 M337V、 FL TDP-43 A382T、 193-414, 193-414 M337V、 193-414 A382T | ThT、TEM | アミロイド様線維
|
古川ほか、2011 |
| 287-322, 287-322 a315t, 287-322 g294a, 287-322 g294v, 287-322 g294p, 287-322 g295s, 292-322, 297-322, 302-322, 307-322. | ThT, TEM, CD, FTIR | アミロイド様フィブリル ペプチド287-322(wtとA315T)はβシートに富んだThT-negative線維を形成する 変異株G294A, G294V, G295SはThT染色性のフィブリルを形成する。アミロイド形成性287-322フラグメントのさらなる解析により、より小さなペプチドがアミロイド形成性を持つことが示された。ペプチド307-322は膜の完全性を破壊する。 全てのペプチドが神経毒性を示す |
Chenら、2010; Liuら、2013; Sunら、2014 |
| 286-331,286-331 A315T | ThT、TEM、AFM | アミロイド様線維
|
Guo et al2011 |
| 103-183, 103-183 C173S, 103-183 C175S | ThT、AFM | アミロイド様フィブリル RRM1ドメインは大きな球状粒子と線維状凝集体を形成する。システインからセリンへの置換はアミロイド形成性を高める
|
正代ほか、2013 |
| 208-265 | ThT, TEM, CD, SAXS | RRM2ドメインが切断されたものは、ThT陰性のフィブリル凝集体を形成する
|
Wang Y.T. et al2013 |
| 307-319, 307-319 A315t, 307-319 A315e | ThT、AFM、CD、FTIR | アミロイド様フィブリル 変異型TDP-43ペプチドは、アルツハイマー病のアミロイドβペプチドの凝集を横断することができる
|
Zhuら、2014年 |
| 246-258,311-323,およびこれらの領域からより小さなペプチドを抽出した
|
ThT, TEM, SLS | アミロイド様フィブリル ThT陽性のアミロイド凝集体
|
Saini and Chauhan,2011,2014 |
| 318-343,311-360,311-360 Q331K、311-360 G335D、311-360 M337V | ThT、CD、AFM | アミロイド様線維
|
Jiangら、2013年、2016年 |
| 341-357 | ThT、CR、TEM、CD、XRD | アミロイド様フィブリル アミロイド形成性コアと推定される341-357ペプチドは幅15-25nm、長さ数百nmのアミロイド線維を形成し、横方向に会合する傾向が顕著である
|
モムペアンら、2015年 |
| 193-414 | ThT、AFM、CD | アミロイド様線維
|
古川ら、2011; プラサドら、2016,2018 |
| 234-273, 274-313, 314-353 | ThS、TEM | アミロイド様フィブリル
274-313および314-353ペプチドは、より長く、まっすぐな、あるいはねじれた線維(直径10-15 nm)を形成し、プリオン様の挙動を示した |
下中ら、2016 |
| 102-269 | ThT、TEM、DLS | アミロイド様フィブリル
|
ガルニエら、2017年 |
| TDP-43 RRM2領域:247-252, 247-255, 247-256, 247-257, 248-253, 248-256, 248-257, 250-259, 252-257, 252-259, 253-259, 253-259. | XRD、MicroED、Cryo-EM | RRMペプチド247-DLIIKGISVHI-257はアミロイド多形の配列を形成し、それらは異なるクラスの立体的ジッパーに適合し、異なるバックボーンコンフォメーションをとる
|
Guentherら、2018b |
| TDP-43の液晶領域
300-306, 321-326, 328-333, 333-343, 370-375, 396-402 |
XRD、MicroED、TEM | これらのセグメントは、アミロイド立体ジッパー構造を形成する
|
Guentherら、2018a |
| TDP-43の液晶領域
312-317, 312-317 A315E, 312-317 A315T |
XRD、MicroED、TEM | これらのセグメントは、キンクしたβシート構造を形成し、膜なし小器官で観察されるのと同様に、ハイドロゲルやタンパク質飛沫の形成に関与している
|
Guentherら、2018a |
| オリゴマー化 | |||
| FL TDP-43 | TEM、AFM、DLS、イムノラベリング | オリゴマー
|
ファングら、2014 |
| FL TDP-43(タンデムダイマー)(アミノ酸残基1-414 x2) | イムノブロッティング | 二量体
|
椎名ほか、2010 |
| FL TDP-43, TDP-43 NTD | 化学架橋、TEM、NMR分光法 | オリゴマー
|
アフロズら、2017 |
| NLS-TDP-25, TDP-25 | FRET, FRAP, FCS, 超解像蛍光顕微法 | オリゴマー
|
北村ら、2017年 |
| FL TDP-43 | 免疫標識、TEM | オリゴマー TDP-43は、ヒトの脳内で神経毒性を持つ球状のオリゴマーを形成している。FTLD-TDPタイプCの脳ではTDP-43のオリゴマーが豊富に存在する
|
Kao et al2015 |
AFM、原子間力顕微鏡;CD、円二色性;CR、コンゴ赤;Cryo-EM、低温電子顕微鏡;FCS、蛍光相関分光法;FL、全長;FRAP、フォトブリーチング後の蛍光回復;FRET、蛍光共鳴エネルギー移動;FTIR、フーリエ変換赤外分光法
LCD、低複雑領域;MicroED、マイクロ電子回折;NLS、核局在シグナル;NMR、核磁気共鳴分光法;NTD、N末端ドメイン;SAXS、小角X線散乱;SLS、静的光散乱;TEM、伝染電子顕微鏡;ThS、チオフラビン-S;ThT、チオフラビン-T;XRD、X線回折。アミロイド様凝集体/凝集体の分析のために使用されるツールは、簡単にボックス1に記載されている。
ボックス1 アミロイド様凝集体の分析のためのツール
AFM(原子間力顕微鏡)。AFMは、分子サイズのカンチレバーを用いて走査することで表面の輪郭を提供し、アミロイド凝集体や繊維の表面トポロジーを提供する。AFM画像は、アミロイド線維/凝集体の高さの特徴を提供することができる。
CD(円形二色性分光法)
円偏光の差動吸光度を測定することにより、CDは広くタンパク質の二次構造要素を特徴付けるために使用されている。アミロイド様凝集体は、可溶性の単量体タンパク質分子に比べて高いβシート構造を示す傾向があり、215nm付近に負のピークを示す。
CR(コンゴレッド複屈折)
アミロイド凝集体と結合すると、CRの吸光度の最大値は490nmから540nmにシフトする。交差偏光下で観察したときにCRに結合した巨視的なアミロイド凝集体は、アップルグリーンの複屈折を表示す。 クライオ電子顕微鏡(Cryo-Electron Microscopy)。
極低温で凍結水和した試料をイメージングするために使用される電子顕微鏡技術で、試料は高分解能での構造決定を可能にする、染料や固定剤を必要とせずに、その本来の状態のままである。低温電子顕微鏡で生成された顕微鏡写真は、アミロイドの様々な構造クラスを区別するために使用されていた。
DLS(動的光散乱)
溶液中の粒子のブラウン運動による散乱光の強度の時間変化を解析し、分子の拡散を検出する。DLSは粒子の流体力学的半径を提供し、アミロイド凝集体の存在を評価したり、粒子の大きさを推定したりするのに使用できる。
FCS(蛍光相関分光法)
FCSは蛍光強度の変動を記録し、拡散係数や流体力学的半径などの情報を提供し、溶液中のモノマーや凝集体の大きさや濃度の指標として使用される。
FRAP(Fluorescence
Recovery After Photobleaching)。フォトブリーチング後の蛍光標識分子の集団の拡散を測定するために使用される分光学的手法。細胞内で凝集した種の移動度を測定することができる。
FRET (Fluorescence
Resonance Energy Transfer)。FRETは、ドナー蛍光体からアクセプター蛍光体へのエネルギー移動を測定し、ミスフォールドされたタンパク質のオリゴマー集合体の小さなサブ集団の存在を検出するために使用することができる。
FTIR(フーリエ変換赤外分光法)
二次構造要素の組成は、分子結合の振動周波数を測定することにより、FTIRによって決定される。FTIRスペクトルは、大きくて硬いアミロイドが1,620cm-1付近で吸収するのに対し、小さくて無秩序な繊維は~1,635cm-1で吸収するタンパク質のミスフォールディング中間体の構造的特徴を提供することができる。
マイクロED(Micro-Electron Diffraction)。極低温下で集束された低線量電子ビームを用いてサブミクロン厚の3次元結晶から回折パターンを収集し、数百ナノメートルの大きさのアミロイド結晶を可視化するために展開する低温電子顕微鏡の新しい方法
NMR分光法(核磁気共鳴分光法)
NMRは、特定の原子核の磁場を測定することで、分子の構造、ダイナミクス、化学環境を決定する分光学的手法である。アミロイドは良好な核スピン緩和を示すため、NMRはクロスβ構造の全体的な対称性の特徴付けに用いられる。
SAXS(小角X線散乱)
小角X線散乱法は、試料を通過したときのX線の小角での弾性散乱を解析することにより、平均粒子径、形状、分布、表面体積比を決定するために使用される。この技術は、アミロイド線維の構造変化を特徴付けるために広く用いられている。
SLS(静的光散乱)
SLS
(Static Light Scattering):散乱光の時間平均強度を利用して溶液中の粒子の分子量を推定し、高分子量のアミロイド様凝集体の存在を特定するのに役立つ。
超解像蛍光顕微鏡
超解像顕微鏡では、励起光または活性化光の時間的または空間的な変調は、試料の高分解能情報を抽出するための解像度の限界を克服するのに役立ち、オリゴマーおよびフィブリル構造の種の形態に関する詳細な情報を提供する。
TEM(伝染型電子顕微鏡)
TEMは、アミロイド凝集体または繊維の形態学的な可視化を提供する。まず、アミロイドサンプルは、イメージングの前に、酢酸ウラニルなどの金属化合物を用いてネガティブに染色されている。
ThS(チオフラビン-S蛍光)
アミロイド凝集体とThSの結合は、440 nmで励起したときに〜520 nmで鋭い蛍光発光ピークを表示す。また、組織切片や細胞培養に存在するアミロイド凝集体を染色するために使用される。
ThT(チオフラビン-T蛍光)
アミロイド様凝集体に平面色素チオフラビン-Tの結合は、445 nmで励起したときに〜485 nmでその蛍光発光強度を増加させる。
XRD(X線回折)
X線にアミロイド繊維をサブジェクトすることは、β線が繊維軸に垂直に実行され、βシートが繊維軸に平行に延びるクロスβパターンとして知られている特定の回折パターンの表示で結果が得られる。
TDP-43タンパク質は、ストレス顆粒のダイナミクスと複雑に関連している(Liu-Yesucevitz et al 2010;Walker et al 2013)。ストレス顆粒に蓄積されたTDP-43レベルの定量化により、ALSにリンクしたD169G変異株およびR361S変異株は、野生型TDP-43よりも大量に蓄積することが明らかにされている(McDonald et al 2011)。
さらに、G348C変異を有するTDP-43は、有意に大きなストレス顆粒を形成し、野生型TDP-43よりも早くストレス顆粒に取り込まれるが、最終的には、野生型TDP-43発現細胞では、顆粒の大きさは変わらないものの、細胞あたりのストレス顆粒の数が多く形成される(Dewey et al 2011)。
さらに、凝集を促進するA315TおよびQ343R変異は、TDP-43含有RNA顆粒の平均サイズを増加させ、その分布密度を減少させ、さらに神経細胞内での移動性を阻害することが示されている(Liu-Yesucevitz et al 2014)。
