
Contents
Melatonin in Alzheimer’s Disease: A Latent Endogenous Regulator of Neurogenesis to Mitigate Alzheimer’s Neuropathology
Md. Farhad Hossain1 & Md. Sahab Uddin2,3 & G. M. Sala Uddin2 & Dewan Md. Sumsuzzman4 & Md. Sumsuzzman4 & Md. Siddiqul Islam 2 & George E. Barreto5,6 & Bijo Mathew7 & Ghulam Md Ashraf8,9
受領した。2019年2月14日 / 改訂:2019年5月20日 / 受理:2019年5月22日
概要
松果体で合成される神経ホルモンであるメラトニンは、加齢に伴う様々な神経変性疾患、特にアルツハイマー病(AD)において幅広い神経保護作用を持つ多機能物質として知られている。アルツハイマー型認知症は、アミロイドβ(Aβ)斑や神経原線維変化(NFT)を含む複数の毒性ペプチドの異常かつ過剰な蓄積によって定義される、進行性の神経変性疾患であり、一般的な認知症の一形態である。
アルツハイマー型認知症は、脳の萎縮が原因で、記憶力の低下、認知機能の低下、シナプスの障害などが起こる。アルツハイマー病は、加齢、概日リズムの乱れ、Aβの蓄積、タウの過リン酸化などが最大の危険因子とされている。現在のところ、ADの進行に対する正確な治療法はない。
この点、メラトニンは、時計遺伝子を制御することで概日リズムの乱れを抑制し、グリコーゲン合成酵素キナーゼ3(GSK3)およびサイクリン依存性キナーゼ5(CDK5)シグナル経路を制御することで、Aβの蓄積およびタウの過リン酸化を抑制するという重要な役割を果たしている。
本総説では、メラトニンが、概日リズムの乱れ、Aβの生成、タウの過剰リン酸化を抑制することで、神経新生に影響を与え、ADの病因となる可能性があることを明らかにした。さらに、メラトニンがADの進行に伴うAβの産生、Aβのオリゴマー化とフィブリル化、タウの高リン化、シナプスの機能障害、酸化ストレス、神経細胞死を阻害することで、神経保護作用を発揮することを見出し、まとめたものである。
キーワード メラトニン . アルツハイマー病 . アミロイドβ . 神経原線維のもつれ 概日リズム
はじめに
アルツハイマー病(AD)は,認知機能や記憶力の低下を特徴とする慢性の神経変性疾患である[1, 2]。アルツハイマー病の病理学的特徴は,主にアミロイドβ(Aβ)からなる細胞外の老人斑[3]と,タウタンパク質からなる細胞内の神経原線維変化(NFT)である[4, 5]。加齢は、ADの最も病理学的な危険因子の一つであり、65歳以上の認知症患者の60~80%を占めると言われている[6, 7]。ADの主な原因はまだ不明であり、リスクの約70%は遺伝的なものであると考えられている[8]。ADは時間の経過とともに悪化し、認知症の症状が徐々に悪化していく[9, 10]。アルツハイマー病の初期段階では、記憶障害は軽度であり、徐々に軽度の症状が出てきて認知機能が低下し、最終的には症状が重くなって認知症になる[11, 12]。国際アルツハイマー病協会(ADI)によると、現在、4,400万人が認知症を患っており、2050年には1億3,500万人に増加すると言われている[13]。そのため、ADの初期段階でコントロールするために必要な措置を講じる必要がある。
メラトニン(N-アセチル-5-メトキシトリプタミン)は、ADの神経変性イベントに対して保護的な役割を果たす循環リズム制御ホルモンである[14]。メラトニンの主な分泌源は脳の松果体で,他にも網膜,骨髄,グリア細胞,膵臓,腎臓,皮膚などの器官が関与しており[15],植物界も関与している[16]。メラトニンは、生まれたときから増加し、10代や思春期にピークを迎えるが[17]、加齢とともに減少し、高齢者では最も低い値となる[12, 13]。メラトニンは、循環器系の調節[18]や、抗炎症作用、細胞保護作用[19, 20]、抗酸化作用[21, 22]など、多機能なホルモンとして知られている。メラトニンは概日時計によって制御されており、ラット[23]やマウス[24]では深夜に血漿メラトニン濃度が最も高くなる。メラトニンは、日中の早い時間帯の12:00〜18:00頃に最も低く、深夜の23:00〜02:00頃に最も高くなる[25]。
加齢に伴い、メラトニンの分泌量は減少し、これはAD発症の重要な要因と考えられている[26, 27]。視交叉上核(SCN)の異常や障害により、松果体ホルモンであるメラトニンが減少し、概日リズムが変化する[1]。脳脊髄液(CSF)中のメラトニンの減少が増えると、メラトニン欠乏はADの進行を助け、ADの脳では酸化的なダメージを受ける[28, 29]。研究によると、アルツハイマー病患者は健常者に比べてメラトニンのレベルが低いことが明らかになった[30, 31]。メラトニンには、抗アミロイド作用やフリーラジカル消去作用があり、ADの進行を抑制する治療薬として期待されている[32, 33]。また、アルツハイマー病患者では、フリーラジカル産生、酸化的DNA損傷、脂質過酸化、ATPの減少、細胞死の発現レベルが健常者に比べて高いことが知られている[34, 35]。メラトニンは、様々な細胞株において、APPの成熟を阻害することで、可溶性アミロイド前駆体プロテイン(APP)の分泌を抑制する能力を持っている[F36u]。さらに、生体内試験モデル[37, 38]および試験管内試験モデル[36, 39-41]において、メラトニンの補充は、Aβの生成および沈着を効率的に抑制する。メラトニンの投与は、Aβの生成とタンパク質の異常なニトロ化を抑制する。興味深いことに、Zhuら[42]は、ラットモデルにおけるメラトニン合成の停止がアルツハイマー病患者の病態に似ていることを生体内試験研究で証明した。さらに、ADトランスジェニック(TG)マウスモデルにおいて、メラトニンを10 mg/kg投与すると、認知機能障害が回復し、Aβの生成が抑制された[43]。
睡眠と覚醒は、Aβの生成に関連する重要な因子であり、Aβレベルが覚醒時に上昇し、睡眠時に低下することも証明されているが、これは昼夜のメラトニンの分泌パターンに起因している可能性がある[44]。日暮れのような時間生物学的障害は、アルツハイマー病患者の精神的衰退[45]や興奮、混乱を高めるのに重要な役割を果たしているが、メラトニン治療は、夕暮れ症候群を軽減するだけでなく、認知機能を高めることもできる[46-48]。ADの実験動物モデルでは、プラークやタングルの形成を含むADの病理学的特徴がメラトニンによって阻害される可能性がある[49-51]。さらに、メラトニンの補充は、ADの進行に関連する睡眠障害の抑制に寄与し、アルツハイマー病患者の記憶と認知の障害を緩和する[52]。本総説では,メラトニンが神経新生に影響を与えることでADの発症を抑制する神経保護機能について議論し,評価する。
メラトニンの発見と合成
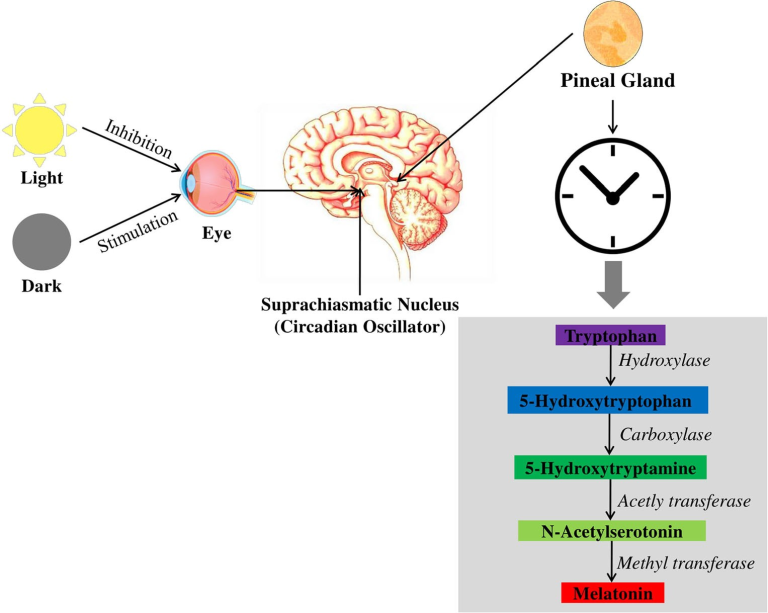
メラトニンは30億年前から存在している古代分子である。また,構造的にも1958年に初めて同定された[53]。メラトニンは,主に松果体から分泌され,体内時計の中枢であるSCNによって活性化され,ヒトを含む哺乳類の概日リズムを制御している。メラトニンは光に感応するホルモンであるため、光周期はメラトニンの合成とその分泌パターンに重要な役割を果たしている。SCNは松果体に神経信号を伝えるための重要な中継センターであり,神経メッセージは中枢および末梢の交感神経系の調節を介して松果体に到達する[54]。まず,目の網膜が光を吸収すると(図1),信号が作られ,網膜視床路を通って視床下部にあるSCNに送られ,視神経に搭載される。その後、この信号は室傍核を経て、脊髄にある中間側細胞柱に進み、最終的に上頸部神経節(SCG)に到達する。SCGはノルエピネフリンの助けを借りて松果体に信号を送り、セロトニンからメラトニンを合成する。ノルエピネフリンは、交感神経節後枝から放出される神経伝達物質で、メラトニンの合成に重要な役割を果たしている。ノルエピネフリンが少なすぎるとメラトニンの合成量が少なくなり、逆に多すぎるとメラトニンの生成量が多くなって目が覚めてしまう[55, 56]。
簡単に言えば,ノルエピネフリンはα-1/βアドレナリン受容体を刺激して膜結合型アデニル酸シクラーゼ-cAMP系を活性化し,アデニル酸シクラーゼ-cAMPの活性化だけでなく,cAMP,カルシウム(Ca2+),ジアシルグリセロール,ホスファチジルイノシトール,プロテインキナーゼCなどの細胞内濃度を上昇させる[57]。セロトニンからのメラトニンの酵素による生合成経路は,Axelrodによって発見された[58]。生化学的には,メラトニンはその前駆体であるトリプトファンから合成される。メラトニンはトリプトファンという前駆体から合成され,その合成過程は主に水酸化,脱炭酸,アセチル化の3つのステップで構成されている。生合成の最初のステップは、トリプトファン水酸化酵素によってトリプトファン(インドール環の5位)が水酸化され、5-ヒドロキシトリプトファン(5-HTP)になることである。次に、側鎖のカルボキシル基が除去され、芳香族アミノ酸脱炭酸酵素(AAD)により脱炭酸されてセロトニンになる。最終段階では、セロトニンはアリールアルキルアミンN-アセチル化酵素(AANAT)によってN-アセチルセロトニンにアセチル化され、その後、ヒドロキシインドール-O-メチル化酵素(HIOMT)の存在によってN-アセチルセロトニンとなる。このアセチル化されたセロトニン、N-アセチルセロトニンはメラトニンに変換され、一般的にメラトニンの生合成に関する律速段階であると考えられている[15, 55]。
図1 松果体によるメラトニン合成の制御と脳内サーカディアンリズムの制御
メラトニンと長生き
いくつかの生物では、概日リズムの回復力が寿命の向上と関連しているが、リズムの乱れは逆の効果をもたらす。興味深いことに、概日リズムは松果体のメラトニンによって同期化され[59-61]、加齢に伴う神経変性の有病率を低下させる効果があると考えられている[26]。このように、メラトニンは老化防止、老化遅延のために働くことが明らかになっている。
メラトニンは、脳において、老化防止、老化遅延[62]、強力な抗酸化物質[63, 64]として作用することが明らかになっている[65]。例えば,メラトニンを補給すると,Paramecium tetraureliaやDrosophila melanogasterなど,いくつかの種で寿命が延びることがわかっている[66-68]。
ある研究では,高齢の動物にメラトニンを投与すると,寿命が20%近く延びることが示されている[69]。しかし、げっ歯類モデルでの実験結果はかなり印象的なものであり、推定された関連性を強調するためには、他の種での拡張研究が必要である。以前、メラトニンが長寿タンパク質であるサーチュイン1(SIRT1)[70]の発現を促進し、自己治癒遺伝子の発現を促すことが明らかになった[1, 71]。SIRT1が細胞培養だけでなく、ADのTGモデルマウスにおいても長寿を促進し、Aβペプチドを減衰させることは驚くべきことではない[72, 73]。アルツハイマー病患者では、SIRT1のRNAおよびタンパク質レベルが顕著に低下しており[74]、SIRT1の過剰発現がADの表現型に対して有益である可能性を示唆している。
さらに,SIRT1 は disintegrin and metalloproteinase domain-containing pro-tein-10 (ADAM10) を介して α-セクレターゼ の活性を促進し,脳内の Aβ含量を減少させた [75] [76].DNAのメチル化[77]やヒストン修飾[78]は,ADと加齢の両方の脳で観察される最もポピュラーなエピジェネティック・スイッチングである。一方、メラトニンは、神経細胞におけるこれらのエピジェネティックな変化をしっかりと制御しており[79]、このようにして、メラトニンは、健康な状態でも病気の状態でも、老化を遅らせ、寿命を延ばすことができるのである。
アルツハイマー病患者のメラトニンと概日リズムの乱れ
睡眠覚醒異常とサーカディアン・ディスラプション(図2)は、ADの進行と密接に関連しており、その結果、環境同調因子や精神的・身体的活動の強さが低下し、サーカディアン・クロックの機能が失われる。不規則な睡眠や睡眠障害は、記憶障害や認知障害のリスクを高め、ADの進行につながる[52]。65歳以上の高齢者の約80%以上に概日リズムの異常や体内脱同期が見られ、睡眠パターン、体温、ホルモン分泌、その他の生体機能に悪影響を及ぼしている[80, 81]。加えて、高齢者はメラトニンの不適切な分泌により早寝早起きとなり、概日システムが変化して、実際の睡眠時間と睡眠効率が大幅に低下し、睡眠潜時が増加する[82]。
睡眠障害と睡眠覚醒パターンの断片化は、夜間の覚醒だけでなく、昼間の昼寝も増加させる[83]。さらに、中等度から重度のアルツハイマー病患者では、日中の睡眠傾向の増加が認知障害の原因となっている[84]。日暮れは、外部刺激に対する注意力の低下、思考の混乱、行動の変化、覚醒と興奮の増加、体温低下に関連する時間生物学的障害であり、痴呆患者の午後遅くまたは夕方早くに周期的に発生する[48-50]。メラトニンは概日時計によって制御されており、ADに対して重要な役割を果たしている。しかし、概日リズムが乱れると、メラトニンの分泌が抑制されるだけでなく、メラトニンリズムのパターンに高度な異常が生じ、その結果、時計の設定や時間の同期ができなくなる。
アルツハイマー型認知症におけるSCNの変化
ヒトを含む哺乳類では,SCNは時計細胞からなる脳の概日時計であり,その機能は視床下部路を制御することで概日時計を同期させることである[85]。神経ペプチドの分類によると、SCNニューロンは、視床下部路からの入力を受けて同調を生成するのに重要な役割を果たす血管作動性腸ペプチド(VIP)ニューロンに分類される[86]。また,SCNの残りの部分には,SCNのニューロンの神経ペプチドであるアルギニンバソプレシン(AVP)が存在し,SCN内だけでなく脳の他の領域でもリズムを調節している[87]。AVP発現ニューロンの概日変動の数は加齢とともに減少し[88]、その減少は深刻であった。
加齢に伴いAVP発現ニューロンの概日変動数は減少し[88]、80-100歳の間に深刻な減少が見られた[89, 90]。同様に、VIP発現ニューロンでも同様の結果が得られ、これらのニューロンの数は若年者に比べて中年者でより減少していた[91]。いくつかの研究では、アルツハイマー病患者では、AVPおよびVIPを発現するニューロンの数が、年齢をマッチさせた健常者に比べて劇的に減少していることが明らかになった。さらに、SCNではバソプレッシンとニューロテンシンのニューロンの発現が減少する一方で、グリア線維酸性タンパク質であるアストロサイトの密度が増加し、ADの病因を進行させるのに役立っている[89, 92, 93]。驚くべきことに、アルツハイマー病患者のAVP mRNAの総量は、年齢をマッチさせた健常者に比べて3倍低く、アルツハイマー病患者にはAVP mRNAの明確で瞬時の日周リズムは見られない[94]。したがって、SCNの変化と加齢は、AVPとVIP経路を介してADの病理に重要な役割を果たしている。
circadian oscillatorは、brain muscle ARNT-like 1 (BMAL1)、circadian locomotor output cycles kaput (CLOCK)、cryptochrome (CRY)、period circadian protein homologue 1 (PER1)、period circadian protein homologue 2 (PER2)などの一連の時計遺伝子から構成されており、これらの遺伝子は細胞内での転写や翻訳のフィードバックループに貢献している[85, 95]。時計遺伝子は、脳領域や末梢組織(松果体など)で広く発現しており、視床下部のSCNで同期している[96, 97]。さらに、SCNは、β-アドレナリンシグナル伝達経路を介して、PER1やCRY2などの時計遺伝子を直接制御している[98]。加齢は、SCNにおけるBMAL1,CLOCK、PER1,およびPER2の発現の緩和に関連する危険因子である[99, 100]。肝臓や心臓などの末梢組織でも、加齢により時計遺伝子の発現が変化していた。老齢ラットの肝臓や心臓では,PER遺伝子のレベルが中年ラットに比べて有意に低下していたが,BMAL1のレベルは夜間に増加していた[101].
