
Contents
- 概要
- 背景
- 本文へ
- ミトコンドリアの構造と機能
- 表1 脳疾患におけるミトコンドリア関連変化のまとめ
- ミトコンドリアの機能障害
- アルツハイマー病におけるミトコンドリア機能障害
- 外傷性脳損傷におけるミトコンドリア機能障害
- 脳卒中におけるミトコンドリア機能障害
- うつ病におけるミトコンドリアの機能障害
- パーキンソン病におけるミトコンドリアの機能障害
- 治療法としてのメチレンブルーと光バイオモデュレーション
- メチレンブルー、フォトバイオモデュレーションとAD
- メチレンブルー、フォトバイオモデュレーション、TBI
- メチレンブルー、フォトバイオモデュレーション、脳卒中
- メチレンブルー、フォトバイオモデュレーションとうつ病
- メチレンブルー、フォトバイオモデュレーション、パーキンソン病
- MBおよびPBMによるミトコンドリア保護の主なメカニズム
- 結論
- 略語

Mitochondria as a target for neuroprotection: role of methylene blue and photobiomodulation
www.ncbi.nlm.nih.gov/pmc/articles/PMC7262767/
オンラインで2020年6月1日公開
概要
ミトコンドリアの機能障害は、神経炎症や酸化ストレスの形成に中心的な役割を果たしており、これらは脳疾患の発症につながる重要な要因である。ミトコンドリアは、神経保護のための有望なターゲットであることが多くの証拠によって示されている。近年、ミトコンドリアを標的とした方法が、炎症や酸化損傷の抑制を通じた脳疾患治療の可能性として考えられている。本レビューでは、脳のミトコンドリア呼吸を改善するために広く研究されている2つのアプローチ、メチレンブルー(MB)と光バイオモジュレーション(PBM)について説明する。MBは広く研究されている薬剤であり、脳疾患の動物モデルにおいて有益な効果が期待できるほか、限られたヒトでの研究も行われている。同様に、PBMは、エネルギー産生を促進し、酸化ストレスと炎症の両方を軽減する非侵襲的な治療法であり、近年注目を集めている。MBとPBMは、ミトコンドリア機能、酸化的損傷、炎症、およびその後の行動症状に対して同様の有益な効果を持つ。しかし、MBとPBMのエネルギー増強作用、抗酸化作用、抗炎症作用のメカニズムは異なっている。本レビューでは、いくつかの異なる脳疾患におけるミトコンドリア機能障害と、MBおよびPBM治療後の病理学的改善に焦点を当てる。
キーワード
ミトコンドリア機能障害、神経保護、メチレンブルー、フォトバイオモデュレーション
背景
脳疾患の原因には、酸化ストレス、炎症、転写変化、興奮毒性など、いくつかの共通した要因がある。ミトコンドリア機能不全は、これらの要因の誘発に中心的な役割を果たし、神経障害を引き起こす[1]。そのため、ミトコンドリア機能障害は、近年、神経疾患の病態生理を研究するいくつかの研究の対象となっている[2-5]。アルツハイマー病(AD)、外傷性脳損傷(TBI)、うつ病、脳卒中、パーキンソン病(PD)などの神経疾患では、ミトコンドリアがエネルギー産生の低下や活性酸素種(ROS)の過剰産生を通じて病態生理に寄与しているとされる[6, 7]。脳疾患におけるミトコンドリア機能障害に関する研究では、ミトコンドリア機能の回復が、神経変性やその他の脳疾患の治療法として期待されている[8-12]。神経疾患の治療後に脳のミトコンドリア機能が改善することを示す研究は数多くある[13-15]。ミトコンドリア呼吸を標的とした最も広く研究されているアプローチであるPBMとMBは、脳疾患に対する有望な治療法として認識されている[16-20]。しかし、MBとPBMのエネルギー増強作用、抗酸化作用、抗炎症作用のメカニズムは異なっている。PBMとMBはそれぞれ異なるメカニズムで効果を発揮するため、これら2つの治療法を組み合わせることで、相乗的に治療効果を高めることが期待される。
本文へ
ミトコンドリアの構造と機能
ミトコンドリアは、真核細胞に存在するオルガネラで、二重膜構造が特徴である[21]。真核細胞では、ミトコンドリアが細胞活動のエネルギー源となるアデノシン三リン酸(ATP)の大部分を生成している。このATPの生成には、ミトコンドリアの電子輸送鎖(ETC)が中心的に関わっている。ミトコンドリアの内膜にある5つのマルチサブユニット複合体が、酸化的リン酸化のための呼吸鎖を形成している[22]。図1,1に示すように 図1,1に示すように、Complex I (NADH-ubiquinone oxidoreductase) とComplex II (succinate-ubiquinone oxidoreductase) は、呼吸鎖への電子の主要な入り口として働いている。NADHとFADH2は、それぞれ電子を複合体Iと複合体IIに転送する。これらの電子は、複合体IIIおよびIVと、ユビキノン(コエンザイムQ10,CoQ)およびシトクロムc(Cyt c)という2つの移動性電子輸送体によってシャトルされながら、電気化学的な勾配を下って複合体間を流れる[23]。移動の過程では、プロトンがミトコンドリア内膜を通って膜間空間に送り込まれることで、プロトンの運動力が確立される。複合体V(ATP合成酵素)はこの過程に依存し、ADPをリン酸化してATPを生成する[24, 25]。
図1 脳疾患における電子漏洩の図
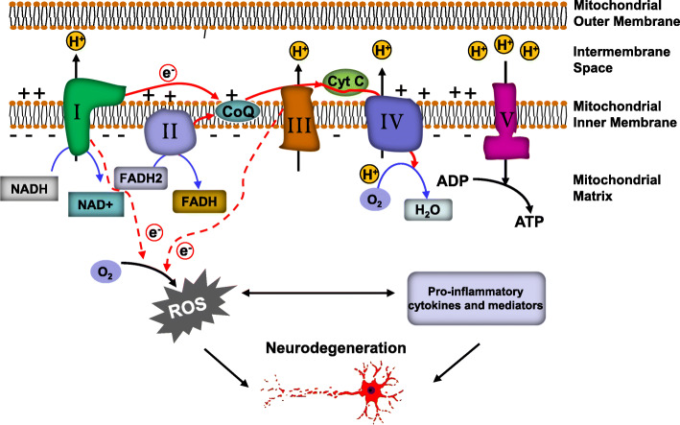
ミトコンドリアETCの電子は、電子輸送体であるNADH、FADH2,ユビキノン(Co-enzyme Q10,CoQ)シトクロムc(Cyt c)の助けを借りて、4つのタンパク質複合体(Complexes I-IV)に沿って移動する。この電子伝達の結果、プロトンは複合体I、III、IVによってミトコンドリアマトリックスから膜間空間に送り込まれ、ミトコンドリア内膜に電気化学的な勾配が発生する。この勾配を利用して、ATP合成酵素(複合体V)を動かし、ATPを生産する。このプロセスは非常に効率的であるが、電子は複合体Iと複合体IIIから逃れてO2に移動し、ラジカルO2–に還元される。この活性酸素の生成は、脳疾患などの病的な状況下では悪化し、炎症プロセスを活性化させるため、活性酸素の生成、炎症、神経細胞の損傷のサイクルが確立される
酸化ストレス、Ca2+の不均衡、電子輸送の機能不全、ミトコンドリア輸送の障害、ミトコンドリアダイナミクスの変化、マイトファジーの障害など、ミトコンドリアによって引き起こされるいくつかの変化が、さまざまな脳疾患に関与していることは、多くの証拠によって証明されている(表(表1)1)[32, 62, 79, 105-108]。これらのミトコンドリアの変化の中でも、酸化ストレスと炎症は、神経細胞の生存に影響を与える最も直接的な関連因子である[109]。
表1 脳疾患におけるミトコンドリア関連変化のまとめ
関心のある症状/ 脳疾患で観察されるミトコンドリア関連の変化
アルツハイマー病 – 活性酸素生成量の増加 [26, 27]
- ミトコンドリアの分裂と融合のバランスが崩れる [8, 28-31]
- ミトコンドリアの酵素の異常 [32-35]
- mtDNAの突然変異の増加 [36]
- ミトコンドリアインポートチャネルの機能異常 [37]
- 炎症の発生 [38, 39]
- ミトコンドリアのインポートチャネルにAPP/Aβが蓄積する [37]
- ミトコンドリア機能不全によるアポトーシス [6, 40, 41]
- Na+/Ca2+交換体の機能低下(ミトコンドリアのCa2+過剰負荷) [42-44]
- ミトコンドリアトラフィッキングの障害 [45-47]
- マイトファジーの欠損 [48, 49]
外傷性脳損傷
- ミトコンドリア膜電位の低下 [50]
- ミトコンドリアのCa2+過剰負荷 [50, 51]の発生
- オキシダーゼ複合体活性の低下 [52]
- ミトコンドリアの融合と分裂の不均衡により、ミトコンドリアの呼吸機能障害、ROS産生の増加、アポトーシス関連因子の放出 [53-57]
- ミトパージの障害 [58]
脳卒中
- 膜イオンポンプの障害、細胞内カリウム流出、ナトリウム流入、膜の脱分極 [59-61]
- ミトコンドリアのCa2+ホメオスタシスの調節障害 [62-64]
- シトクロムcの放出によるアポトーシス誘導 [65, 66]
- ミトコンドリアの過剰なスーパーオキシド生成 [67-69]
- ミトコンドリアダイナミクスの異常 [70-74]
- マイトファジーの異常 [75-77]
うつ病
- ミトコンドリアOXPHOS活性の阻害 [78]
- ミトコンドリア酵素の含有量の減少 [79, 80]
- ミトコンドリアの呼吸鎖の複合体やNa+, K + -ATPaseの活性の阻害 [81-85]
- mtDNA変異の増加 [78, 86, 87]
- ミトコンドリアETCの障害 [78, 88, 89]
パーキンソン病
- ミトコンドリア呼吸障害 [90, 91]
- 遺伝子変異によるミトコンドリア機能不全 [92-95]
- 過剰なROS産生 [90, 96, 97]
- ミトコンドリアの動態異常 [98-100]
- DAニューロンにおけるミトコンドリアのCa2+過剰負荷 [101, 102]
- 損傷したミトコンドリアの不適切なトラフィッキング [103]
- マイトファジーの機能低下 [104]
ミトコンドリアの機能障害
ミトコンドリアは、エネルギー生産に酸素を使用するため、活性酸素の主な発生源となる。前述のように、ミトコンドリアのETCは、ミトコンドリア内膜に存在する5つのタンパク質複合体から構成されている。補酵素であるNADHとFADH2は、ETCに沿って電子輸送を担う。このプロセスは比較的効率的であるが、約0.4~4%の酸素が不完全に還元され、活性酸素が生成される[10]。通常の代謝条件では、活性酸素の主な生成部位は複合体IIIである[110]。