突然変異、D169G、G294A、Q343R、N390D、Q331K、およびM337Vは、神経細胞株SH-SY5Yにおいて、TDP-43陽性包接体の形成を増強することが見出された(Nonakaka et al 2009a)。
* *
もっともらしい病理学的メカニズムは、突然変異によるTDP-43タンパク質の安定性の変化である。ある研究では、突然変異G298S、Q331K、およびM337Vを有するALSリンク型TDP-43は、等原性細胞株において、野生型TDP-43よりも長い半減期と高い安定性を示した(半減期:〜24〜48時間、野生型TDP-43は12時間)(Ling et al 2010)。
(Watanabe et al 2013)およびAustin et al 2014)の研究からのさらなる証拠は、TDP-43が野生型TDP-43の半減期であることを示している。 A315T、 Q343R、 N352S、 M337V、 G298S、 G348C、 A382T、 D169G、 および K263E)が、タンパク質の半減期および凝集傾向の増加を介して関連している可能性があり、それが自身のmRNAの処理にさらに影響を与え、TDP-43の翻訳のミスレギュレーションを引き起こす可能性があることが示されている(Watanabe er al)。 、 2013; Austin er al)。
* *
特定の変異もまた、TDP-43のプロテアーゼ媒介分解に対する感受性の増加をもたらしている(Nonaka et al 2009b)。カルパイン-Iは、組換えTDP-43のA315TおよびM337V変異株タンパク質を野生型TDP-43よりも迅速に断片化することができ、一方、D169G変異株TDP-43は、試験管内試験(試験管内試験)でカスパーゼ-3によってより効率的に切断された(Yamashitato et al 2012;Change et al 2016)。興味深いことに、TDP-43における別の変異A90Vは、カスパーゼ-3による消化に対する部分的な抵抗性を付与する(Wobst et al 2017)。
TDP-43の核枯渇および細胞質蓄積
ALSやFTLD-TDPの顕著な特徴の一つは、核内での機能的なTDP-43の喪失と、脳や脊髄ニューロンでの細胞質包接体への沈着の増加である(Arai er al 2006; Neumann er al 2006)。TDP-43は主に核内に存在するが、核と細胞質の間をシャトルすることで多様な機能に関与している(Ayala er al 2008)。
実際、TDP-43は、核内でmRNAスプライシングやその他のRNA代謝に関与するいくつかのタンパク質と相互作用し、また、mRNA翻訳に関与するタンパク質などのいくつかの細胞質タンパク質とも相互作用する(Freibaum er al 2010; Ling er al 2013)。
したがって、TDP-43の細胞内濃度は、ネガティブフィードバック機構を介してその安定したレベルを維持するように厳密に自動制御される(Ayala er al 2011)。病的なTDP-43の誤局在化を助長するイベントの正確な順序は議論されているが、核内TDP-43の枯渇は包接体形成に先行しているようである(Lee er al 2011; Xu、 2012)。
しかし、注目すべきは、TDP-43に関連したmRNA代謝の障害が、TDP-43の細胞質への蓄積や凝集に比べて、ALSやFTLD-TDPの病因の中心的なものである可能性があることだ。TDP-43の細胞質蓄積および包接体への凝集は、機能喪失および毒性機能の獲得の両方をもたらすことが認められている(Vanden Broeck et al 2015; EderleおよびDormann 2017)。
数多くの研究が、神経細胞におけるTDP-43の細胞質凝集の有害な影響を支持してきた(Igaz et al 2009;Pesiridis et al 2011;Yang et al 2011;Wang Y.T. et al 2013)。
包接体への細胞質蓄積は、mRNA輸送に必要なTDP-43の量を減少させる。最近、TDP-43はまた、リボソームタンパク質である活性化プロテインCキナーゼ1(RACK1)の受容体と相互作用することにより、トランスレーショナルリプレッサーとして機能し、それにより、グローバルなタンパク質合成の阻害をもたらすことが明らかにされている(Russo er al)。
興味深いことに、RACK1はまた、ALS患者の運動ニューロンのTDP-43包接体にも隔離されていることが判明した(Russo er al 2017)。細胞質TDP-43もまた、ミトコンドリア障害に関与していることが提案されており、これについては後述する(Wang er al 2016)。
* *
TDP-43のシグナル配列、核局在化シグナル(NLS)および核輸出シグナル(NES)は、TDP-43の核細胞質シャトルを調節する(Winton et al 2008a)。核局在化シグナル(NLS)(または核輸出シグナル(NES)配列の欠失は、TDP-43の機能を損なう。
予想されるように、核局在化シグナル(NLS)欠失を有するTDP-43は、細胞質凝集体として蓄積し、ネイティブTDP-43を封鎖することさえ可能であり、それによってTDP-43プールをさらに枯渇させ、その結果、クロマチンアセンブリおよびヒストン処理を調節する転写物を変化させる(Wittonon et al 2008a;Amlli-Wolf et al 2015)。
同様に、核輸出シグナル(NES)欠失を有するTDP-43変異株は、核凝集体を形成する傾向を示した(Winton et al 2008a)。核局在化シグナル(NLS)の家族性ALS関連変異は、これまでに1つの家族性ALS関連変異、すなわちA90Vのみが同定されているが、いくつかのC末端変異は細胞質局在の増加を促進することもあるが、どのようにしてそれが起こるのかは完全に解明されていない(Barmada er al 2010; Mutihac er al 2015)。
したがって、核内インポーチンの役割、輸送パートナー、およびトランジット中のTDP-43コンフォメーションに対する変異の影響など、核細胞質輸送に影響を及ぼす因子は、さらなる調査が必要である(Archbold et al 2018)。
TDP-43のC末端断片化
カスパーゼおよびカルパインプロテアーゼによるタンパク質分解的切断を介したTDP-43のC末端断片の生成は、以前に「C末端ドメイン」のセクションで議論したように、著名な毒性生成メカニズムの一つであるように思われる(Zhang er al 2007, 2009; Dormann er al 2009; Igaz er al 2009; Johnson er al 2009; Yang er al 2011; Xu、 2012; Buratti、 2015)。
翻訳後修飾
TDP-43における2つの最も病理学的に重要な共通の翻訳後修飾(PTM)は、リン酸化とユビキチン化である(Arai et al 2006; Neumann et al 2006,2009; Hasegawa et al 2008; Inukai et al 2008)。
脳サンプル中のTDP-43陽性介在物からのTDP-43のリン酸化は、異なる部位でのTDP-43のリン酸化を検出するための高特異性抗体が入手可能であることから、よく知られているが、TDP-43のユビキチン化については、現在、盛んに研究が行われている。
最近では、アセチル化、ポリADPリボシル化、システイン酸化などの他のPTMもALS患者から同定されている。これらのPTMを詳細に解析することで、ALSにおける新たなTDP-43の毒性メカニズムが明らかになる可能性がある(Kametani er al 2016)(図4)。
図4 TDP-43タンパク質の翻訳後修飾
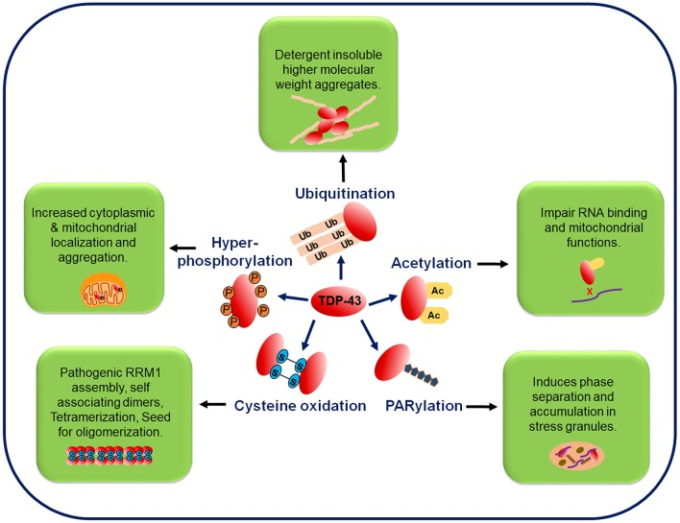
TDP-43は、リン酸化、ユビキチン化、アセチル化、PARylation、システイン酸化などの翻訳後修飾を受けている。TDP-43の全長およびC末端フラグメントのリン酸化はALSの病理学的特徴であり、細胞質の誤局在化の増加と関連している
FTLDおよびALSの脳内包物では、病的なTDP-43はユビキチン化状態で見られ、ユビキチン化部位での変異はTDP-43の凝集を減少させる。アセチル化は、不溶性で高リン酸化されたTDP-43凝集体の蓄積を促進する。PARylationはTDP-43のストレス顆粒への相分離を促進する。酸化ストレスを媒介とするシステイン酸化は、オリゴマー化と凝集を促進する。Acはアセチル化、Pはリン酸化、PARylationはポリADPリボシル化、Ubはユビキチン化
リン酸化
TDP-43は、41個のセリン残基、15個のスレオニン残基、および8個のチロシン残基を有し、これらは潜在的なリン酸化部位として作用する可能性がある。カゼインキナーゼ、CK1およびCK2は、Ser-379,Ser-403,Ser-404,特にSer-409/Ser-410でのリン酸化を媒介することが示されており、これは、現在、ALS病理学のシグネチャーと考えられている(Neumann et al 2006,2009)。
別のキナーゼであるグリコーゲン合成酵素キナーゼ(GSK3)もまた、TDP-43のリン酸化に関与していることが判明している(Sreedharan et al 2015)。TDP-43のリン酸化は、神経細胞における細胞質の誤局在化および凝集の増加と関連している(Nonaka et al 2009a、 2016; Barmada et al 2010; Liachko et al 2010; Choksi et al 2014)。
注目すべきことに、明らかにリン酸化されたTDP-43包接体が、ALS患者およびFTLD患者の脳皮質と脊髄細胞の間で報告されている。罹患した脳皮質ではリン酸化されたC末端断片の蓄積が見られるのに対し、脊髄細胞ではリン酸化された全長のTDP-43の蓄積が優勢であることが報告されている(Neumann et al 2009)。リン酸化TDP-43に対して開発された抗体は、TDP-43包接体の迅速な検出ツールとしての可能性を示している。
ユビキチン化
TDP-43はまた、ALSおよびFTLD脳内包物においてもユビキチン化状態で発見されている(Neumann et al 2006,2007b)。E3ユビキチンリガーゼ(Parkin)は、ユビキチンリジン、K-48,およびK-63を介してTDP-43をユビキチン化することが示されている。
これは、そのタンパク質分解の検出可能な証拠なしに、TDP-43の包接体への細胞質蓄積を容易にする(Seyfried et al 2010;Hebron et al 2013)。ユビキチン共役酵素UBE2E3およびユビキチン-イソペプチダーゼY(UBPY)は、酵母の2ハイブリッドスクリーンにおいて、TDP-43と相互作用することが同定され、この相互作用は、その不溶性高分子量凝集体のユビキチン化および蓄積を促進することが提案されている(Hans et al 2014)。
注目すべきことに、K263E変異を有するFTLD関連TDP-43は、過剰にユビキチン化されることが観察されたが、これは、おそらく、正に荷電したリジン残基がRRM2ドメイン内の負に荷電したアスパラギン酸残基で置換されることによる、そのミスフォールディングの結果であると考えられる(Hans er al 2014)。
驚くべきことに、Scotterらは、完全長TDP-43凝集体が、K-48-およびK-63-連結ポリユビキチン鎖の両方によって標識され、その後、異なる運命に向けられることを実証した:K-48-連結ポリユビキチン鎖に対するTDP-43のユビキチンプロテアソーム媒介分解、およびK-63-連結ポリユビキチン鎖によるTDP-43のオートファジー的除去(Scotter et al 2014)。
さらに、プロテオミクスを用いて、いくつかのユビキチン化部位もTDP-43のRRM1ドメインの近傍で同定されており、RNA結合タンパク質rasGAP SH3ドメイン結合タンパク質1(G3BP)、ポリ(A)結合タンパク質細胞質1(PABPC1)、および真核生物開始因子4A1(eIF4A1)を含む約35のタンパク質が、ユビキチン化されたTDP-43を含む洗剤不溶性画分中に見出された(Dammer er al 2012)。
2012). さらに、これらのユビキチン化部位での変異もまた、TDP-43の蓄積を減少させることが発見され、それによってユビキチン化がTDP-43の凝集を調節することが示唆された(Dammer er al 2012)。