図2 夜間に光を浴びるとメラトニン合成が急性的に抑制され,アルツハイマー病を引き起こす概日リズムが乱れる
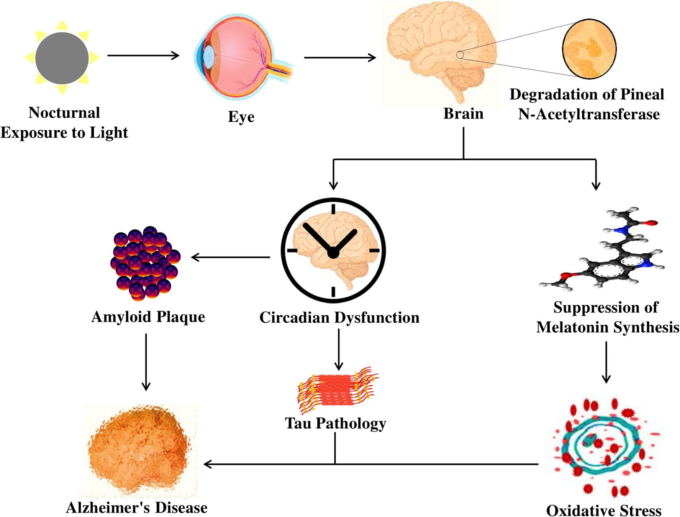
加齢によってアルツハイマー病の病態が進行することはよく知られている。Wuら[102]は、時計遺伝子とアルツハイマー病の病態との相関関係を明らかにしようと試みた。彼らは、対照群、前臨床群、臨床群のアルツハイマー病患者における時計遺伝子(BMALl、CRY1,PER1など)のリズミカルな発現を調べた。その結果、前臨床アルツハイマー病患者(Braak I-II)と臨床アルツハイマー病患者(Braak V-VI)では、BMALlと呼ばれるこれらの時計遺伝子のリズム発現が低下していた。
MT1遺伝子の発現はSCNで検出されるが、MT2は視床下部での発現が非常に低いため検出されない[106, 107]。これまでの研究では,死後の視床下部SCNにおいてMT1受容体の免疫反応が観察されており[108, 109],これは,SCNのリズムに対するメルアトニンのフィードバック役割と一致している[110]。さらに、MT1受容体は、SCNに位置するAVP発現ニューロンの一部と同居しているように見えたことから、このニューロンのサブポピュレーションがSCNにおけるメラトニン作用を構成していることが示唆された。しかし、SCNへのMT1発現ニューロンは、若年者よりも高齢者の方が強く減少しており、同じ結果がADの最終ステージ(すなわちBraakステージV-VI)にも見られたが、正常なコントロール(すなわちBraakステージ0)には見られなかった[109]。
CSF中のメラトニンレベルは、ADの進行とともに減少し、これはBraakステージの検査で明らかになった[111]。さらに、別の研究では、アルツハイマー病患者のCSFメラトニンレベルは、年齢をマッチさせた正常対照者と比較すると、わずか5分の1であることが証明された[112]。アポリポ蛋白(APOE)e4/4型の患者のメラトニンレベルは、APOEe3/4型の患者よりもさらに瞬間的に低かった[112]。ADの神経病理が進行すると、Braak病期で決定されるように、CSF中のメラトニンレベルが低下する[111]。さらに注目すべきは、ADの神経病理を持たない老齢の対照群(すなわち、Braakステージ0)と比較して、ADの初期症状を呈している前臨床アルツハイマー病患者(すなわち、BraakステージI-II)では、CSFのメラトニンレベルがすでに瞬間的に低下していることであり[111]、これはヒトの松果体の研究でも確認されている[30]。前臨床AD(Braak段階I-II)および後期臨床AD(Braak段階V-VI)の松果体メラトニン濃度は、対照群(Braak段階0)と比較して低下しており、日周リズムにおける松果体メラトニンの含有量が失われている。さらに、松果体メラトニンの含有量は、CSFメラトニンのレベルと非常に大きな相関関係があることがわかった[30]。
加齢に伴い、メラトニンの分泌量とSCNのMT1受容体のレベルは急激に減少し、ADの病理ではさらに減少する。興味深いことに、メラトニンレベルの低下は、PER1であるnCdRY1であり、一方、対照被験者(すなわちBraak 0)は、ADに関連するだけでなく、ADにおいてもこれらの時計遺伝子の正常な日周リズムの発現を発揮することができる。さらに、ADモデルマウスでは、AβがBMAL1とPER2の概日時計遺伝子の発現を直接低下させ、概日リズムを変化させていた[103]。この結果は、松果体における時計遺伝子の発現の乱れが、ADの進行に関係していることを示している。
アルツハイマー病におけるメラトニン濃度の低下
メラトニンは、SCNと松果体の両方で概日リズムを調節しており、MT1とMT2の受容体によって広く交渉されている[104]。MT1メラトニン受容体はニューロパソロジー。メラトニンは、生体内試験および試験管内試験で強力な抗酸化剤および神経保護剤であることが示されている[113-115]。以前の研究では、TG ADモデルマウスでは、酸化的病理やアミロイド病理を抑制し、生存率を高めることが示されている[37]。また、神経芽細胞腫の細胞やラットにおいて、メラトニンはタウやニューロフィラメントの過リン酸化を有意に減少させる[116]。ラットでは、メラトニンを補充することで、記憶保持障害が大幅に改善され、タウのリン酸化と酸化ストレスが停止し、メラトニン合成によるプロテインホスファターゼ-2A(PP-2A)の活性が回復した[42]。したがって,メラトニンの欠乏がADの発症に関与している可能性がある。
アルツハイマー病におけるSCNへの入力の減少
内因性の概日時計は,環境の指標,特に明暗サイクルによって,24時間の環境精神サイクルに合わせて駆動される。光はSCNを最も強く刺激するものと考えられている。さらに、メラトニンは、メラトニン受容体の存在下で、SCNに対して強力な活性を発揮する[104, 117, 118]。メラトニンを補充すると、光とSCNの間の同期入力が不足している盲目の人の睡眠障害が著しく改善され、自由に動く概日リズムが得られる[119, 120]。メラトニンによる自走式リズム活動を再教育するためには、メラトニンの投与と被験者の概日位相という2つの要因が関与する[121]。メラトニンの概日作用は、時差ぼけや交代勤務、および概日的な睡眠障害の治療に重要な役割を果たしている[122, 123]。加齢やADでは、SCNの変性が起こり、Zeitgebersの強度が低下したり、神経経路が機能しなくなったり、反応性が低下したりすることがある。これらの変化は、これらの状況における概日リズムの乱れを後押しする可能性がある[122]。光は、光入力経路(すなわち、視床下部路)を介して作用し、SCNの入力に不可欠であると考えられている。照度とメラトニンリズムの位相シフトの間には、顕著な用量反応関係がある。加齢に伴い、適度な光量に対する位相シフトの反応が低下し、特に短波長に対する反応が低下することで、加齢に伴う概日リズム疾患の進行に寄与する可能性がある[124, 125]。加齢に伴い、概日リズムシステムへの光の投射が減少し、ADの場合はさらに顕著になる理由が数多く証明されている。夜間の覚醒と昼間の眠気が増えるため、高齢者、特にアルツハイマー病患者は、若い人よりも昼間の生活で低いレベルの照度にさらされることになる[126, 127][128, 129]。また、水晶体の光を伝染する能力は、加齢とともに徐々に低下する。さらに、高齢者やアルツハイマー病患者では、メラトニン含量が減少し、SCNのMT1受容体が著しく減少しているため、SCNにおけるメラトニンの概日作用が影響を受けている可能性がある[109]。さらに、身体活動や社会的接触は、加齢や特にADにおいて著しく減少することがよく知られており、これも概日リズムの乱れの一因となりうる[130, 131]。これらを総合すると、SCNへの入力の減少は、ADの間のSCNの不活性化に明らかに寄与していると考えられる。
メラトニンとアルツハイマー病の特徴
Aβ毒性におけるメラトニン
APPは、39〜43個のアミノ酸残基からなるAβの前駆体である。図3には、Aβペプチド生成の最も危険な因子の一つであるアミロイド原性のβアミロイド前駆体タンパク質(βAPP)を示している。興味深いことに,Aβ42は,Aβの中でも最も神経毒性の高い形態である[132]。このAβ42は,γセクレターゼの存在下で適切なタンパク質分解を受けて生成され,βプリーツ状のシートの形成を助け,その結果,脳内に老人斑が形成されることになる。脳内で生成されたこれらのプラークは、シナプスの破壊やニューロンの機能異常の原因となるペプチドの凝集やアミロイド線維の形成を及ぼし、ニューロンの完全性を失わせる[33]。さらに,Aβのクリアランスと産生の不均衡は,Aβの蓄積の原因となり,脳の病態や細胞のホメオスタシスの乱れに重要な役割を果たしている。Aβは、遺伝子のフリーラジカル、酸化ストレス、auのリン酸化、GSK3βやCDK5の活性化によるDNAやタンパク質の破壊、脂質の酸化、エネルギー代謝の低下などを引き起こし、ADの病態を引き起こす[1, 133]。
先に述べたように、メラトニンは脳内で神経保護、抗酸化、抗アミロイド生成の機能を持ち、脳内でのAβ形成の可能性を減少させる[134]。メラトニンは、Aβの毒性を積極的に低下させ、βシートの形成を阻害し、線維性アミロイドに対する予防効果を示す。メラトニンは,生体内試験 [37]および試験管内試験モデル [40, 135]の両方で,Aβ誘発性の脂質過酸化,酸化ストレス,概日変化,細胞死,およびDNA損傷に対する予防効果を示す。
カスパーゼ-3/Bcl-2/GSK-3β/PP2Aの制御
カスパーゼ-3,B細胞リンパ腫-2(Bcl-2),PP2A,グリコーゲン合成酵素キナーゼ3β(GSK3β)を含むいくつかの主要酵素は,発現量を増加または減少させることによって,ADの病因と密接に関連している[136-138]。これらの酵素は,Aβやタウの過分極のシグナル伝達経路も制御している可能性がある。中でも,カスパーゼ-3の活性化は,AD脳におけるAβ産生の増加につながるβセクレターゼ活性を改善し,アルツハイマー病患者のカスパーゼ-3レベルが,健常者や年齢をマッチさせた人よりも高いことがよく知られている[139]。同時に、GSK3はタウの過リン酸化を進行させ、PP2Aが減少するとタウタンパク質の過リン酸化を抑制することができる[140, 141]。
カスパーゼ-3の亢進は、ADの細胞死に直接関与しているが [142]、メラトニンは、AD TGマウスモデルにおいて、カスパーゼ-3をダウンレギュレートし、抗アポトーシス作用を持つBcl2レベルを上昇させる能力を持っている [143, 144]。一方,メラトニンは,Aβ誘発動物モデルにおいて,カスパーゼ-3の活性化[145]を緩和するだけでなく,アポトーシス[146]を抑制する。さらに、メラトニンを投与すると、Aβ1-42誘発マウスにおいて、GSK-3βおよびカスパーゼ-3の発現レベルが有意に低下し、PP2AおよびBcl-2のレベルが上昇した。以上のことから、メラトニンは、PP2AやGSK-3β、およびカスパーゼ-3やBcl-2を制御して細胞のアポトーシスを抑制することで、タウの過リン酸化や有害物質を減少させる候補となり得ると考えられる。
図3 アミロイド斑の形成につながるAPPとその切断産物の処理におけるメラトニンの役割
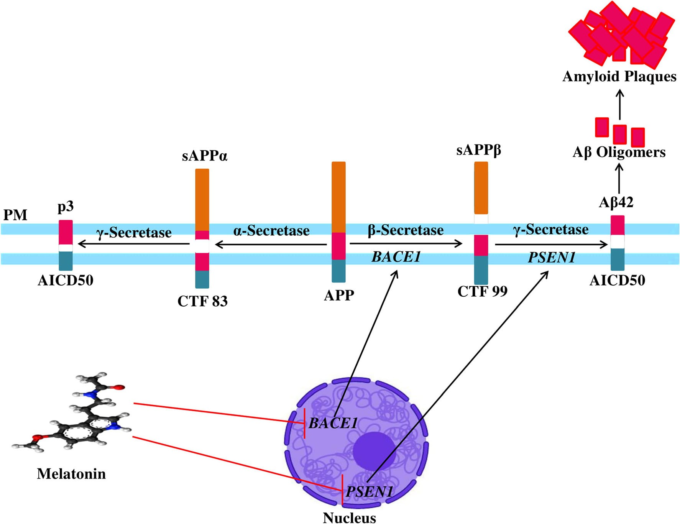
PMは細胞膜、APPはアミロイド前駆体タンパク質、sAPPαは可溶性アミロイド前駆体タンパク質-α、sAPPβは可溶性アミロイド前駆体タンパク質-β、CTFはC末端フラグメント、AICDはアミロイド前駆体タンパク質の細胞内ドメイン
記憶障害とAβ蓄積の抑制
ADは,主にエピソード記憶の障害によって発症し,その結果,言語,注意,作業記憶などの多くの分野で障害が進行する[147, 148]。Gongら[149]は、Aβ1-42誘発マウスモデルの認知機能を評価するために、行動学的な試験を行った。空間学習能力と記憶能力を評価するために、コントロール、Aβ1-42注射群、メラトニン投与群で指向性ナビゲーション・トレイルを実施した。その結果、メラトニン投与群はAβ1-42投与群に比べて潜伏時間が短いことがわかった。認知・記憶機能を向上させるためには、マウスはIII象限に長く滞在し、中央のアリーナを頻繁に横切る必要がある。メラトニン投与群は、Aβ1-42投与群と比較して、III象限に長く留まり、中央の正方形のアリーナでの線の横断が増加し、有意な結果を示した。また、運動量を測定するためにopen-field testを行い、自発的な活動を評価するためにstep-through testとstep-down testを行った。メラトニン投与群では、Aβ1-42投与群と比較して、ミスが減少し、潜伏時間も大幅に改善された。これらの結果から、メラトニンには、認知機能と自発的活動を効率的に回復させる能力があることがわかった[149]。さらに、メラトニンを投与すると、2×ADマウスのMorris Water Maze(プローブテスト)とBarnes Maze(円形プラットフォームテスト)による学習能力が向上する[150]。
タウの病理におけるメラトニン
タウは微小管関連タンパク質と呼ばれ、微小管の組み立ての進行に関与し、微小管の安定性を維持している[151]。細胞内NFTの主要成分であるリン酸化されたタウは、アルツハイマー病患者の記憶力や認知力の低下に重要な役割を果たしている[10, 152]。リン酸化により、タウタンパク質はその生物学的活性を維持することができなくなり[153]、微小管を破壊することでタウオパチーを引き起こす[154, 155]。このように、アルツハイマー病患者ではタウの過リン酸化によって神経変性が起こり、その期間中、アルツハイマー病患者では健常者と比較して3-4倍もの過リン酸化タウが検出される。
この時期、アルツハイマー病患者は健常者と比較して3-4倍もタウの高リン化が進んでいる[156-158]。メラトニンは、水[159]や脂質[158]への溶解性など、血液脳関門を通過しやすい特徴を持っており、この特徴は他の抗酸化剤とは全く異なる。メラトニン自体は、フリーラジカルスカベンジャーとして機能し、図4に示すような抗酸化酵素を活性化する。メラトニンは、イソプロテレノールで誘発されたラット脳におけるタウの過リン酸化とプロテインキナーゼA(PKA)の過剰活性化を抑制する[33]。メラトニンは,ワートマニン[160],オカダイン酸[161],カリクリンA[42]によって誘発されたN2aおよびSH-SY5Y神経芽細胞におけるタウの過リン酸化を有意に減少させる。興味深いことに,メラトニンは,海馬の神経細胞におけるGSK3およびCDK5の活性を調節することで,海馬の変性の進行を抑制し,認知機能を高めるという極めて重要な効果を発揮している[48]。さらに,メラトニンは,カスパーゼ-3,BCL2関連X(Bax),前立腺アポトーシス応答-4(Par-4)の発現レベルを抑制し,神経細胞のアポトーシスの進行を抑制することにも寄与している[163]。
PI3K/Akt/GSK3の制御
プロテインキナーゼやプロテインホスファターゼの不適切な制御は、タウの過リン酸化に重要な役割を果たしている。その中でもGSK3は、ADの発症に関わる重要なキナーゼの一つである[164]。GSK3を過剰に発現させると、生体内試験および試験管内試験の方法でタウの過リン酸化を引き起こす[165-167]。ワートマニンはホスファチジルイノシトール3キナーゼ(PI3K)阻害剤であり,PI3K経路を阻害することでGSK3の発現を活性化する[168]。Dengら[160]は,アルツハイマー病の病態解明に広く用いられている神経芽細胞腫N2a細胞株にワートマニンを投与すると,GSK3の活性化を介してタウの過リン酸化が起こることを明らかにした。ワートマニンを投与すると、タウの過リン酸化が活性化されるだけでなく、脂質過酸化、酸化ストレス、c-JUN N-terminal kinase (JNK)、extracellular signal-regulated kinase (ERK)、p38などの活性化がN2a細胞株で起こった。また、メラトニンを投与すると、GSK3の過剰活性化によって引き起こされたタウの過リン酸化を抑制する効果があった。また、メラトニンは、スーパーオキシドディスムターゼ(SOD)を中心とした抗酸化作用の活性化を通じて、酸化ストレス、脂質過酸化、JNK、p38のレベルを抑制した。さらに,メラトニンは,PI3K/Akt/GSK3経路を調節することで,タウの過リン酸化を抑制する[169, 170]。
タウの過リン酸化と記憶障害の緩和
カイニン酸(KA)は、記憶喪失や神経変性疾患を引き起こすグルタミン酸のアナログとして知られている[171]。Shiら[172]は、海馬におけるタウのリン酸化(Ser199およびSer396)の発現が、KAによる生体内試験および試験管内試験の両方の方法で増加することを明らかにした。記憶障害を評価するために、彼らはKA誘発マウスの学習能力をモリス水迷路試験で調べた。脱出潜時の平均値は、KA投与群が対照群よりも高く、メラトニン投与群(Mel+KA)はKA投与群に比べて脱出潜時を再減少させた。また、メラトニン投与群(Mel+KA)は、KA投与群と比較して逃避潜時が短縮された。これらの結果を総合すると、KA投与マウスは対照群と比較して目標の象限に到達するまでの時間が短く、Mel+KA群はKA投与群と比較して時間が短縮された。これらの結果から、メラトニン投与により、KA誘発動物モデルの記憶障害が減少し、タウのリン酸化が緩和されることが証明された。これは、記憶障害がタウの過リン酸化と効率的に関係していることから、メラトニンの保護効果が証明された。また,メラトニンは,KA誘発海馬ニューロンにおけるタウのリン酸化(Ser396およびSer199)を顕著に軽減した。さらに、メラトニンの不十分な補給は、認知機能障害の発生と強く関連しているが、メラトニンの投与は、神経細胞の変性の進行を抑制し、認知機能を高める効果がある[111, 112]。
小胞体(ER)ストレスは、ADの進行と密接に関係しており、その間、ERシャペロンタンパク質であるGRP78のレベルは、アルツハイマー病患者の側頭角と海馬で増加していた[174, 175]。KA処理は、海馬の細胞死につながる小胞体ストレスの活性化を誘導する[176, 177]。さらに、KAによって誘発されたERストレスは、GSK3βとCDK5を含む2つの主要なキナーゼを誘発し、タウの超リン酸化を引き起こす。SB216763とロスコビチンは、GSK3βとCDK5を特異的に阻害する薬剤である。これらの阻害剤は、タウの過リン酸化を抑制する候補であるKA誘発腰椎ポカンパー神経細胞に投与された。これらの結果は、小胞体ストレスがGSK3とCDK5の発現を誘発し、それがタウの過リン酸化を助けることを示唆している。興味深いことに、メラトニンを投与すると、GSK3とCDK5の活性化が緩和され、KAによる小胞体ストレスを抑制することができる。さらに、メラトニンは、KAによって誘発された海馬のGRP78レベルの上昇を有意に軽減した[172]。このように、メラトニンは、GSK3およびCDK5を制御し、小胞体ストレスを緩和することで、神経細胞障害やタウの過リン酸化を予防する有望な候補であると考えられる。
図4 アルツハイマー病の原因となる酸化ストレスと神経原線維変化に対するメラトニンの役割
アルツハイマー型認知症の神経炎症におけるメラトニンの役割
Rosales-Corralら[28]は,メラトニンが,線維性アミロイドβ(fAβ)によって誘発されるインターロイキン-1-β(IL1-β),インターロイキン-6(IL-6),腫瘍壊死因子-α(TNF-α)などの炎症促進分子を顕著に抑制することを報告した。興味深いことに,メラトニンがNF-κBのDNA結合活性を抑制するという実験データがある[178, 179]。最近の研究では,メラトニンが,Aβ処理した脳切片において,濃度依存的にNF-κBを介したIL-6を抑制することが明らかになった[180]。また,ラットにメラトニンを投与すると,Aβによる学習・記憶障害が抑制され,海馬では補体1q(C1q)に加えて,NF-κB関連のIL-1βが有意に減少することが示されている[181]。
IL-1,IL-6,TNF-αなどの炎症性サイトカインは、ADのような慢性炎症性疾患や神経変性疾患の進行に極めて重要な役割を果たしている[182-185]。ミクログリアやアストロサイトで合成されるIL-1やIL-6は、Aβや老人斑を生成する原因となり、TGマウスでは神経変性を引き起こしている[186, 187]。炎症、反応性酸素種(ROS)の生成、Aβおよび老人斑は、AD発症の主要な危険因子であり、IL-1およびIL-6の存在によって加速される[183, 188, 189]。さらに、酸化ストレスは、生体内試験 [190]および試験管内試験 [191]モデルでのAβ産生を改善し、ADの進行に寄与する。このような状況の中、メラトニンはSOD活性を示すことで、間葉系幹細胞のIL-1β、IL-6,TNF-αを抑制する効果を発揮している[192, 193]。
メラトニンは、抗酸化酵素の活性を発揮することで、酸化ストレスを軽減する[194, 195]。また,メラトニンの外因性投与(500μg/kg)は,雌のシリアン・ハムスターにおいて,銅-亜鉛スーパーオキシドディスムターゼ(CuZnSOD)だけでなく,マンガンスーパーオキシドディスムターゼ(MnSOD)のmRNAの発現を増強し[196],メラトニンの投与(5mg/kg)は,ラットの腎臓,肝臓,脳におけるSOD活性を増強した[196]。Aβペプチド(25-35)[197]やD-ガラクトース[198]で誘発されたげっ歯類(ラットおよびマウス)が脳に酸化損傷を起こしても、メラトニン(0.1〜10 mg/kg)はSODおよびグルタチオンペルオキシダーゼ(GPx)の活性を回復させる。同様に、メラトニン(10 mg/kg)も、胎児の脳でATP産生を増加させることによって酸化的ミトコンドリア損傷から保護し、ラットの脳でGPx活性を刺激することが示されている[199]。これらの証拠から、メラトニンは、抗酸化作用と抗炎症作用を示すことで、ADのリスクを抑制する可能性がある。