あるレベルの活性酸素は、正常な細胞機能および健康的な生理学的プロセスに必要であり、これには、無酸素への反応、細胞シグナル伝達経路、および分裂反応の誘導が含まれる[111, 112]。報告されているように、活性酸素シグナルは、細胞間コミュニケーションのカスケードにつながり、シグナルが異なる組織を介して長距離を伝搬することを可能にする[113]。さらに、生理学的レベルの活性酸素が重要な役割を果たすことは、肉芽腫患者の免疫系で例示されている。NADPHオキシダーゼシステムの欠陥により、これらの患者は、多くの症例で見られる持続的な感染症から身を守るために十分な活性酸素を産生することができない[112]。前述したように、ミトコンドリアで生成された活性酸素は、ストレスに応答してシグナル分子として働き、それによって核内で転写変化が開始される[114]。これらの転写変化は、全身防御の向上に寄与するタンパク質の発現増加を誘導する。
ある程度の活性酸素は有益であるが、過剰な活性酸素の生成を特徴とする酸化ストレスは、いくつかの神経変性疾患の病態生理の主要な要因であると認識されている [10]。ミトホルミシスは、低レベルの活性酸素は保護的で有益であるが、高レベルの活性酸素は有害であるという現象を表している[111, 112]。
様々な疾患状態での活性酸素の生成は、防御機構の最大の能力を圧倒し、過剰な活性酸素の生成は、細胞内の脂質、タンパク質、核酸の酸化的損傷を誘発する[115, 116]。電気化学的勾配の形成やETCの機能には、ミトコンドリア内膜の完全性が不可欠であるため、膜を構成する脂質やタンパク質が酸化的に損傷を受けると、ETCの機能がさらに低下することになる[117]。さらに、ETCの機能が低下すると、活性酸素の過剰生成が促進されるため、酸化ストレスと呼吸鎖の欠陥という破壊的なサイクルがますます確立されることになる[118]。
ETCへの影響に加えて、高レベルの活性酸素は、ミトコンドリア機能の他の側面にも悪影響を及ぼす可能性がある。Ca2+は、ピルビン酸デヒドロゲナーゼ、NAD結合イソクエン酸デヒドロゲナーゼ、2-oxoglutarteデヒドロゲナーゼなどのミトコンドリア酵素の活性化に重要な役割を果たしている[119]。ミトコンドリアは、ミトコンドリアカルシウムユニポーター複合体(MCU)を介して、ミトコンドリアマトリックスにCa2+を取り込むことができる[120]。このプロセスは、ATPの加水分解とETCによって生成される電気化学的勾配によって駆動され、ミトコンドリアの代謝、細胞質のCa2+シグナル、および細胞死を制御する[121]。正常な状態では、カルシウムの取り込みは、ミトコンドリアの呼吸機能を高め、ミトコンドリアの機能をシナプスの活動に合わせて調整する [122, 123]。しかし、過剰な活性酸素の発生は、Ca2+のホメオスタシスを乱し、Ca2+オーバーロードを誘発する。Ca2+レベルの上昇は、ミトコンドリアの電位の変化を引き起こし、さらにROSの生成を誘発する[124]。この過程で、ミトコンドリアは、膜電位の崩壊、ミトコンドリア伝染性の増加、およびミトコンドリア外膜の破裂を起こす可能性がある [125]。膜の伝染性が高まると、最終的にシトクロムcが放出され、それによって細胞のアポトーシスが始まる [109, 121, 126]。
活性酸素の過剰生産は、ミトコンドリアの損傷を引き起こすことによって、マイトファジーを誘導することもできる[127]。マイトファジーとは、ミトコンドリアのストレスや損傷に反応して、オートファジーによってミトコンドリアが選択的に退化することである [128]。この過程で、内膜の持続的な脱分極により、PTEN誘導キナーゼ1(PINK1)のミトコンドリア外膜への蓄積が誘導される[129]。PINK1の蓄積は、ミトフシン2(Mfn2)のリン酸化、パーキンのミトコンドリア外膜へのリクルート、ミトコンドリア外膜タンパク質上でのリン酸化ユビキチン鎖の形成、オートファジー受容体(OPTN、NDP52など)のリクルートを引き起こす[130]。これらの受容体は、ユビキチンとLC3に結合し、オートファゴソームを形成する[128]。マイトファジーが阻害されると、ミトコンドリアの機能障害が生じることを示唆する証拠が増えている[128]。マイトファジーの障害は、AD、PD、筋萎縮性側索硬化症(ALS)、ハンチントン病など、ほとんどの神経変性疾患で実証されている [131, 132]。マイトファジーの機能が低下すると、損傷したミトコンドリアを効率的に除去することができず、その結果、活性酸素の過剰産生が促進され、過剰な活性酸素によって引き起こされる炎症の亢進が悪化する[132, 133]。
マイトファジーに加えて、ミトコンドリアの融合と分裂のバランス(すなわち、ミトコンドリアダイナミクス)も品質管理メカニズムとして認識されている。その結果、ミトコンドリアダイナミクスの低下は、ミトコンドリア機能障害の本質的な原因の一つと考えられている[134]。酸化ストレスは、ミトコンドリアの過剰な分裂を引き起こし、ミトコンドリア機能不全の原因となることが示唆されている[135, 136]。さらに、分裂を促進するタンパク質Drp1をノックダウンすると、炎症性因子の生成や活性酸素の生成が抑制されることがわかっており、細胞の損傷からの保護においてミトコンドリアのダイナミクスが重要な役割を果たしていることが示されている[137]。
さらに、ミトコンドリアの機能障害と炎症の間には、周期的な関係があることを裏付ける証拠が数多く存在する[138]。ミトコンドリアの擾乱とミトコンドリア成分の細胞質への放出は、自然免疫応答の活性化を誘発し、炎症性メディエーターの放出を増加させることがよく知られている[139-141]。Zhouらは、ROSを生成するミトコンドリアが、NLRP3フラマソーム依存性の炎症経路を介して炎症を誘発することを報告している[142]。さらに、NLRP3インフラマソーム複合体は、ミトコンドリア機能不全のセンサーとして働くことができる[142]。このような自然免疫の活性化は、ミトコンドリアの機能障害により、損傷関連分子パターン(DAMPs)の放出が促されることで起こる可能性がある[143]。
ミトコンドリアDAMPの一種であるミトコンドリアDNA(mtDNA)は、細菌のDNAと類似しているため、強い炎症反応を引き起こす可能性がある[144]。前述のように、正常な状態では、マイトファジーは欠陥のあるミトコンドリアの分解を担っている[128]。しかし、脳疾患では、活性酸素の過剰生産によりマイトファジーが障害され、損傷したミトコンドリアが野放しになることがある[145]。これらの欠陥のあるミトコンドリアは、最終的に膜の完全性が失われ、mtDNAが細胞質に逃げ込む可能性がある [146]。その結果、mtDNAは、I型インターフェロンの放出や他のインターフェロン駆動型遺伝子の発現を誘発する可能性がある [147, 148]。
逆に、炎症はミトコンドリアの機能にも影響を与える。例えば、IL-1βは、ミトコンドリアの分裂関連タンパク質Drp1を介して、ミトコンドリアの断片化を誘導し、ミトコンドリアの呼吸数を減少させることが示されている[149]。さらに、腫瘍壊死因子α(TNF-α)で処理されたミトコンドリアは、より小さく、より凝縮しており、内膜のひだ(すなわち、クリスタ)が少なく、中空であることが示されている。さらに、これらのミトコンドリアは、膜電位が低下し、細胞内のATP産生量が減少し、ROS発生量が著しく増加した[150].さらに、神経芽腫細胞ではTNF処理後にマイトファジーが増加したことから、炎症とマイトファジーの相互作用が示唆された[151]。これらの証拠を総合すると、炎症とミトコンドリア機能障害の間には周期的な関係があることが強く裏付けられる。
アルツハイマー病におけるミトコンドリア機能障害
アルツハイマー病(AD)は、加齢に依存した進行性の神経変性疾患であり、学習・記憶障害の進行、神経原線維変化(NFT)の形成、アミロイドβ(Aβ)の細胞外への沈着を特徴とする[152, 153]。これらの病理学的特徴 [153, 154]に加えて、多くの研究が、ミトコンドリアの異常がADの発症に関与していることを示している。例えば、アルツハイマー病患者では、代謝低下、脳血流低下、酸素抽出量の低下、酸素利用量の低下が観察されている[155-157]。さらに、アルツハイマー病患者の脳では、酸化ストレスと活性酸素生成の増加も観察されている[158]。観察された活性酸素生成の増加は、DNA、タンパク質、脂質の損傷を誘発する能力があり、これは、ADの神経組織における典型的な病理学的特徴である[26, 27]。
アルツハイマー病患者の脳では、局所血流と酸素利用率の低下に加えて、ミトコンドリアの分裂と融合のバランスが損なわれていることが観察されている[28, 29]。アルツハイマー病患者では、Opa1,Mfn1,Mfn2などのミトコンドリア融合タンパク質の発現が低下している一方で、分裂タンパク質であるDrp1,FiS1,Mff、Miefのレベルが上昇しており、融合タンパク質に対する分裂タンパク質の比率が上昇していることが、ミトコンドリアや神経細胞の機能障害に寄与している可能性が示唆されている[8, 28, 30, 31]。