アセチル化
TDP-43には20個のリジン残基があり、そのうちのいくつかは、K-145およびK-192のようなアセチル化しやすい残基である(Cohen et al 2015; Wang P. et al 2017)。リジンをグルタミン残基に変異させたアセチル化模倣体を用いて、TDP-43のアセチル化は、RNA結合を障害し、ミトコンドリア機能を妨害し、神経細胞培養物中の不溶性および高リン酸化TDP-43凝集体の蓄積を促進することが示された(Cohen et al 2015)。
別の研究では、ヒ素誘発性酸化ストレスは、TDP-43のアセチル化および〜75〜250kDaの凝集体の形成を誘発し得る(Cohen et al 2015;Wang P. et al 2017)。さらに、リジン145でのアセチル化に対して上昇した抗体Ac-K145は、実際に、ALS患者の脊髄におけるアセチル化TDP-43陽性病変を同定することができた(Cohen er al 2015; Wang P. er al 2017)。
他のリジンが生体内でアセチル化しやすいかどうか、もしそうであるならば、TDP-43の凝集にどのような影響があるのかを調べることが残されている。理解できるように、非特異的なマルチサイト生体内試験(生体内試験またはアスピリンのようなアセチル化剤を介して媒介される試験管内試験(試験管内試験)でのアセチル化でさえ、TDP-43の正味の電荷を劇的に変化させ、これは静電反発を介してその凝集傾向に影響を与え得る(Abdolvahabi et al 2015; Ayyadevara et al 2017; Prasad et al 2018)。
ポリADPリボシル化
ポリADP-リボシル化(またはPARylation)は、DNA損傷部位に急速に出現する翻訳後修飾であり、癌、細胞周期制御、DNA修復経路、クロマチン再編成などに意味を持つ。Bai、 2015)。ポリ(ADP-リボース)ポリメラーゼ(PARP)酵素は、標的タンパク質上のグルタミン酸やアスパラギン酸などの酸性残基のカルボキシル基にエステル結合を介してADP-リボース単位を結合させる。
重合性PAR鎖は、サブユニットがリボース-リボース結合を介して互いに連結されたときに形成される(Leung、 2014)。PAR上の負の電荷は、標的タンパク質の構造を変化させ、タンパク質-DNA/RNAおよびタンパク質-タンパク質相互作用を修飾することができる。
実際、PARのニル化は、ALSに関与する本質的に障害されたタンパク質の相分離を誘導することが見出されている(Altmeyer et al 2015)。予備的データ(Duan et al 2018)は、hnRNPA1およびTDP-43がともにPARyl化され、PARyl化されたタンパク質に結合し得ることを示唆している。
PARP酵素であるタンキラーゼは、TDP-43核局在化シグナル(NLS)配列中に存在するPAR結合モチーフを介して非共有結合的にPARを付着させることにより、TDP-43の凝集を減少させることが示された。PAR結合は、試験管内試験(試験管内試験)でTDP-43の相分離を促進することが明らかになり、また、哺乳類の細胞およびニューロンにおけるストレス顆粒へのTDP-43の蓄積に必須であることが示された(Mcgurk et al 2018)。
システインの酸化
システイン残基は、タンパク質を適切に折り畳むためのジスルフィド架橋に加えて、細胞の酸化還元状態を維持するために重要な役割を果たしている。細胞の酸化還元バランスの変化と酸化ストレスは、ALSの病態に寄与する要因として提案されている。
したがって、システイン酸化は、ALSにおける重要な経路を表す可能性がある(Valle and Carri、 2017; Buratti、 2018)。試験管内試験(試験管内試験)および細胞ベースの研究を用いて、Cohenらは、酸化ストレスがシステイン酸化によるジスルフィド結合形成を介してTDP-43の架橋を促進することを報告している。
TDP-43タンパク質に存在する6つのシステイン残基(C39,C50,C173,C175,C198,およびC244)のうち、173,175,198,および244の位置にある4つのシステイン残基は、高度に保存されており(ヒト、マウス、ショウジョウバエ、およびゼブラフィッシュにおいて酸化およびジスルフィド結合形成を受けることができる(Cohen et al 2012)。
重要なことに、システインを生成するALS-linkedミスセンス変異(G358C、S379C、およびG295C)は、潜在的に異常なTDP-43ジスルフィド架橋を増強する可能性のある追加のシステインを導入する。注目すべきことに、分子間および分子内架橋はまた、TDP-43の細胞内局在性および溶解性の変化をもたらし得る(Cohen et al 2012)。
RRM1ドメインの構造機能解析により、このドメイン内のシステイン(C-173,C-175)はTDP-43のコンフォメーションに重要であり、これらは病原性RRM1の自己組織化にも関与していることが示唆されている(Shodai er al)。
別の研究では、RRM2ドメインのシステイン(C-198およびC-244)は、酸化時に自己凝集性ジスルフィド結合二量体を形成し、凝集種に組み立てられ得る(Rabdano et al 2017)。注目すべきことに、2つのN末端システイン(C39およびC50)の酸化は、おそらくプロセスをプライミング(播種)することによって、オリゴマー化に寄与し得る。オリゴマー形成の有意な減少は、これらの位置に変異が導入されたときに、観察された(Bozzo et al 2017)。別の研究では、分子間のN末端システインジスルフィドが、最初にNTDホモ二量体を形成することにより、TDP-43の四量体化をもたらし、二量体および四量体の両方がTDP-43の凝集を阻害することが明らかにされている(Jiangian et al 2017)。システイン残基は、NTDドメイン、RRM1ドメインおよびRRM2ドメインに存在し、すべて酸化され得、試験管内試験(試験管内試験)および生体内試験(生体内試験)の両方の条件下でTDP-43の機能喪失および凝集をもたらす。提案的には、RRM1の酸化に起因する構造変化は、NTDおよびRRM2ドメインのシステイン酸化よりも、TDP-43の凝集およびALSの病理学に重要であるように思われる(Chang er al 2013)。最近、我々は、RRM2ドメインを包含する組換え精製TDP-43 C末端フラグメントが自発的にシステイン結合ホモダイマーを形成し、アミロイド様凝集体に変換できることを示した(Prasad et al 2018)。
TDP-43の凝集
TDP-43のアミロイド様会合
神経細胞に沈着したTDP-43がアミロイドの集合体のような特徴を持っているかどうかは、まだ議論の余地がある。初期の報告では、ALS患者の脳で見られるTDP-43のフィラメント様構造は、アミロイド特異的色素であるチオフラビン-T(ThT)およびコンゴレッドでは染色されないことが示唆されていた(Neumann et al 2006年;Johnson et al 2009)。
いくつかのALS症例から、チオフラビン-S(ThS)/ThT染色アミロイド凝集体が報告されている(Bigio et al 2013年;Robinson et al 2013)。したがって、TDP-43の潜在的にアミロイド原性を有する挙動を生体内試験(生体内試験)および試験管内試験(試験管内試験)の両方で解読することには、かなりの関心が存在する。
* *
組換え的に発現した全長TDP-43は、試験管内試験(試験管内試験)において、ALSやFTLD患者の変性ニューロンに見られるものと同様の、滑らかな顆粒状糸状のThT陰性凝集体を形成することが示されている(Johnson et al 2009年;Furukawa et al 2011)。
TEMでは、細い繊維が太い束に積み重なっていることが明らかになっており、これもサルコシル不溶性を示している(Furukawa er al 2011)。これらの完全長TDP-43フィブリル凝集体をプロテアーゼ処理した後、質量分析を行ったところ、フィブリルコア構造は、RRM1からC末端までの異なるC末端フラグメントから構成されていることがわかった(Furukawa er al)。 別の研究では、細菌細胞内でのTDP-43の過剰発現に続いて、形成されたTDP-43包接体もまた、ThT陰性であることが判明した(Capitini er al 2014)。
* *
しかしながら、特定の他の研究において、野生型およびALS関連変異株TDP-43のペプチドの両方が、それらのアミロイド様の性質を示唆するβシートに富んだThT陽性フィブリラー凝集体を効率的に形成することが示されている(Chen et al 2010; Guo et al 2011; Sun et al 2011; Zhu et al 2014)(表2)。
TDP-43の凝集のための異なるアミロイド原性コアは、そのC末端領域から、配列を含めて定義されている。286-331,311-360,および342-366(Chen et al 2010;Guo et al 2011;SainiおよびChauhan 2011;Mompean et al 2015;Jiang et al 2016)。
アミロイド様凝集体を形成することが示されているTDP-43からの最短ペプチドは、DLIII(247〜250)およびNFGAF(312〜316)であり、これらは、ヒト膵島アミロイドポリペプチド(IAPP)のアミロイド原性コア配列との類似性を有する(Furkurawa et al 2011;SainiおよびChauhanan 2011,2014;Prasad et al 2016)。
特筆すべきことに、A315TおよびG335DのようなALSリンク変異を含むTDP-43ペプチドは、自己播種および交差播種能力を有するアミロイド様凝集を増強することが見出されている(Guo et al 2011年;Jiangian et al 2016)。C末端領域の家族性突然変異が、短いαヘリックスのβシート構造転移への傾向を増加させることが議論されてきた(Sun and Chakrabartty、 2017)。
* *
TDP-43のRRM2ドメインおよび低複雑性ドメイン(LCD)からのアミロイド原性ペプチドの高分解能構造が得られており、これは特徴的なアミロイド立体ジッパー構造を採用し得る(Guenther et al 2018a、b)。
RRM2ペプチド(aa 247-257)は、立体ジッパー構造の異なるクラスに適合するアミロイド凝集体の異なるタイプを形成することが示された。この多形能力は、異なるバックボーンコンフォメーションを採用する能力に起因していた(Guenther et al 2018b)。
さらに、LCD領域からのペプチド、aa312〜317およびそのALSリンク変異株、A315EおよびA315Tは、このLCD領域の他のいくつかのペプチドとは異なり、相分離された飛沫およびヒドロゲルの形成を促進するキンク状のβシート構造を形成することも示された(Guenther et al 2018a)。
* *
アミロイドβ(Aβ)-42ペプチドのアミロイド凝集について以前に報告されたものと同様に、TDP-43タンパク質上の低いネット電荷は、その溶解性を低下させ、その凝集を改善するのに対し、高いネット電荷では静電反発が支配的であり、これはTDP-43の凝集を阻害し得る(Mompean et al 2016a)。
実際、我々は最近、異なるホフマイスター系列アニオンの存在下でのTDP-43のC末端フラグメント(aa 193-414)の試験管内試験(試験管内試験)でのアミロイド原性凝集を探索した。我々は、コスモトロピックアニオンがそのアミロイド様凝集率を阻害するのに対し、カオストロピックアニオンは大幅に加速することを見出した(Prasad er al)。
アミロイドフィブリルの形態学的特徴もまた、コスモトロピックアニオン対カオストロピックアニオンの存在下で変化した。さらに、リジンの電荷をマスクするであろう、試験管内試験(試験管内試験)でのアスピリン媒介の非特異的リジンアセチル化は、TDP-43のC末端フラグメントのアミロイド様凝集を有意に減少させた(Prasad et al 2018)。
TDP-43の生理学的対病理学的オリゴマー化
