
Contents
Mechanisms of Insulin Action and Insulin Resistance
www.ncbi.nlm.nih.gov/pmc/articles/PMC6170977/
オンラインでの公開 2018 Aug 1
マックス・C.ピーターセン、ジェラルド I. シュルマン
概要
1921年のインスリンの発見はビッグバンであり、そこからインスリンの作用と抵抗性に関する研究の広大な宇宙が広がっていった。この1世紀の間に、いくつかの発見は成熟し、臨床応用のための堅固で肥沃な土壌を形成したが、他の発見はまだ未解明であり、科学的に議論の余地がある。ここでは、2型糖尿病(2型糖尿病)の新しい治療法を開発するために、これらの研究を統合し、さらなるメカニズムの解明に役立てようとしている。このような治療法を合理的に開発するためには、2型糖尿病の主要な病態生理プロセスの1つであるインスリン抵抗性に関する詳細な知識が必要である。インスリン抵抗性を理解するためには、正常なインスリン作用についての知識が必要である。この総説では、インスリンの標的となる骨格筋、肝臓、白色脂肪組織の3つの組織に焦点を当てて、インスリン作用の生理学とインスリン抵抗性の病態生理学の両方を解説する。我々は、インスリンに対する細胞の自律的な反応を担う複雑なシグナル伝達エフェクターを、協調的な生体反応を生み出す組織特異的な機能と重ね合わせて、統合的な生理学的視点を構築することを目指している。まず、セクションIIでは、筋肉、肝臓、白色脂肪組織におけるインスリンの細胞自律的な直接作用のエフェクターと効果について、インスリン受容体から下流に向かってレビューしている。第3章では、全身のインスリン作用における組織間のクロストークの重要な役割、特に脂肪分解と肝臓の糖新生の間の不可欠な相互作用について考察する。続いてIV章では、インスリン抵抗性の病態生理について述べる。特に、慢性的な栄養過多の状況下で、どのようなシグナル伝達経路や機能がインスリン抵抗性になるのかに注目し、「選択的肝インスリン抵抗性ˮ」という現象の別の説明を示す。セクションV、VI、VIIでは、インスリン抵抗性のメディエーターと考えられるいくつかの証拠を批判的に検証している。セクションVでは、生理活性脂質であるジアシルグリセロール、セラミド、アシルカルニチンとインスリン抵抗性との関連性について検討し、セクションVIでは、小胞体やミトコンドリアにおける栄養ストレスがインスリン抵抗性に与える影響について検討し、セクションVIIでは、炎症性メディエーター、分岐鎖アミノ酸、アディポカイン、ヘパトカインなど、インスリン抵抗性を誘発するとされる非細胞自律的因子について検討する。最後に第VIII節では、これらのメディエーターをメタボリックに起因する糖新生と異所性脂質の蓄積という最終的な共通経路に結びつける、インスリン抵抗性の統合モデルを提案する。
I. 序論
2型糖尿病(2型糖尿病)は、21世紀を代表する医療課題の一つである(960)。先進国では、比較的安価でカロリー密度が高く、満腹感が不十分で嗜好性の高い食品を過剰に摂取しているため、かつてないほど肥満が増加している。米国では、糖尿病と糖尿病予備力を合わせた有病率が50%を超えている(538)。2型糖尿病を発症するのは肥満した人の一部に過ぎないが、肥満は2型糖尿病の主要な危険因子であり、2型糖尿病の罹患率は肥満の罹患率に比例している(381)。2型糖尿病の特徴である空腹時高血糖は、主なグルコース低下ホルモンであるインスリンの作用不足によるものである。そのため、インスリン作用のメカニズムを理解することは、2型糖尿病に対する効果的な治療戦略を継続的に開発するために不可欠である。
インスリンは内分泌系のペプチドホルモンであり、標的細胞の細胞膜に結合した受容体に結合して、栄養摂取に対する統合的な同化反応を組織化する。すべての動物において、インスリンまたはインスリン様ペプチド(ILP)が同定されている(120)。無脊椎動物では、ILPは分裂促進シグナルを提供するが、代謝プロセスや燃料選択への影響はそれほど大きくない(917)。哺乳類は、進化の過程で遺伝子の重複を利用して、関連するペプチドホルモンであるインスリン、インスリン様成長因子(IGF)-1,IGF-2に特化した機能を発達させた(120)。IGF-1とIGF-2は、哺乳類の細胞の成長と分化を促進し、一方、インスリンは主に代謝フラックスを制御する(204)。しかし、インスリン受容体とIGF-1受容体の間には高い相同性があり、多くの細胞種でハイブリッドヘテロダイマーを形成し、多くの下流エフェクターを共有していることから、これらの機能的な区別が曖昧であることが明らかになった(41, 770)。インスリンとIGF-1のシグナル伝達機能の重複は、高インスリン血症といくつかの癌との間に確立された関係にも寄与していると考えられる(631)。この総説では、哺乳類のインスリンがインスリン受容体に結合することによる生理学的効果と、2型糖尿病の前兆であるインスリン抵抗性の状態でインスリンの効果が減弱する分子メカニズムに焦点を当てている。