さらに、これまでの研究では、複合体IV、ピルビン酸脱水素酵素複合体、α-ケトグルタル酸脱水素酵素複合体などのミトコンドリア酵素の異常が、ADの発症に寄与していることがわかっている[32-35]。ミトコンドリア酵素の発現異常のメカニズムの一つは、これらの酵素のサブユニットをコードするmtDNA配列に変異が蓄積することである。Coskunらは、mtDNAの変異が、健常者よりもアルツハイマー病患者に多く見られることを示している[36]。また、アルツハイマー病患者ではmtDNA L鎖ND6の転写が低下し、mtDNA/核DNA比が低下することで、酸化的リン酸化が低下することが示唆されており、ADで観察されるミトコンドリア異常の一部を説明していると考えられている[36]。
さらに、ミトコンドリアのカルシウム(mCa2+)シグナルは、ADに伴うミトコンドリア酵素による代謝機能障害の異常における重要な調節因子である。ADのマウスモデルでは、Na+/Ca2+交換体を欠損させると、記憶障害が先行し、代謝機能障害と過剰なスーパーオキシドの産生を介してアミロイドーシスとタウの過リン酸化が有意に増加することから、mCa2+シグナルの欠損がADの進行に寄与していることが示唆されている[42, 43]。Na+/Ca2+交換体は、神経末端からCa2+を除去するため、神経の正常な発火に不可欠である[45]。
そのため、軸索末端のミトコンドリアの数と機能は非常に重要である。ADではミトコンドリアの輸送障害が観察されており、この輸送障害がシナプス前神経終末におけるミトコンドリアの不在に寄与し、これらの領域におけるATP供給の減少をもたらすと考えられている[46]。局所的なATP供給の低下は、ATP依存性の小胞神経伝達物質の負荷やシナプス小胞輸送の減衰を引き起こす可能性がある[46, 47]。
さらに、先に述べたように、マイトファジーは、損傷したミトコンドリアを除去することで、正常な神経機能を維持するのに不可欠である。多くの研究が、ADにおいてマイトファジーが欠損していることを明らかにし、ADの病理を改善するための戦略として、マイトファジーを回復させるアプローチを提案している[48, 49]。
より直接的には、ミトコンドリアの機能不全と、アミロイドおよびタウの異常処理との間の相互作用を支持する研究がある。アミロイド前駆体タンパク質(APP)トランスジェニックマウスモデルでは、APPがミトコンドリア膜に捕捉され、ミトコンドリアインポートチャネルにAPP/Aβが蓄積することでミトコンドリア機能に影響を与える[159]。ミトコンドリアインポートチャネルが阻害されると、ETC酵素の機能が損なわれ、毒性のある過酸化水素種の蓄積が誘発される[37]。さらに、ミトコンドリアの機能障害は、異常なAβ産生を引き起こすことが示されている[160]。ミトコンドリアのETCを化学的に阻害すると、タンパク質分解によるAPPの処理が促進され、Aβのレベルが上昇することが示されている[160]。別の研究では、AD由来の細胞内Aβレベルの増加は、酸化ストレスと神経細胞のアポトーシスの増加を伴うことが分かっている[161]。
外傷性脳損傷におけるミトコンドリア機能障害
TBIは、転倒、スポーツ事故、自動車事故、暴力、その他の物理的外傷源によってしばしば後天的に生じる傷害である[162]。物理的な傷害に続いて、ミトコンドリアの損傷、過剰な活性酸素の放出、ATP生成の低下、神経細胞の損失、神経炎症、血液脳関門(BBB)の機能不全、浮腫の形成など、細胞の微小環境における分子変化が二次的な傷害の原因となる[163, 164].中でもミトコンドリア機能障害は、TBIによる神経細胞のアポトーシスや壊死細胞死に重要な役割を果たしている。例えば、ミトコンドリア代謝の研究では、Complex IV活性の低下がTBI後14日まで持続するという証拠が示されている[52]。さらに、TBI後、アポトーシスタンパク質(Bak/Bax)は、膜孔形成を通じてミトコンドリアの膨らみを誘発する。この過程で、小さなミトコンドリア由来の活性化因子カスパーゼが放出され、続いてアポプトソームが形成され、神経細胞のアポトーシスが起こる[163]。
TBIは、興奮性神経伝達物質の継続的な放出と、神経伝達物質の取り込み機構の障害、あるいは逆転の両方を引き起こし、その結果、シナプス間隙に神経伝達物質が過剰に存在し、その後、興奮毒性が発生する[165]。電位依存性Ca2+チャネルは、興奮毒性を媒介する主要なメカニズムの一つである[165]。これらのチャネルからのCa2+の流入を緩衝するために、ミトコンドリアは過剰な細胞質Ca2+を取り込む。しかし、これは、ミトコンドリアの膜電位(ΔΨm)を犠牲にして達成される[165]。結果として生じる膜の脱分極は、ATPの枯渇、ミトコンドリア膜の伝染、そして最終的にはアポトーシスまたはネクローシスにつながる可能性がある[165]。さらに、ミトコンドリアのCa2+過剰負荷は、膜伝染性遷移の活性化、シトクロムcの放出、ピリジンヌクレオチドの放出、および呼吸阻害を介して、ROSの過剰生産を刺激する可能性がある[51, 166]。
また、ミトコンドリアの分裂と融合のバランスも、TBIによる脳損傷の一因となる.ミトコンドリアの融合と分裂の不均衡、特に過剰なミトコンドリアの分裂は、ミトコンドリアの呼吸機能障害、ROS産生の増加、アポトーシス関連因子の放出を誘発することが示されている[53-57]。この融合と分裂の不均衡は、TBI後24時間で海馬のミトコンドリアの長さが有意に増加し、その後、損傷後72時間で長さが有意に減少することで証明されている[53]。さらに、ミトコンドリア分裂関連タンパク質Drp1のレベルがTBI後に有意に上昇したことから、ミトコンドリア分裂の増加が、観察されたミトコンドリアの長さの増加を説明すると考えられた[53].興味深いことに、タンパク質阻害剤であるMdivi-1を用いてミトコンドリアの分裂を阻害すると、その後のミトコンドリア長の減少が阻止され、海馬で新たに形成された神経細胞の損失が減少した[53]。
最後に、TBIではマイトファジーが障害されていることが報告されている。損傷を受けたミトコンドリアは神経細胞内に蓄積され、高レベルの活性酸素を生成して他のミトコンドリアに損傷を与え、最終的には神経細胞死を誘発する[53]。
脳卒中におけるミトコンドリア機能障害
虚血性脳卒中は最も一般的な脳卒中で、87%以上の症例で発症する[167]。通常、脳内の1つまたは複数の脳動脈の血栓による閉塞が原因となる[168, 169]。この閉塞により、細胞のホメオスタシスに必要な酸素、グルコース、その他の栄養素が、虚血した脳内の目的地に到達できなくなる.その結果、エネルギーホメオスタシスとATP合成が阻害される[168]。実際、酸素とグルコースの供給不足は、虚血後数分以内にミトコンドリアの機能障害を引き起こし、ATPの枯渇とROSの過剰生産を引き起こす[168]。ATPの枯渇は、膜のイオンポンプの故障、細胞内のカリウム流出、ナトリウム流入、膜の脱分極など、細胞に悪影響を及ぼす事象のカスケードを誘発することが示されている[59-61]。ある研究では、ATP依存性Ca2+チャネルの障害が、ミトコンドリアマトリックスにおけるCa2+の過負荷を誘発し、これがROS産生とミトコンドリア機能障害をさらに悪化させると報告している[62, 65]。
ミトコンドリアの損傷によって引き起こされる過剰な活性酸素の生成と膜伝染性の増加は、内在性アポトーシス経路の開始につながる [66, 170]。このアポトーシスのプロセスは、ミトコンドリアから細胞質へのシトクロムcの放出に依存している[66]。いったん放出されると、シトクロムc、プロカスパーゼ-9,アポトーシス・プロテアーゼ活性化因子1(APAF-1)dATPは、細胞質内でアポトソームと呼ばれる複合体を形成する [66]。アポプトソームが形成されると、カスパーゼ-9の活性化が誘導され、続いてカスパーゼ-3やその他の実行系カスパーゼが活性化されて、アポトーシスのプロセスが達成される[66]。
活性酸素の発生とアポトーシスの開始に加えて、ミトコンドリアの動態も虚血性脳卒中の病理に関与している[168]。ミトコンドリアの分裂と融合の不均衡は、虚血誘発性の神経細胞死に必要な初期事象としてよく知られている[70-72]。虚血性脳卒中の中大脳動脈閉塞(MCAO)モデルマウスでは、再灌流後3時間という早い段階でミトコンドリアの分裂が認められている[73]。さらに、ミトコンドリア分裂タンパク質であるDrp1とOpa1のレベルがMCAO後に上昇していることも確認されている[73]。興味深いことに、虚血に耐性のある神経細胞は、ミトコンドリアの動態を融合に近づける(すなわち、分裂を減らす)ことができることが示されており、ミトコンドリアの動態のバランスが虚血傷害に対する神経細胞の反応に重要な役割を果たしていることが示唆されている[74]。
一方、虚血性脳卒中におけるマイトファジーの役割については、いまだに議論の余地がある。蓄積されたデータは、マイトファジーが脳卒中後に保護的にも破壊的にもなりうることを示している[75, 171-173]。ほとんどの研究では、マイトファジーは障害を受けたミトコンドリアを除去し、細胞死シグナルカスケードを抑制することができるため、マイトファジーの活性化は虚血性脳卒中の有望な治療標的であると支持されている[76, 77]。しかし、マイトファジーの阻害がMCAO後の神経保護効果を与えることや、過剰なマイトファジーが脳卒中後の神経細胞死を引き起こすことを見出した研究もある[75]。
出血性脳卒中は、脳卒中の中で2番目に多いタイプである。出血性脳卒中は、弱くなった血管が破裂し、周囲の脳に出血することで起こる。脳内出血の患者や実験モデルでは、脳血流の低下が認められている[174-176]。この血流低下は、ミトコンドリア呼吸の異常やミトコンドリアの基礎的なATP産生の低下と密接に関連している[177]。また、脳内出血後にNMDA受容体が活性化され、それによって大量のCa2+が流入し、NADPHオキシダーゼやミトコンドリアETCを介してスーパーオキシドが過剰に産生されることが報告されている[63, 64].