アルツハイマー病のコリン作動性システムに対するメラトニンの作用
コリン作動性システムの障害は、ADの初期段階の病因としても考えられている[200, 201]。大脳皮質や海馬のコリン作動性神経支配の主要な起源である基底核の神経細胞は、ADの脳では圧倒的かつ急速に変性している[202-204]。驚くべきことに、ADの初期段階では、ADのアセチルコリン(ACh)レベルは減少しているように見えるが、逆に、合成酵素と加水分解酵素であるコリンアセチルトランスフェラーゼ(ChAT)とアセチルコリンエステラーゼ(AChE)の活性は、ADの後期段階まで変化しなかった[205-207]。生検と剖検の両方で、アルツハイマー病患者の大脳新皮質におけるChAT活性が著しく低下し、認知症の予後と相関していることが観察された[208]。AChの欠乏に関与する分子メカニズムはまだ不明であるが、AChE阻害剤は軽度から中等度のADの治療法として使用されている[209]。
メラトニンは、コリン作動性システムに防御的な作用を及ぼす。Guermonprezら[210]は、メラトニンがペルオキシナイト依存性のコリン輸送阻害およびいくつかの神経系タンパク質のChAT活性をわずかに低下させることを報告した。また、APP695トランスジェニックマウスにメラトニンを4ヶ月間投与したところ、Aβの沈着による学習・記憶障害が顕著に改善され、メラトニン投与により前頭葉皮質と海馬でのChAT活性が向上したことが示された[211]。同様に、別の研究では、メラトニンが、卵巣摘出成人ラットの空間記憶障害を有意に改善し、前頭皮質と海馬のChAT活性を低下させたことが示された[212]。しかし、メラトニンはC h AT活性を高めることはできなかったが、脳室内に注入したAβ誘発ChAT活性は著しく低下した[213]。また,メラトニンは,リポ多糖(LPS)誘発AChE活性の上昇に対してのみ抑制効果を示したが,CSF投与ラットでは変化が認められなかった。これらの結果は、ストレプトゾトシン(STZ)誘発の認知症モデルにおけるAChE活性に対するメラトニンの抑制効果と一致している[33, 214]。
コリンエステラーゼ阻害剤(ドネペジル、タクリン、リバスチグミン、ガランタミンなど)は、アセチルコリンを遮断することで、認知機能や行動、グローバルな変化を安定させたり、減少を遅らせたりすることが可能であることが、プラセボ対照試験の系統的レビューとメタ分析で明らかになっている[215]。ADでは、概日リズムの乱れがメラトニンの分泌低下につながり、その結果、睡眠の質の低下や認知機能の障害が生じる。メラトニンの補充は、サンダウン症候群、軽度認知障害(MCI)認知症やその他の睡眠覚醒障害に先行する病的に異質な症候群の治療に有効であることが明らかになった。メラトニンは,AChE活性を阻害するだけでなく,活性酸素や窒素種の強力な消去剤として,ADの睡眠障害の回復やAβ毒性の軽減にも貢献している[33, 216]。
AChE阻害剤とメラトニンのどちらがアルツハイマー病患者にとって最良の選択であるかは、まだ未解決の問題であり、したがって、この臨床的証拠を解明するためにさらなる研究が必要である。しかし、これらの2つの分子を組み合わせることで、相乗効果が得られる可能性がある。最近、ハイブリッド手法を用いて、タクロン-メラトニンハイブリッドをADの新規多機能薬として設計・合成した[217, 218]。これらの化合物を組み合わせることで、コリン作用と抗酸化作用の両方が改善され、個々の化合物よりも優れていることが示唆された。さらに、これらの化合物の組み合わせは、低毒性を示し、中枢神経系に浸透することができるかもしれない。驚くべきことに、これらのハイブリッドの1つであるN-(2-(1H-インドール-3-イル)エチル)-7-(1,2,3,4-テトラヒドロアクリジン-9-イルアミノ)ヘプタンアミドを直接静脈内投与すると、APP/PS1マウスの脳におけるAβ誘発性プログラム細胞死とアミロイド負荷が減少した。さらに、Aβ病理の減少は、認知機能の回復と関連していた[218]。
アルツハイマー病におけるメラトニンと神経新生
典型的な加齢は神経新生を変化させ、Aβの蓄積は動物モデルや細胞培養において、前駆細胞の増殖や神経細胞の分化を阻害する[219-221]。神経細胞の機能障害により、神経細胞の脆弱性が記憶障害となり、ADを引き起こす[222]。TGの動物モデルを用いたこれまでの研究では、Aβペプチド[225, 226]やAPPの発現が進行すると、脳室下帯(SVZ)や顆粒下帯(SGZ)における海馬の神経新生が低下し[220, 223, 224]、海馬における前駆細胞の増殖だけでなく、細胞の生存率も自然に低下することが示されている[227]。
オリゴマーAβは細胞増殖を抑制し[228]、β-カテニンのダウンレギュレーションとアポトーシスを介して神経新生を促進し、GSK3βを活性化してAβの産生とタウのリン酸化を促進する[229]。臨床研究によると,ADの脳では,β-カテニン[231, 232]とWnt/β-カテニンの発現が対照群に比べて低下していることが明らかになった[230]。興味深いことに、Wnt/β-カテニンは、神経細胞の毒性やアポトーシスを抑制するだけでなく、タウのリン酸化も抑制する[233]。その結果、新しいニューロンの生成やニューロンの維持が、ADを保護するための有効な治療基準として考えられるかもしれない。
興味深いことに、メラトニンは、神経新生を制御するために肯定的な役割を果たしており、また、いくつかの実験的研究では、メラトニンが、ADで損なわれている細胞増殖と神経新生を増加させることが証明されている[234]。先に述べたように、MT1とMT2は成人の脳だけでなく、神経細胞にも存在している[235]。メラトニンには、樹状突起形成[236]、樹状突起の成熟を促進する作用があり、メラトニンと運動を組み合わせることで、神経新生が促進される[237, 238]。アルツハイマー病患者では、睡眠不足がメラトニンの減少に直接関係しており、これがAβの生成を増加させ、記憶機能障害を引き起こす。メラトニンは、神経幹細胞の増殖および分化を促進し[239]、新しい神経細胞の生存を促進することが動物実験で証明されている[240]。また、メラトニンは錐体神経細胞の減少を防ぎ[241]、松果体切除ラットモデルにおいて神経新生を促進する[242]。メラトニンは、ERKシグナル経路を介して成人の海馬[243]および脳室下帯[244]の細胞増殖を促進し、デキサメタゾンによる海馬の変化を防ぐこともできる[244]。メラトニンは、β-カテニンタンパク質の発現を増加させ、PLC/DAG、PI3K/Akt、PKCを刺激して、フォスフォリル化とGSK-3βの不活性化をもたらす[245]。メラトニンは、AChE活性を低下させ、抗疲労効果を示し[245]、また、α-セクレターゼ[246]や可溶性アミロイド前駆体タンパク質-αの産生を刺激し、神経前駆細胞(NPC)の増殖[247]や、神経細胞の生存を促進する役割を担っている[248]。また,加齢に伴う神経細胞の死には酸化ストレスが関与しているが[249],メラトニンを投与すると酸化ストレスの障害を防ぎ,α-セクレターゼによって増殖や分化を促進することができる[250]。
アルツハイマー病患者におけるメラトニン濃度
アルツハイマー病患者のメラトニン濃度は、年齢をマッチさせた対照群と比較して低下していることが明らかになっている[112, 129, 251-253]。アルツハイマー病患者では髄液中のメラトニン濃度が低下しており、主流のメラトニン産生が減少していることが示唆されている。興味深いことに、患者が認知障害を伴わない前臨床段階(BraakステージI-II)では、CSFメラトニンレベルの低下さえも観察されており、CSFメラトニンの低下がADの初期段階を検出するための早期バイオマーカーとなる可能性が示唆されている[30, 111]。メラトニンレベルの低下の背後にある分子メカニズムを探るために、Wuら[30]は研究を行った。その結果、ノルアドレナリン制御の乱れ、モノアミン酸化酵素A(MAO-A)活性の上昇による5-ヒドロキシトリプタミンの枯渇が、メラトニンリズムの不均衡をもたらしていることがわかった。メラトニンの振幅が減少し、それに対応して概日システムが変化する理由としては、複屈折装置の物理的特性から、網膜視床路(RHT)やSCNと松果体の関連性の不具合まで、光の伝達経路の変化が考えられている[254]。
しかし、松果体ホルモンは光によって抑制されるため [255, 256]、光の伝達の障害がメラトニンレベルの低下として解釈されることは容易ではない。いずれにしても、メラトニンの分泌が変化すれば、アルツハイマー病患者に見られる睡眠障害、夜間の落ち着きのなさ、サンダウン症候群の一因となるであろう[257]。もう一つの理由は、アルツハイマー病患者の代謝の変化にあると考えられる。APOE-ε4/4の出現は、Aβの毒性の増大とより迅速な疾患の進行に関連しているが、この特定のAD亜集団におけるメラトニンの減少は、複数のAPOE亜型を持つ患者よりも明らかに強いものである[258]。この見解によると、メラトニンの不足は、ADの原因の一つではなく、結果として目に見えるようになるかもしれないが、メラトニンの不足は病気を悪化させるかもしれない。夜間のメラトニンレベルの低下は、痴呆患者の精神障害と関連していることも実証されている[259]。
アルツハイマー病治療のターゲットとしてのメラトニン
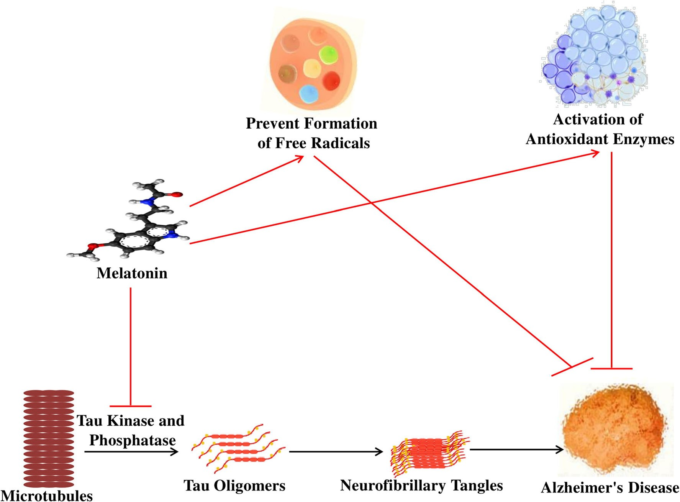
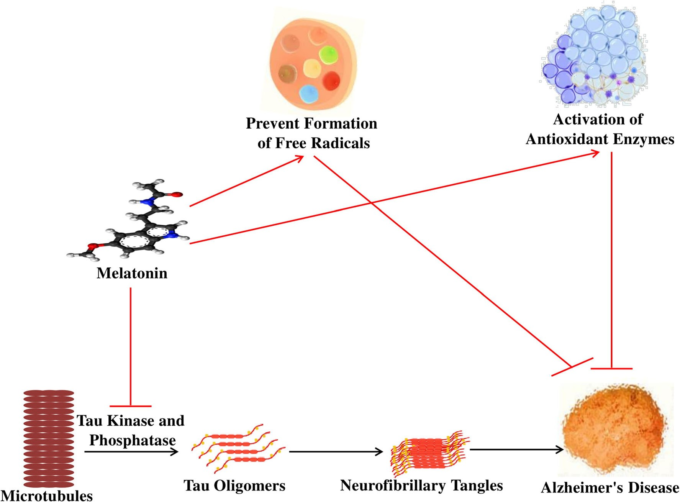
多くの生体内試験研究[37]、試験管内試験研究[40, 135]、症例報告[46]、パイロット研究[260]、小規模臨床研究[261]において、メラトニンは、AD-認知症患者におけるAβの毒性や線維形成の抑制、フリーラジカルの消去、ミトコンドリアの損傷の防止、睡眠障害や日暮れなどの概日リズムの乱れに有効であると考えられている(図5)。メラトニンは,バランスのとれた両親媒性,血液脳関門通過能力,任意の細胞区画(ミトコンドリア)への進入,抗酸化特性など,他の類似成分に比べて7つの利点があることが注目されている[115, 262]。
Aβの産生を阻害する
APPに由来するAβは,老人斑を形成することでADの発症メカニズムに関連する最も研究されている危険因子である[134]。メラトニンは,Aβと直接相互作用し,Aβの凝集を阻止することが,チオフラビン蛍光法によって証明されている[263]。APP遺伝子のプロモーターにはcAMP応答性領域があり、これがAPPの合成を誘導し、プロスタグランジンの合成も促進する。プロスタグランジンの増加は、APP mRNAの過剰発現を促し、その結果、神経炎症や神経変性を引き起こす[264]。興味深いことに、メラトニンおよび/またはその代謝物は、PEG/COX-2経路を阻害することでプロスタグランジンを抑制し、さらにアデニルシクラーゼを阻害することでcAMPの産生を阻害し、最終的にAPPの合成を緩和する[32]。
メラトニンは、MT2受容体を介してホスホリパーゼC(PLC)を刺激し、プロテインキナーゼC(PKC)を活性化して、αセクレターゼを介したAPPの切断を促進し、GSK-3の発現をリン酸化して不活性化する[245, 265]。さらに、メラトニンは、GSK-3活性化の予防メカニズムを豊かにするJNK産生を抑制する[266]。そのため、GSK-3の活性化を阻害することは、APPの合成を中断し、タウの過リン酸化を抑えるための重要な要素であると考えられる。
さらに,AβはAChE活性を上昇させ,それが細胞内Ca2+の増加につながり,酸化ストレスや活性酸素の生成を引き起こすが,メラトニンは,Aβ誘発マウスモデルにおいて,活性酸素の生成と細胞内Ca2+レベルを低下させることで,AChE発現の増加を抑制する[263]。さらに,メラトニンは,Aβ25-35およびAβ40誘発ラットPCL2褐色細胞腫およびマウスN2a神経芽細胞腫細胞モデルにおいて,5-50μMの濃度で細胞死および酸化ストレスを予防した[267]。メラトニンを投与すると,Aβ1-42誘発マウスモデルにおいて,認知機能が改善し,アポトーシスが減少する[149]。
図5 メラトニンを標的としたアルツハイマー病発症抑制のための治療戦略
PM 細胞膜, APP アミロイド前駆体タンパク質, AICD50
アミロイド前駆体タンパク質の細胞内ドメイン50
Aβのオリゴマー化とフィブリル化の阻害
Aβの集合体には、モノマー、可溶性オリゴマー、不溶性フィブリルの3つの主要なグループがあり、これらはAβプールと呼ばれている[268]。それぞれのプールは、毒性のあるオリゴマー(すなわち8-24マー)α-シノリゴマー(すなわち6-18マー)タウオリゴマー(すなわち3-15マー)などのマーの範囲に基づいて、Aβの複数の凝集構造を包んでおり、凝集体を作る過程で神経細胞に毒性を与える[269]。これらの凝集体はフィブリルに変化する。可溶性のAβオリゴマーは,2量体,3量体,4量体,5量体,10量体など様々な構造を示す[270-272]。最も毒性の強いAβはAβ1-42として知られており,アミロイド斑を形成し,神経死を引き起こす。興味深いことに、Aβ1-42は、フィブリル化する前に、単量体、三量体、四量体が混在した状態でその一貫性を維持している。Aβ1-42は、N-メチル-D-アスパラギン酸受容体(NMDAR)の脱感作を緩和し、細胞内のCa2+を過剰に発現させることで神経細胞死を促進し[273]、AMPAシナプス受容体の密度を低下させる[274]。
メラトニンは、活性酸素の発生を防ぐプロトフィブリルと自発的に結合し、その抗酸化作用を発揮して神経細胞を保護する。メラトニンを投与すると,Aβプロトフィブリルを誘導したマウスの酸化ストレスが軽減される。また、メラトニンは、Aβペプチドとの親和性により、アミロイド線維形成の予防効果があることが、試験管内試験線維化アッセイで証明されている[263]。Pappollaら[275]は、メラトニンがAβ1-40およびAβ1-42と相互作用する能力を持ち、βシート構造のペプチドの量を減少させることで、Aβ1-40およびAβ1-42のフィブリル形成を阻害することを証明している。メラトニンは、Aβ1-40の抑制につながる抗線維形成特性を示すことで、AD進行の発症リスク因子であるAPOEと直接相互作用する[49, 276]。
別の研究では,メラトニンがアミロイドペプチドの疎水性領域(17-28)に関与して非共有結合を形成し,Aβの線維形成を抑制することが明らかにされている[277]。さらに,可溶性Aβはアストロサイトで過剰な活性酸素を生成し,前脳基底部の神経細胞の破壊に寄与する[278]。興味深いことに,メラトニンは,活性酸素の発生を抑制するだけでなく,Aβ1-42誘発マウスモデルにおける抗酸化酵素(カタラーゼやGPx)の活性低下を回復させる。
シナプス機能障害の抑制
シナプス障害は、認知症の引き金となる最も身近なADの兆候の1つである[279]。アルツハイマー病患者では,過剰なAβが,海馬や新皮質におけるシナプス刺激因子である長期増強(LTP)を抑制し,長期抑圧(LTD)の誘導を誘発し[280],NMDARを活性化することで,シナプス伝達,可塑性,さらには神経細胞死を阻害する[281, 282]。さらに、アルツハイマー病患者ではシナプスの総数が正常な人よりも少なくなっている[283]。メラトニンは、LTPを著しく向上させ、シナプスの伝達を促進する[284]。Corralesら[285]は、メラトニンがグルタミン酸シナプスの密度を高め、活性化することで、シナプスの抑制を減らし、海馬のLTPを回復させることを明らかにした。また、メラトニンはシナプス前のタンパク質であるシナプトフィシンの活性を高めることで、シナプスの数を増やし[286]、海馬の神経細胞の樹状突起の構造を改善する[287]。Rudnitskayaら[288]は、OXYSラットにおいて、メラトニンの投与(0.04 mg/kg)により、前頭葉皮質および海馬におけるAβ蓄積の発現が緩和されるだけでなく、損傷した神経細胞や死んだ神経細胞が減少することを証明した。また、メラトニン投与により、シナプスの総数が増加し、シナプスの病理学的変化も抑制されたことが明らかになった。さらに、夜間にCSF中のメラトニンのレベルがピークに達すると、樹状突起の数やその長さ、厚さが改善されることも分かった[236]。Benleulmi-Chaachouaら[289]は、MT1受容体がシナプス前膜に存在し、海馬、視床下部、大脳皮質のシナプス前タンパク質ネットワークの一部であることを確認している。一方,MT2受容体は,3β/CRMP-2シグナル伝達経路を活性化することで,中枢神経細胞の機能的な軸索形成に寄与している[290]。
Aliら[291]は,D-ガラクトースを投与したマウスモデルでは,記憶に関連するシナプス前タンパク質であるシナプトフィシン,SNAP25,シナプス後タンパク質であるPSD95,およびp-GluR1(845番セル)の発現がウェスタンブロット法により,ビヒクル群と比較して減少していることを明らかにした。興味深いことに、これらの減少した効果は、メラトニン処理(10 mg/kg)によって回復し、タンパク質マーカーが有意に増加した。P-CREB Ser 133は,シナプスの形成や記憶機能に関連する転写因子である[292]。D-ガラクトース投与マウスではp-CREB Ser 133のレベルが低下しているが、メラトニン投与ではp-CREB Ser 133のレベルが上昇し、記憶障害も予防される[291]。
酸化ストレスと神経細胞死の阻止
酸化ストレスは、ADを含む加齢に伴う神経細胞の減少と関連している[293]。酸化ストレスはアミロイド生成経路によってAβを蓄積し、ミトコンドリアの損傷、神経炎症、活性酸素の発生、およびAGEs(Advanced glycation end products)の生成を引き起こす[294-296]。これらの結果は、記憶力や認知力の低下と関連しており、ADの脳では炎症性メディエーターの活性化も関与している[297]。さらに、アルツハイマー病患者では、Aβの蓄積によりフリーラジカルが生成され、これが酸化的損傷を変化させ、神経細胞内のCa2+レベルを上昇させ、神経細胞内でフリーラジカルが生成される。フリーラジカルの増加は、ミトコンドリアの機能不全と関連し、ATPの枯渇と神経細胞死を引き起こす[298]。
メラトニンは、大脳皮質における過酸化水素(H2O2)と一酸化窒素(NO)の進行を直接解毒する[299]。酸化ストレスマーカーである8-オキソグアニンは,ADおよび認知症の患者に分布している。Aliら[291]は,酸化ストレスを評価するために,D-ガラクトース誘発マウスモデルで免疫蛍光染色を行った。アリら[291]は,D-ガラクトース誘発マウスモデルにおいて,8-オキソグアニンレベルが,DG,CA1,CA3を含む海馬の領域で,ビヒクル投与マウスと比較して高いことを発見した。メラトニン投与は、DG、CA1,CA3領域における8-オキソグアニンの過剰発現を抑制するだけでなく、D-ガラクトース誘発モデルにおける酸化ストレスを克服した。また、D-ガラクトース誘発モデルでは、p-IKKβ、NF-κB、シクロオキシゲナーゼ-2(COX-2)一酸化窒素合成酵素2(NOS2)IL-1β、TNF-αの発現量が自動車群と比較して増加していたが、メラトニン投与によりその発現量の増加が抑えられた[291]。
さらに,メラトニンは,AβモデルにおけるNF-κBの発現を抑制し[300],p300 HAT経路を阻害することでp52のアセチル化を抑制する[263]。JNK経路は、ADの進行に顕著に関連するGSK-3の活性化に関与している。メラトニンは、酸化ストレス条件下でJNKの活性化を抑制し、また、抗酸化作用を発揮して抗GSK活性を示する[301]。進行性グリケーションエンドプロダクツ受容体(RAGE)、グリア線維酸性タンパク質(GFAP)、イオン化カルシウム結合アダプター分子1(Iba1)は、ミクログリア症やアストロサイトーシスの原因となる。しかし、メラトニンは、RAGE、GFAP、Iba1の発現レベルを低下させる重要な役割を果たし、また、シトクロム(Cyt)c、カスパーゼ-9,カスパーゼ-3,切断されたポリ(ADP-リボース)ポリメラーゼ-1(PARP1)などのいくつかのアポトーシスメディエーターを促進し、D-ガラクトース誘発マウスモデルの神経細胞死に寄与している[291]。さらに,メラトニンは,抗酸化作用を発揮してフリーラジカルを中和し,アルツハイマー病患者におけるAβ形成の可能性を低減する[302]。メラトニンを10mg/kgの用量で投与すると、KA誘発成人ラットモデルにおける神経細胞死、ミクログリアの活性化、および脂質過酸化が減少する[302]。一方、メラトニンは、試験管内試験モデルにおいて、フリーラジカルの形成やAβの生成を抑制することで、神経細胞死を保護した[303]。従って、メラトニンは、ADの進行中の有望な治療ターゲットになるかもしれない。
結論
松果体産物であるメラトニンは、強力な抗酸化物質であり、フリーラジカルを消去する力を持っている。メラトニンは、酸化ストレスを抑制することにより、AD脳における炎症性サイトカインの発現、ミトコンドリア機能障害、神経炎症、および神経細胞死を抑制することが明らかになった。加齢に伴い、メラトニン濃度が徐々に低下すると、ADの進行が促進されることがしばしば証明されている。従って、メラトニンレベルの低下は、ADの病理のもう一つの危険因子である可能性がある。