アルツハイマー病、パーキンソン病、プリオン病などの神経変性疾患では、神経細胞毒性は凝集性タンパク質/ペプチドのオリゴマー形態を介して発揮されると提案されている(Kayed er al 2003; Haass and Selkoe、 2007)。
最近、いくつかの研究では、TDP-43のオリゴマー化とその潜在的な神経毒性特性についても検討されている(表2)。証拠は、正常な脳では、TDP-43が二量体形態で主に神経細胞核に存在していることを示唆している(Kuo et al 2009;椎名 et al 2010;Afroz et al 2017)。
NTD領域、特にその最初の10アミノ酸は、二量体化に不可欠であるように思われる(Chang et al 2012;Zhang Y. J. et al 2013;Mompean et al 2017)。最近、架橋実験により、正常なヒトの脳において、TDP-43は、二量体としてだけでなく、二量体、三量体、四量体および多量体というオリゴマー種のスペクトルで存在し得ることが明らかにされた(Afroz et al 2017)。
このオリゴマー化は、おそらく、そのRNA標的に対する親和性および特異性の増大によって、および/または他のRNAスプライシング因子の最適なリクルートを介して、RNA結合におけるTDP-43の機能的役割にとって重要であることが提案されている。
* *
対照的に、TDP-43オリゴマーの病理学的形態も報告されており(表2これは核内TDP-43オリゴマーとは構造的に異なる可能性がある。椎名らは、細胞内で過剰発現した86kDaのTDP-43二量体において、N末端領域(aa 3-183)が分子間相互作用ドメインとして機能していることを報告している。
したがって、彼らは、二量体のTDP-43が、病理学的な高分子量のTDP-43凝集体の形成の種となる可能性を提案している(Shiina et al 2010)。実際、HEK293細胞でTDP-43リピートを86 kDaのタンパク質として発現させたタンデムTDP-43構築物を発現させると、TDP-43凝集体の蓄積が誘導された。さらに、ALSの死亡脳からの抽出物の免疫ブロットでも86kDaの種が観察された(Shiina er al)。
* *
Fangらは、完全長TDP-43がスフェロイド状およびリング状のオリゴマー構造を形成し、神経細胞への細胞毒性を有することを報告している(Fang et al 2014)。組換え的に発現した全長TDP-43をサイズ排除クロマトグラフィーで精製した後、DLSおよびTEM分析により、オリゴマー性TDP-43を含む画分が40〜400nmのサイズ分布を有することが示された。
TDP-43オリゴマーはまた、Aβ-42ペプチドとクロスシードする傾向を示し、それにより、一般的なアミロイドオリゴマー構造間の構造的相互変換性を実証している(Kayed er al)。 金免疫標識されたFTLD-TDP脳画分のTEM分析は、〜50 nmの直径を有するTDP-43オリゴマーを明らかにした(Fang et al 2014;Kao et al 2015)。
さらに、TDP-43オリゴマーに対して発現したポリクローナル抗体(TDP-O)は、試験管内試験(試験管内試験)で得られたオリゴマー凝集体だけでなく、より重要なことに、TDP-43マウスモデルの脳切片から得られたオリゴマー、およびFTLD-TDP感染患者から得られたオリゴマーも検出することができた。これは、ALSのバイオマーカーとしてのTDP-43オリゴマー検出法の開発に向けた一歩である。
* *
最近の研究では、TDP-43オリゴマーの有益な形態が骨格筋において同定されている(Vogler et al 2018)。これらのSDS耐性オリゴマーは、ストレス顆粒で観察されるものとは異なることが発見され、ミオ顆粒と呼ばれた。
さらに、ミオ顆粒はアミロイド様の特性を示した。凍結乾燥したミオグラニュールのX線回折は、4.8Åの反射を持つ回折パターンを示したが、10Åの反射を持たないことから、典型的なクロスβシート配列のミオグラニュールではないことが示唆された。
TDP-43ミオグラニュールは、サルコメアの形成に関与するタンパク質をコードするmRNAを含むため、機能的に重要であると思われる(Becker and Gitler、 2018; Vogler er al)。
TDP-43凝集体のプリオン様挙動
致死的なヒト神経変性疾患であるクロイツフェルト・ヤコブ病(CJD)およびクルは、感染性プリオンタンパク質PrPの、罹患した脳内でのアミロイド様構造の凝集体への沈着を伴う(Aguzzi et al 2008;AguzziおよびCalella 2009)。
プリオンは、Stanley Prusinerによって、新規な「タンパク質だけの」感染剤であると最初に提案された(Prusiner、 1982)。酵母およびポドスポラなどの真菌もまた、プリオン様要素を保有することが発見されている(Wickner、 1994; Derkatchら、 2001; Maddeleinら、 2002; Patelら、 2009; LiebmanおよびChernoff、 2012)。
いくつかの真菌プリオンは、「タンパク質のみ」で感染することが鮮明に示されている(King and Diaz-Avalos、 2004; 田中ら、 2004; Patel and Liebman、 2007)。感染性プリオン集合体の伝達性は、それらの並外れたプロテアーゼおよび洗浄剤耐性、およびより多くの病理学的集合体を誘導するための「播種」によって細胞から細胞へ、および器官から器官へと伝播する能力に起因する(Caughey et al 2009;CobbおよびSurewickz 2009)。
実際、いくつかの酵母プリオンはまた、異種交配を介して、またはシャペロンの利用可能性に影響を及ぼすことによって、提案されているように、ポリグルタミン、トランススチレチンおよびTDP-43等のような特定のヒトアミロイド原性タンパク質の凝集および/または毒性に影響を及ぼすことができる(Derkatch et al 2001年;Meriin et al 2002年;Park et al 2017年;Verma et al 2018)。
* *
蓄積された証拠は、Aβ-42,α-シヌクレイン、およびTDP-43等を含む、以前は非プリオンタンパク質と考えられていたいくつかの他のタンパク質が、試験管内試験(試験管内試験)および疾患モデルの両方でプリオン様の挙動を示し得ることを示唆している(Brundin er al 2010; HockおよびPolymenidou、 2016)。
TDP-43の場合、古川らは、組換え的に発現した全長TDP-43の予め形成されたサルコシル不溶性のフィブリラー凝集体をTDP-43を発現するHEK293T細胞に導入すると、内因性TDP-43の凝集が、ALS患者で観察されたものと同様の、洗剤不溶性およびユビキチン化された介在物へと誘導されることを報告している(古川 et al 2011)。
別の重要な研究では、野中らは、ALS/FTLD患者の脳からTDP-43凝集体の異なる株を同定した(Nonaka er al 2013)。これらのTDP-43凝集体をTDP-43を発現するSH-SY5Yヒト神経芽腫細胞に導入すると、種子依存性の不溶性TDP-43介在物の形成が観察され、使用した親TDP-43種子の病理学的プロファイルに類似していた。
また、TDP-43の凝集体は、細胞間で連続的に伝播することができ、それによってプリオン様の挙動をさらに支持することができた。さらに、不溶性TDP-43の播種能は、熱やプロテイナーゼ処理の影響を受けなかったが、ギ酸によって阻害されており、これらの凝集体のβシート構造が播種能に重要であることが示された(Nonaka er al)。
* *
別の研究では、HEK293細胞におけるソルビトール誘導浸透圧ストレスから得られたオリゴマーTDP-43,およびALS脳溶解物から得られたオリゴマーは、播種能力およびマイクロベシクル/エクソソソームを介した細胞間伝達を示すことが明らかになった(Feiler et al 2015)。
さらに、Smethurstらは、異なるALS患者の脳からの病理学的TDP-43凝集体を完全長TDP-43を発現する細胞に播種すると、スキーン、ドット状、粒状などの多様な範囲のTDP-43包接体を同定しており、おそらくプリオンに特徴的なストレイン様伝播を示すものと思われる。
実際、TDP-43を含む不溶性画分を繰り返し接種すると、蓄積されたTDP-43の一貫した増加が示され、それによってプリオン様のシード化された凝集体と細胞間伝播の能力が支持された(Smethurst et al 2016)。
* *
最近の研究では、石井らは、電子密度の高い顆粒におけるリン酸化およびユビキチン化されたTDP-43凝集体の形成、およびこれらのTDP-43陽性顆粒の、MG-132誘発ストレスを受け、TDP-43の野生型およびC末端領域を発現する細胞への細胞間伝播の時間経過顕微鏡観察を実証した(石井 et al 2017)。
別の研究では、40アミノ酸長ペプチド(aa 274-313および314-353にまたがる)は、試験管内試験(試験管内試験)でアミロイド様フィブリルを形成することができ、これらのアミロイド様フィブリルの導入は、野生型TDP-43を発現するSH-SY5Y細胞において、リン酸化されたC末端TDP-43フラグメントを含む包接体形成を誘導した(Shimonaka et al 2016)。
さらに、TDP-43陽性のサルコシル不溶性画分をトリプシンで消化すると、プリオン株様の挙動も観察され、274-313および314-353フィブリル処理した細胞とは異なるTDP-43バンドパターンを示し、テンプレート依存性の凝集を示唆していた(Shimonaka et al 2016)。TDP-43オリゴマーは、シード化された凝集とプリオン様伝達に関与しており、それらがどのようにして伝播するのか、詳細な解明が必要である。
TDP-43の相分離
いくつかの神経変性疾患に関与していることがますます認識されているプロセスは、液体-液体相分離(LLPS)と呼ばれるプロセスを介して、プリオン様ドメインを含むタンパク質による膜なしの飛沫様オルガネラの形成である(図5)(Shin and Brangwynne、 2017)。
TDP-43,FUS、hnRNPA1およびhnRNPA2/B1などのようないくつかのRNA結合タンパク質は、本質的に乱れた領域を含み、一過性の分子間相互作用を介して相分離を受けることができる(Burke er al 2015; Lin et al 2015; Molliex et al 2015; Patel et al 2015; Conicella et al 2016; Batlle et al 2017; Gopal et al 2017; Li et al 2017; SunおよびChakrabartty 2017; Uversky 2017)。
) プリオン様低複雑性ドメイン(LCD)を有するタンパク質は、この領域において、アルギニン、リジン、グルタミン、セリン、グルタミン酸、時折グリシン、アラニンおよびプロリンを含む極性および荷電アミノ酸の過剰発現を示し、芳香族残基、特にチロシンおよびフェニルアラニンが散在している(Shin and Brangwynne、 2017)。
LLPSの挙動は、疎水性、カチオン-πおよびπ-π相互作用、ならびにプリオン様LCDドメイン内の極性および荷電アミノ酸の電荷パターニングなどの過渡的な分子間相互作用によって駆動されるように見える(Shin and Brangwynne、 2017; Simonら、 2017)。
図5 TDP-43の液-液相分離(LLPS)と液-固相分離(LSPS)
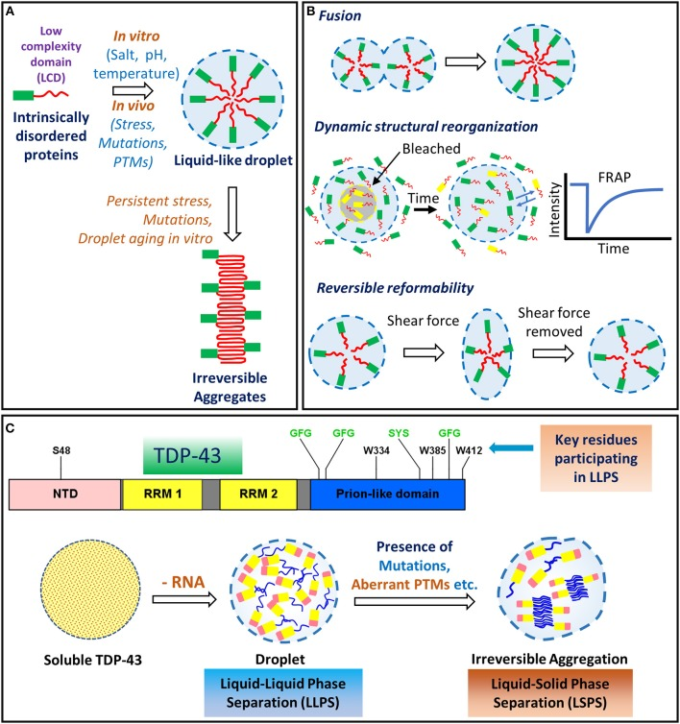
(A) 低複雑性/プリオン様ドメインを含むタンパク質は、塩の存在、pHの変化、温度変化などの影響を受けて、膜のない球状のコンパートメントへと相分離を行う。持続的なストレス、突然変異、飛沫老化は、アミロイド様凝集体のような病理学的構造への不可逆的な凝集を誘発する可能性がある
(B) 飛沫のような特性は、本質的に乱れたタンパク質によって示される:小さな飛沫がより大きな飛沫に自由に融合する能力、内部構造成分の動的な再配置を可能にする過渡的な分子間相互作用、および外部からのせん断力を除去したときに可逆的な改質性