うつ病におけるミトコンドリアの機能障害
うつ病の発症にミトコンドリアの機能障害が関与していることを示す証拠が増えている[21]。神経細胞は、主にミトコンドリアの酸化的リン酸化からエネルギーを得ており、酸化的リン酸化が低下すると、ATPの産生が細胞のエネルギー需要を満たせなくなることはよく知られている[178]。慢性的な軽度のストレスは、マウスモデルの海馬、大脳皮質、視床下部において、酸化的リン酸化を阻害し、ミトコンドリアの膜電位を低下させ、ミトコンドリアの超微細構造の損傷を誘発することが示されており、ミトコンドリアの機能障害とうつ病との間に密接な関係があることが示されている[78, 179]。また、別の研究では、うつ病患者では、前頭前野、帯状回、尾状核におけるグルコースの利用率が低下しており、ATPの産生が低下していることが示唆されている[80, 180, 181]。この知見と同様に、うつ病患者では健常者に比べてミトコンドリアのATPの産生やミトコンドリア酵素の発現が低下していることが他の報告で明らかになっている[79, 80]。これらの結果は、うつ病患者の筋肉組織でも示されている[182]。さらに、うつ病の病態は、ミトコンドリア呼吸鎖の複合体の阻害や、Na+, K + -ATPaseの活性と密接な関係があることを示すいくつかの証拠がある[81-85]。
さらに、mtDNAとうつ病の間に興味深い関連性があることを示唆する研究が数多くある。以前の研究では、うつ病患者の68%がmtDNAの変異を有しているのに対し、対照群では35%であった[80]。同様に、うつ病患者の白血球では、mtDNAのコピー数変異が正常な対照群に比べて有意に少なく、一方でmtDNAの酸化的損傷が有意に多いことがわかった[183]。興味深いことに、酸化されたmtDNAは、炎症性サイトカインを活性化し、うつ病に重要な役割を果たすことが知られている炎症を誘発することがわかった[86, 184]。さらに、神経細胞のATP産生や酸化ストレスを制御することが知られている16のミトコンドリア遺伝子(TIMM8B、SLC25A23、SFN、SLC25A30、UCP2など)が、うつ病患者と健常者の間で発現量が異なることが判明した[185]。
うつ病における酸化ストレスの重要な役割は、いくつかの研究で証明されている[186-188]。ミトコンドリアのETCは、活性酸素によって誘発される酸化ストレスの主要な発生源であるため、ETCの機能不全がうつ病の発症に関与しているという仮説が立てられている[78]。この酸化ストレスは、ETC内の複合体が電子を提供して酸素を不完全に還元し、その後、過酸化物やスーパーオキシドを含む酸素ラジカルを生成することで生じる[88].過剰なラジカルは、脂質やタンパク質を損傷したり、mtDNAを酸化させたり、DNAの切断を引き起こしたりする[88, 89]。このプロセスは、うつ病の発症に重要な役割を果たしている可能性がある。
パーキンソン病におけるミトコンドリアの機能障害
パーキンソン病は、2番目に多い神経変性疾患として認識されており、ドパミン作動性(DA)ニューロンの進行性消失とレビー小体の存在によって特徴づけられる[189].PDの病因の1つとして、ミトコンドリアの機能障害を支持する証拠が増えている[90]。特発性パーキンソン病患者の単一神経細胞を対象とした研究では、複合体IおよびIIの存在量が典型的に減少していることが明らかになった[91].複合体Iを阻害すると、ヒト、ハエ、げっ歯類においてドーパミン神経変性が誘導されることが示されており、PDにおけるミトコンドリア機能不全の重要な役割が示されている[90, 190, 191].これに関連して、パーキンソン病患者の死後の脳サンプルでは、酸化的な損傷が見つかっている[96].
さらに、ミトコンドリア機能障害を引き起こすことができる特定のミトコンドリア関連遺伝子の変異が、家族性形態のPDの原因となることが示されている[92].SNCA、LRRK2、Parkin、PINK1、ATP13A2などの多数の遺伝子の変異は、家族性PDの単発性原因として認識されている[93].これらの変異は、ミトコンドリアの機能障害と直接関連している[93]。さらに、特発性パーキンソン病患者の単一神経細胞のmtDNAでは、共通の欠失を背景に、複数の欠失が増加していた[93].同様に、散発性パーキンソン病患者の黒質では、mtDNA変異の蓄積とmtDNAコピー数の減少が認められた[94, 95].しかし、対照群で加齢とともに見られたmtDNAコピー数の増加は、パーキンソン病患者では見られなかった[93].