メラトニンはまた、抗Aβ凝集作用を発揮し、ヒトを含む様々な生物種のAβ誘導において、タウの高リン化や神経毒性を抑制する。メラトニンは、SCNおよびCLOCK遺伝子を制御することにより、概日変動を保護し、ADを抑制することが期待される。メラトニンは、シナプスの構造とその可塑性の安定化に寄与し、記憶や認知機能をさらに向上させる。したがって、メラトニンは、安全で親和性の高い抗酸化物質として、将来的にADの強力な治療標的となる可能性がある。
略語
AD(アルツハイマー病)Aβ(アミロイドβ)APP(アミロイド蛋白質前駆体)AANAT(アリールアルキルアミンN-アセチルトランスフェラーゼによるN-アセチルセロトニン)AVP(アルギニンバソプレッシン)ACh(アセチルコリン)AChE(アセチルコリンエステラーゼ)AGE(advanced glycation end products)ADAM10(a disintegrin and metalloproteinase domain-containing pro- tein-10)の略語である。AAD(芳香族アミノ酸脱炭酸酵素)BMAL1(brain mus-cle ARNT-like 1)CSF(脳脊髄液)CRY(クリプトクローム)CLOCK(概日運動量サイクルカプト)COX2(シクロオキシゲナーゼ-2)ChAT(コリンアセチルトランスフェラーゼ)C1q(補体1q)CuZnSOD(共役亜鉛スーパーオキシドディスムターゼ)CDK5(サイクリン依存性キナーゼ5)。ER(小胞体)GPx(グルタチオンペルオキシダーゼ)H2O2(過酸化水素)HIOMT(ヒドロキシインドール-O-メチル基転移酵素)IL1-β(インターロイキン-1-β)IL-6(インターロイキン-6)KA(カイニン酸)LPS(リポポリサッカライド)LTP(長期増強)LTD(長期抑圧)MCI(軽度認知機能障害)MnSOD(マンガンスーパーオキシドディスムターゼ)。NFT(神経原線維変化)NO(一酸化窒素)NOS2(一酸化窒素合成酵素2)NF-κB(核内因子カッパベータ)PLC(ホスホリパーゼC)PKC(プロテインキナーゼC)。PI3K、ホスファチジルイノシトール3キナーゼ;PER、周期サーカディアンタンパク質ホモログ;PSEN1,プレセニリン-1;PSEN2,プレセニリン-2;ROS、活性酸素種;RHT、網膜視床路;SOD,
スーパーオキシドディスムターゼ、SCN(視交叉上核)SCG(上殿神経節)SIRT1(サーチュイン1)TNF-α(腫瘍壊死因子-α)VIP(血管作動性腸ペプチド)。5-HTP(5-ヒドロキシトリプトファン)Bcl2(B細胞リンパ腫2)PP2A(プロテインホスファターゼ2A)GSK3β(グリコーゲンシンターゼキナーゼ3β)PKA(プロテインキナーゼA)Bax(BCL2関連X)。Par-4(前立腺アポトーシス応答-4)JNK(c-JUN N-terminal kinase)ERK(ex-tracellular signal-regulated kinase)MAO-A(モノアミン酸化酵素A)RAGE(receptor for advanced glycation end products)GFAP(glial fibril- lary acidic protein)Iba1(ionized calcium binding adaptor molecule 1)APOE(apolipoprotein E)PARP1(poly(ADP-ribose) polymase-1)。
文献
1. Shukla M, Govitrapong P, Boontem P et al (2017) Mechanisms of melatonin in alleviating Alzheimer ’s disease. Curr Neuropharmacol 15:1010–1031. doi.org/10.2174/ 1570159X15666170313123454
2. Uddin MS, Al Mamun A, Asaduzzaman M et al (2018) Spectrum of disease and prescription pattern for outpatients with neurolog- ical disorders: an empirical pilot study in Bangladesh. Ann Neurosci 25:25–37. doi.org/10.1159/000481812
3. Uddin MS, Stachowiak A, Mamun AA et al (2018) Autophagy and Alzheimer’s disease: from molecular mechanisms to thera- peutic implications. Front Aging Neurosci 10:1-18. doi. org/10.3389/fnagi.2018.00004
4. Uddin MS, Mamun AA, Hossain MS et al (2016) Exploring the effect of Phyllanthus emblica L. on cognitive performance, brain antioxidant markers and acetylcholinesterase activity in rats: promising natural gift for the mitigation of Alzheimer’s disease. Ann Neurosci 23:218–229. doi.org/10.1159/000449482
5. Uddin MS, Mamun AA, Labu ZK et al (2018) Autophagic dys- function in Alzheimer’s disease: cellular and molecular mechanis- tic approaches to halt Alzheimer’s pathogenesis. J Cell Physiol 234(6):8094–8112. doi.org/10.1002/jcp.27588
6. Uddin MS, Al Mamun A, Kabir MT, et al (2018) Nootropic and anti-Alzheimer’s actions of medicinal plants: molecular insight into therapeutic potential to alleviate Alzheimer’s neuropathology. Mol Neurobiol 1–20. doi.org/10.1007/s12035-018-1420-2
7. Uddin MS, Mamun AA, Takeda S et al (2018) Analyzing the chance of developing dementia among geriatric people: a cross- sectional pilot study in Bangladesh. Psychogeriatrics. 19(2):87– 94. doi.org/10.1111/psyg.12368
8. Uddin MS, Amran MS (eds) (2018) Handbook of research on critical examinations of neurodegenerative disorders, 1st ed. USA: IGI Global. doi.org/10.4018/978-1-5225-5282-6
9. Mamum AA, Uddin MS, Wahid F et al (2016) Neurodefensive effect of Olea europaea L. in alloxan-induced cognitive dysfunc- tion and brain tissue oxidative stress in mice: incredible natural nootropic. J Neurol Neurosci 7:1-12. doi.org/10.21767/ 2171-6625.1000126
10. Uddin MS, Mamun AA, Iqbal MA et al (2016) Analyzing nootropic effect of Phyllanthus reticulatus Poir. on cognitive functions, brain antioxidant enzymes and acetylcholinesterase activity against aluminium-induced Alzheimer’s model in rats: applicable for con- trolling the risk factors of Alzheimer’s disease. Adv Alzheimer’s Dis 05:87–102. doi.org/10.4236/aad.2016.53007
11. Grandy JK (2013) Melatonin: therapeutic intervention in mild cognitive impairment and Alzheimer disease. J Neurol Neurophysiol 04:1–6. doi.org/10.4172/2155-9562. 1000148
12. Uddin MS, Kabir MT, Al Mamun A et al (2018) APOE and Alzheimer’s disease: evidence mounts that targeting APOE4 may combat Alzheimer’s pathogenesis. Mol Neurobiol 56(4): 24506–25.41 –16. doi.org/10.1007/s12035-018-1237-z
13. ADI G8 (2050) Policy Briefing reveals 135 million people will live with dementia by Alzheimer’s Disease International
14. Savaskan E, Ayoub MA, Ravid R et al (2005) Reduced hippocampal MT2 melatonin receptor expression in Alzheimer’s disease. J Pineal Res 38:10–16. doi.org/10.1111/j.1600-079X.2004.00169.x
15. Naseem M, Parvez S (2014) Role of melatonin in traumatic brain injury and spinal cord injury. Sci World J 2014:1–13. doi. org/10.1155/2014/586270
16. Wang L, Feng C, Zheng X et al (2017) Plant mitochondria syn- thesize melatonin and enhance the tolerance of plants to drought stress. J Pineal Res 63:e12429. doi.org/10.1111/jpi.12429
17. Payne JK (2006) The trajectory of biomarkers in symptom man- agement for older adults with cancer. Semin Oncol Nurs 22:31– 35. doi.org/10.1016/j.soncn.2005.10.005
18. Sugden D (1983) Psychopharmacological effects of melatonin in mouse and rat. J Pharmacol Exp Ther 227:587–591
19. Bilici D, Akpinar E, Kiziltunç A (2002) Protective effect of mel- atonin in carrageenan-induced acute local inflammation. Pharmacol Res 46:133–139
20. Carrillo-Vico A, Lardone P, Álvarez-Sánchez N et al (2013) Melatonin: buffering the immune system. Int J Mol Sci 14: 8638–8683. doi.org/10.3390/ijms14048638
21. Srinivasan V, Pandi-Perumal SR, Spence DW et al (2010) Potential use of melatonergic drugs in analgesia: mechanisms of action. Brain Res Bull 81:362–371. doi.org/10.1016/j. brainresbull.2009.12.001
22. Tan D-X, Manchester LC, Sanchez-Barcelo E et al (2010) Significance of high levels of endogenous melatonin in mamma- lian cerebrospinal fluid and in the central nervous system. Curr Neuropharmacol 8:162–167. doi.org/10.2174/ 157015910792246182
23. Benot S, Molinero P, Soutto M et al (1998) Circadian variations in the rat serum total antioxidant status: correlation with melatonin levels. J Pineal Res 25:1–4. doi.org/10.1111/j.1600-079X. 1998.tb00378.x
24. Kasahara T, Abe K, Mekada K et al (2010) Genetic variation of melatonin productivity in laboratory mice under domestication. Proc Natl Acad Sci 107:6412–6417. doi.org/10.1073/ pnas.0914399107
25. Semenova NV, Madaeva IM, Bairova TA et al (2018) Association of the melatonin circadian rhythms with clock 3111T/C gene poly- morphism in Caucasian and Asian menopausal women with in- somnia. Chronobiol Int 35:1–11. doi.org/10.1080/ 07420528.2018.1456447
26. Reiter RJ (1995) The pineal gland and melatonin in relation to aging: a summary of the theories and of the data. Exp Gerontol 30:199–212
27. Reiter RJ, Tan DX, Poeggeler B et al (1994) Melatonin as a free radical scavenger: implications for aging and age-related diseases. Ann N Y Acad Sci 719:1–12
28. Rosales-Corral S, Tan D-X, Reiter RJ et al (2003) Orally admin- istered melatonin reduces oxidative stress and proinflammatory cytokines induced by amyloid-beta peptide in rat brain: a compar- ative, in vivo study versus vitamin C and E. J Pineal Res 35:80–84
29. Smith MA, Hirai K, Hsiao K et al (1998) Amyloid-beta deposition in Alzheimer transgenic mice is associated with oxidative stress. J Neurochem 70:2212–2215
30. Wu Y-H, Feenstra MGP, Zhou J-N et al (2003) Molecular changes underlying reduced pineal melatonin levels in Alzheimer disease: alterations in preclinical and clinical stages. J Clin Endocrinol Metab 88:5898–5906. doi.org/10.1210/jc.2003-030833
31. Wu Y-H, Swaab DF (2005) The human pineal gland and melato- nin in aging and Alzheimer’s disease. J Pineal Res 38:145–152. doi.org/10.1111/j.1600-079X.2004.00196.x
32. Rosales-Corral SA, Acuña-Castroviejo D, Coto-Montes A et al (2012) Alzheimer’s disease: pathological mechanisms and the
beneficial role of melatonin. J Pineal Res 52:167–202. https:// doi.org/10.1111/j.1600-079X.2011.00937.x
33. Lin L, Huang Q-X, Yang S-S et al (2013) Melatonin in Alzheimer’s disease. Int J Mol Sci 14:14575–14593. doi. org/10.3390/ijms140714575
34. Devi L, Prabhu BM, Galati DF et al (2006) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochon- drial dysfunction. J Neurosci 26:9057–9068. doi.org/10. 1523/JNEUROSCI.1469-06.2006
35. Butterfield DA, Drake J, Pocernich C, Castegna A (2001) Evidence of oxidative damage in Alzheimer’s disease brain: cen- tral role for amyloid beta-peptide. Trends Mol Med 7:548–554
36. Lahiri DK, Ghosh C (1999) Interactions between melatonin, reactive oxygen species, and nitric oxide. Ann N Y Acad Sci 893:325–330
37. Matsubara E, Bryant-Thomas T, Pacheco Quinto J et al (2003) Melatonin increases survival and inhibits oxidative and amyloid pa- thology in a transgenic model of Alzheimer’s disease. J Neurochem 85:1101–1108. doi.org/10.1046/j.1471-4159.2003.01654.x
38. Lahiri DK, Chen D, Ge Y-W et al (2004) Dietary supplementation with melatonin reduces levels of amyloid beta-peptides in the murine cerebral cortex. J Pineal Res 36:224–231. doi.org/ 10.1111/j.1600-079X.2004.00121.x