C)TDP-43の液-液相分離(LLPS)は、親水性残基と疎水性残基の両方の影響を受ける。(G/S)-(F/Y)-(G/S)モチーフ(緑色で強調表示)は、いくつかの本質的に無秩序なタンパク質において、過渡的な相互作用を介して相分離を促進する(Li er al 2018)
トリプトファン残基は、疎水性相互作用によってLLPSを促進する(Li et al 2018)。TDP-43のRNA分子との相互作用の枯渇は、高い蛋白質に起因する。RNAの比率は、液体-固体相分離(LSPS)を介して不可逆的な凝集につながる可能性がある(Maharana et al 2018)
ALSに関連した変異もまた、不可逆的凝集体の形成を導くことが提案されている。FRAP、フォトブリーチング後の蛍光回復;LCD、低複雑性ドメイン;LLPS、液-液相分離;LSPS、液-固相分離;NTD、N末端ドメイン;PTM、翻訳後修飾;RRM、RNA認識モチーフ
ALSにリンクしたFUS変異株の相分離された飛沫は、アミロイド様フィブリル凝集体に成熟する傾向を示すことが判明した(Patel et al 2015)。したがって、LLPSは、ストレス条件下での飛沫への本質的に乱れたタンパク質の一過性の局在化は、病的な不可逆的な凝集体への液体コンパートメント内でのそれらのコンフォメーションの遷移の危険性を持っているように巨大な危険因子であるように思われる。
RNA結合タンパク質の相分離挙動は、密接にストレス顆粒を形成する彼らの傾向と関連しているようである(Molliex et al 2015; ProtterおよびParker 2016; Riback et al 2017)。
* *
ある研究では、変異株TDP-43飛沫は不規則な形態を示したが、ThT染色はアミロイド様の特徴を示さなかった(Conicella et al 2016)。Conicellaらは、プリオン様TDP-43のC末端領域(aa 276-414)が塩およびRNAの存在下で試験管内試験(試験管内試験)で相分離を受けることを報告している。
興味深いことに、特定のALS関連TDP-43変異、例えばA321G、Q331K、およびM337Vは、相分離能力を減少させ、不規則な形態で凝集する傾向を増加させることが見出されている(Conicella et al 2016)。
構造解析により、α-ヘリカルセグメント(aa:320〜340)中のトリプトファン残基W334が、TDP-43のプリオン様ドメインの相分離に決定的に重要であることが示されている(Li et al 2017,2018)。Wangらは、NTDのS48におけるリンホミメティック置換がTDP-43のLLPSを破壊し、NTDの重合を減少させることを示唆しており、したがって、それは、生体内のALSモデルにおいて低レベルでリン酸化されていることが発見された保存されたリン酸化部位である(Wang et al 2018)。
印象的なことに、ポリ(ADP-リボース)ポリメラーゼであるタンキラーゼは、その核局在化シグナル配列に負に帯電したポリ(ADP-リボース)ポリメラーゼを付加することによってTDP-43を修飾することが発見され、これは、LLPSを促進し、神経細胞におけるストレス顆粒へのTDP-43の蓄積を促進することがわかった(Mcgurk et al 2018)。
* *
最近、Gopalらは、軸索細胞内のRNP輸送顆粒を含むTDP-43が、球状の形状、融合、せん断力による変形性、TDP-43の内部再分布の迅速さ、1,6-ヘキサンジオール処理による弱い疎水性相互作用の破壊に対する感受性などの飛沫状の性質を示すことを示している。
また、M337VおよびG298SのようなALS-linked TDP-43変異は、顆粒粘度の増加および軸索輸送機能の破壊を示すことが見出された(Gopal er al 2017)。印象的には、高いタンパク質:RNAの比率に応じて、細胞内のRNA分子とTDP-43の相互作用の枯渇は、最近、液体-固体相分離(LSPS)を介してTDP-43の不可逆的な凝集を引き起こすことが発見された(Maharana et al 2018)。したがって、相分離のモジュレーターを見つけることは、途方もない治療の可能性を秘めているかもしれない。
TDP-43誘導細胞毒性の新たなメカニズム
TDP-43タンパク質ターンオーバーの調節障害
細胞内のタンパク質の恒常性は、ユビキチン-プロテアソームシステム(UPSオートファジー、およびERストレスで活性化されたアンフォールドタンパク質応答(UPR)を介して維持される。誤局在化と凝集によるTDP-43のターンオーバー異常はALSの重要なイベントとして現れ、神経細胞のプロテオスタシスの異常はALSで確認されている(Braun、 2015; Budiniら、 2017; Ramesh and Pandey、 2017)(図6)。
図6 TDP-43誘導病理学の概略図
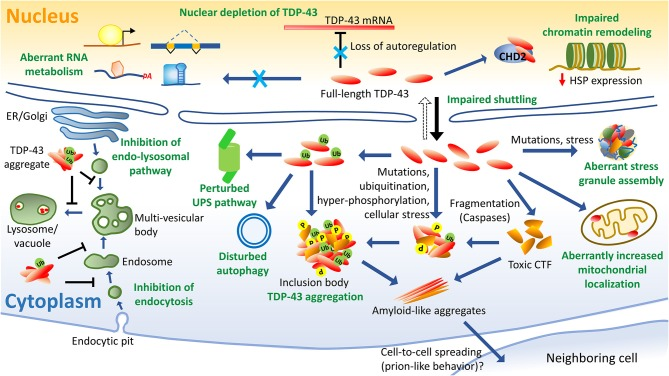
TDP-43に関連した細胞機能障害のいくつかの側面がALSで確認されている。例えば、異常なRNA代謝をもたらす核の枯渇や、TDP-43レベルの自己調節の喪失などが挙げられる。高リン酸化およびユビキチン化されたTDP-43の細胞質への蓄積は、ALSの特徴である
TDP-43の断片化は、有毒で凝集しやすいC末端断片(CTF)の形成をもたらする。TDP-43の突然変異は、異常なストレス顆粒の集合および放出につながる可能性がある。TDP-43のミトコンドリア局在が異常に増加すると、TDP-43の機能が損なわれる
TDP-43はまた、誤って制御されたオートファジーおよびプロテオソームプロセスと関連している。TDP-43の発現は、エンドサイトーシスの主要な構成要素の発現を変化させることにより、エンドサイトーシスのプロセスに影響を与える可能性がある
また、TDP-43の凝集体はエンドリソソーム経路の阻害剤として同定されている。TDP-43はクロマチンリモデリング蛋白質CHD2と相互作用してクロマチンダイナミクスを阻害し、ヒートショックプロテインの発現を抑制することが明らかになった
また、TDP-43の洗浄剤耐性を持つβシートに富んだ集合体がプリオン様に細胞間を伝播することが、神経細胞モデルで実証されている。CHD2,クロモドメインヘリカーゼDNA結合タンパク質2,CTF、C末端断片、ER、小胞体、HSP、熱ショックタンパク質、P、リン酸化、Ub、ユビキチン化、UPS、ユビキチンプロテアソームシステム
TDP-43は、主要なオートファジー関連タンパク質ATG7(オートファジー関連7)のmRNAと結合することにより、オートファジーの調節に関与していることが分かっているが、ALSに関連したTDP-43の変異の中には、ATG7のmRNA結合能力を廃止するものもある(Bose er al 2011)。
TDP-43はまた、神経細胞におけるいくつかのオートファジーリソソーム経路タンパク質の発現を調節する転写因子TFEB(転写因子EB)の局在に影響を与えることができる(Xia et al 2016)。LC3およびp62/SQSTM1のようなオートファジーマーカーに陽性の包接体が、ALSおよびFTLD患者の脊髄において同定されており、ALS疾患の進行におけるオートファジーの関与を示唆している(King er al 2010a; Budini er al 2017)。
UBQLN2におけるALS関連変異は、オートファジーの障害を引き起こし、全体的なTDP-43レベルの上昇を誘導し、神経細胞におけるTDP-43の凝集を促進する(Osaka er al 2016)。荒木らは、G298SやA382Tのような疾患関連のTDP-43変異株は、ユビキチン-プロテアソームシステムを介して野生型タンパク質よりも迅速にターンオーバーされることを発見しており、したがって、TDP-43のタンパク質分解およびクリアランスの病理学的関連性を強調している(Araki er al 2014)。
* *
TDP-43関連毒性の救済におけるオートファジーの役割は、オートファジーが疾患の進行を促進するか、または遅らせることができることを示す相反するデータによって示唆されるように、複雑なプロセスであるかもしれない(Barmada er al 2014)。
TDP-43を発現する酵母細胞を用いた系統的な遺伝子スクリーニングでは、液胞融合機構とエンドリソソーム経路がTDP-43のクリアランスと細胞生存の維持に重要であることが明らかになった。印象的なことに、TDP-43クリアランスに寄与するオートファジー経路もまた、細胞毒性を高めることが判明した(Leibiger et al 2018)。
Filimonenkoらは、オートファジー過程が欠損した細胞では、TDP-43の蓄積が増加することを報告している。輸送に必要なエンドソームソーティング複合体(ESCRT)は、オートファジー経路に関与する重要なタンパク質である。
ESCRTサブユニットが欠乏すると、異常な形態を持つ多節体(MVB)が形成される。ESCRT欠損細胞では、TDP-43がユビキチン陽性介在物に蓄積していることが確認された(Filimonenko et al 2007)。
* *
完全長TDP-43およびその断片は、ユビキチン-プロテアソームシステム(UPS)またはオートファジーを介して分解されるように指示されているユビキチン基質としても知られている。初期の研究では、可溶性および凝集したTDP-43は、ユビキチン-プロテアソームシステム(UPS)およびオートファジーの両方によってクリアされることが示唆された(Urushitani et al 2009年;Wang et al 2010年;Zhang et al 2010)。
最近、Scotterらは、可溶性TDP-43が主にユビキチン-プロテアソームシステム(UPS)によって分解されるのに対し、TDP-43の細胞傷害性凝集体はオートファジーによって優先的に除去されることを示している(Scotter et al 2014)。
Barmadaらは、ニューロンのオートファジーを有意に刺激し、TDP-43のターンオーバーを促進し、それによって一次ニューロン、ヒトiPSC由来のニューロンおよびアストロサイトの成長を改善することができる強力な化合物をファーマコフォアライブラリーから同定した(Barmada et al 2014)。
エンドサイトーシスの障害
エンドサイトーシスの異常がALSにおけるTDP-43の毒性の一因である可能性が示唆された。TDP-43の異常なレベルは、酵母細胞および細胞モデルにおいてエンドサイトーシス関連タンパク質と共局在化することでエンドサイトーシスを阻害し、そのような共局在化はALS患者の前頭皮質組織においても観察された(Liu er al 2017)。
エンドサイトーシスの障害は、TDP-43凝集の増加と関連しており、一方、エンドサイトーシスの増強は、TDP-43毒性および運動ニューロン機能障害を逆転させることが見出された(Liu et al 2017)。別の研究では、TDP-43ノックダウンは、ヒトiPSC由来のニューロンにおいて、リサイクルエンドソームの数と運動性を特異的に減少させることが見出されたが、TDP-43過剰発現は逆の効果をもたらした(Schwenk et al 2016)。
さらに、TDP-43のノックダウンは、神経細胞の成長および生存に重要な細胞表面受容体の発現を減少させることにより、樹状突起の成長に影響を及ぼすことも見られた(Schwenk et al 2016)。Leibigerらはまた、エンドサイトーシスとエンドリソソーム経路がTDP-43発現によって著しく障害されることを示している(図6)。
エンドリソソーム経路もまた、オートファジーとは無関係にTDP-43クリアランスに寄与している可能性がある。TDP-43病理におけるエンドリソソーム経路の重要性は、特定のALS関連遺伝子がこの経路の構成要素、例えば荷電多胞体タンパク質2B(CHMP2B)をコードしているという観察によって強調される(Leibiger et al 2018)。
ミトコンドリアへのTDP-43の異常に増加した局在化および関連する毒性
ミトコンドリアの機能不全は、ミトコンドリアの機能不全を介して引き起こされることがよく知られている(Saxena and Caroni、 2011)。ミトコンドリア後のニューロンは、細胞膜間のイオン勾配の維持と細胞内コミュニケーションのためのATPに対する要求が高い(Kann and Kovacs、 2007; Verkhratsky er al 2014)。