遺伝子の損傷や突然変異に加えて、ミトコンドリアの分裂と融合の誤作動が、PDにおけるドーパミン神経細胞の死を引き起こす可能性がある[98].以前の研究では、PD関連遺伝子(PINK1やParkinなど)が、ミトコンドリアの分裂と融合のバランスを調節する上で極めて重要な役割を果たしていることが報告されている[99]。散発性PDの神経毒モデルを用いた最近の研究では、一酸化窒素レベルの上昇によりパーキンのニトロシル化が誘導され、その結果、パーキンがDrp1を抑制する能力が低下し、それによってミトコンドリアの過断片化が引き起こされた[100]。
PDではミトコンドリアのカルシウム過剰症も見られた。ドパミン神経細胞では、小胞体から放出される過剰なCa2+がミトコンドリアのCa2+ホメオスタシスに影響を与え、ミトコンドリアの機能障害とアポトーシスカスケードを引き起こしていた[101]。さらに、PDのゼブラフィッシュモデルにおいて、ミトコンドリアのCa2+過剰負荷を抑制することで、神経保護効果が得られることがわかった[102]。
他の脳疾患と同様に、損傷したミトコンドリアの不適切な輸送とマイトファジーの低下が、ミトコンドリアの機能障害とPDの病因に寄与していることが示唆されている[103, 104]。
治療法としてのメチレンブルーと光バイオモデュレーション
多くの証拠が、いくつかの脳疾患の発症にミトコンドリア機能障害が関与していることを示唆している。そのため、ミトコンドリアや細胞呼吸を標的とした治療法の開発に注目が集まっている。そのような治療法として、メチレンブルーと光バイオモジュレーションがある。
メチレンブルー(3,7-ビス(ジメチルアミノ)-フェノチアジン-5-ニウムクロリド、MB)は、FDA(米国食品医薬品局)に認可された医薬品で、マラリア治療、メトヘモグロビン血症、シアン化物中毒などに有効な薬剤として使用されてきた[192, 193]。最近では、神経変性疾患、虚血性脳障害、TBIの治療におけるMBの潜在的な役割が研究者の注目を集めている[9, 17, 22, 194-197]。さらに、前臨床試験および臨床試験において、精神病に対するMBの有益な効果が報告されている[9, 17, 22, 195-197]。アルツハイマー病患者およびAD動物モデルにおいて、MB投与後、認知機能が有意に改善した[198, 199]。無作為化二重盲検プラセボ対照臨床試験によると、低用量のMBは、短期記憶課題中の機能的MRI活動を増加させることができ、また、記憶の検索を改善した[200]。さらに、MBはヒトの臨床試験でも検証されており、軽度から中等度のアルツハイマー病患者がMB治療後に認知機能と脳血流の両方の改善を示した[201]。神経疾患に対するMBの治療的役割の可能性は、MBの還元型と酸化型の間の変化に起因すると考えられる[193]。この過程で、MBは血液脳関門を容易に通過し、還元型からミトコンドリアの電子輸送鎖(ETC)に電子を提供することで、酸素消費量とATP生成量を増加させることができる[22, 202]。
MBの静脈内投与は、経口投与に比べて薬剤の利用可能な濃度が高くなるため、最適な投与方法である[201]。投与後、MBは様々な組織に高濃度で蓄積され、脳組織のMB濃度は注射後1時間の血清濃度の10倍にもなる[201]。脳内にかなり蓄積されることで、MBは容易にBBBを通過し、神経細胞のミトコンドリアに優先的に侵入することができるが、ミトコンドリアへの侵入のメカニズムは不明である[202]。MBはBBBを容易に通過し、ミトコンドリアに強い親和性を持ち、強力な抗酸化剤として作用することから、FDAはMBを毒物によるメトヘモグロビン血症の治療のための解毒剤として日常的に処方することを承認した[203-205]。
フォトバイオモジュレーション(PBM)は、もともと「低レベルレーザー療法(LLLT)」として知られており、約50年前に初めて報告された。これは、赤いビーム(400〜720 nm)または近赤外(700〜1000 nm)レーザーを生体組織に適用することを意味する[206]。PBM療法は、さまざまな生物学的プロセスを調節し、組織の損傷や細胞の損傷に対する保護効果を与えることができる[8, 207, 208]。例えば、LLIは低温損傷後の腓腹筋の再生を促進し、損傷後の血管新生を促進することが分かっており、筋損傷後の再生と血管新生に対するPBMの潜在的な役割を示している[209, 210]。さらに、PBMの有益な効果は、再発性アフタ性口内炎、皮膚の熱傷、糖尿病、スポーツ傷害、変形性関節症に対して実験的に実証されている[211-216]。最も重要なのは、PBMが熱や毒性の影響を受けずに細胞レベルで光生物学的効果を付与できることである[217]。興味深いことに、ミトコンドリアがPBMの標的であると考えられており、低レベルのレーザーが複合体IVに光子を供与することで、複合体IVの活性を高め、それに伴って酸素消費量が増加すると考えられている[202, 218]。
メチレンブルー、フォトバイオモデュレーションとAD
ADでは、明らかなプラークの沈着や記憶障害が起こる前に、ミトコンドリアの機能不全が起こることが多いことがわかっている[219]。これまでの研究では、ミトコンドリアの機能障害とAβの蓄積との関連が示されている[31, 220, 221]。蓄積されたAβは、ミトコンドリアの酵素であるアミロイド結合型アルコールデヒドロゲナーゼ(ABAD)に結合できることが示されている。ABADは、ミトコンドリアの保護に重要なプロセスであるエストラジオールのエストロンへの変換を担う酵素である[222-224]。このAβとABADの結合は、ABADの構造変化を誘発し、Aβ-ABAD複合体の形成をもたらし、ミトコンドリアの膜伝染性の変化や呼吸酵素活性の低下を引き起こすことが示されている[225]。しかし、興味深いことに、MBはABADの過剰発現を減少させ、Aβレベルを低下させ、したがってAβ-ABAD結合を減少させることにより、ABADの機能を維持し、ミトコンドリアの機能障害を軽減し、Aβの蓄積に対してプラスの効果を与えることが示されている[226]。
ADに対するMBのさらなる有益な効果は、キモトリプシンおよびトリプシン様プロテアソームの活性増加によるAβのクリアランスである[199]。3xTg-ADおよびAPP/PS1マウスモデルにMBを投与した過去の研究では、キモトリプシンおよびトリプシン様プロテアソームの活性化およびβ-セクレターゼ活性の減衰を通じて、Aβの沈着が減少し、学習および記憶の改善が認められた[198, 199, 227]。同様の結果がトランスジェニックPSAPPマウスにも見られ、MBはβ-セクレターゼの活性と発現を弱めることができると報告されている[198]。このように、MBは神経毒性のあるオリゴマーAβの形成を抑制し、行動学的な結果を改善することが示されている[19, 20]。
リン酸過多のタウタンパク質(p-tau)によって凝集されたNFTは、ADのもう一つの病理学的特徴である[228]。MBは、タウとタウの相互作用を弱めることで、この凝集を抑制することが報告されている[229]。さらに、MBは、微小管結合ドメインに作用することで、タウフィラメントの形成を防ぐことも可能である[230]。以前の試験管内試験の研究では、MBのN-非置換フェノチアジン環がフィラメント形成の阻害に必要であることが判明した[230]。臨床試験では、軽度および中等度のアルツハイマー病患者にMBを1日3回、60mgを24カ月間経口投与した[229]。その結果、MBを服用した患者は、対照群と比較して50週後の認知機能低下率が81%減少したことが示された[229]。著者らは、アルツハイマー病患者に対するMBの有益な効果は、タウ凝集の防止に関連していることを示唆した[229]。
さらに、MBは、NADHからComplex IVへ電子を運ぶ触媒的なレドックスサイクラーとして、ミトコンドリアのETCと直接相互作用することが示唆されている[17, 22]。MBとその還元型であるロイコMBとの間で変換が行われる間、Complex IとIIIの病的な閉塞をバイパスして、過剰なROSが減少する[17, 22]。MBはまた、Complex IVの活性を高め、ヘム合成をアップレギュレートすることで、AD脳のミトコンドリア機能を促進することができることが知られている[231-233]。
PBMのADに対する神経保護効果も、過去数十年にわたって確立されてきた。アミロイドβタンパク質前駆体(AβPP)のトランスジェニックマウスモデルでは、Aβプラークとアミロイドの量が有意に減少し、行動の改善を伴っていた[234]。別のトランスジェニックマウスモデル(K369I tauトランスジェニックモデル)では、リン酸化されたタウと神経原線維のもつれの量が有意に減少し、酸化ストレスマーカーの発現も減少した[235]。最近、AD治療におけるPBMのメカニズムに関する新たな視点が提案された。それは、PBMによって刺激された間葉系幹細胞が単球系へと成熟し、それによって試験管内試験でのAβ貪食能力が向上することが示されたというものである[236]。さらに、PBMは、脳内のAβ負担を大幅に軽減することで、空間学習や記憶能力を向上させることが示されている[236]。また、試験管内試験の研究では、PBMがERK/CREB経路を活性化し、BDNFの発現をアップレギュレートすることで、Aβ毒性に対する神経保護作用を発揮し、神経細胞における樹状突起棘の消失を抑制することが報告されている[237]。