39. Song W, Lahiri DK (1997) Melatonin alters the metabolism of the β-amyloid precursor protein in the neuroendocrine cell line PC12. J Mol Neurosci 9:75–92. doi.org/10.1007/BF02736852
40. Zhang Y, Wang Z, Wang Q et al (2004) Melatonin attenuates beta- amyloid-induced inhibition of neurofilament expression. Acta Pharmacol Sin 25:447–451
41. Olivieri G, Hess C, Savaskan E et al (2001) Melatonin protects SHSY5Y neuroblastoma cells from cobalt-induced oxidative stress, neurotoxicity and increased beta-amyloid secretion. J Pineal Res 31:320–325
42. Zhu LQ, Wang SH, Ling ZQ et al (2004) Effect of inhibiting melatonin biosynthesis on spatial memory retention and tau phos- phorylation in rat. J Pineal Res 37:71–77. doi.org/10.1111/ j.1600-079X.2004.00136.x
43. Corpas R, Griñán-Ferré C, Palomera-Ávalos Vet al (2018) Melatonin induces mechanisms of brain resilience against neurodegeneration. J Pineal Res 65:e12515. doi.org/10.1111/jpi.12515
44. Kang J-E, Lim MM, Bateman RJ et al (2009) Amyloid-β dynam- ics are regulated by orexin and the sleep-wake cycle. Science (80-) 326:1005–1007. doi.org/10.1126/science.1180962
45. Moe V, Larsen P (1995) Symposium: Cognitive processes and sleep disturbances: sleep/wake patterns in Alzheimer’s disease: relationships with cognition and function. J Sleep Res 4:15–20
46. Brusco LI, Márquez M, Cardinali DP (1998) Monozygotic twins with Alzheimer’s disease treated with melatonin: case report. J Pineal Res 25:260–263
47. Brusco L, Fainstein I, Márquez M, Cardinali D (1999) Effect of melatonin in selected populations of sleep-disturbed patients. Neurosignals 8:126–131. doi.org/10.1159/000014580
48. Brusco LI, Márquez M, Cardinali DP (2000) Melatonin treatment stabilizes chronobiologic and cognitive symptoms in Alzheimer’s disease. Neuro Endocrinol Lett 21:39–42
49. Poeggeler B, Miravalle L, Zagorski MG et al (2001) Melatonin re- verses the profibrillogenic activity of apolipoprotein E4 on the Alzheimer amyloid Abeta peptide. Biochemistry 40:14995–15001
50. Feng Z, Chang Y, Cheng Y et al (2004) Melatonin alleviates be- havioral deficits associated with apoptosis and cholinergic system dysfunction in the APP 695 transgenic mouse model of Alzheimer’s disease. J Pineal Res 37:129–136. doi.org/10. 1111/j.1600-079X.2004.00144.x
51. Wang X-C, Zhang J, Yu X et al (2005) Prevention of isoproterenol-induced tau hyperphosphorylation by melatonin in the rat. Sheng Li Xue Bao 57:7–12
52. McCurry SM, Reynolds CF, Ancoli-Israel S et al (2000) Treatment of sleep disturbance in Alzheimer’s disease. Sleep Med Rev 4:603–628. doi.org/10.1053/smrv.2000.0127
53. Lerner AB, Case JD, Takahashi Y et al (1958) Isolation of mela- tonin, the pineal gland factor that lightens melanocytes 1. J Am Chem Soc 80:2587–2587. doi.org/10.1021/ja01543a060
54. Ueck M, Wake K (1977) The pinealocyte–a paraneuron? A review Arch Histol Jpn 40(Suppl):261–278
55. Stehle JH, Saade A, Rawashdeh O et al (2011) A survey of mo- lecular details in the human pineal gland in the light of phylogeny, structure, function and chronobiological diseases. J Pineal Res 51: 17–43. doi.org/10.1111/j.1600-079X.2011.00856.x
56. Yonei Y, Hattori A, Tsutsui K, et al (2010) Effects of melatonin: basics studies and clinical applications
57. Stokkan K-A, Reiter RJ (1994) Melatonin rhythms in Arctic urban residents. J Pineal Res 16:33–36. doi.org/10.1111/j.1600- 079X.1994.tb00079.x
58. Sayler A, Wolfson A (1969) Hydroxyindole-o-methyl transferase (HIOMT) activity in the Japanese quail in relation to sexual mat- uration and light. Neuroendocrinology 5:322–332. doi.org/ 10.1159/000121892
59. Pévet P (2002) Melatonin. Dialogues Clin Neurosci 4:57–72
60. Armstrong SM, Redman JR (1991) Melatonin: a chronobiotic with anti-aging properties? Med Hypotheses 34:300–309
61. Kunz D (2004) Chronobiotic protocol and circadian sleep propen- sity index: new tools for clinical routine and research on melatonin and sleep. Pharmacopsychiatry 37:139–146. doi.org/10. 1055/s-2004-827167
62. Maestroni GJ, Conti A (1989) Beta-endorphin and dynorphin mimic the circadian immunoenhancing and anti-stress effects of melatonin. Int J Immunopharmacol 11:333–340
63. Benot S, Goberna R, Reiter RJ et al (1999) Physiological levels of melatonin contribute to the antioxidant capacity of human serum. J Pineal Res 27:59–64
64. Yon J-H, Carter LB, Reiter RJ, Jevtovic-Todorovic V (2006) Melatonin reduces the severity of anesthesia-induced apoptotic neurodegeneration in the developing rat brain. Neurobiol Dis 21: 522–530. doi.org/10.1016/j.nbd.2005.08.011
65. Hardeland R (2013) Melatonin and the theories of aging: a critical appraisal of melatonin’s role in antiaging mechanisms. J Pineal Res 55:N/a. doi.org/10.1111/jpi.12090
66. Thomas JN, Smith-Sonneborn J (1997) Supplemental melatonin increases clonal lifespan in the protozoan Paramecium tetraurelia. J Pineal Res 23:123–130. doi.org/10.1111/j.1600-079X. 1997.tb00344.x
67. Pierpaoli W, Dall’Ara A, Pedrinis E, Regelson W (1991) The pineal control of aging. The effects of melatonin and pineal grafting on the survival of older mice. Ann N Y Acad Sci 621: 291–313
68. Bonilla E, Medina-Leendertz S, Díaz S (2002) Extension of life span and stress resistance of Drosophila melanogaster by long- term supplementation with melatonin. Exp Gerontol 37:629–638
69. Masana MI, Dubocovich ML (2001) Melatonin receptor signal- ing: finding the path through the dark. Sci Signal 2001:pe39– pe39. doi.org/10.1126/stke.2001.107.pe39
70. Cristòfol R, Porquet D, Corpas R et al (2012) Neurons from senescence-accelerated SAMP8 mice are protected against frailty by the sirtuin 1 promoting agents melatonin and resveratrol. J Pineal Res 52:271–281. doi.org/10.1111/j.1600-079X. 2011.00939.x
71. Chang H-M, Wu U-I, Lan C-T (2009) Melatonin preserves lon- gevity protein (sirtuin 1) expression in the hippocampus of total sleep-deprived rats. J Pineal Res 47:211–220. doi.org/10. 1111/j.1600-079X.2009.00704.x
72. Wang J, Fivecoat H, Ho L et al (2010) The role of Sirt1: at the crossroad between promotion of longevity and protection against
Alzheimer’s disease neuropathology. Biochim Biophys Acta, Proteins Proteomics 1804:1690–1694. doi.org/10.1016/j. bbapap.2009.11.015
73. Albani D, Polito L, Batelli S et al (2009) The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by α-synuclein or amyloid-β (1-42) pep- tide. J Neurochem 110:1445–1456. doi.org/10.1111/j. 1471-4159.2009.06228.x
74. Julien C, Tremblay C, Émond V et al (2009) Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol 68:48–58. doi.org/10.1097/ NEN.0b013e3181922348
75. Qin W, Yang T, Ho L et al (2006) Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem 281: 21745–21754. doi.org/10.1074/jbc.M602909200
76. Tippmann F, Hundt J, Schneider A et al (2009) Up-regulation of the α-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J 23:1643–1654. doi.org/10.1096/fj.08-121392
77. West RL, Lee JM, Maroun LE (1995) Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J Mol Neurosci 6:141–146. doi.org/10. 1007/BF02736773
78. Ding H, Dolan PJ, Johnson GVW (2008) Histone deacetylase 6 in- teracts with the microtubule-associated protein tau. J Neurochem 106: 2119–2130. doi.org/10.1111/j.1471-4159.2008.05564.x
79. Jenwitheesuk A, Nopparat C, Mukda S et al (2014) Melatonin regu- lates aging and neurodegeneration through energy metabolism, epige- netics, autophagy and circadian rhythm pathways. Int J Mol Sci 15: 16848–16884. doi.org/10.3390/ijms150916848
80. Van Someren EJ (2000) Circadian and sleep disturbances in the elderly. Exp Gerontol 35:1229–1237
81. Foley DJ, Monjan AA, Brown SL et al (1995) Sleep complaints among elderly persons: an epidemiologic study of three commu- nities. Sleep 18:425–432
82. Huang Y-L, Liu R-Y, Wang Q-S et al (2002) Age-associated dif- ference in circadian sleep-wake and rest-activity rhythms. Physiol Behav 76:597–603
83. McCurry SM, Logsdon RG, Teri L et al (1999) Characteristics of sleep disturbance in community-dwelling Alzheimer’s disease pa- tients. J Geriatr Psychiatry Neurol 12:53–59. doi.org/10. 1177/089198879901200203
84. Bonanni E, Maestri M, Tognoni G et al (2005) Daytime sleepiness in mild and moderate Alzheimer’s disease and its relationship with cognitive impairment. J Sleep Res 14:311–317. doi.org/10. 1111/j.1365-2869.2005.00462.x
85. Reppert SM, Weaver DR (2002) Coordination of circadian timing in mammals. Nature 418:935–941. doi.org/10.1038/ nature00965
86. Haugh RM, Markesbery WR (1983) Hypothalamic astrocytoma. Syndrome of hyperphagia, obesity, and disturbances of behavior and endocrine and autonomic function. Arch Neurol 40:560–563
87. Kalsbeek A, van Heerikhuize JJ, Wortel J, Buijs RM (1996) A diurnal rhythm of stimulatory input to the hypothalamo-pituitary-adrenal sys- tem as revealed by timed intrahypothalamic administration of the vasopressin V1 antagonist. J Neurosci 16:5555–5565
88. Hofman MA, Swaab DF (1995) Influence of aging on the seasonal rhythm of the vasopressin-expressing neurons in the human su- prachiasmatic nucleus. Neurobiol Aging 16:965–971. doi. org/10.1016/0197-4580(95)02016-0
89. Swaab DF, Roozendaal B, Ravid R et al (1987) Suprachiasmatic nucleus in aging, Alzheimer’s disease, transsexuality and Prader- Willi syndrome. Prog Brain Res 72:301–310