インスリン受容体は多くの細胞が発現しているが、グルコースのホメオスタシスにおけるインスリンの役割は、骨格筋、肝臓、白色脂肪細胞に対するインスリンの直接的な作用に代表される。これらの組織では、代謝の恒常性維持において異なる役割を果たしており、組織特異的なインスリンのシグナル伝達経路が必要となる。例えば、骨格筋では、インスリンはグルコースの輸送とグリコーゲンの合成を促進し、グルコースの利用と貯蔵を促進する。肝臓では、インスリンはグリコーゲン合成を活性化し、脂肪生成遺伝子の発現を増加させ、グルコン生成遺伝子の発現を減少させる。白色脂肪組織(WAT)では、インスリンは脂肪分解を抑制し、グルコース輸送と脂肪生成を増加させる。これらの多様な作用にもかかわらず、インスリンのシグナル伝達に関与する近位成分は、すべてのインスリン応答性細胞において驚くほど類似している。インスリンの生理的反応が細胞種によって異なるのは、主に遠位のエフェクターが異なるためである。骨格筋、肝臓、WAT癌におけるインスリンの細胞自律的作用を、代謝フラックスの生理的調節に関連するシグナル伝達現象に重点を置いて、セクションIIで検討する。
インスリンは、これらの直接作用に加えて、標的組織に重要な間接作用を及ぼす。これらの間接作用は、統合された状況特異的な性質を持つため、培養細胞でモデル化することは難しく、結果的にインスリンの細胞自律的な直接作用に比べて理解が進んでいない。インスリンの間接作用の一例として、インスリンが白色脂肪組織脂肪分解を抑制することで、肝のアセチル-CoA含量が減少し、ピルビン酸カルボキシラーゼ活性がアロステリックに低下することが挙げられる。このメカニズムは、グリセロール回転の抑制と相まって、インスリンが白色脂肪組織の脂肪分解を抑制することで、肝の糖新生を抑制することを可能にする(684, 903)。インスリンの膵島におけるパラクラインシグナルによるグルカゴン分泌の抑制や、中枢神経系(CNS)におけるインスリン作用は、インスリンの間接的な作用の他の重要な経路である。これらの生理学的プロセスについては、第III部で検討する。
上記のような総合的なグルコース低下作用を得るために、より高い血中インスリン濃度が必要となる場合、被験者はインスリン抵抗性であると考えられる。糖尿病、脂肪異栄養症(642)、多嚢胞性卵巣症候群(202)、非アルコール性脂肪性肝疾患(520)などの様々な臨床症状は、空腹時の血漿インスリン濃度の上昇を伴っている。このような内分泌系膵臓の仕事量の増加と、その結果としてのβ細胞の分解は、顕性2型糖尿病発症の主要なメカニズムである(380, 389, 750)。しかし、2型糖尿病の発症におけるインスリン抵抗性の重要性は、インスリン抵抗性が将来の2型糖尿病診断の最良の予測因子であることを明らかにした前向きなヒトの研究によって強調されている(481, 884)。インスリンの作用は、細胞の種類によって異なる機能を果たすため、インスリン抵抗性は、様々なインスリン標的組織において多様な機能的影響を及ぼす。インスリン抵抗性の細胞・分子生理学については、第IV章で説明するが、その中で特に注目したいのは、遮断される特定の分子部位、間接的なインスリン作用の寄与、インスリン受容体の下流にあるシグナル伝達経路の中にインスリン反応を維持するものとインスリン抵抗性を示すものがあるという、経路選択的な肝インスリン抵抗性の存在である(99, 921)。
第4章でインスリン抵抗性の現象を説明したので、次にそのメカニズムを検討する。インスリン抵抗性のメカニズムは、関与する分子メディエーター、経路、ネットワークを用いて分類するのが最も有用である。本レビューの残りの部分では、このパラダイムを用いて、提案されているいくつかの細胞性インスリン抵抗性のメカニズムの実験的裏付けを検討する。
肝臓や骨格筋のインスリン抵抗性の発症には、ジアシルグリセロール(DAG)セラミド、アシルカルニチンなどの脂質が関与していると考えられている(127, 561, 724)。解明されたメカニズムの経路は、実験的な裏付けの程度に差はあるものの、あるメディエーターの関与が他のメディエーターの関与を排除するものではないというように、ほとんどが互いに平行している。脂質によって誘発される肝臓や筋肉のインスリン抵抗性に関与すると考えられるメディエーター、経路、ネットワークについては第V章で述べる。