MBと同様に、PBMのADに対する効果は、ミトコンドリア機能の改善を介してメカニズム的に起こる[40, 238]。以前の研究では、ストレプトゾトシン(STZ)誘発ADモデルに対するPBMの有益な効果が示された[8]。この効果は、いくつかのミトコンドリア関連のメカニズムによってもたらされた。まず、PBMがミトコンドリアの分裂関連タンパク質の発現を抑制し、融合関連タンパク質の発現を改善することで、ミトコンドリアの動態を直接改善することを示した[8]。次に、ミトコンドリアダイナミクスの回復は、STZ注射によって上昇したBax/Bcl-2比を減少させることで、ミトコンドリアのホメオスタシスを促進した[8]。さらに、我々の研究では、PBMがADに見られる酸化的損傷や炎症を抑制することができることが示された[8]。
メチレンブルー、フォトバイオモデュレーション、TBI
TBIではミトコンドリア機能障害がよく研究されているため、神経保護の手段としてMBがミトコンドリア機能障害を治療できるかどうかが検討されている。他の神経疾患で見られる有益な効果に加えて、MBはTBIによる浮腫を軽減し、病変体積を減衰させ、行動スコアを改善できることがわかっている[163]。これまでの研究で、MBは血液脳関門を通過し、静脈内注射後に循環血液中の10〜20倍の濃度で脳内に蓄積されることが報告されている[163]。低用量のMB(0.5-5mg/kg)を投与した動物では、副作用は報告されていない[239, 240]。しかし、高用量のMB(>10 mg/kg)では、ETCが阻害されるため、副作用が生じる可能性があることに留意しなければならない[239]。
TBIの初日に低用量のMB(1.5mg/kg)を使用した研究では、損傷した半球で浮腫が有意に減少することが示された[241, 242]。このような浮腫の減少は、TBI関連の死亡率を低下させる上で重要である。生き残ったニューロンの数に対するMBの効果も、以前の研究で判明している。TBI後24時間および72時間の時点で、MBを投与した後の生存ニューロン数は、ビヒクル注射を投与した群に比べて有意に増加した。また、MB投与群ではBelin 1の発現が増加し、LC3-IIとIL3-Iの比率が上昇したことから、オートファジーの誘導が亢進していることがわかった[241]。
最近、試験管内試験の細胞モデル(酸素グルコース遮断/再酸素化損傷、OGD)とTBI動物モデルを用いた研究では、MBの投与が神経細胞のミトコンドリア機能障害に有益な効果を与えることができるかどうかを検討した。OGDモデルでは、MBの投与により神経細胞の過剰な活性酸素産生が抑制され、ミトコンドリアの膜電位が維持され、ATP生成量が増加したことから、MBがOGDによるミトコンドリア機能障害を軽減することが示された[243]。このことは、MB治療後のTBI動物モデルにおいて、血液脳関門の保護、シトクロムc放出および神経細胞アポトーシスの減少によってさらに確認された[243]。
MBに加えて、PBMはミトコンドリア機能を高め、血流を改善し、腫れを減らし、酸化ストレスを減らし、炎症を抑制し、アポトーシスを減衰させることが報告されているため、TBIの治療戦略としての可能性が示唆されている[244]。傷害を受けたマウスの頭部に近赤外レーザー(808 nm)を用いたPBMを照射すると、神経学的なパフォーマンスが向上し、病変サイズが減少することが示されている[245]。興味深いことに、波長665nmと810nmのレーザーを用いたPBMでは神経学的に有意な改善が見られたのに対し、波長730nmと980nmのレーザーを用いたPBMでは改善が見られなかったことから、レーザーの波長がPBMの効果に関与しているようである[246]。この現象は、レーザー光のターゲットによって説明できるかもしれない。複合体IVは665nmから810nmのレーザー光を吸収することが提案されており、PBM治療のターゲットであることが示唆されている[246]。軽度の頭部TBIを受けたマウスのPBM治療後のATP産生の上昇は、PBMの細胞標的としてミトコンドリアETCの役割が提案されていることを裏付けるものである[247]。
TBIに対するPBMの有効性は、TBI患者においても示されている。学習記憶、数学的能力、および神経心理学的テストの結果は、9ヶ月間のPBM治療後に有意に改善された[248]。さらに、重度のTBIを受けた患者は、PBM治療の5日後に腕と手を自然に動かすことができた[249]。
メチレンブルー、フォトバイオモデュレーション、脳卒中
MBやPBMが提案されているもうひとつの神経疾患は、脳卒中である[16, 250]。局所脳虚血ラットモデルでは、MBを慢性的に経口投与することで、行動が有意に改善された[16]。さらなる研究では、総病変容積、脳浮腫、灰白質および白質の損傷を減少させるMBの有益な役割が示され、これらすべてが行動結果の改善に寄与した[16, 18]。Linらは、神経細胞株であるHT22細胞において、MBを投与することでミトコンドリア複合体I、II、IIIの活性が向上し、酸素消費量とグルコース取り込み量が有意に増加することを示した[251]。彼らの生体内試験の実験結果によると、MBを投与した動物では、脳全体のグルコース取り込み量と血流が有意に増加した[251]。さらに、試験管内試験の研究では、複合体I、III、V阻害剤の阻害効果下でも、MB投与後に酸素消費率が上昇したことが明らかになった[252]。また、神経細胞のATP産生量は、MB投与により改善することが示されている[252]。MCAOラットモデルでは、虚血・再灌流傷害後のComplex I, III, IVの活性低下がミトコンドリア機能の低下に寄与していたが、低用量のMBで回復した[252]。
我々が以前に行った研究では、MBがミトコンドリア機能を高めることで、脳卒中による行動障害を減弱させ、梗塞周辺部の神経新生を改善することがわかった[9]。この回復の根本的なメカニズムは、生まれたばかりの神経細胞を取り巻くミトコンドリア依存性の微小環境の改善であった[9]。
さらに、急性脳虚血傷害モデルでは、神経機能の改善にマイトファジーの増強が関係していることが判明し、MBは虚血によるミトコンドリア構造の崩壊を抑制することができた[253]。しかし、脳卒中におけるマイトファジーの効果については、まだ議論の余地がある。また、脳虚血におけるMBの神経保護作用は、マイトファジーの亢進とミトコンドリア膜電位の維持に起因することが示唆されている[253]。
MBと同様に、PBM療法は、脳卒中の有望な治療法として提案されている[254]。動物を用いたこれまでの研究では、660〜808nmの低レベルレーザーを用いたPBM治療は、熱的影響を受けることなく神経学的評価スコアを改善することが示されている[4, 255, 256]。同様に、脳卒中発症後24時間以内に赤外線レーザー治療を行うことの安全性と有効性は、ヒトの患者を対象とした研究で検証されている[257]。これらの研究と同様に、試験管内試験の細胞アッセイを用いたLED-PBMに関する多くの研究が発表されている[258, 259]。ヒト上皮細胞(HEP-2)とマウス皮下結合組織細胞(L-929)の両方において、PBM処理は代謝と増殖を増加させた[258]。虚血性脳卒中と認知症との関連は、これまでの研究でよく調べられている[260, 261]。興味深いことに、PBMは脳神経発生を促進することで、虚血性脳卒中後の認知症患者の保護効果も示している[262]。虚血性脳卒中の光血栓症(PT)モデルを用いた研究では、PBMはミトコンドリア機能と神経新生を促進することで行動成績を改善することが観察された[4]。
MBと同様に、PBMの標的は神経細胞のミトコンドリアであると考えられる。GCIの6時間後に開始したPBMは、健全なミトコンドリアの動態を維持し、ミトコンドリアの断片化を抑制することで、CA1神経細胞の死を防ぐことが示された[263]。この過程において、PBMによるミトコンドリアの完全性の維持は、ミトコンドリアの酸化的損傷と過剰なマイトファジーを弱め、ミトコンドリア依存性のアポトーシスを抑制した[263]。新生児動物モデルを用いた研究では、低酸素虚血(HI)によって誘発された大脳皮質と海馬の脳病変が、PBMの投与後に有意に減少した[254]。さらに、そのメカニズムを調べたところ、PBMはHIによって誘発されたミトコンドリアの断片化を抑制し、ミトコンドリアのダイナミクスを著しく回復させることがわかった[254]。さらに、PBMはHIに誘発されたミトコンドリアの膜崩壊を抑制し、ATP産生を改善し、タンパク質のカルボニル化、DNAの酸化的損傷、脂質の過酸化を減少させた[254]。
メチレンブルー、フォトバイオモデュレーションとうつ病
1983年に行われた初期の研究では、MBが躁鬱病の治療法として認識されていた[197]。彼らの研究では、MBの経口投与により、標準的な治療法が効かなかった19人の躁鬱病患者のうち14人の症状が有意に改善された[197]。これに伴い、二重盲検臨床試験においても、MBはうつ病の長期治療において有効な追加薬であることが示された[264]。さらに、MBは重度のうつ病にも効果的に作用する。重度のうつ病を対象とした対照試験に参加した患者の症状は、15mg/日のMB治療を受けた後、有意に改善された[265]。これまでの研究では、双極性障害の治療に対するMBの有効性も確認されている[266]。さらに、ヒトの患者と動物モデルの両方で、TBI後に抑うつ様の行動が観察されることがある[267, 268]。しかし、MBはこの抑うつ様行動を抑制することが示されている[269]。