90. Zhou J-N, Swaab DF (1999) Activation and degeneration during aging: a morphometric study of the human hypothalamus.
Microsc Res Tech 44:36–48. doi.org/10.1002/(SICI)1097- 0029(19990101)44:1<36::AID-JEMT5>3.0.CO;2-F
91. Zhou JN, Hofman MA, Swaab DF VIP neurons in the human SCN in relation to sex, age, and Alzheimer’s disease. Neurobiol Aging 16:571–576
92. Swaab DF, Fliers E, Partiman TS (1985) The suprachiasmatic nucleus of the human brain in relation to sex, age and senile dementia. Brain Res 342:37–44
93. Swaab DF, Grundke-Iqbal I, Iqbal K et al (1992) Tau and ubiquitin in the human hypothalamus in aging and Alzheimer’s disease. Brain Res 590:239–249
94. Liu RY, Zhou JN, Hoogendijk WJ et al (2000) Decreased vaso- pressin gene expression in the biological clock of Alzheimer dis- ease patients with and without depression. J Neuropathol Exp Neurol 59:314–322
95. Lowrey PL, Takahashi JS (2000) Genetics of the mammalian cir- cadian system: photic entrainment, circadian pacemaker mecha- nisms, and posttranslational regulation. Annu Rev Genet 34:533–
562. doi.org/10.1146/annurev.genet.34.1.533
96. Yoo S-H, Yamazaki S, Lowrey PL et al (2004) PERIOD2:: LUCIFERASE real-time reporting of circadian dynamics reveals per- sistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci 101:5339–5346. doi.org/10.1073/pnas.0308709101
97. Steeves TDL, King DP, Zhao Yet al (1999) Molecular cloning and characterization of the HumanCLOCKGene: expression in the suprachiasmatic nuclei. Genomics 57:189–200. doi.org/ 10.1006/geno.1998.5675
98. Simonneaux V, Poirel V-J, Garidou M-L et al (2004) Daily rhythm and regulation of clock gene expression in the rat pineal gland. Brain Res Mol Brain Res 120:164–172
99. Kolker DE, Fukuyama H, Huang DS et al (2003) Aging alters circadian and light-induced expression of clock genes in golden hamsters. J Biol Rhythm 18:159–169. doi.org/10.1177/ 0748730403251802
100. Yamazaki S, Straume M, Tei H et al (2002) Effects of aging on central and peripheral mammalian clocks. Proc Natl Acad Sci 99: 10801–10806. doi.org/10.1073/pnas.152318499
101. Claustrat B, Brun J, Chazot G (2005) The basic physiology and pathophysiology of melatonin. Sleep Med Rev 9:11–24. https:// doi.org/10.1016/j.smrv.2004.08.001
102. Wu Y-H, Fischer DF, Kalsbeek A et al (2006) Pineal clock gene oscillation is disturbed in Alzheimer’s disease, due to functional disconnection from the Bmaster clock^. FASEB J 20:1874–1876. doi.org/10.1096/fj.05-4446fje
103. Song H, Moon M, Choe HK et al (2015) Aβ-induced degradation of BMAL1 and CBP leads to circadian rhythm disruption in Alzheimer’s disease. Mol Neurodegener 10:13. doi.org/ 10.1186/s13024-015-0007-x
104. Reppert SM, Weaver DR, Ebisawa T (1994) Cloning and charac- terization of a mammalian melatonin receptor that mediates repro- ductive and circadian responses. Neuron 13:1177–1185
105. Ekmekcioglu C (2006) Melatonin receptors in humans: biological role and clinical relevance. Biomed Pharmacother 60:97–108. doi.org/10.1016/j.biopha.2006.01.002
106. Weaver DR, Reppert SM (1996) The Mel1a melatonin receptor gene is expressed in human suprachiasmatic nuclei. Neuroreport 28:109–11
107. Thomas L, Purvis CC, Drew JE et al (2002) Melatonin receptors in human fetal brain: 2-[(125)I]iodomelatonin binding and MT1 gene expression. J Pineal Res 33:218–224
108. Wu Y-H, Zhou J-N, Balesar R et al (2006) Distribution of MT1 melatonin receptor immunoreactivity in the human hypothalamus and pituitary gland: colocalization of MT1 with vasopressin, oxy- tocin, and corticotropin-releasing hormone. J Comp Neurol 499: 897–910. doi.org/10.1002/cne.21152
109. Wu Y-H, Zhou J-N, Van Heerikhuize J et al (2007) Decreased MT1 melatonin receptor expression in the suprachiasmatic nucle- us in aging and Alzheimer’s disease. Neurobiol Aging 28:1239– 1247. doi.org/10.1016/j.neurobiolaging.2006.06.002
110. Liu C, Weaver DR, Jin X et al (1997) Molecular dissection of two distinct actions of melatonin on the suprachiasmatic circadian clock. Neuron 19:91–102
111. Zhou J-N, Liu R-Y, Kamphorst W et al (2003) Early neuropatho- logical Alzheimer’s changes in aged individuals are accompanied by decreased cerebrospinal fluid melatonin levels. J Pineal Res 35: 125–130
112. Liu R-Y, Zhou J-N, van Heerikhuize J et al (1999) Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer’s disease, and apolipoprotein E-ε4/4 genotype 1. J Clin Endocrinol Metab 84:323–327. doi.org/10.1210/ jcem.84.1.5394
113. Reiter RJ, Tan DX, Manchester LC, El-Sawi MR (2002) Melatonin reduces oxidant damage and promotes mitochondrial respiration: im- plications for aging. Ann N Y Acad Sci 959:238–250
114. Reiter RJ, Tan D, Osuna C, Gitto E (2000) Actions of melatonin in the reduction of oxidative stress. J Biomed Sci 7:444–458. https:// doi.org/10.1159/000025480
115. Pappolla MA, Chyan Y-J, Poeggeler B et al (2000) An assessment of the antioxidant and the antiamyloidogenic properties of mela- tonin: implications for Alzheimer’s disease. J Neural Transm 107: 203–231. doi.org/10.1007/s007020050018
116. Wang J, Wang Z (2006) Role of melatonin in Alzheimer-like neu- rodegeneration1. Acta Pharmacol Sin 27:41–49. doi.org/ 10.1111/j.1745-7254.2006.00260.x
117. Dubocovich ML, Rivera-Bermudez MA, Gerdin MJ, Masana MI (2003) Molecular pharmacology, regulation and function of mam- malian melatonin receptors. Front Biosci 8:d1093–d1108
118. Reppert SM, Godson C, Mahle CD et al (1995) Molecular char- acterization of a second melatonin receptor expressed in human retina and brain: The Mel1b melatonin receptor. Proc Natl Acad Sci 92:8734–8738. doi.org/10.1073/pnas.92.19.8734
119. Lockley SW, Skene DJ, James K et al (2000) Melatonin adminis- tration can entrain the free-running circadian system of blind sub- jects. J Endocrinol 164:R1–R6
120. Sack RL, Brandes RW, Kendall AR, Lewy AJ (2000) Entrainment of free-running circadian rhythms by melatonin in blind people. N Engl J Med 343:1070–1077. doi.org/10.1056/ NEJM200010123431503
121. Skene DJ (2003) Optimization of light and melatonin to phase- shift human circadian rhythms. J Neuroendocrinol 15:438–441
122. Swaab DF (Dick F (2003) The human hypothalamus : basic and clinical aspects. Elsevier
123. Arendt J, Skene DJ, Middleton B et al (1997) Efficacy of melato- nin treatment in jet lag, shift work, and blindness. J Biol Rhythm 12:604–617. doi.org/10.1177/074873049701200616
124. Duffy JF, Zeitzer JM, Czeisler CA (2007) Decreased sensitivity to phase-delaying effects of moderate intensity light in older subjects. Neurobiol Aging 28:799–807. doi.org/10.1016/j. neurobiolaging.2006.03.005
125. Herljevic M, Middleton B, Thapan K, Skene D (2005) Light- induced melatonin suppression: age-related reduction in response to short wavelength light. Exp Gerontol 40:237–242. doi. org/10.1016/j.exger.2004.12.001
126. Shochat T, Martin J, Marler M, Ancoli-Israel S (2000) Illumination levels in nursing home patients: effects on sleep and activity rhythms. J Sleep Res 9:373–379
127. Mishima K, Okawa M, Shimizu T, Hishikawa Y (2001) Diminished melatonin secretion in the elderly caused by insuffi- cient environmental illumination 1. J Clin Endocrinol Metab 86: 129–134. doi.org/10.1210/jcem.86.1.7097
128. Ancoli-Israel S, Klauber MR, Jones DW et al (1997) Variations in circadian rhythms of activity, sleep, and light exposure related to dementia in nursing-home patients. Sleep 20:18–23
129. Ohashi Y, Okamoto N, Uchida K et al (1999) Daily rhythm of serum melatonin levels and effect of light exposure in patients with dementia of the Alzheimer’s type. Biol Psychiatry 45: 1646–1652
130. Laurin D, Verreault R, Lindsay J et al (2001) Physical activity and risk of cognitive impairment and dementia in elderly persons. Arch Neurol 58:498–504
131. Barnes LL, Mendes de Leon CF, Wilson RS et al (2004) Social resources and cognitive decline in a population of older African Americans and whites. Neurology 63:2322–2326
132. Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurode- generation: lessons from the Alzheimer’s amyloid β-peptide. Nat Rev Mol Cell Biol 8:101–112. doi.org/10.1038/nrm2101
133. Selkoe DJ (2002) Alzheimer’s disease is a synaptic failure. Science (80- ) 298:789–791. doi.org/10.1126/science. 1074069
134. He H, Dong W, Huang F (2010) Anti-amyloidogenic and anti- apoptotic role of melatonin in Alzheimer disease. Curr Neuropharmacol 8:211 –217. doi.org/10.2174/ 157015910792246137
135. Wang X-C, Zhang Y-C, Chatterjie N et al (2008) Effect of mela- tonin and melatonylvalpromide on β-amyloid and neurofilaments in N2a cells. Neurochem Res 33:1138–1144. doi.org/10. 1007/s11064-007-9563-y
136. Jin N, Yin X, Yu D et al (2015) Truncation and activation of GSK- 3β by calpain I: a molecular mechanism links to tau hyperphosphorylation in Alzheimer’s disease. Sci Rep 5:8187. doi.org/10.1038/srep08187
137. van Eersel J, Ke YD, Liu X et al (2010) Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc Natl Acad Sci 107:13888– 13893. doi.org/10.1073/pnas.1009038107
138. Shimohama S, Tanino H, Fujimoto S (1999) Changes in caspase expression in Alzheimer’s disease: comparison with development and aging. Biochem Biophys Res Commun 256:381–384. https:// doi.org/10.1006/bbrc.1999.0344
139. Tesco G, Koh YH, Kang EL et al (2007) Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron 54:721–737. doi.org/10.1016/j.neuron.2007.05.012
140. Oh S, Gwak J, Park S, Yang CS (2014) Green tea polyphenol EGCG suppresses Wnt/β-catenin signaling by promoting GSK- 3β- and PP2A-independent β-catenin phosphorylation/degrada- tion. BioFactors 40:586–595. doi.org/10.1002/biof.1185
141. Yang C-C, Kuai X-X, Li Y-L et al (2013) Cornel iridoid glycoside attenuates tau hyperphosphorylation by inhibition of PP2A de- methylation. Evid Based Complement Alternat Med 2013: 108486. doi.org/10.1155/2013/108486
142. Louneva N, Cohen JW, Han L-Y et al (2008) Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer’s disease. Am J Pathol 173:1488–1495. doi.org/10.2353/ ajpath.2008.080434
143. Espino J, Bejarano I, Redondo PC et al (2010) Melatonin reduces apoptosis induced by calcium signaling in human leukocytes: ev- idence for the involvement of mitochondria and Bax activation. J Membr Biol 233:105–118. doi.org/10.1007/s00232-010- 9230-0
144. Ling X, Zhang LM, Lu SD et al (1999) Protective effect of mel- atonin on injuried cerebral neurons is associated with bcl-2 protein over-expression. Zhongguo Yao Li Xue Bao 20:409–414
145. Jang M-H, Jung S-B, Lee M-H et al (2005) Melatonin attenuates amyloid beta25–35-induced apoptosis in mouse microglial BV2 cells. Neurosci Lett 380:26–31. doi.org/10.1016/j.neulet. 2005.01.003
146. Harada J, Sugimoto M (1999) Activation of caspase-3 in beta- amyloid-induced apoptosis of cultured rat cortical neurons. Brain Res 842:311–323
147. Sahab Uddin M, Nasrullah M, Hossain MS et al (2016) Evaluation of nootropic activity of Persicaria flaccida on cognitive perfor- mance, brain antioxidant markers and acetylcholinesterase activity in rats: implication for the management of Alzheimer’s disease. Am J Psychiatry Neurosci 4:26. doi.org/10.11648/j.ajpn. 20160402.12
148. Uddin MS, Asaduzzaman M, Mamun AA, Iqbal MA, Wahid FRR (2016) Neuroprotective activity of Asparagus racemosus Linn. Against ethanol- induced cognitive impairment and oxidative stress in rats brain: auspicious for controlling the risk of Alzheimer’s disease. J Alzheimer’s Dis Park 6:1–10. doi. org/10.4172/2161-0460.1000245
149. Gong Y-H, Hua N, Zang X et al (2018) Melatonin ameliorates Aβ 1-42-induced Alzheimer’s cognitive deficits in mouse model. J Pharm Pharmacol 70:70–80. doi.org/10.1111/jphp.12830
150. O’Neal-Moffitt G, Delic V, Bradshaw PC, Olcese J (2015) Prophylactic melatonin significantly reduces Alzheimer’s neuro- pathology and associated cognitive deficits independent of anti- oxidant pathways in AβPPswe/PS1 mice. Mol Neurodegener 10: 27. doi.org/10.1186/s13024-015-0027-6
151. Cleveland DW, Hwo SY, Kirschner MW (1977) Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol 116:227–247
152. Shipton OA, Leitz JR, Dworzak J et al (2011) Tau protein is required for amyloid {beta}-induced impairment of hippocampal long-term potentiation. J Neurosci 31:1688–1692. doi.org/ 10.1523/JNEUROSCI.2610-10.2011
153. Goedert M, Spillantini MG, Cairns NJ, Crowther RA (1992) Tau proteins of Alzheimer paired helical filaments: abnormal phos- phorylation of all six brain isoforms. Neuron 8:159–168
154. Ferrer I, Gomez-Isla T, Puig B et al (2005) Current advances on different kinases involved in tau phosphorylation, and implica- tions in Alzheimer’s disease and tauopathies. Curr Alzheimer Res 2:3–18
155. Moreira PI, Honda K, Zhu X et al (2006) Brain and brawn: paral- lels in oxidative strength. Neurology 66:S97–S101. doi. org/10.1212/01.wnl.0000192307.15103.83
156. Lee VM, Balin BJ, Otvos L, Trojanowski JQ (1991) A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science 251:675–678
157. Khatoon S, Grundke-Iqbal I, Iqbal K (1992) Brain levels of microtubule-associated protein tau are elevated in Alzheimer’s disease: a radioimmuno-slot-blot assay for nanograms of the pro- tein. J Neurochem 59:750–753
158. Khatoon S, Grundke-Iqbal I, Iqbal K (1994) Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett 351:80–84
159. Costa EJ, Lopes RH, Lamy-Freund MT (1995) Permeability of pure lipid bilayers to melatonin. J Pineal Res 19:123–126
160. Deng Y, Xu G, Duan P et al (2005) Effects of melatonin on wortmannin-induced tau hyperphosphorylation. Acta Pharmacol Sin 26:519–526. doi.org/10.1111/j.1745-7254.2005. 00102.x
161. Pandi-Perumal SR, Cardinali DP (2007) Melatonin : from mole- cules to therapy. Nova Biomedical Books