脂質毒性とは無関係と考えられるインスリン抵抗性の細胞内メカニズムについては、かなりの文献がある。これらには、小胞体ストレスとアンフォールドタンパク質反応(481)、様々な細胞内コンパートメントで作用する活性酸素中間体(37)、グルコースと脂肪酸の間の基質競争(397, 677)などが含まれる。セクションVIでは、典型的な肥満に伴うインスリン抵抗性に、これらの経路がそれぞれ関与していることを示す実験的証拠を、統合的な生理学的枠組みにおける役割を考慮しながら検討している。
最後に、代謝生理が統合されたものであるという認識が高まっていることから、インスリン抵抗性のメカニズムとして、インスリンに反応する組織間のクロストークが関与しているかどうかを調べることになった。例えば、活性化した脂肪組織のマクロファージは、メタボリックな機能障害と強く関連している(331, 446, 594)。炎症がインスリン抵抗性を促進するメカニズムについては、現在鋭意研究中である。さらに、この20年間で、インスリン感受性に影響を及ぼすと考えられる数十種類の内因性循環生理活性ペプチドホルモンが同定され、また、循環分岐鎖アミノ酸がインスリン抵抗性の予測バイオマーカーとなりうることも明らかになってきた(577)。セクションVIIでは、これらの循環因子のカタログを提供するのではなく、インスリン作用や抵抗性の細胞メカニズムとの関連性が確立されているものに焦点を当てている:レチノール結合タンパク質-4(RBP4)アディポネクチン、フェツイン-A、および線維芽細胞成長因子21(FGF21)。
今回の総説では、インスリン作用と非作用の両方を包括的に扱うことで、健康と病気において極めて重要なこのシグナル伝達軸の生理機能を理解するための統一的な枠組みを提示し、2型糖尿病の予防と治療を促進するための将来の発見につながることを期待している。第8章では、このような統一的なまとめを試みている。
II. インスリンの直接作用
A. 近位のインスリンシグナル伝達 インスリン受容体とその直接基質
インスリンは、標的細胞の細胞膜上のインスリン受容体(INSR)に結合することにより、既知の生理作用のすべてを発揮する(297)。INSRは、インスリンと結合する2つの細胞外αサブユニットと、膜上に広がる2つのβサブユニットからなるヘテロ4量体の受容体チロシンキナーゼであり、それぞれにチロシンキナーゼドメインが存在する(343)。INSRにはAとBの2つのアイソフォームがあるが、Bアイソフォームはインスリンに対する特異性が高く、分化した肝臓、筋肉、白色脂肪組織に発現する主要なアイソフォームであり、したがってインスリンの代謝作用のほとんどを媒介していると考えられている(44)。Aアイソフォームは、エクソン11のスプライシングによって分化したもので、胎児期に多く発現し、IGF-2に対する高い親和性が特に有効である(44)。INSRには2つのインスリン結合部位があるが、負の協同性を示す。つまり、一方の部位にインスリンが結合すると、もう一方の部位のインスリン結合親和性が低下する(186)。つまり、1つの部位にインスリンが結合すると、もう1つの部位のインスリン結合親和性が低下する(186)。誘導されたβサブユニットの構造変化は、キナーゼ活性化ループのシス-オートインヒビションを解除し、活性化ループのチロシンTyr1162,Tyr1158,Tyr1163の順にトランス-オートリン酸化を可能にする(344, 889)。三回のリン酸化で活性化されたβサブユニットは、さらに膜貫通部のTyr972を含む残基でチロシンリン酸化を受ける。これらの追加的なイベントは、INSRの基質のリクルートに重要である(941)。INSR活性化の下流にあるシグナルイベントは、機能的に大きく分けて有糸分裂シグナルと代謝シグナルに分けられる。有糸分裂シグナルは主に、多くの受容体チロシンキナーゼに共通するマイトジェン活性化プロテインキナーゼ(MAPK)経路の活性化に関与しており、このシグナル軸については多くのレビューがなされている(41, 400, 535, 616)。代謝反応を促すのに必要なインスリン濃度は、有糸分裂反応に必要な濃度よりも低い。この関係はIGF-1受容体では逆になる(41)。この総説では、代謝を制御するINSR活性化経路に焦点を当てる。
すべての細胞型において、活性化されたINSRは、まずリン酸化チロシン結合スカフォールドタンパク質をリクルートし、そのスカフォールドタンパク質が下流のエフェクターを活性化することにより、下流の代謝シグナルを開始する(図1)(826)。これは、他の多くの受容体チロシンキナーゼが細胞質の基質を直接リン酸化するのとは対照的である。INSRに様々なリン酸化チロシン結合タンパク質が結合することで、インスリンシグナルの初期段階で複数の機能モジュールが活性化される。INSRは複数のリン酸化チロシン結合タンパク質と結合することができる。SHCはそのリン酸化チロシン結合(PTB)ドメインを介して、INSRのpTyr972と相互作用する(343)。SH2B1,SH2B2/APS、GRB10,GRB14は、Src homology 2 (SH2)ドメインを介して、活性化された三リン酸化INSR活性化ループと相互作用する(190, 343)。