これらの改善は、気分障害における型破りなガス状神経伝達物質である一酸化窒素(NO)の役割と、一酸化窒素合成酵素(NOS)の選択的阻害によって、メカニズム的に説明できるかもしれない[264, 265, 270, 271]。MBは、脳内のNOSを阻害するだけでなく、様々なシトクロムを含む他のヘム含有酵素にも影響を与える[272, 273]。
PBMはまた、抗うつ剤としての役割を果たす可能性も示唆されている。レセルピン誘発性うつ病の研究では、PBMの治療後、運動能力をテストする強制水泳テストの結果が改善した[274]。また、レーザー鍼治療を数週間行ったところ、うつ病患者の症状が大幅に改善した[275, 276]。さらに、76歳女性の大うつ病性障害の症例報告では、経頭蓋(t-PBM)および経鼻(i-PBM)治療後に抑うつ症状が改善した[277]。うつ病に対するPBMの有益な効果は、空間制限によるうつ病の動物モデルや、Abelson helper integrationsite-1 (Ahi1)KOマウスでも実証されている[208]。これらの2つのうつ病モデルマウスでは、いずれもPBMによって抑うつ行動が効果的に改善された[208]。さらにメカニズムの調査では、ATP産生、ミトコンドリアComplex IVの活性と発現がPBM投与後に有意に改善されることが示唆された[208]。したがって、PBMはエネルギー代謝を改善することで、うつ病の行動障害を軽減する可能性がある。
メチレンブルー、フォトバイオモデュレーション、パーキンソン病
前述のように、最も一般的な運動障害であるパーキンソン病は、ミトコンドリア機能障害を特徴とするが、ドパミン神経細胞の損傷に寄与する根本的なメカニズムは不明である[90]。しかし、ミトコンドリア機能障害によって誘発される酸化ストレスは、PDにおける神経細胞の消失と密接に関連している[278, 279].6-OHDA誘発PDモデルでは、ドーパミンの枯渇を誘発するために6-OHDA投与が行われ、ミトコンドリア機能の崩壊と活性酸素の過剰産生が引き起こされた[280-283]。MBには抗酸化作用があることから、ある研究グループは、MBの投与によって黒質部(SNc)のドーパミン細胞に神経保護作用がもたらされると予想した[284]。行動学的な改善は伴わなかったものの、低用量のMBを経口投与することで、6-OHDAによるSNcドーパミン細胞の減少が緩和された[284]。
PDのロテノンモデルでは、MBがロテノンによるミトコンドリア複合体I、II、IIIの阻害を弱め、フリーラジカルの生成を減少させ、行動の結果を改善することができ、同様の結果が得られた[252, 285]。MPTP(1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine)誘発PD動物モデルを用いてドーパミン細胞に対するMBの効果を評価した研究では、MBが脳由来向神経性因子(BDNF)のアップレギュレーションを引き起こし、その下流のシグナル伝達経路の活性化を誘導することがわかり、BDNFがMBによる神経保護の要因の一つである可能性が示された[286]。
6-OHDAおよびMPTP誘発PD動物モデルも、PDのPBM治療の研究に広く使用されている。これらのモデルでは、PBM治療は15%の細胞損失の病変を完全に救済することができた[287, 288]。しかし、50%までの細胞損失を伴う病変は回復しなかった[289]。これらの結果は、リポポリサッカライド(LPS)誘発ドーパミン神経細胞減少ラットモデルで確認された[290]。PBMは、炎症性アメーバ状ミクログリアを阻害することで、15%のLPS誘発細胞損失からラットを完全に救済することができたが、50%の細胞損失のグループには有意な神経保護は見られなかった[290]。
PBMはまた、パーキンソン病患者の頭部を赤色発光ダイオード(LED)に9週間暴露するという無作為化臨床試験でも使用されている[291]。9週間の治療後、偽薬群には有意な差がなかった[291].しかし、PBM群では有意な歩行の改善が見られた[291]。別の臨床報告では、2週間のPBM療法を行ったパーキンソン病患者において、言語、認知、歩行、凍結エピソードの改善が認められた[292]。他の神経変性疾患に対するPBM治療の研究から得られた証拠を考慮すると、PDに対するPBMの有益な効果の根底にあるメカニズムは、ミトコンドリア機能の改善と酸化ストレスの減少に起因する可能性があることはもっともである[293]。
MBおよびPBMによるミトコンドリア保護の主なメカニズム
報告されているように、MBとPBMは共にエネルギー産生を高め、酸化ストレスを減少させることが知られている[294-296]。しかし、これらの効果の根本的なメカニズムは、2つの治療法の間で異なっている。図2,2に示すように 図2,2に示すように、MBは触媒的なレドックスサイクラーとして働き、NADHから供与された電子によってミトコンドリアマトリックス内で還元される[297, 298]。この結果、ロイコMBとも呼ばれるMBH2が形成される。BH2は、複合体Iと複合体IIIの間で損なわれたETCをバイパスすることができ、酸化還元電位が低いため、還元型と酸化型の間で容易にリサイクルすることができる[297, 298]。
図2 MBによるミトコンドリア保護を支える主要なメカニズム
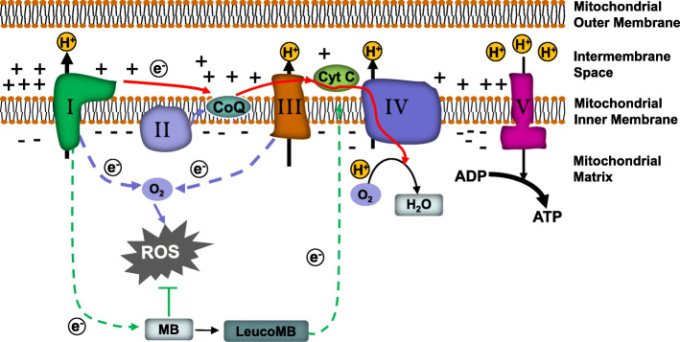
MBは、代替電子輸送体として働くことで、電子伝達の経路を迂回する。MBは、Complex IとComplex IIIの間のETCをバイパスすることで、電子漏洩とそれに伴う活性酸素の発生を効率的に抑制する
前述のように、ミトコンドリアは活性酸素の主要な発生場所である。病的な状況下では、Complex IとComplex IIIが、ミトコンドリアにおける電子漏洩の主要な部位として機能する[22]。Complex IとComplex IIIから漏れた電子はO2に移動し、O2- -に還元される[299]。過剰な活性酸素は、主にComplex IとComplex IVにダメージを与え、さらなるミトコンドリアの機能障害を誘発することになる[299, 300]。さらに、高レベルの活性酸素と酸化されたミトコンドリアDNAは、炎症性サイトカインの放出を通じて自然免疫反応を誘発するNLRP3インフラマソームの活性化を誘発する可能性がある[142, 301-303]。しかし、MBの酸化/還元は、ミトコンドリアの代替的な電子伝達経路として作用することができる。MBは、Complex IおよびComplex IIIをバイパスすることで、電子漏洩とそれに続くROSの発生を効率的に抑制する[22]。活性酸素の減少により、MBは炎症の抑制にも一役買うことができる。MBの神経保護作用のメカニズムとしては、カスパーゼ-6活性の阻害[304]、PMCAポンプ機能の回復[305]、PI3K/Akt/GSK3β経路の活性化[306]、炎症性サイトカインの減少[286]などが報告されている。シナプス毒性およびMARK4/PAR1を介したタウリン酸化の抑制 [286]、Nrf2/AREシグナル経路の活性化およびMEF2D関連生存経路の誘導 [307]などがあり、これらのプロセスにミトコンドリアが関与している可能性を排除できない [304]。
しかし、ミトコンドリア機能に対するPBMの有益な効果のメカニズムは全く異なり、細胞内に存在する発色団に依存している。光生物学の第一法則によれば、PBMが生物学的効果をもたらすためには、対象となる組織内の発色団によって吸収される必要があり、ミトコンドリアはこの光吸収において重要な役割を果たしている[308]。
ETCの複合体IV(シトクロムc酸化酵素、CCO)は、2つのヘム部位(ヘムαとヘムα3)、2つの酸化還元活性を持つ銅中心(CuAとCuB)、1つの亜鉛中心と1つのマグネシウム中心を持つ13個の独立したタンパク質サブユニットを含んでおり、これらすべてが可視光の吸収発色団であると考えられている[295, 309]。ATP生成の過程で、複合体IVは、還元型シトクロムcから電子とともに4つのプロトンを酸素に移動させ、2つのH2O分子を形成する[22, 295]。この過程は、ATP合成酵素の活性を促進するプロトン勾配の形成に寄与する[22, 295]。
これまでの研究によると、PBMはComplex IVの活性を高め、それに伴ってATPの生成量を増加させると考えられている(図(3)3)[295]。この活性向上は、PBMを介したComplex IVからのNOの光解離によるものと考えられている[310]。NOは、ヘムα3やCuBと非共有結合することで、Complex IVを阻害する分子である[310, 311]。NOがComplex IVから光解離すると、Complex IVの活性が高まり、エネルギー生産が向上する[310]。正常な状態では、活性酸素は正常なミトコンドリアの代謝によって低レベルで生成される[310]。PBMが正常な細胞のComplex IV活性を刺激すると、ミトコンドリアの膜電位が通常のベースラインレベルよりも上昇し、その結果、短時間でやや控えめな活性酸素生成量の増加が生じる [312]。活性酸素の短いバーストは、細胞質内のNF-kBを活性化することができる[312]。放出されたNF-kBは、細胞質から核に輸送され、そこでミトコンドリアの動態、炎症、抗酸化作用に関連する遺伝子を含む150以上の遺伝子の発現を誘導する[217]。