162. Peng C-X, Hu J, Liu D et al (2013) Disease-modified glycogen synthase kinase-3β intervention by melatonin arrests the patholo- gy and memory deficits in an Alzheimer’s animal model. Neurobiol Aging 34:1555–1563. doi.org/10.1016/j. neurobiolaging.2012.12.010
163. Feng Z, Qin C, Chang Y, Zhang J (2006) Early melatonin supple- mentation alleviates oxidative stress in a transgenic mouse model
of Alzheimer’s disease. Free Radic Biol Med 40:101–109. https:// doi.org/10.1016/j.freeradbiomed.2005.08.014
164. Bhat RV, Budd Haeberlein SL, Avila J (2004) Glycogen synthase kinase 3: a drug target for CNS therapies. J Neurochem 89:1313– 1317. doi.org/10.1111/j.1471-4159.2004.02422.x
165. Eldar-Finkelman H (2002) Glycogen synthase kinase 3: an emerg- ing therapeutic target. Trends Mol Med 8:126–132
166. Li X, Bijur GN, Jope RS (2002) Glycogen synthase kinase-3beta, mood stabilizers, and neuroprotection. Bipolar Disord 4:137–144
167. Bhat RV, Budd SL (2002) GSK3beta signalling: casting a wide net in Alzheimer’s disease. Neurosignals 11:251–261. doi.org/ 10.1159/000067423
168. Krasilnikov MA (2000) Phosphatidylinositol-3 kinase dependent pathways: the role in control of cell growth, survival, and malig- nant transformation. Biochemistry (Mosc) 65:59–67
169. Hoppe JB, Frozza RL, Horn AP et al (2010) Amyloid-β neuro- toxicity in organotypic culture is attenuated by melatonin: involve- ment of GSK-3β, tau and neuroinflammation. J Pineal Res 48: 230–238. doi.org/10.1111/j.1600-079X.2010.00747.x
170. Wang J, Xiao X, Zhang Yet al (2012) Simultaneous modulation of COX-2, p300, Akt, and Apaf-1 signaling by melatonin to inhibit proliferation and induce apoptosis in breast cancer cells. J Pineal Res 53:77–90. doi.org/10.1111/j.1600-079X.2012.00973.x
171. Xue F, Shi C, Chen Q et al (2017) Melatonin mediates protective effects against kainic acid-induced neuronal death through safeguarding ER stress and mitochondrial disturbance. Front Mol Neurosci 10:49. doi.org/10.3389/fnmol.2017.00049
172. Shi C, Zeng J, Li Z et al (2018) Melatonin mitigates kainic acid- induced neuronal tau hyperphosphorylation and memory deficits through alleviating ER stress. Front Mol Neurosci 11(5). https:// doi.org/10.3389/fnmol.2018.00005
173. Medeiros R, Baglietto-Vargas D, LaFerla FM (2011) The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci Ther 17:514–524. doi.org/10.1111/j.1755-5949.2010. 00177.x
174. Salminen A, Kauppinen A, Suuronen T et al (2009) ER stress in Alzheimer’s disease: a novel neuronal trigger for inflammation and Alzheimer’s pathology. J Neuroinflammation 6:41. https:// doi.org/10.1186/1742-2094-6-41
175. Hoozemans JJM, Veerhuis R, Van Haastert ES et al (2005) The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol 110:165–172. doi.org/10.1007/s00401- 005-1038-0
176. Kim JS, Heo RW, Kim H et al (2014) Salubrinal, ER stress inhib- itor, attenuates kainic acid-induced hippocampal cell death. J Neural Transm 121:1233–1243. doi.org/10.1007/s00702- 014-1208-0
177. Zhang X-M, Zhu J (2011) Kainic acid-induced neurotoxicity: targeting glial responses and glia-derived cytokines. Curr Neuropharmacol 9:388–398. doi.org/10.2174/ 157015911795596540
178. Fernández-Bachiller MI, Pérez C, Campillo NE et al (2009) Tacrine-melatonin hybrids as multifunctional agents for Alzheimer’s disease, with cholinergic, antioxidant, and neuropro- tective properties. ChemMedChem 4:828–841. doi.org/10. 1002/cmdc.200800414
179. Spuch C, Antequera D, Isabel Fernandez-Bachiller M et al (2010) A new Tacrine–melatonin hybrid reduces amyloid burden and behavioral deficits in a mouse model of Alzheimer’s disease. Neurotox Res 17:421–431. doi.org/10.1007/s12640-009- 9121-2
180. Lau WWI, Ng JKY, Lee MMK et al (2012) Interleukin-6 autocrine signaling mediates melatonin MT 1/2 receptor-induced STAT3 Tyr 705 phosphorylation. J Pineal Res 52:477–489. doi.org/10. 1111/j.1600-079X.2011.00965.x
181. Shen Y, Zhang G, Liu L, Xu S (2007) Suppressive effects of melatonin on amyloid-β-induced glial activation in rat hippocam- pus. Arch Med Res 38:284–290. doi.org/10.1016/j.arcmed. 2006.10.007
182. Paouri E, Tzara O, Kartalou G-I et al (2017) Peripheral tumor necrosis factor-alpha (TNF-α) modulates amyloid pathology by regulating blood-derived immune cells and glial response in the brain of AD/TNF transgenic mice. J Neurosci 37:5155–5171. doi.org/10.1523/JNEUROSCI.2484-16.2017
183. McGeer PL, McGeer EG (1995) The inflammatory response sys- tem of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev 21:195–218
184. Uddin MS, Kabir MT, Jakaria M, et al (2019) Endothelial PPARγ is crucial for averting age-related vascular dysfunction by stalling oxidative stress and ROCK. Neurotox Res doi.org/10. 1007/s12640-019-00047-5
185 . Beg um MM, Islam A , Begum R e t a l ( 2019 ) Ethnopharmacological inspections of organic extract of Oroxylum indicum in rat models: a promising natural gift. Evidence-Based Complement Altern Med 2019:1–13. https:// doi.org/10.1155/2019/1562038
186. Hüll M, Berger M, Volk B, Bauer J (1996) Occurrence of interleukin-6 in cortical plaques of Alzheimer’s disease patients may precede transformation of diffuse into neuritic plaques. Ann N Y Acad Sci 777:205–212
187. Campbell IL, Abraham CR, Masliah E et al (1993) Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc Natl Acad Sci U S A 90:10061–10065
188. Strauss S, Bauer J, Ganter U et al (1992) Detection of interleukin-6 and alpha 2-macroglobulin immunoreactivity in cortex and hippo- campus of Alzheimer’s disease patients. Lab Investig 66:223–230
189. Selkoe DJ (1996) Amyloid beta-protein and the genetics of Alzheimer’s disease. J Biol Chem 271:18295–18298
190. Zhang L, Zhao B, Yew DT et al (1997) Processing of Alzheimer’s amyloid precursor protein during H2O2-induced apoptosis in hu- man neuronal cells. Biochem Biophys Res Commun 235:845– 848. doi.org/10.1006/BBRC.1997.6698
191. Hsiao K, Chapman P, Nilsen S et al (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274:99–102
192. Liu X, Xu Y, Chen S et al (2014) Rescue of proinflammatory cytokine-inhibited chondrogenesis by the antiarthritic effect of melatonin in synovium mesenchymal stem cells via suppression of reactive oxygen species and matrix metalloproteinases. Free Radic Biol Med 68:234–246. doi.org/10.1016/j. freeradbiomed.2013.12.012
193. Anuthakoengkun A, Itharat A (2014) Inhibitory effect on nitric oxide production and free radical scavenging activity of Thai me- dicinal plants in osteoarthritic knee treatment. J Med Assoc Thail 97(Suppl 8):S116–S124
194. Guenther AL, Schmidt SI, Laatsch H et al (2005) Reactions of the melatonin metabolite AMK (N1-acetyl-5-methoxykynuramine) with reactive nitrogen species: formation of novel compounds, 3-acetamidomethyl-6-methoxycinnolinone and 3-nitro-AMK. J Pineal Res 39:251–260. doi.org/10.1111/j.1600-079X. 2005.00242.x
195. Tan D, Reiter RJ, Manchester LC et al (2002) Chemical and phys- ical properties and potential mechanisms: melatonin as a broad spectrum antioxidant and free radical scavenger. Curr Top Med Chem 2:181–197
196. Antolín I, Rodríguez C, Saínz RM et al (1996) Neurohormone melatonin prevents cell damage: effect on gene expression for antioxidant enzymes. FASEB J 10:882–890
197. Shen YX, Xu SY, Wei W et al (2002) The protective effects of melatonin from oxidative damage induced by amyloid beta- peptide 25-35 in middle-aged rats. J Pineal Res 32:85–89
198. Shen Y-X, Xu S-Y, Wei Wet al (2002) Melatonin reduces memory changes and neural oxidative damage in mice treated with D-ga- lactose. J Pineal Res 32:173–178
199. Wakatsuki A, Okatani Y, Shinohara K et al (2001) Melatonin protects fetal rat brain against oxidative mitochondrial damage. J Pineal Res 30:22–28
200. Smith MA, Sayre LM, Monnier VM, Perry G (1995) Radical AGEing in Alzheimer’s disease. Trends Neurosci 18:172–176
201. Uddin MS, Mamun AA, Kabir MT, et al (2017) Neurochemistry of neurochemicals: messengers of brain functions. Journal of Intellectual Disability – Diagnosis and Treatment 5:137-151. doi.org/10.6000/2292-2598.2017.05.04.6
202. Mecocci P, MacGarvey U, Beal MF (1994) Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol 36:747–751. doi.org/10.1002/ana.410360510
203. Weldon DT, Rogers SD, Ghilardi JR et al (1998) Fibrillar beta- amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo. J Neurosci 18:2161–2173
204. Du Yan S, Zhu H, Fu J et al (1997) Amyloid-beta peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci U S A 94:5296–5301
205. Yan SD, Roher A, Chaney M et al (2000) Cellular cofactors po- tentiating induction of stress and cytotoxicity by amyloid beta- peptide. Biochim Biophys Acta 1502:145–157
206. Perini G, Della-Bianca V, Politi V et al (2002) Role of p75 neurotrophin receptor in the neurotoxicity by beta-amyloid pep- tides and synergistic effect of inflammatory cytokines. J Exp Med 195:907–918
207. Walker DG, Lue L-F, Beach TG (2002) Increased expression of the urokinase plasminogen-activator receptor in amyloid beta peptide-treated human brain microglia and in AD brains. Brain Res 926:69–79
208. Brandt R, Hundelt M, Shahani N (2005) Tau alteration and neu- ronal degeneration in tauopathies: mechanisms and models. Biochim Biophys Acta Mol basis Dis 1739:331–354. doi. org/10.1016/j.bbadis.2004.06.018
209. Takuma K, Yan SS, Stern DM, Yamada K (2005) Mitochondrial dysfunction, endoplasmic reticulum stress, and apoptosis in Alzheimer’s disease. J Pharmacol Sci 97:312–316
210. Guermonprez L, Ducrocq C, Gaudry-Talarmain YM (2001) Inhibition of acetylcholine synthesis and tyrosine nitration in- duced by peroxynitrite are differentially prevented by antioxi- dants. Mol Pharmacol 60:838–846
211. Hensley K, Carney JM, Mattson MP et al (1994) A model for beta- amyloid aggregation and neurotoxicity based on free radical gen- eration by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci U S A 91:3270–3274
212. Cardoso SM, Santana I, Swerdlow RH, Oliveira CR (2004) Mitochondria dysfunction of Alzheimer’s disease cybrids en- hances Aβ toxicity. J Neurochem 89:1417–1426. doi.org/ 10.1111/j.1471-4159.2004.02438.x
213. Sheehan JP, Swerdlow RH, Miller SW et al (1997) Calcium ho- meostasis and reactive oxygen species production in cells trans- formed by mitochondria from individuals with sporadic Alzheimer’s disease. J Neurosci 17:4612–4622
214. Swerdlow RH, Parks JK, Cassarino DS et al (1997) Cybrids in Alzheimer’s disease: a cellular model of the disease? Neurology 49:918–925
215. Beal MF (1998) Mitochondrial dysfunction in neurodegenerative diseases. Biochim Biophys Acta 1366:211–223
216. Beal MF (2000) Oxidative metabolism. Ann N Y Acad Sci 924: 164–169
217. Thiffault C, Bennett JP (2005) Cyclical mitochondrial deltapsiM fluctuations linked to electron transport, F0F1 ATP-synthase and
mitochondrial Na+/ca+2 exchange are reduced in Alzheimer’s disease cybrids. Mitochondrion 5:109–119. doi.org/10. 1016/j.mito.2004.12.002
218. Trimmer PA, Keeney PM, Borland MK et al (2004) Mitochondrial abnormalities in cybrid cell models of sporadic Alzheimer’s dis- ease worsen with passage in culture. Neurobiol Dis 15:29–39
219. Haughey NJ, Liu D, Nath A et al (2002) Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid β-peptide by amyloid β-peptide. NeuroMolecular Med 1:125– 136. doi.org/10.1385/NMM:1:2:125
220. Donovan MH, Yazdani U, Norris RD et al (2006) Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer’s disease. J Comp Neurol 495:70–83. doi.org/ 10.1002/cne.20840
221. Wen PH, Hof PR, Chen X et al (2004) The presenilin-1 familial Alzheimer disease mutant P117L impairs neurogenesis in the hip- pocampus of adult mice. Exp Neurol 188:224–237. doi. org/10.1016/j.expneurol.2004.04.002
222. Mu Y, Gage FH (2011) Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener 6:85. doi. org/10.1186/1750-1326-6-85
223. Hamilton A, Holscher C (2012) The effect of ageing on neurogenesis and oxidative stress in the APPswe/PS1deltaE9 mouse model of Alzheimer’s disease. Brain Res 1449:83–93. doi.org/10.1016/j.brainres.2012.02.015
224. Rodríguez JJ, Jones VC, Tabuchi M et al (2008) Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS One 3:e2935. doi. org/10.1371/journal.pone.0002935
225. Martinez-Canabal A (2014) Reconsidering hippocampal neurogenesis in Alzheimer’s disease. Front Neurosci 8:147. doi.org/10.3389/fnins.2014.00147
226. Haughey NJ, Nath A, Chan SL et al (2002) Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural pro- genitor cell homeostasis, in models of Alzheimer’s disease. J Neurochem 83:1509–1524
227. Ghosal K, Stathopoulos A, Pimplikar SW (2010) APP intracellu- lar domain impairs adult neurogenesis in transgenic mice by in- ducing neuroinflammation. PLoS One 5:e11866. doi.org/ 10.1371/journal.pone.0011866
228. Porayette P, Gallego MJ, Kaltcheva MM et al (2009) Differential processing of amyloid-β precursor protein directs human embry- onic stem cell proliferation and differentiation into neuronal pre- cursor cells. J Biol Chem 284:23806–23817. doi.org/10. 1074/jbc.M109.026328
229. Phiel CJ, Wilson CA, Lee VM-Y, Klein PS (2003) GSK-3α reg- ulates production of Alzheimer’s disease amyloid-β peptides. Nature 423:435–439. doi.org/10.1038/nature01640
230. Zhou F, Zhang L, Wang A et al (2008) The association of GSK3β with E2F1 facilitates nerve growth factor-induced neural cell dif- ferentiation. J Biol Chem 283:14506–14515. doi.org/10. 1074/jbc.M706136200
231. Lie D-C, Colamarino SA, Song H-J et al (2005) Wnt signalling regulates adult hippocampal neurogenesis. Nature 437:1370– 1375. doi.org/10.1038/nature04108
232. Anderton BH, Dayanandan R, Killick R, Lovestone S (2000) Does dysregulation of the notch and wingless/Wnt pathways underlie the pathogenesis of Alzheimer’s disease? Mol Med Today 6:54–59
233. Alvarez AR, Godoy JA, Mullendorff K et al (2004) Wnt-3a over- comes β-amyloid toxicity in rat hippocampal neurons. Exp Cell Res 297:186–196. doi.org/10.1016/j.yexcr.2004.02.028