したがって、ミトコンドリア輸送、ミトコンドリア長、細胞内Ca2+レベル、ミトコンドリア呼吸およびATP産生の欠陥は、ニューロンの適切な機能を著しく阻害し、神経変性を加速し得る[(Lin and Beal、 2006; Reddy、 2009; Johri and Beal、 2012; Lezi and Swerdlow、 2012; Smith et al、 2017)でレビューされている]。
ミトコンドリアの機能不全は、野生型TDP-43またはその変異株を発現する生体内試験(生体内試験)および試験管内試験(試験管内試験)モデルの両方で記録されており、したがって、ミトコンドリアは、TDP-43毒性を媒介する経路として暗示されている(Braun et al 2011年;Wang W. et al 2013年;Stribl et al 2014)(図7)。
図7 TDP-43病理におけるミトコンドリアの役割
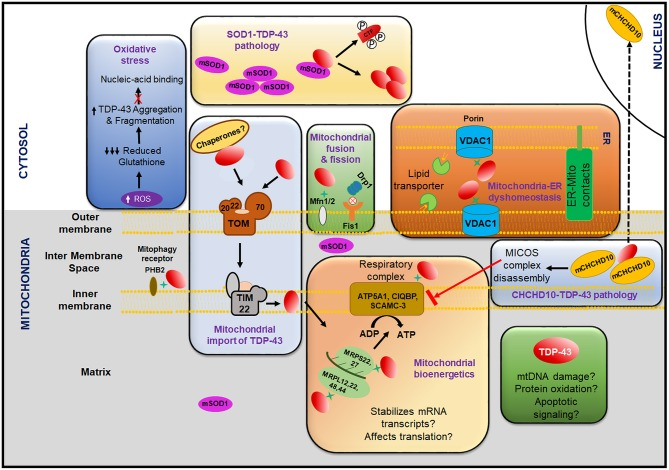
TDP-43を媒介とするミトコンドリアの機能不全は、活性酸素の産生を増加させ、その結果、減少したグルタチオンレベルを低下させ、TDP-43の凝集を増加させ、またTDP-43が核酸に結合することを阻害する。変異型SOD1は、TDP-43の細胞質の誤局在化、断片化、リン酸化および凝集を引き起こす可能性がある
ミトコンドリア分裂タンパク質Drp1とFiS1の相互作用を阻害することにより、TDP-43の過剰発現/凝集によって引き起こされるミトコンドリア機能不全が大幅に減少する。TDP-43はまた、カルシウムシグナル伝達、ATP産生、脂質輸送に潜在的な影響を与える可能性のあるERとミトコンドリアの接触を破壊する
TDP-43は、外膜複合体(TOM70)を介してミトコンドリアに、またTIM22を介して内膜を介してミトコンドリアに取り込まれる。TDP-43の輸入には、シャペロンやミトコンドリア膜電位などのいくつかの因子が関与していると考えられている
内在化後、TDP-43は、ミトコンドリアの翻訳機械に関与するいくつかのタンパク質(MRPS22, 27およびMRPL12, 22, 48, 44)やミトコンドリア呼吸器複合体(ATPA、 CIQBP、 SCAMC-3)と相互作用していることがわかっている
TDP-43はまた、呼吸器複合体IのND3/6の翻訳を阻害するため、ミトコンドリアのバイオエネルギーを著しく阻害し、ATP産生を低下させる。TDP-43の過剰発現はCHCHD10のミトコンドリアから核への局在を変化させ、CHCHCHD10の機能喪失変異はMICOS複合体の分解と関連しており、呼吸器複合体のアセンブリをネガティブに制御している可能性がある
TDP-43はまた、ミトコンドリア融合タンパク質Mfn2およびマイトファジー受容体PHB2を含む他の重要なミトコンドリアタンパク質と相互作用する。TDP-43は、ここでは赤い楕円形の構造で描かれている。TDP-43とミトコンドリアタンパク質との相互作用は、緑の星で示されている。阻害は赤の十字マークで示されている
ATPA、ATP合成酵素サブユニットA;CHCHD10,コイル状コイルヘリックス-コイルヘリックス-コイルヘリックスドメイン含有10;CIQBP、補体成分1Q結合タンパク質;CTF、C末端断片;Drp1,ダイナミン関連タンパク質1。ER-ミトコンタクト、小胞体(ER)-ミトコンドリアコンタクト;FiS1,フィッション1(ミトコンドリア);Mfn2,マイトフーシン-2;MICOS、ミトコンドリアコンタクトサイトおよびクリスタエ組織化システム。MRPL、ミトコンドリアリボソームタンパク質(大サブユニット);MRPS、ミトコンドリアリボソームタンパク質(小サブユニット);ND、NADH脱水素酵素;P、リン酸化;PHB2,禁止酵素-2;ROS、活性酸素;SCAMC-3,小カルシウム結合ミトコンドリアキャリアタンパク質3;SOD1,スーパーオキシドジスムターゼ1;TIM、内膜トランスロカーゼ;TOM、外膜トランスロカーゼ
初代運動ニューロンにおいて、TDP-43またはその変異株を過剰発現させると、ミトコンドリア長が減少し、ミトコンドリア運動が障害されることがわかっているが、これはミトコンドリア融合タンパク質であるmitofusin-2 (Mfn2)を共発現させることで反転させることができる(Wang W. er al)。
また、マウスモデルでは、変異型TDP-43の発現により、ミトコンドリアの輸送と分布に異常が生じた(Magrane et al 2014)。さらに、ハエモデルでのTDP-43の発現は、ミトコンドリアの分裂と断片化を促進し、ミトコンドリアのダイナミクスの障害を示唆することが示された(Altanbyek er al 2016)。
酵母モデルでは、TDP-43発現は、酸化ストレスの増加およびミトコンドリア周囲TDP-43凝集体の形成を引き起こすことが判明した。重要なことは、機能的なミトコンドリアの存在がTDP-43の劇症効果を悪化させることが観察され、ミトコンドリアがTDP-43の毒性を媒介する標的であることを示唆していることだ(Braun er al 2011; Braun、 2012)。
最近、TDP-43およびその疾患関連変異株は、様々なモデルにおいてミトコンドリア異常を有意に増強することが明らかになり、それによってALS患者で観察されるミトコンドリア機能障害を反映していることがわかってきた(Wang W. er al 2013; Wang er al 2016)。
* *
ミトコンドリアは活性酸素種(ROS)の産生の主要な場所として知られており、また、活性酸素による損傷の主要な標的としても知られている。タンパク質の酸化/カルボニル化、脂質の過酸化、グルタチオンのような抗酸化物質の枯渇、細胞内の遊離鉄含有量の増加、およびDNAへの損傷は、酸化ストレスの広く使用されているマーカーである(FarrugiaとBalzan 2012)(図7)。
印象的なことに、変異型 TDP-43 は酸化的損傷を誘発し、抗酸化応答モジュレーターである Nrf2 の核内蓄積を増加させることがわかっている(Duan er al)。 その後、TDP-43はNrf2の核内局在を増加させたが、実際にはNrf2の全発現は著しく低下し、Nrf2/ARE経路はNSC-34細胞株において障害され、その結果、神経突起の減少と脂質過酸化産物の増加をもたらすことが明らかになった(Tian et al 2017)。
また、ショウジョウバエにおけるTDP-43の発現は、タンパク質のカルボニル化およびグルタチオンS-トランスフェラーゼD1のレベルを増加させることが記録された(Carri et al 2015; Zhan et al 2015)。最近、簡単な赤/白色アッセイを開発することにより、酵母TDP-43凝集モデルにおいて、TDP-43凝集も酸化ストレスを誘導することを確認した(Bharathi er al 2016, 2017)。
* *
TDP-43凝集と酸化ストレスは相互に助長し合っているようである。エタクリニン酸を用いたグルタチオンの枯渇は、TDP-43の不溶性を増加させ、また、一次皮質ニューロンにおけるTDP-43の断片化を促進する(Iguchi et al 2012)(図7)。
これと一致するように、脂質過酸化の産物である4-ヒドロキシノネナールでTDP-43を修飾すると、COS-7細胞におけるTDP-43の不溶性および細胞質局在がかなり増加することが観察されている(Kabuta et al 2015)。
最近、TDP-43変異株発現細胞をGSHモノエチルエーテルで処理することにより細胞内還元グルタチオン(GSH)を増加させることは、凝集体形成、活性酸素の発生および細胞死を減少させることが示されている(Chen et al 2018)。
さらに、TDP-43発現細胞を様々な酸化剤に曝露すると、システインの酸化とジスルフィド結合の形成がTDP-43の凝集を促進することが明らかになった(Cohen et al 2012)。これと一致して、RRM1ドメインのシステイン残基の酸化はタンパク質の凝集を促進し、TDP-43の核酸結合能を阻害した(Chang er al 2013)。要約すると、TDP-43の凝集と酸化ストレスの相互作用は、ミトコンドリアへの劇症的な影響と同様に、TDP-43の毒性を誘発する。
* *
興味深いことに、スーパーオキシドジスムターゼ1(SOD1)は、SOD1がミトコンドリア局在化シグナルを欠いているにもかかわらず、外膜トランスロカーゼ(TOM)複合体を介してミトコンドリアに輸送されている。変異型SOD1は、ミトコンドリアの膜間空間(IMS)およびマトリックスに蓄積し、毒性を誘発する(Zeineddine et al 2017)。
ミスフォールドSOD1はまた、ミトコンドリア外膜(OMM)上に凝集し、ミトコンドリア依存性のアポトーシスに関与する。注目すべきは、外因性変異型SOD1凝集体の添加は、TDP-43の細胞質的な誤局在化を引き起こし、その凝集を増強することが報告されている(Zeineddine et al 2017)(図7)。
また、変異型SOD1の発現は、TDP-43のC末端断片化およびリン酸化を増加させることが見出されており、変異型SOD1とTDP-43断片との相互作用は、アポトーシスを介した毒性を媒介すると推測されている(Jeon et al 2018)。
* *
TDP-43がどのようにしてミトコンドリアの機能にダメージを与えるのか、そのメカニズムの詳細が現在明らかにされつつある。変異型TDP-43の発現は、ERタンパク質ベシクル関連膜タンパク質(VAPB)とミトコンドリアタンパク質チロシンホスファターゼ相互作用タンパク質(PTPIP51)の相互作用を阻害することにより、ERとミトコンドリアの接続を混乱させ、また、ミトコンドリアによるカルシウムの取り込みを減少させ、これはCa2+依存性ATP合成経路およびニューロン内のミトコンドリアの輸送に悪影響を及ぼす(Stoica et al 2014)。
特筆すべきことに、VAPB-PTPIP51接触の損失を介したミトコンドリア-ER接触の損失は、オートファジーを刺激する(Gomez-Suaga et al 2017)。融合の減少および同時に増加したミトコンドリアの融合は、ミトコンドリア後のニューロンに有害な影響を与え得ることが知られている。
注目すべきは、TDP-43の過剰発現もまた、ミトコンドリア分裂因子であるダイナミン関連タンパク質1(Drp1)および分裂1(FiS1)のレベルの同時増加を伴うミトコンドリア分裂を促進することだ(Xu et al 2010)。
TDP-43変異を有するALS患者由来の線維芽細胞は、ミトコンドリアへのDrp1のリクルートが有意に増加し、ミトコンドリアの断片化が亢進したことが報告されている。実際、P110と呼ばれるFiS1/Drp1の選択的ペプチド阻害剤は、このミトコンドリア機能不全を大幅に減少させ、それによってミトコンドリア毒性に高レベルのDrp1を直接的に関与させることが判明した(Joshi et al 2018)(図7)。
* *
ALSの病理学的特徴であるTDP-43の細胞質蓄積は、様々な細胞小器官、主にミトコンドリアとの非自発的な相互作用をもたらす(Scotter et al 2015)。2012年には、運動ニューロン様細胞NSC-34細胞で発現した全長および切断されたTDP-43タンパク質がミトコンドリアに局在し、その機能不全を引き起こすことが明らかになった(Hong et al 2012)(図7)。
2009年には、TDP-43の相互作用相手を特定することを目的としたプロテオミクス研究が行われ、いくつかのミトコンドリアリボソームタンパク質(小サブユニットと大サブユニットの両方)や、ミトコンドリア呼吸器複合体タンパク質(ミトコンドリアF1複合体(ATP5A1)のαサブユニット-1を含む)との相互作用が明らかになった(Freibaum et al 2010)。
最近では、プロテオミクススクリーニングにより、以下のようないくつかのTDP-43と相互作用するミトコンドリアタンパク質も明らかにされている。ミトコンドリアポリン、重要なマイトファジー受容体である電圧ゲートアニオンチャネル1(VDAC1禁止物質2(PHB2ミトコンドリア融合タンパク質、ミトフシン-2(MFN2およびミトコンドリア呼吸複合体タンパク質、ユビキノール-シトクロムクレダクターゼコアタンパク質IおよびII(UBQCRC1およびUBQCRC2)を含む、いくつかのTDP-43と相互作用するミトコンドリアタンパク質が明らかにされた(Davis er al)。
、 2018).