図3 PBMによるミトコンドリア保護を支える主要なメカニズム
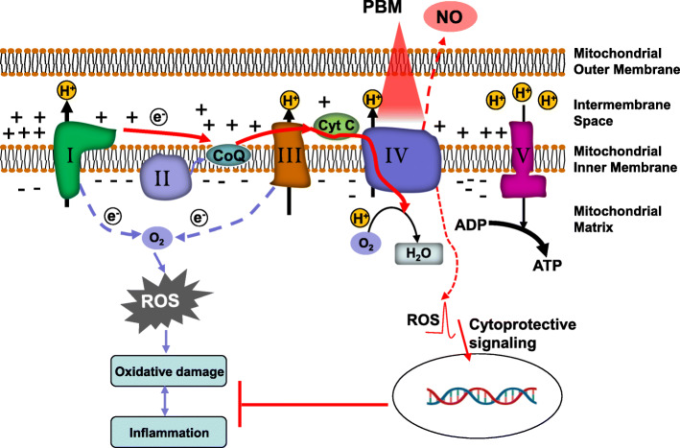
PBM処理により、NOがComplex IV(シトクロムc酸化酵素、CCO)から解離し、Complexの活性が上昇する。これにより、電子の移動、プロトンの汲み上げ、ATPの合成が活発になり、細胞のエネルギーレベルが向上する。また、PBMが正常な細胞のComplex IV活性を刺激すると、ミトコンドリアの膜電位が通常の基準値よりも上昇し、その結果、短時間ではあるが活性酸素の生成が増加する。この活性酸素は、細胞保護シグナルを活性化し、活性酸素による酸化的損傷や神経炎症を抑制する。
病的な状態では、PBMによるミトコンドリアの膜電位の上昇は、活性酸素の生成を低下させることができる[217, 295]。その結果、炎症を引き起こすNF-kBの活性が低下する。PBMが、活性化した炎症細胞から産生される炎症性サイトカインのレベルを低下させることができることが実証されている[313-315]。このようなPBMの抗炎症作用は、正確なメカニズムは不明であるが、ミクログリアのM1「炎症性」表現型とM2「抗炎症性」表現型との間の変化を仲介する能力に起因すると考えられる[4, 206]。
結論
MBとPBMは、脳疾患に対する2つの有望な治療法である。MBとPBMは、その根本的なメカニズムは異なるものの、ともにミトコンドリアを標的としている(表(表2).2)。これらの研究は、ミトコンドリアの機能障害が、いくつかの脳疾患の神経保護のための潜在的な標的であることを示唆する説得力のある証拠を提供している。ミトコンドリア機能障害に関連するプロセスとしては、ETC機能の低下、過剰なROS産生、酸化的損傷、Ca2+過剰、異常なマイトファジー、ミトコンドリアダイナミクスの変化、ミトコンドリアトラフィッキングの障害、そしてその後の神経炎症などが挙げられ(図4)これらのほとんどは、最近報告された脳老化の10の特徴に含まれている[316, 317]。MBとPBMは、それぞれ異なるメカニズムでミトコンドリアを標的としているため、これらを組み合わせた治療法は、どちらか一方の治療法の能力を超えて、脳疾患の症状を改善することができるかもしれない。このような治療法は、今後、動物実験や臨床試験で真剣に検討する必要がある。もし効果が証明されれば、MBとPBMの併用は、複数の脳疾患の治療に新たな道を開く可能性がある。
表2 神経変性疾患および脳損傷に対するMBおよびPBMの効果の要約
| 疾患 | MBによる治療の効果 | PBMによる治療の効果 |
|---|---|---|
| AD | •機能的MRI活動を増加させ、記憶の検索を改善する[ 200 ] | •は過リン酸化タウ、神経原線維変化、および酸化ストレスを軽減し、[ 234、235 ] |
| •AβレベルとAβ-ABAD結合を減少させる[ 226 ] | •Aβ食作用の能力を高める[ 236 ] | |
| •減衰活性およびβセクレターゼの発現、行動結果を神経毒性のオリゴマーAβの形成を阻害し、改善する[ 19、20、198 ] | •Aβの負担を大幅に軽減することにより、空間学習と記憶を改善する[ 236 ]。 | |
| •を活性化することにより神経保護を発揮する | ||
| •阻害p-タウ凝集およびタウ-タウ相互作用[ 229、230 ] | ERK / CREB経路とBDNFの発現のアップレギュレーション[ 237 ] | |
| •は、過剰なROS産生を削減[ 17、22 ] | ||
| •アップレギュレート複合体IVを活性化し、ヘム合成およびミトコンドリア機能[ 226、231 – 233 ] | •ミトコンドリアのダイナミクスを復元する[ 8 ] | |
| TBI | •浮腫と病変の体積を減らし、行動スコアを改善する[ 163 ] | •神経学的改善[ 246 ] |
| •オートファジーを増加させる[ 242 ]。 | •は、ミトコンドリアの機能を高め、血流を改善し、腫れ減少[ 244、247 ] | |
| •過剰なROS産生を抑制し、ミトコンドリア機能障害、シトクロムc放出、およびニューロンアポトーシスを軽減する[ 243 ]。 | ||
| •は、酸化ストレス、炎症阻害を減少させ、減衰アポトーシス[ 244、247 ] | ||
| 脳卒中 | •限局性脳虚血後の行動結果を改善する[ 16 ] | •[神経学的評価スコアを改善4、255、256 ] |
| •減少病変体積、脳浮腫、グレーと白-質のダメージ[ 16、18 ] | •を刺激する神経発生とは、ミトコンドリア機能を改善する[ 4、262 ] | |
| •脳のグローバルグルコース取り込みと血流を増加させ、[ 251、252 ] | •ミトコンドリアの完全性を維持する[ 263 ] | |
| •は、ミトコンドリア機能を増やし、[ 9、252 ] | •ミトコンドリアの断片化を弱め、ミトコンドリアのダイナミクスを回復する[ 254 ] | |
| •ミトコンドリアの構造と機能を維持する[ 253 ] | ||
| •マイトファジーを増加させ、ミトコンドリア膜電位を維持する[ 253 ] | •タンパク質のカルボニル化、DNAの酸化的損傷、脂質の過酸化を減少させる[ 254 ]。 | |
| うつ | •重度のうつ病の患者の症状を改善する[ 265 ] | •は、抑うつ症状改善[ 274 – 277 ] |
| •選択的に阻害する一酸化窒素合成酵素(NOS)[ 264、265、270、271 ] | •ATP産生を改善し、ミトコンドリア複合体IVの活性と発現を増加させる[ 208 ]。 | |
| PD | •ドーパミンの喪失を軽減し、ミトコンドリア機能の破壊とROSの過剰産生を減らす[ 280 – 284 ] | •細胞の喪失を減らし、炎症性アメーバ様ミクログリアを抑制する[ 287 – 290 ] |
| •は、複合体I、II、およびIIIの活動を改善し、フリーラジカルの生産が減少し、改善行動の結果[ 252、285 ] | •スピーチ、認知、歩行、およびPD患者のエピソードを凍結[改善291、292 ] | |
| •脳由来神経栄養因子(BDNF)の発現をアップレギュレートする[ 286 ]。 | •ミトコンドリア機能を改善し、酸化ストレスを軽減する[ 293 ] |
図4 ミトコンドリア機能不全に伴うミトコンドリア関連の変化
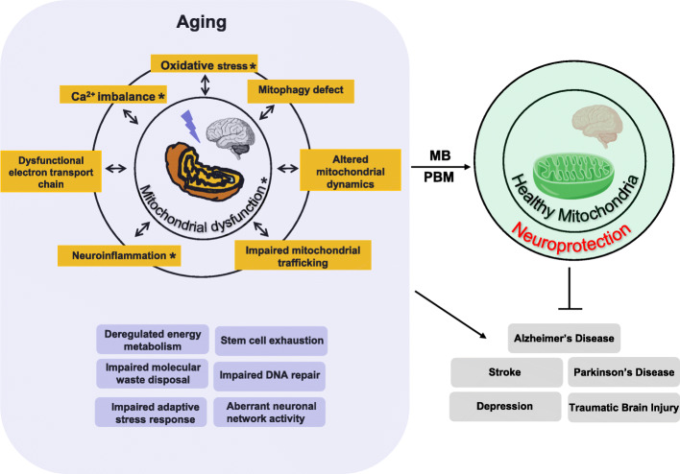
ミトコンドリア機能不全、酸化的損傷、神経細胞のCa2+制御異常、神経炎症は、脳の老化の特徴である(他の6つの特徴は、エネルギー代謝の乱れ、幹細胞の消耗、分子の廃棄物処理の障害、DNA修復の障害、適応ストレス応答の障害、神経細胞のネットワーク活動の異常である)。脳の老化の特徴が、いくつかの脳疾患の病因に影響を与えていることを示唆する証拠が増えている[316, 317]。MBとPBMは、ミトコンドリア機能障害の様々な側面を軽減することで、いくつかの脳疾患の病理学的症状を軽減し、神経保護につながる可能性がある。
略語
- ABAD アミロイド結合型アルコール脱水素酵素
- AD アルツハイマー型認知症
- Ahi1 Abelson helper integrationsite-1
- ALS 筋萎縮性側索硬化症
- APE-1 アポトーシス・プロテアーゼ活性化因子1
- APP アミロイド前駆体タンパク質
- ATP アデノシン三リン酸
- Aβ Amyloid-β
- AβPP アミロイドβ蛋白質前駆体
- BBB 血液-脳関門
- BDNF 脳由来神経栄養因子(Brain-derived neurotrophic factor)
- CAT カタラーゼ
- Cyt c シトクロムc
- DA ドーパミン作動性
- DAMPs Damage-associated molecular patterns 損傷関連分子パターン
- ETC 電子輸送チェーン
- GPx グルタチオンペルオキシダーゼ
- GR グルタチオンレダクターゼ
- GSH グルタチオン
- HEP-2 ヒト上皮細胞
- HI 低酸素性虚血
- LED 発光ダイオード
- LPS リポポリサッカライド(Lipopolysaccharide
- MB メチレンブルー
- MCAO 中大脳動脈閉塞症
- MCU ミトコンドリア・カルシウム・ユニポーター複合体
- Mfn2 ミトフシン2
- MPTP 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- mtDNA ミトコンドリアDNA
- NFT 神経原線維のもつれ
- NO 一酸化窒素
- NOS 一酸化窒素合成酵素(Nitric oxide synthase
- PBM フォトバイオモデュレーション
- PD パーキンソン病
- PINK1 PTEN誘導性キナーゼ1
- PT 光血栓症
- p-tau 高リン酸化タウタンパク質
- ROS 活性酸素種
- SNc 黒質部分のコンパクト化
- SOD スーパーオキシドディスムターゼ
- STZ ストレプトゾトシン
- TBI 外傷性脳損傷
- TNFα 腫瘍壊死因子α