234. Ramírez-Rodríguez G, Vega-Rivera NM, Benítez-King G et al (2012) Melatonin supplementation delays the decline of adult hip- pocampal neurogenesis during normal aging of mice. Neurosci Lett 530:53–58. doi.org/10.1016/j.neulet.2012.09.045
235. Lacoste B, Angeloni D, Dominguez-Lopez S et al (2015) Anatomical and cellular localization of melatonin MT 1 and MT 2 receptors in the adult rat brain. J Pineal Res 58:397–417. https:// doi.org/10.1111/jpi.12224
236. Domínguez-Alonso A, Ramírez-Rodríguez G, Benítez-King G (2012) Melatonin increases dendritogenesis in the hilus of hippo- campal organotypic cultures. J Pineal Res 52:427–436. doi. org/10.1111/j.1600-079X.2011.00957.x
237. Liu J, Somera-Molina KC, Hudson RL, Dubocovich ML (2013) Melatonin potentiates running wheel-induced neurogenesis in the dentate gyrus of adult C3H/HeN mice hippocampus. J Pineal Res 54:222–231. doi.org/10.1111/jpi.12023
238. Hong Y, Palaksha KJ, Park K et al (2010) REVIEW ARTICLE: Melatonin plus exercise-based neurorehabilitative therapy for spi- nal cord injury. J Pineal Res 49:201–209. doi.org/10.1111/j. 1600-079X.2010.00786.x
239. Moriya T, Horie N, Mitome M, Shinohara K (2007) Melatonin influences the proliferative and differentiative activity of neural stem cells. J Pineal Res 42:411–418. doi.org/10.1111/j. 1600-079X.2007.00435.x
240. Ramírez-Rodríguez G, Klempin F, Babu H et al (2009) Melatonin modulates cell survival of new neurons in the hippocampus of adult mice. Neuropsychopharmacology 34:2180–2191. https:// doi.org/10.1038/npp.2009.46
241. De Butte M, Pappas BA (2007) Pinealectomy causes hippocampal CA1 and CA3 cell loss: reversal by melatonin supplementation. Neurobiol Aging 28:306–313. doi.org/10.1016/j. neurobiolaging.2005.12.004
242. Rennie K, De Butte M, Pappas BA (2009) Melatonin promotes neurogenesis in dentate gyrus in the pinealectomized rat. J Pineal Res 47:313–317. doi.org/10.1111/j.1600-079X.2009. 00716.x
243. Tocharus C, Puriboriboon Y, Junmanee T et al (2014) Melatonin enhances adult rat hippocampal progenitor cell proliferation via ERK signaling pathway through melatonin receptor. Neuroscience 275:314–321. doi.org/10.1016/j.neuroscience.2014.06.026
244. Sotthibundhu A, Phansuwan-Pujito P, Govitrapong P (2010) Melatonin increases proliferation of cultured neural stem cells obtained from adult mouse subventricular zone. J Pineal Res 49: 291–300. doi.org/10.1111/j.1600-079X.2010.00794.x
245. McArthur AJ, Hunt AE, Gillette MU (1997) Melatonin action and signal transduction in the rat suprachiasmatic circadian clock: ac- tivation of protein kinase C at dusk and Dawn 1. Endocrinology 138:627–634. doi.org/10.1210/endo.138.2.4925
246. Shukla M, Htoo HH, Wintachai P et al (2015) Melatonin stimu- lates the nonamyloidogenic processing of β APP through the pos- itive transcriptional regulation of ADAM10 and ADAM17. J Pineal Res 58:151–165. doi.org/10.1111/jpi.12200
247. Agrawal R, Tyagi E, Shukla R, Nath C (2008) Effect of insulin and melatonin on acetylcholinesterase activity in the brain of amnesic mice. Behav Brain Res 189:381–386. doi.org/10.1016/j. bbr.2008.01.015
248. Ishida A, Furukawa K, Keller JN, Mattson MP (1997) Secreted form of beta-amyloid precursor protein shifts the frequency depen- dency for induction of LTD, and enhances LTP in hippocampal slices. Neuroreport 8:2133–2137
249. Yuan T-F, Gu S, Shan C et al (2015) Oxidative stress and adult neurogenesis. Stem Cell Rev Rep 11:706–709. doi.org/10. 1007/s12015-015-9603-y
250. Sarlak G, Jenwitheesuk A, Chetsawang B, Govitrapong P (2013) Effects of melatonin on nervous system aging: neurogenesis and neurodegeneration. J Pharmacol Sci 123:9–24
251. Uchida K, Okamoto N, Ohara K, Morita Y (1996) Daily rhythm of serum melatonin in patients with dementia of the degenerate type. Brain Res 717:154–159
252. Skene DJ, Vivien-Roels B, Sparks DL et al (1990) Daily variation in the concentration of melatonin and 5-methoxytryptophol in the human pineal gland: effect of age and Alzheimer’s disease. Brain Res 528:170–174
253. Mishima K, Tozawa T, Satoh K et al (1999) Melatonin secretion rhythm disorders in patients with senile dementia of Alzheimer’s type with disturbed sleep–waking. Biol Psychiatry 45:417–421. doi.org/10.1016/S0006-3223(97)00510-6
254. Skene DJ, Swaab DF Melatonin rhythmicity: effect of age and Alzheimer’s disease. Exp Gerontol 38:199–206
255. Reiter RJ, Tan D-X (2002) Role of CSF in the transport of mela- tonin. J Pineal Res 33:61
256. Reiter RJ (1993) The melatonin rhythm: both a clock and a calen- dar. Experientia 49:654–664
257. Cardinali DP, Brusco LI, Liberczuk C, Furio AM (2002) The use of melatonin in Alzheimer’s disease. Neuro Endocrinol Lett 23(Suppl 1):20–23
258. Cardinali DP, Furio AM, Brusco LI (2010) Clinical aspects of melatonin intervention in Alzheimer’s disease progression. Curr Neuropharmacol 8:218–227. doi.org/10.2174/ 157015910792246209
259. Magri F, Locatelli M, Balza G et al (1997) Changes in endocrine orcadian rhythms as markers of physiological and pathological brain aging. Chronobiol Int 14:385–396. doi.org/10.3109/ 07420529709001459
260. Mahlberg R, Kunz D, Sutej I et al (2004) Melatonin treatment of day-night rhythm disturbances and sundowning in Alzheimer dis- ease: an open-label pilot study using actigraphy. J Clin Psychopharmacol 24:456–459
261. Asayama K, Yamadera H, Ito T et al (2003) Double blind study of melatonin effects on the sleep-wake rhythm, cognitive and non- cognitive functions in Alzheimer type dementia. J Nippon Med Sch 70:334–134
262. Hardeland R (2005) Antioxidative protection by melatonin: mul- tiplicity of mechanisms from radical detoxification to radical avoidance. Endocrine 27:119–130
263. Gunasingh Masilamoni J, Philip Jesudason E, Dhandayuthapani S et al (2008) The neuroprotective role of melatonin against amyloid β peptide injected mice. Free Radic Res 42:661–673. doi. org/10.1080/10715760802277388
264. Husson I, Mesplès B, Bac P et al (2002) Melatoninergic neuro- protection of the murine periventricular white matter against neo- natal excitotoxic challenge. Ann Neurol 51:82–92
265. Zhu G, Wang D, Lin Y-H et al (2001) Protein kinase C ϵ sup- presses Aβ production and promotes activation of α-secretase. Biochem Biophys Res Commun 285:997–1006. doi.org/ 10.1006/bbrc.2001.5273
266. Luo Y, Packer L (2006) Oxidative stress and age-related neurode- generation. CRC/Taylor & Francis
267. Pappolla MA, Sos M, Omar RA et al (1997) Melatonin prevents death of neuroblastoma cells exposed to the {Alzheimer} amyloid peptide. J Neurosci 17:1683–1690
268. Guerrero-Muñoz MJ, Castillo-Carranza DL, Kayed R (2014) Therapeutic approaches against common structural features of toxic oligomers shared by multiple amyloidogenic proteins. Biochem Pharmacol 88:468–478. doi.org/10.1016/j.bcp. 2013.12.023
269. Sengupta U, Nilson AN, Kayed R (2016) The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 6:42–49. doi.org/10.1016/j.ebiom.2016.03. 035
270. Walsh DM, Tseng BP, Rydel RE et al (2000) The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry 39:10831–10839
271. Glabe CG (2006) Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging 27:570–
Alzheimer’s disease. J Alzheimers Dis 47:103–6.11 org/10.3233/JAD-150161
doi.
575. doi.org/10.1016/j.neurobiolaging.2005.04.017
272. Chen Y-R, Glabe CG (2006) Distinct early folding and aggrega- tion properties of Alzheimer amyloid-β peptides Aβ40 and Aβ42. J Biol Chem 281:24414–24422. doi.org/10.1074/jbc. M602363200
273. Stys PK, You H, Zamponi GW (2012) Copper-dependent regula- tion of NMDA receptors by cellular prion protein: Implications for neurodegenerative disorders. J Physiol 590:1357–1368. https:// doi.org/10.1113/jphysiol.2011.225276
274. Gu Z, Liu W, Yan Z (2009) β-Amyloid impairs AMPA receptor trafficking and function by reducing ca 2+ /calmodulin-dependent protein kinase II synaptic distribution. J Biol Chem 284:10639– 10649. doi.org/10.1074/jbc.M806508200
275. Pappolla M, Bozner P, Soto C et al (1998) Inhibition of Alzheimer beta-fibrillogenesis by melatonin. J Biol Chem 273:7185–7188
276. Poirier J, Davignon J, Bouthillier D et al (1993) Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet (London, England) 342:697–699
277. Bazoti FN, Tsarbopoulos A, Markides KE, Bergquist J (2005) Study of the non-covalent interaction between amyloid-β- peptide and melatonin using electrospray ionization mass spec- trometry. J Mass Spectrom 40:182–192. doi.org/10.1002/ jms.738
278. Johnstone M, Gearing AJ, Miller KM (1999) A central role for astrocytes in the inflammatory response to beta-amyloid; chemokines, cytokines and reactive oxygen species are produced. J Neuroimmunol 93:182–193
279. Scheff SW, Price DA (2003) Synaptic pathology in Alzheimer’s disease: a review of ultrastructural studies. Neurobiol Aging 24: 1029–1046
280. Li S, Hong S, Shepardson NE et al (2009) Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62:788–801. doi.org/10.1016/j.neuron.2009.05.012
281. Palop JJ, Mucke L (2010) Amyloid-β–induced neuronal dysfunc- tion in Alzheimer’s disease: from synapses toward neural net- works. Nat Neurosci 13:812–818. doi.org/10.1038/nn. 2583
282. Liu D, Yang Q, Li S (2013) Activation of extrasynaptic NMDA receptors induces LTD in rat hippocampal CA1 neurons. Brain Res Bull 93:10–16. doi.org/10.1016/j.brainresbull.2012. 12.003
283. Scheff SW, Price DA, Schmitt FA, Mufson EJ (2006) Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 27:1372–1384. https:// doi.org/10.1016/j.neurobiolaging.2005.09.012
284. Cardinali DP, Golombek DA, Rosenstein RE et al (1997) Melatonin site and mechanism of action: single or multiple? J Pineal Res 23:32–39. doi.org/10.1111/j.1600-079X.1997. tb00332.x
285. Corrales A, Vidal R, García S et al (2014) Chronic melatonin treatment rescues electrophysiological and neuromorphological deficits in a mouse model of Down syndrome. J Pineal Res 56: 51–61. doi.org/10.1111/jpi.12097
286. Thiel G (1993) Synapsin I, synapsin II, and synaptophysin: marker proteins of synaptic vesicles. Brain Pathol 3:87–95
287. Ikeno T, Nelson RJ (2015) Acute melatonin treatment alters den- dritic morphology and circadian clock gene expression in the hip- pocampus of Siberian hamsters. Hippocampus 25:142–148. doi.org/10.1002/hipo.22358
288. Rudnitskaya EA, Muraleva NA, Maksimova KY et al (2015) Melatonin attenuates memory impairment, amyloid-β accumula- tion, and neurodegeneration in a rat model of sporadic
289. Benleulmi-Chaachoua A, Chen L, Sokolina K et al (2016) Protein interactome mining defines melatonin MT 1 receptors as integral component of presynaptic protein complexes of neurons. J Pineal Res 60:95–108. doi.org/10.1111/jpi.12294
290. Liu D, Wei N, Man H-Yet al (2015) The MT2 receptor stimulates axonogenesis and enhances synaptic transmission by activating Akt signaling. Cell Death Differ 22:583–596. doi.org/10. 1038/cdd.2014.195
291. Ali T, Badshah H, Kim TH, Kim MO (2015) Melatonin attenuates D-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF- K B/JNK signaling path- way in aging mouse model. J Pineal Res 58:71–85. doi.org/ 10.1111/jpi.12194
292. Yoo DY, Kim W, Lee CH et al (2012) Melatonin improves d- galactose-induced aging effects on behavior, neurogenesis, and lipid peroxidation in the mouse dentate gyrus via increasing pCREB expression. J Pineal Res 52:21–28. doi.org/10. 1111/j.1600-079X.2011.00912.x
293. Olanow CW (1993) A radical hypothesis for neurodegeneration. Trends Neurosci 16:439–444
294. Melo JB, Agostinho P, Oliveira CR (2003) Involvement of oxidative stress in the enhancement of acetylcholinesterase activity induced by amyloid beta-peptide. Neurosci Res 45: 117–127
295. Wang J-Y, Wen L-L, Huang Y-N et al (2006) Dual effects of antioxidants in neurodegeneration: direct neuroprotection against oxidative stress and indirect protection via suppression of glia- mediated inflammation. Curr Pharm Des 12:3521–3533
296. Lu J, Zheng Y-L, Wu D-M et al (2007) Ursolic acid ameliorates cognition deficits and attenuates oxidative damage in the brain of senescent mice induced by d-galactose. Biochem Pharmacol 74: 1078–1090. doi.org/10.1016/j.bcp.2007.07.007
297. Mallidis C, Agbaje I, Rogers D et al (2007) Distribution of the receptor for advanced glycation end products in the human male reproductive tract: prevalence in men with diabetes mellitus. Hum Reprod 22:2169–2177. doi.org/10.1093/humrep/dem156
298. Chetsawang B, Govitrapong P, Ebadi M (2004) The neuroprotec- tive effect of melatonin against the induction of c-Jun phosphory- lation by 6-hydroxydopamine on SK-N-SH cells. Neurosci Lett 371:205–208. doi.org/10.1016/j.neulet.2004.08.068
299. Feng Z, Zhang J (2005) Long-term melatonin or 17beta-estradiol supplementation alleviates oxidative stress in ovariectomized adult rats. Free Radic Biol Med 39:195–204. doi.org/10. 1016/j.freeradbiomed.2005.03.007
300. León J, Escames G, Rodríguez MI et al (2006) Inhibition of neu- ronal nitric oxide synthase activity by N 1-acetyl-5- methoxykynuramine, a brain metabolite of melatonin. J Neurochem 98:2023–2033. doi.org/10.1111/j.1471-4159. 2006.04029.x
301. Deng W-G, Tang S-T, Tseng H-P, Wu KK (2006) Melatonin sup- presses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood 108:518–524. doi.org/10.1182/blood-2005-09-3691
302. Wang X, Zheng W, Xie J-W et al (2010) Insulin deficiency exac- erbates cerebral amyloidosis and behavioral deficits in an Alzheimer transgenic mouse model. Mol Neurodegener 5:46. doi.org/10.1186/1750-1326-5-46
303. Chung S-Y, Han S-H (2003) Melatonin attenuates kainic acid- induced hippocampal neurodegeneration and oxidative stress through microglial inhibition. J Pineal Res 34:95–102
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.