これらの基質は、重要な制御機能を果たすことができる(190, 343)。例えば、インスリンシグナルによって活性化されるmTORC1によってGRB10がリン酸化され安定化することで、INSRの活性がフィードバック阻害される(339)。GRB2やSHCのような他のINSR基質は、インスリンシグナルの分裂促進作用に関与し(41)、一方、SH2B2/APSは、少なくとも一部の細胞種では、代謝性インスリン反応の開始に役立つ(471)。このような近位のリン酸化チロシンに基づくインスリンシグナルの減衰は、受容体の内在化と脱リン酸化によって部分的に行われる。INSRの内在化を制御する重要な因子の一つがCEACAM1であり、CEACAM1自体がINSRの基質である(568, 660)。INSRの脱リン酸化は、タンパク質チロシンホスファターゼ(PTPase)特にPTP1Bによって行われる。しかし、この減弱はおそらく、INSRが体内に取り込まれた後、時間的に遅れて起こる(808)。INSRは活性化直後、NAD(P)Hオキシダーゼ4(NOX4)を活性化してPTP1Bの活性を阻害する。NOX4由来のH2O2がPTP1Bの活性を阻害することで、インスリンシグナルの初期段階でフィードフォワード増幅が行われる(515, 921)。
図1. 近位のインスリンシグナル伝達
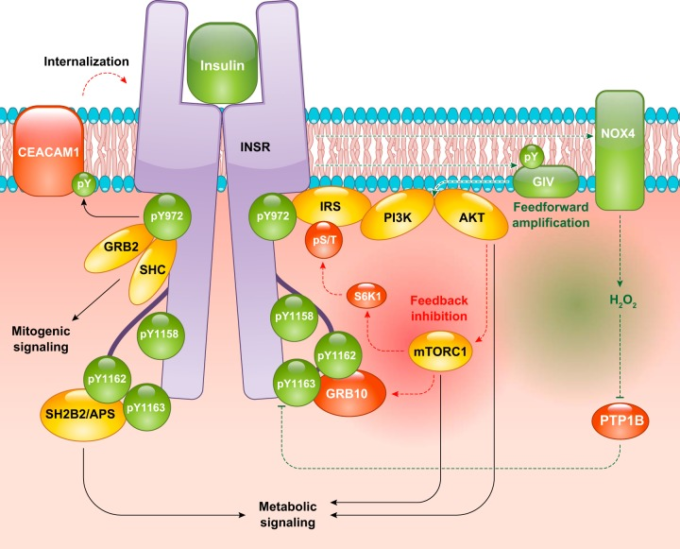
インスリンが結合すると、インスリン受容体(INSR)は自己リン酸化し、多様な基質を取り込む。インスリンシグナルの2つの主要なアームは、分裂促進(GRB2とSHCによって開始される)と代謝(インスリン受容体基質(IRS)タンパク質とSH2B2/APSによって開始される)である。インスリンシグナルはまた、正のフィードバック機構(GIVによるホスホイノシド-3-キナーゼ(PI3K)-AKTシグナルの増強、NAD(P)Hオキシダーゼ4(NOX4)由来のH2O2によるホスファターゼ阻害)と負のフィードバック機構(GRB10の安定化とINSRへのリクルート、S6キナーゼ1(S6K1)の活性化によるIRSタンパク質のリン酸化と阻害)によって特徴づけられる。緑の丸と矢印が活性化イベント、赤の丸と矢印が抑制イベントを表す。
前述のINSR基質は重要な役割を果たしているが、INSRの足場として最もよく知られているのは、インスリン受容体基質(IRS)ファミリーである。IRSには6つのアイソフォーム(IRS1-6)があるが、IRS1とIRS2がINSR活性化による代謝作用の大部分を担っていると考えられており、筋肉特異的または肝臓特異的にIRS1とIRS2の両方を欠損させたマウスでは、これらの組織でInsrを欠損させた場合と同様の現象が起こる(83, 196, 498)。IRSタンパク質には、NH2-末端にプレックストリンホモロジー(PH)ドメインとPTBドメインがあり、活性化されたINSRに結合する。IRSのPTBドメインがINSRのpTyr972に結合すると、INSRは複数のIRSのチロシン残基をリン酸化し、その結果、下流のシグナル伝達エフェクターがリクルートされてインスリン反応が伝播・増幅される(343)。IRSタンパク質のCOOH末端にある70以上のセリン/スレオニンリン酸化部位は、IRSの活性とタンパク質の安定性に影響を与え、S6キナーゼ(S6K)によるインスリンシグナルのフィードバック阻害を媒介することができる(169, 554, 899)。後の章で検討するように、IRSのリン酸化は、いくつかの刺激がインスリン抵抗性を引き起こすと考えられている主要なメカニズムでもある。
チロシンリン酸化されたIRSタンパク質は、制御サブユニットであるp85サブユニットと触媒サブユニットであるp110サブユニットを含むホスホイノシチド-3-キナーゼ(PI3K)ヘテロ二量体に結合する。具体的には、チロシンリン酸化されたIRSのYXXMモチーフが、PI3K制御サブユニットのSH2ドメインをリクルートする(826)。5種類のPI3K制御サブユニットのアイソフォームは、3つの遺伝子(Pik3r1,Pik3r2,Pik3r3)によってコードされ、3つのPI3K触媒サブユニットのアイソフォームは、3つの遺伝子(Pik3ca、Pik3cb、Pik3cd)によってコードされている。当然のことながら、PI3Kのヘテロ二量体の組成の組み合わせは、アイソフォーム特異的な機能の研究を複雑にしている。しかし、インスリン作用におけるPI3K活性の重要性は確立されている。PI3Kを薬理学的に阻害すると、インスリンによるグルコース輸送とDNA合成の刺激が消失することや、PI3Kサブユニットをノックアウトした様々なモデルから、PI3Kはインスリンシグナルの必須ノードであるという分類が一般的に支持されている(88, 128, 150, 240, 790, 826)。