* *
TDP-43のミトコンドリア局在の阻害は、TDP-43媒介毒性を救済することが示されている(Wang et al 2016)。ほとんどがミトコンドリアの機能不全に起因する抗酸化酵素SOD1媒介ALS病理とは異なり、TDP-43は、そのRNA/DNA結合領域によっても毒性を引き起こすと考えられている(Bozzo er al 2017)。
しかしながら、内側のミトコンドリア膜画分におけるTDP-43の存在が観察され、呼吸器複合体IサブユニットをコードするミトコンドリアND3およびND6 mRNAに優先的に結合していることから、TDP-43毒性におけるミトコンドリア経路の役割に再び注目が集まっている(Wang er al 2016)。
実際、TDP-43 M337V変異株を発現するトランスジェニックマウスモデルにおいて、ミトコンドリア局在の阻害は、認知機能障害を緩和し、ミトコンドリア機能を回復させることができた(Wang W. et al 2017)。このことは、毒性を誘発する重要なメカニズムの一つとしてのミトコンドリアとTDP-43の相互作用を強化している。
* *
コイル状ヘリックスドメイン含有10(CHCHD10)タンパク質の変異は、ALSと関連しており、変異CHCHD10タンパク質分子は、ミトコンドリアの膜間空間に局在し、TDP-43と相互作用することも見出されている(Lehmer et al 2018)。
CHCHCHD10タンパク質は、クリスタエの形態の組織化に関与し、それによってミトコンドリアの完全性に重要な役割を果たしている(Woo er al 2017)。CHCHCHD10の機能変異の損失は、呼吸鎖複合体のアセンブリに負の影響を与えるミトコンドリア接触部位とクリスタエ組織化システム(MICOS)の分解に関連している(Genin et al 2016)(図7)。
TDP-43過剰発現は、ミトコンドリアから核へのCHCHD10局在を変化させ、CHCHCHD10の機能喪失変異は、TDP-43の細胞質蓄積を誘導する(Woo et al 2017)。興味深いことに、CHCHD10における変異によって引き起こされるミトコンドリア完全性の損失は、TDP-43のミトコンドリア局在とは独立していることが示されている(Genin et al 2018)。
* *
最近、マウスモデルでA315T TDP-43変異株を発現させたところ、ミトコンドリア局在は検出されたものの、ミトコンドリアの生体エネルギー、特に酸化的リン酸化には大きな変化は見られなかった(Kawamata et al 2017)。
逆に、ミトコンドリアのカルシウム取り込みの増加が観察されたが、その潜在的な意味合いについては、さらなる調査が必要である。TDP-43は、電子輸送鎖転写物を含むミトコンドリア転写物の中間体に結合して安定化することが示されており、また、通常の条件下でもかなりの量のTDP-43がミトコンドリアに輸送されることから、ミトコンドリアとの関連におけるTDP-43の機能と毒性の分子機構の詳細を明らかにするための研究がさらに必要である(泉川 et al 2017)。
* *
金属イオンのホメオスタシスの調節障害
金属イオンのホメオスタシスの調節異常は、多くの神経変性疾患に関与している(Gaeta and Hider、 2005; Lovejoy and Guillemin、 2014; Chen P. er al 2016)。金属イオンレベルの増加は、酸化ストレス、ミトコンドリア機能不全、タンパク質のミスフォールディング、DNA損傷、ERストレスなどのような生理的障害を与える可能性がある(Roos er al 2006)。
(Roos er al 2006; Wright and Baccarelli、 2007; Dang er al 2014)。印象的なことに、増加した鉄および鉄関連タンパク質レベルは、ALS患者の脳皮質および血中血清において見出されている(Veyrat-Durebex et al 2014年;Yu et al 2014)。
最近、TDP-43 A315T変異株を発現するTDP-43マウスモデルにおいて、野生型TDP-43を発現する対照マウスと比較して、亜鉛、マンガンおよび銅イオンのレベルの有意な増加が観察された(Dang et al 2014)。
この突然変異株によって引き起こされる金属調節障害のメカニズムおよび脊髄細胞の関与の理由は明らかではないが、これらの金属イオンのレベルの増加は、脊髄における酸化タンパク質の上昇量によって観察されるように、酸化ストレスおよびミトコンドリア機能不全に起因する可能性がある(Dang er al 2014)。
別の研究では、亜鉛イオンはまた、試験管内試験(試験管内試験)でTDP-43凝集性を増加させることが示されている(Caragounis et al 2010)。反対に、CuII(atsm)およびCuII(gtsm)のような特定の銅ベースの錯体は、トランスジェニックマウスおよび神経細胞モデルにおけるTDP-43およびSOD1関連毒性の表現型を有意に改善する可能性を示している(Parker er al 2012; Roberts er al 2014; Williams er al 2016)。
特筆すべきことに、亜鉛イオンは、神経細胞培養物において包接体形成および凝集を誘導することができ、この効果は、銅または鉄では観察されず、亜鉛特異的な効果を示した(Caragounis et al 2010)。別の研究では、通常亜鉛イオンに対して親和性を示すそのヒスチジン、システイン、およびグルタミン酸残基を介したRRM1-2ドメインを有するTDP-43フラグメントは、亜鉛イオンの存在下で、ThT染色ロープ状の凝集体(流体力学的直径を有する:300〜1000nm)に凝集し、さらに小さなオリゴマー構造(20〜30nm)にも凝集することが示された(Garnier et al 2017)。
最近、Ashらは、鉛、水銀およびスズなどの重金属が、PC12細胞株においてTDP-43の凝集および核内包物の形成を誘発し得ることを実証した(Ash et al 2018)。鉛やメチル水銀への曝露は、神経細胞におけるTDP-43の恒常性を乱し、そのスプライシング活性を調節できないことがわかった。また、鉛は用量依存的に試験管内試験(試験管内試験)でTDP-43の溶解度を低下させ、TDP-43の相分離を促進する可能性があった(Ash et al 2018)。このように、金属イオン含有量とTDP-43の機能や凝集との関係については、十分な調査が必要である。
クロマチンリモデリングへの干渉
注目すべきは、クロマチンリモデリング、ヒストン修飾、DNAメチル化などのエピジェネティックなプロセスが、神経細胞の機能および発生のいくつかの側面に関与していることだ(Bastle and Maze、 2019)。実際、クロマチン調節の変化は、アルツハイマー病、ハンチントン病、ALSなどの神経変性疾患の病態にも関与している可能性がある(Berson et al 2018; Bastle and Maze 2019)。
重要な研究では、TDP-43はヌクレオソームダイナミクスを損なうことが判明した(Berson et al 2017)。ここでは、ショウジョウバエにおけるヌクレオソームリモデリング因子であるクロモドメインヘリカーゼDNA結合タンパク質1(CHD1)のノックダウンは、ストレス顆粒の数およびサイズの増加、ならびに目に見えるストレス顆粒を示す細胞の割合の増加と関連していることが示された。
TDP-43は熱ショック応答蛋白質の発現低下と関連しており、生存率を低下させていたが、CHD1のアップレギュレーションにより生存率を回復させることができた。また、ヒストンクリアランス異常によるTDP-43によるクロマチン動態の変化は、CHD1を過剰発現させることで緩和されることがわかった。
実際、共免疫沈降法により、TDP-43はCHD1と物理的に相互作用し、CHD1が凝集しやすい性質のためにクロマチン機械系へのリクルートを制限していることが明らかになった。印象的なことに、ALSおよびFTLD脳皮質におけるTDP-43の細胞質蓄積はまた、そのヒトオルソログCHD2のレベルの低下と関連していることが判明し、それにより、異常なクロマチンリモデリングがALSに関与している可能性があることを示唆している(Berson et al 2017)(図6)。
さらに、神経細胞の分化、樹状突起の伸長およびシナプス機能に重要な別のクロマチンリモデリングタンパク質である神経細胞Brg1-associated factor(nBAF)の機能的サブユニットのレベルは、培養された運動ニューロンおよびALSの脊髄運動ニューロンにおいて、変異株TDP-43によって低下した(Tibshirani et al 2017)。
また、TDP-43の核喪失、すなわちFUSは、nBAF複合体の形成に関与するタンパク質をコードするmRNAの核保持と同時に関連している。さらに、ALSにリンクしたTDP-43 M337V変異株は、神経細胞における重要なエピジェネティックマーカーであるH3S10Ph-K14Acのグローバルな発現レベルを減少させることが示されたのに対し、野生型TDP-43の過剰発現は、ヒストンH3K9Me3レベルの増加を引き起こした(Masala et al 2018)。まとめると、エピジェネティクスにおけるTDP-43の役割は、ALSの発症に重要な意味を持つ可能性がある。
他の神経変性疾患におけるTDP-43陽性の包摂
病理的にユビキチン化されリン酸化されたTDP-43包接体は、ALSやFTLD-TDP患者の神経変性と一般的に関連しているが(Arai er al 2006; Neumann er al 2006; Kwong er al 2008)、TDP-43陽性包接体交差免疫反応性病理は、レビー小体型認知症(Thiashi er al 2007; Nakashima-Yasuda er al 2007; Lin and Dickson、 2008; Corticobasal degeneration (Uryu er al 2008)、進行性核上性麻痺(PSP)などの他の神経疾患の患者でも観察されている。
2007年;中島安田 et al 2007年;Lin and Dickson 2008年大脳皮質基底変性症(瓜生 et al 2008年進行性核上性麻痺(PSP)(横田 et al 2010年;古賀 et al 2017年認知症パーク型認知症(横田 et al 2010年;古賀 et al 2017)などの他のいくつかの神経疾患の患者でも観察されている。
2017グアムの認知症パーキンソニズムALS複合体(G-PDC)(長谷川 et al 2007;Geser et al 2008ピック病(Freeman et al 2008;LinおよびDickson et al 2008;Dardis et al 2016海馬硬化症(Amador-Ortiz et al 2007)。
しかしながら、注目すべきことに、脳内でのTDP-43凝集体の局在は、これらの異なる病理学的状態の間で異なっている。ALS、FTLD、G-PDC患者では、TDP-43包接体は脳内に広く分布しているが、アルツハイマー病、パーキンソン病、PSP患者では大脳辺縁部に多く分布している(Baloh、 2011)。
* *
免疫組織化学的には、ハンチントン病患者の大部分にTDP-43が存在することが明らかにされており、ジストロフィー性神経突起や細胞内介在物にはハンチンチンタンパク質と共局在しているが、核内領域には存在しないことが明らかにされている(Schwab er al 2008)。
Schwabらは、リン酸化特異的TDP-43抗体を用いた免疫染色を用いて、ハンチントン病サンプル中に病理学的にリン酸化されたTDP-43が存在することを示した(Schwab et al 2008)。別の研究では、TDP-43は細胞培養モデルにおいてポリグルタミン凝集体と共局在することが判明しており、アミノ酸320と367の間に存在するTDP-43のC末端領域のグルタミン/アスパラギンリッチ(Q/N)領域がポリグルタミン凝集体と相互作用することが判明した(Fuentealba et al 2010)。
線虫および細胞培養モデルからのさらなる研究により、ポリグルタミンの毒性は、実際には、下流のエフェクタータンパク質であるプログラニューリン(PGRN)によるTDP-43発現の抑制によって低減され得ることが明らかになった(Tauffenberger et al 2013)。ハンチンチン毒性に対するTDP-43およびPGRN媒介の効果については、哺乳動物モデルおよびハンチントン病患者を用いた更なる調査が必要である(Tauffenberger et al 2013)。
* *
特筆すべきことに、TDP-43の免疫反応性は、アルツハイマー病患者の約75%の脳においても検出されている(Amador-Ortiz et al 2007;東 et al 2007;Uryu et al 2008;King et al 2010b;Josephs et al 2014)。
免疫組織化学的分析では、TDP-43介在物がタウ陽性の神経線維芽細胞タングル(NFTs)と共存していることが判明しており、これはアルツハイマー病におけるAβ-42の独立した役割を示唆している(Thiashi er al 2007)。
* *
さらに、TDP-43蛋白質障害はパーキンソン病患者やトランスジェニックマウスのパーキンソン病モデルにおいても検出されており、α-シヌクレインのドーパミン神経ニューロンに対する毒性は、TDP-43の過剰発現に伴って引き起こされることが明らかになっている(Arai er al)。
驚くべきことに、TDP-43は、早期発症パジェット病および前頭側頭型認知症を伴う包摂体ミオパチー(IBMPFD散発性IBM、筋原線維性ミオパチー、眼球咽頭筋ジストロフィー(OPMDおよび縁付き液胞を伴う遠位ミオパチー(DMRV)などの他のいくつかの疾患においても、細胞質および球質包摂体を形成することが発見されている(Weihl er al)。
2008;Kusters et al 2009;Olive et al 2009;Salajegheh et al 2009)。) また、TDP-43陽性の介在物は、散発性介在体筋炎(sIBM)患者の骨格筋や、バロシン含有タンパク質(VCP)に変異のあるIBMでも報告されている(Weihl et al 2008;Baloh 2011)。
TDP-43陽性の介在物を持つ疾患のスペクトルを考えると、TDP-43介在物が本当に疾患の引き金となっているのか、それともAβ-42やα-シヌクレインなどの他の一次凝集タンパク質の影響を受けた誘導副産物に過ぎないのかを明らかにするためには、さらなる研究が必要であると考えられる。
ALSの治療戦略
ALSに関連した一般毒性メカニズムを標的とする
ALSの治療法は、グルタミン酸介在性興奮毒性、タンパク質凝集、酸化ストレスの増加、小胞体ストレス、ミトコンドリア機能不全、神経炎症および遺伝子異常などを含む、進行性の運動ニューロン変性に関連する多数のメカニズムを含む複雑な疾患であるため、非常に困難である。