PI3Kは、ホスファチジルイノシトール-4,5-二リン酸(PIP2)からホスファチジルイノシトール-3,4,5-三リン酸(PIP3)の生成を触媒する。この逆反応は、第10染色体上で削除されたホスファターゼ・アンド・テンシン・ホモログ(PTEN)によって触媒される。PTENの活性は、PTENとPIP3-Rac交換体2(P-REX2)との相互作用が関与していると考えられる不完全なメカニズムにより、インスリンによって阻害される(319)。このようにPI3Kの活性化とPTENの阻害が協調して行われることで、PIP3が正味で蓄積され、インスリンシグナルが伝播し増幅される。PIP3は、PHドメインを持つタンパク質を細胞膜にリクルートし、下流のシグナル伝達エフェクターを共役させる。そのようなエフェクターとしては、ホスホイノシド依存性キナーゼ1(PDK1)とAKTがある。PIP3と結合したAKTは、PDK1によって活性化ループ(AKT1ではThr308)で、またmTORC2によって疎白色脂肪組織性モチーフ(AKT1ではSer473)でリン酸化されて活性化される(729)。重要なことは、AKT Ser473のリン酸化は、おそらく細胞内インスリン作用の最も一般的な情報であるにもかかわらず、INSRの活性化とAKT Ser473のリン酸化を結びつける正確なシグナルカスケードは不明であるということである。mTORC2によるAKT Ser473のリン酸化は、IRSに部分的に依存しない。活性化されたAKTは、多様な機能経路において多くの下流基質をリン酸化するため、AKTはインスリンシグナルの波及効果における重要なノードとなっている。AKTが正常なインスリン作用に重要であることは、AKT2の部分的な機能喪失変異がフィンランドの人口の約1%に認められ、筋肉や脂肪組織におけるインスリン刺激によるグルコース取り込みが損なわれ、内因性のグルコース産生が増加することからも明らかになっている(454)。PI3K-AKTシグナルは、グアニン交換因子GIV/GirdinのINSRによるチロシンリン酸化によって増強され、近位のインスリン作用をフィードフォワードで増幅する(500, 514)。
これらの近位のインスリンシグナル伝達イベント-インスリン受容体の活性化とシグナル伝達タンパク質(特にIRS、PI3K、AKTアイソフォーム)のリクルート/リン酸化-は、インスリン標的組織でほぼ保存されており、インスリン反応は細胞膜で開始される。ここでは、インスリンに反応する主要な細胞を個別に検討し、特定の下流のエフェクターがどのようにして組織特異的な生理的反応を引き起こすかをより詳しく説明する。
B. 骨格筋のインスリンシグナル エフェクターと効果
骨格筋はエネルギーを消費する組織である。筋細胞が蓄えたエネルギーは、解糖によって生成された3炭素単位(乳酸、アラニン)を除いて、ほとんどが自分自身で使用するためのものであり、骨格筋によって放出され、ほとんどが肝臓に循環される。インスリンは、グルコースが豊富にあることを骨格筋に伝える。したがって、心筋細胞のインスリンシグナルカスケードは、グルコースの取り込みとグリコーゲンの純合成を促進するように特化されている。これらのプロセスには筋細胞のインスリン受容体が絶対的に必要であることは、筋特異的INSRノックアウト(MIRKO)マウスを用いた高インスリン血症-高血糖クランプ試験で証明されている(407)。筋特異的にGrb10をノックアウトしたマウスでは、前述のようにINSRへのフィードバック阻害が失われるため、筋細胞のインスリン感受性が高まり、筋サイズが増大する(329)。骨格筋にはIRS1とIRS2の両方が発現しているが、筋肉における主なINSR基質はIRS1であると考えられる。IRS2ではなくIRS1をノックダウンすると、L6ラット筋管およびヒト初代筋管において、インスリン刺激によるグルコース輸送に障害が生じる(87, 340, 837)。また、Irs2-/-マウスのヒラメ筋では、用量依存的なインスリンによるグルコース取り込み刺激が正常に行われる(316)。Irs2は、インスリンによる心筋細胞の脂質代謝の制御に重要であると考えられる(87)。骨格筋には、PI3K触媒サブユニットの主要なアイソフォームであるp110αとp110βの両方が発現している。5種類のPI3K調節サブユニットのスプライスアイソフォームのうち、p85α、p85β、p55αは骨格筋に最も関連していると考えられている(826)。これらのアイソフォームを筋肉特異的に欠失させたマウスでは、インスリン刺激によるグルコース取り込みやグリコーゲン合成に障害が見られる(505)。膜のPIP3含量が増加すると、PHドメインを含むキナーゼであるPDK1とAKTが膜に呼び込まれる(471)。骨格筋にはAKT1とAKT2の両方が存在するが、インスリン刺激による糖代謝にはAKT2の方が重要であるようだ。ヒト初代筋管におけるAkt2のRNA干渉は、インスリンによるグルコース取り込みとグリコーゲン合成の刺激を無効にしたが、Akt1のノックダウンはこれらのパラメータに影響を与えなかった(87)。このパラダイムを裏付けるように、Akt2-/-マウスは重度の耐糖能異常を示し(141)、一方、Akt1-/-マウスは正常な耐糖能を示すが、Akt1-/-マウスでは重度の成長障害があるため、代謝の表現型が複雑になっている(142)。