Dugger and Dickson、 2017; Tan er al)。 何十年にも及ぶ広範な研究にもかかわらず、ALSにおける神経細胞機能障害に関与する事象のシーケンスは、ほとんど明らかにされていないままである。ALSに対する治療法の選択肢は非常に限られており、これまでのところ、ALSに対して有効な治療法または診断バイオマーカーは開発されていない(Mitsumoto er al)。 ALSの治療法の開発に向けたいくつかの取り組みが進行中である。これらには、細胞機能障害を引き起こす特定のメカニズムを標的とした低分子の同定が含まれており、そのいくつかは以下で議論される。
グルタミン酸介在性興奮毒性
グルタミン酸は哺乳類の神経系における重要な神経伝達物質である。グルタミン酸受容体を過剰に刺激すると、Na+およびCa2+イオンの流入が増加し、これが興奮毒性を引き起こし、神経細胞の損傷または神経細胞死につながる(Heath and Shaw、 2002; Miller er al)。 また、神経細胞の過剰興奮性を抑制するナトリウムチャネル遮断薬メキシレチンや、グルタミン酸拮抗薬メマンチンは、現在、ALS治療のための臨床試験が行われている(De Carvalho er al)。
酸化ストレス
酸化ストレスは、ALSにおける運動ニューロンの変性に寄与し、また、ミトコンドリア機能障害やタンパク質凝集などの他の細胞病理学的メカニズムにも影響を及ぼす(Barber er al)。 バーバー et al 2006)。2017年には、新しい抗酸化剤であるエダラボン(別名:ラディカバ)が、リルゾール以来20年以上ぶりにALS治療薬としてFDAに承認された新薬となった。これは、フリーラジカルスカベンジャーであり、神経や血管内皮細胞への酸化ストレスを緩和する強力な抗酸化剤である(吉野・木村 2006年;武井 et al 2017)。
神経炎症
ALS患者の神経細胞の進行性変性には、神経炎症反応が関与していることが示されている。マスト細胞の数の増加は、セリンプロテアーゼ、ヒスタミン、セロトニンなどの混合物を放出するマスト細胞の脱顆粒による神経筋接合部の変性と関連している。
マシチニブは、主にc-Kit、Lyn、FynキナーゼなどのIII型成長因子受容体を標的とする選択的チロシンキナーゼ阻害剤であり、特にマスト細胞の生存、分化、脱顆粒化の制御に有効である。マシチニブは、退化する神経軸索周辺に蓄積するマスト細胞の脱顆粒を標的とし、炎症性サイトカインの放出を減少させることで、中枢神経系の神経炎症を予防することが明らかになっている(Trias et al 2016年;Hammamam et al 2017)。
脳の常在マクロファージであるミクログリア細胞を標的とすることもできる。マシチニブは、リルゾールのアドオンとして 2017年にALS治療薬の第III相試験に入った(Mora and Hermine、 2017)。細胞内クロラミンを産生することで活性化したマクロファージのプロ炎症反応を逆転させる別の低分子免疫調節薬NP001もまた、ALS治療薬の第II相試験に入っている(Miller et al 2014年 2015)。
筋トロポニン活性化
筋萎縮と筋力の低下、特に呼吸筋は、ALSの病理学的な重要な障害の一つである。筋の筋膜の機能は、ミオシンとアクチンの結合に依存しており、この結合はアクチン関連タンパク質であるトロポミオシン、トロポニンによって制御されている。
筋小胞体からのカルシウムイオンの放出とトロポニン複合体への結合は、骨格筋の高速収縮に重要な役割を果たしている。低分子のティラセムチヴは、速骨格筋のサルコメアトロポニンを選択的にカルシウムイオンに感作し、調節性トロポニン複合体からのカルシウムイオンの放出を遅らせることができ、それによって筋機能および筋力を改善する神経筋入力に対する筋肉の応答を増幅させることができる(Russell et al 2012年;Hansen et al 2014年;Hwee et al 2014)。
Tirasemtivは、ALS治療のための第III相試験が行われたが、ALS患者の忍容性の低さが報告されたため、結果は残念なものであった。最近、まだ別の次世代の高速骨格筋活性化剤であるCK-2127107が第III相試験に入っており、ティラセムチブの限界に対処する可能性がある(Andrews er al 2017; Nace、 2017)。
熱ショック応答活性化
ヒートショックプロテイン、またはシャペロンは、誤って折り畳まれたタンパク質を本来の機能的な構造にリフォールディングすることにより、細胞の生存を促進する。ヒートショック転写因子1(HSF1)は、ストレス条件下でのいくつかのヒートショックプロテインの発現のマスターレギュレーターである(Neef er al 2011)。
低分子のアリモクロモールは、HSF1の強力な活性化剤であり、Hsp70およびHsp90の発現も増幅する。最近の研究では、アリモクロモールは、TDP-43凝集体レベルのHSF1介在性の低下を示した(Kieran et al 2004年;Kalmar et al 2014年;Wang P. et al 2017)。アリモクロモールは、ALSに対する第II相試験においても有望な結果を示している。
オートファジー誘導
細胞内のタンパク質分解機構とオートファジー経路は、誤って折り畳まれたタンパク質や凝集したタンパク質をクリアするために重要な役割を果たしている。哺乳類のラパマイシン標的(mTOR)キナーゼは、細胞シグナル伝達、タンパク質合成、オートファジー経路の制御に関与する重要なタンパク質である。
トレハロースやラパマイシンのようないくつかの低分子は、保護的なオートファジーを誘導し、神経細胞の健康を改善することができる。mTORの低分子阻害剤であるラパマイシンは、ファゴフォアからのオートファゴソームの形成を介してオートファジーを刺激し、タンパク質分解を促進する(Ravikumar et al 2004年;Bachar-Wikstrom et al 2013)。
ラパマイシンは、TDP-43マウスモデルにおいて、オートファジーを誘導し、記憶力を改善し、運動機能障害を救済することが示されたが、これは、カスパーゼ-3レベルおよび細胞質のTDP-43包接体の量の減少を明らかにした(Wang et al 2012)。ALS治療に対するラパマイシンの有効性は、第II相臨床試験でモニターされている(Mandrioli et al 2018)。
TDP-43の凝集およびクリアランスの標的化
TDP-43凝集体の低分子阻害剤
TDP-43に関連する病態に対する低分子の介入は、その凝集挙動、ストレス顆粒のダイナミクス、核内細胞質のシャットリング、カスパーゼ抵抗性などを目的とする必要がある。アミロイド様凝集体の低分子阻害剤や、非病原性のネイティブモノマーやオリゴマーの安定化剤は、神経細胞の毒性を緩和する可能性がある。
タファミディスは、トランススチレチンアミロイド症の治療に使用される唯一のFDA承認の抗アミロイド薬であり、トランススチレチンをホモ四量体種へと停止させることによって作用する(Bulawa er al 2012)。我々は最近、試験管内試験(試験管内試験)および酵母モデルを用いて、アクリジンイミダゾール誘導体(AIM4)であるTDP-43凝集阻害分子を同定した(Prasad et al 2016;Raju et al 2016)。
別の研究では、ハイスループットスクリーニングを用いて、TDP-43の包接体への凝集を減少させ、ラットPC12細胞における毒性を救済するいくつかの化合物が同定された(Boyd et al 2014)。さらに、4-アミノキノリン誘導体は、TDP-43の核酸結合特性を変化させ、そのカスパーゼ媒介切断を増強することが示されている(Cassel et al 2012)。
また、銅錯体CuII(btsc)やCuII(atsm)で処理したALSモデルでは、ストレス顆粒のような特定の細胞内コンパートメント内でCuIIイオンをゆっくりと放出することにより、TDP-43のストレス顆粒への蓄積を抑制し、C末端フラグメントの凝集を抑制することが報告されている(Donnelly er al)。
ヒートショックプロテイン
酵母において、シャペロンHsp104の過剰発現は、特定の酵母プリオン凝集体を効率的に溶解することが鮮やかに示されている(Chernoff et al 1995; ShorterおよびLindquist 2005; LiebmanおよびChernoff 2012)。
Hsp104は、最近、いくつかの神経変性疾患の酵母モデルにおいて、潜在的なディスアグリガーゼとして使用されている(JackrelおよびShorter 2014; Jackrel et al 2014; Sweeny et al 2015; SweenyおよびShorter 2016; Torrente et al 2016)。
ランダム突然変異誘発を用いて、TDP-43,FUS、およびα-シヌクレインの凝集体を溶解する能力を示す遺伝子組換えHsp104変異株が得られた(Jackrel et al 2014)。実際、Hsp104の変異株であるA503V/S/CおよびV426Lは、野生型TDP-43およびALSリンク型TDP-43 M337V変異株の毒性を低減し、凝集を抑制し、核局在化を促進することができた。
また、野生型のHsp104ではなく、Hsp104 A503VおよびA503S変異株は、試験管内試験(試験管内試験)で短いTDP-43フィラメントおよび非晶質構造を溶解する傾向を示し、同様の観察は、FUSおよびα-シヌクレインフィブリル凝集体についても文書化された(Jackrel and Shorter、 2014; Jackrelら、 2014)。
六量体Hsp104の低温電子顕微鏡構造が利用可能になり、これにより、基質の結合および会合解除の機構的側面が明らかになり、これは、その会合解除活性のさらなる最適化に役立つかもしれない(Gates er al 2017)。酵母Hsp40シャペロンであるSiS1の過剰発現に続いて、TDP-43凝集の劇症的効果の減少が観察された(Park et al 2017)。
実際、SiS1は、SiS1とTDP-43との間に直接的な物理的関連性を示す証拠はなかったが、酵母細胞をTDP-43に関連した毒性から救い出し、成長および生存を改善し、異常な細胞形態から保護することができた。さらに、哺乳類のSiS1ホモログであるDNAJB1を齧歯類の初代神経細胞で過剰発現させると、TDP-43媒介毒性も緩和され、SiS1およびその関連ホモログがALSに対する神経保護効果を有する可能性が示唆された(Park er al 2017)。
以前、ヒートショックは、TDP-43の可逆的な核凝集を誘導することが示されている(Udan-Johns et al 2014)。興味深いことに、別のHsp40タンパク質であるDNAJB6の過剰発現は、ヒートショック誘発性TDP-43核凝集体の形成を抑制することが見出された。
DNAJB6はTDP-43の無秩序なC末端プリオンドメインと関連していることが示され、おそらくその凝集挙動だけでなく、他のRNA結合パートナーとの相互作用も調節している可能性がある(Udan-Johns et al 2014)。
核内インポート受容体
核内インポート受容体(NIR)は、核内細孔機械の一部であり、シャペロンやディスアグリゲラーゼとして作用することが示されている(Chook and Suel、 2011)。実際、カリオフェリン-β1は、その古典的な核局在化シグナル(cNLS)配列と結合することにより、TDP-43フィブリル化を減少させたり、逆にしたりする能力を示している。
同様に、カリオフェリン-β2はまた、それらのプロリン-チロシン核局在化シグナル(PY-NLS)配列と相互作用することによって、いくつかの他のRNA結合タンパク質-FUS、TATA-box結合タンパク質関連因子15(TAF15EWS RNA結合タンパク質1(EWSR1hnRNPA1,およびhnRNPA2のフィブリル化を減少させ得る(Guo et al 2018)。
また、Karyopherin-β2は、FUSおよびhnRNPA1によって形成された相分離された飛沫および異常なフィブリル含有ヒドロゲルを溶解する傾向を示すことが見出された。また、カリオフェリン-β2は、これらのRNA結合タンパク質のストレス顆粒への蓄積を抑制し、その核内局在性と細胞機能を回復させることができた。このように、有望な治療標的としての核内インポーチンティングには、コンディゼラブルな関心が存在する(Hofweber et al 2018; Yoshizawa et al 2018)。
ヒートショック因子
ヒートショック転写因子は、細胞のプロテオスタシスを維持する上で重要な役割を有する(Neef et al 2011年;San Gil et al 2017)。エレガントな研究では、ヒートショック因子1(HSF1)は、細胞培養およびマウスALSモデルにおいて、不溶性TDP-43のレベルを低下させることが示された(Chen H. J. er al)。
HSF1媒介のTDP-43クリアランスは、シャペロンHsp70およびそのコ・シャペロンDNAJB2aと密接に関連していることが判明した。Chenらは、DNAJB2aが不溶性TDP-43凝集体を認識し、リフォールディングおよび可溶化のためにHsp70に向かってそれらを指示することを示唆している(Chen H. J. er al 2016)。
印象的には、HSF1を活性化することは、Hsp40およびHsp70シャペロンの高い転写誘導を示し、これは、介在物体へのTDP-43凝集を有意に抑制した。このように、HSPおよびHSF、ならびにHSF1の低分子活性化剤の解離ポテンシャルを利用することは、TDP-43の抗凝集および治療薬のためのエキサイティングな展望であるように思われる(Wang P. et al 2017)。
結論
TDP-43タンパク質は、RNA代謝とホメオスタシスにおけるその多彩な機能により、細胞の健康に不可欠なタンパク質として浮上していた。その重要性を裏付けるように、核細胞質分布の不均衡、遺伝子変異、異常な翻訳後修飾や凝集によるTDP-43のホメオスタシスの異常は、RNAホメオスタシスの誤制御や細胞毒性の原因として、ますます受け入れられてきている。
TDP-43が引き起こす毒性のいくつかの側面、例えば分裂毒性やプロテオソーム過負荷などが明らかにされているが、神経変性につながる毒性メカニズムは、どのように、どのような順序で発生するのかについては、まだ十分に理解されていないのが現状である。
ALSやFTLD以外にも、いくつかの神経変性疾患にTDP-43陽性の包接体が存在することから、神経変性の全般的な過程においてTDP-43がより広範で重要な役割を果たしていることが示唆されている。このように、TDP-43の機能異常を標的とすることは、多くの神経変性疾患に適用可能な共通の治療法を見つける鍵を握っていると考えられる。