心筋細胞のインスリンシグナルカスケードの機能的効果として、最もよく研究されているのは、グルコース輸送活性の増加であろう。これは、GLUT4貯蔵小胞(GSV)にパッケージされたグルコーストランスポーターGLUT4の細胞膜への移動と融合が高度に調整されることによって達成される(471)。筋肉におけるこのプロセスは、PI3K依存性およびPI3K非依存性の経路が報告されている脂肪細胞におけるプロセスとは、やや対照的である。今後の研究により、インスリン刺激による筋肉のグルコース取り込みに不可欠なPI3K非依存性のメカニズムが明らかになるかもしれないが、現在のところ、主にPI3K依存性の制御が関与していることがわかっている(505, 818)。意外なことに、Pik3r1-/-マウスでは、インスリン刺激によるグルコース輸送が逆説的に増加したが、この効果は他のPI3K制御サブユニットによる代償によるものと考えられる(833)。骨格筋のPik3r1とPik3r2の両方を欠損したマウス(Pik3r1 mKO Pik3r2-/-マウス)では、インスリン刺激によるグルコース輸送に障害が見られた(505)。Pik3r1 mKO Pik3r2-/-マウスにおけるこの障害の大きさは、インスリン刺激によるグルコース輸送が完全にPI3K依存性である場合に予想されるものよりも小さかった。また、PI3Kの活性化自体は、L6筋管においてGLUT4のトランスロケーションを引き起こすのに十分ではない可能性があり、PI3K非依存性のメカニズムが作用している可能性が示唆された(352, 471, 505)。しかし、PI3K阻害剤であるワートマニンは、インスリン刺激による筋のグルコース取り込みを完全に消失させることができる(274, 818)。GLUT4の移動に対するPI3Kの制御は、AKTとRho GTPase RAC1を介した並行したシグナル伝達によって媒介され、GSVの移動と融合に関わる多くのタンパク質の協調的な作用が関与している(140, 471, 818)。
AKTは、心筋細胞のグルコース取り込みに関与するいくつかのタンパク質をリン酸化する。これらのAKT基質の中で最もよく知られているのは、GTPase-activating protein (GAP) AKT substrate of 160 kDa (AS160) (TBC1D4としても知られている)と、関連するGAP TBC1D1である(384, 727, 831)。AKTによるリン酸化は、小胞輸送を制御する小さなRab GTPaseタンパク質スイッチのTBC1D4/TBC1D1の不活性化を阻害し、その結果、GSVの転位が促進される(413)。RAB8,RAB10,RAB14は、TBC1D4/TBC1D1の標的として様々に示唆されている(471)。TBC1D4のThr649は、生理的に重要なAKT基質であり、Thr649Alaノックイン変異のホモ接合マウスは、インスリン刺激による筋細胞のGLUT4トランスロケーションが損なわれ、耐糖能異常を示す(131)。AS160の生理的な重要性は、TBC1D4に切断型の突然変異を持つ家族の存在によってさらに確認された(178)。TBC1D1は、AKTよりもAMP-activated kinase (AMPK)の標的としてよく知られているが(831)、筋肉特異的にTBC1D1を欠失させたマウスでは、インスリン刺激による筋肉のグルコース取り込みに障害が見られる(194)。ヒトの筋肉におけるインスリン刺激によるGSVのトランスロケーションに対するTBC1D4とTBC1D1の相対的な生理的重要性はまだ不明であり、筋線維の種類によっても異なる可能性がある(118, 540)。Tbc1d1とTbc1d4の両方を生殖細胞で欠損させたマウスでは、インスリン刺激による筋肉の糖取り込みが完全に阻害され、Tbc1d1-/-やTbc1d4-/-の単独欠損マウスよりも重度の耐糖能異常が生じる(118)。AKTはまた、GSVの膜のターゲティングと融合に関与する標的タンパク質をリン酸化するが、これらのプロセスは筋細胞よりも脂肪細胞でよく理解されているので、後述することにする。一般に、AKTによるTBC1D1/TBC1D4のリン酸化は、インスリンがGLUT4のトランスロケーションに「ブレーキをかける」と考えることができる(413)。
Rho GTPase RAC1は、骨格筋におけるインスリン刺激によるグルコース取り込みの第2のPI3K依存性シグナル伝達機構を調整する。RAC1のシグナルは、皮質のアクチンの再編成を誘導することによってGLUT4の移動を促進する(47, 140, 818)。RAC1の直接の標的はp21関連キナーゼ(PAK)であり、インスリンはGTP結合型のRAC1を促進し、PAKの自己抑制を解除してPAKのリン酸化を促進する(140, 818)。筋特異的にRAC1をノックアウトすると、AKTの活性化が維持されるにもかかわらず、インスリン刺激によるグルコース取り込みが著しく損なわれ(817)、構成的に活性なRAC1を筋に強制的に過剰発現させると、インスリン刺激がなくてもGLUT4の移動が起こる(858)。RAC1による皮質アクチンの再編成がGLUT4の移動を促進する具体的なメカニズムについては今後も調査が必要であるが、GSVが細胞膜の下に繋ぎ止められたり、膜の張力が変化したりすることが関係しているのかもしれない(413)。
インスリンの刺激により心筋細胞に入ったグルコースは、解糖とグリコーゲン合成という2つの主要な経路をとる。健康なヒトと2型糖尿病患者の筋肉におけるインスリン刺激によるグルコース処理の主な経路はグリコーゲン合成(約75%)であり、エネルギー貯蔵ホルモンとしてのインスリンの一般的な目的的役割と一致している(182, 768)。空腹時のラットヒラメ筋では、インスリン単独で相対的なグルコース酸化(VPDH/VTCA)が5〜60%に増加し、残りは脂肪酸酸化に起因する(D. Song, T. Alves, R. Perry, and G. Shulman, unpublished data)。インスリン刺激による骨格筋の解糖系フラックスとグリコーゲン合成量の増加は、主にグルコース輸送活性の増加とそれに続くグルコース代謝物によるアロステリックな制御の結果であるが、インスリンは解糖系とグリコーゲン合成の両方を独立して制御している(152, 689, 769)。インスリンは、骨格筋の第一解糖系酵素のアイソフォームであるヘキソキナーゼIIの転写を正に制御し、解糖能を比較的ゆっくりと粗く制御する(589)。
一方、グリコーゲン合成は、インスリンによって、同化作用(グリコーゲン合成酵素(GS))と異化作用(グリコーゲンホスホリラーゼ(GP))の両方のフラックスが急激に制御される(158)。この急性制御は、共有結合による修飾(インスリンはGSとGPの両方の脱リン酸化を促進する)と(グルコース-6-リン酸による)アロステリーの両方によって行われる。最初に、1960年にインスリンによって制御されることが示された最初の酵素であるグリコーゲン合成酵素について考察する(872)。インスリンによるリン酸化に基づくグリコーゲン合成酵素の制御は、一部、グリコーゲン合成酵素キナーゼ3(GSK3)のαおよびβアイソフォームのそれぞれのSer21およびSer9におけるAKTのリン酸化と不活性化によって行われる(157, 171, 172, 214, 813)。このように不活性化されたGSK3のGSに対するキナーゼ活性は低下し、脱リン酸化されたGSはより活性化される。同時に、インスリンがプロテインホスファターゼ1(PP1)を活性化すると、GSの脱リン酸化が促進される(578, 613)。細胞はPP1のホスファターゼ活性を様々な経路の多くの標的に利用しているが、GSに対する特異性はPP1の4つのグリコーゲン標的制御サブユニットによって与えられる(578)。これらの制御サブユニットには、PP1,GS、グリコーゲンの結合ドメインが含まれており、代謝の足場としての役割を果たしている(374)。骨格筋では、GMが最も高発現しており、GMを欠損したマウスでは筋グリコーゲンの貯蔵量が減少する(90, 185, 815)。インスリンは、PP1のグリコーゲン粒子へのターゲティングを促進し、GSに対するPP1の活性を高めるが、この活性の具体的な分子機構はまだ解明されていない(374)。定説的には、不活性なGSK3と活性なPP1の組み合わせは、活性で脱リン酸化された筋GSの形成を促進し、その結果、グリコーゲン合成が促進される(159)。しかし、GSK3α/βのSer21/Ser9をアラニンに変異させ、GSK3がインスリンに対して感受性を失うようにしたノックインマウスの研究から、グリコーゲン合成におけるGSK3の重要性が疑われるようになった(84, 85)。これらのマウスはインスリン刺激によるグリコーゲン合成が正常で、筋グリコーゲン量も正常である(84)。興味深いことに、グルコース-6-リン酸によるアロステリックな活性化を受けないように筋グリコーゲン合成酵素を操作したマウスは、インスリン刺激によるグリコーゲン合成が著しく低下し、筋グリコーゲン含量も低かった(85)。これらのデータから、インスリンによるGSの急性制御は主にグルコース-6-リン酸のアロステリックな制御によって行われ、インスリン刺激によるグルコース取り込みとインスリン刺激によるグリコーゲン合成が機能的に結合していることが示唆される。GSのリン酸化状態は、グルコース-6-リン酸に対する酵素の親和性を調節する役割を果たしている。リン酸化が解除されたGSは、グルコース-6-リン酸のアロステリーに対する感受性が高くなり、インスリン刺激によるグリコーゲン合成の活性化が促進される(85, 769)。
しかし、インスリンが正味の糖新生を促進するには、GSの活性を高めるだけでは不十分である。グリコーゲンの循環を防ぐためには、グリコーゲンホスホリラーゼ活性を同時に低下させる必要がある(640)。グリコーゲン代謝の異化側では、グリコーゲンホスホリラーゼ活性がインスリンによって制御されるが、そのメカニズムはGSとほぼ同様で、リン酸化とアロステリーである(97)。古典的なメカニズムでは、活性化されたホスホリラーゼキナーゼがSer15のリン酸化を介してグリコーゲンホスホリラーゼを活性化する。インスリンはホスホリラーゼキナーゼの脱リン酸化と不活性化を促進し、結果的にグリコーゲンホスホリラーゼの脱リン酸化と不活性化を促進する(433, 949)。さらに、インスリンがPP1をグリコーゲン粒子にターゲティングすると、PP1のグリコーゲンホスホリラーゼに対する活性が高まり、Ser15を脱リン酸化してホスホリラーゼ活性を低下させる(949, 951)。この2つのメカニズムにより、インスリンはグリコーゲン分解を減少させ、グリコーゲンの純合成を促進することができる。また、グリコーゲン合成酵素と同様に、グルコース-6-リン酸の阻害によるホスホリラーゼのアロステリック制御は、インスリンによるグリコーゲン分解の制御に不可欠なメカニズムである(97)。今後、グリコーゲンホスホリラーゼのインスリン制御をグリコーゲン合成酵素と同様に変化させた研究を行い、生体内試験での筋グリコーゲン代謝を完全に理解することが必要である。
このように、筋のインスリン作用は、グルコースの利用と貯蔵を促進するための緊密な連携リレーである(図2)。グルコースの取り込みとグリコーゲンの合成というこれらの生理的結果は古くから知られているが、その分子基盤はまだ解明されていない。特に、インスリン刺激によるグルコース取り込みには、さまざまなタンパク質メディエーターが関与していると考えられているので、ここではその概要を紹介するにとどめる。筋グリコーゲン代謝の研究は、生化学の起源にまで遡る歴史を持っている。現代のツールを用いて、その制御について新たな驚くべき洞察を得ようとしている研究者は、まさに巨人の肩の上に立っているのである。
図2 骨格筋におけるインスリンシグナル伝達のカスケード
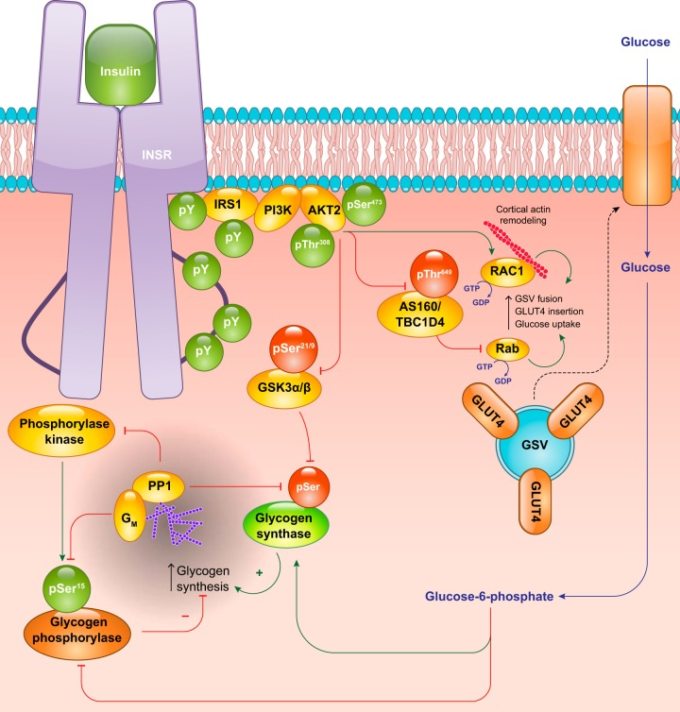
インスリン受容体(INSR)の活性化は、骨格筋細胞においてグルコースの取り込みとグリコーゲンの貯蔵という2つの主要な代謝機能を持つ。インスリンによるグルコース取り込みの促進は、GLUT4を含む貯蔵小胞(GSV)の細胞膜への移動を介して起こる。その結果、細胞内のグルコース-6-リン酸生成量が増加し、グリコーゲン代謝タンパク質の脱リン酸化が協調して行われることで、正味のグリコーゲン合成が可能になる。緑の丸と矢印は活性化イベントを、赤の丸と矢印は抑制イベントを表す。GSK3はグリコーゲン合成酵素キナーゼ3,PI3Kはホスホイノシチド-3-キナーゼ、PP1はプロテインホスファターゼ1である。
C. 肝臓のインスリンシグナル エフェクターと効果
内分泌膵臓からのインスリンは門脈に分泌されるため、肝臓は一般循環に比べて2〜3倍高いインスリン濃度にさらされる(136)。門脈のインスリン濃度の測定は、特にげっ歯類では困難であり、ほとんど行われていないが、インスリンを末梢から注入して肝のインスリン作用を研究する研究者は、末梢から測定した血漿インスリン濃度の増加分は、肝臓で「見た」門脈のインスリン濃度の増加分とは一致しないことを念頭に置く必要がある。
インスリン作用の多様な同化作用は、肝のインスリンシグナルカスケードに例示されている。インスリンは、グリコーゲン、脂質、タンパク質など、すべての主要な代謝性高分子の合成を促進する。さらに、インスリンは肝グルコース産生(HGP)を迅速かつ強力に減少させる(136)。空腹時のHGPが増加し、このパラメータがインスリンに対して鈍感になることは、2型糖尿病の特徴であるため、インスリンによるHGPの抑制を測定することは、肝のインスリン感受性を示す生理学的な指標として一般的に報告されている。しかし、インスリンによる即時的なHGPの抑制には、直接的な要素と間接的な要素の両方があることが、ここと第III部で検討される(105, 136, 504, 661, 704)。HGPの肝外制御は定量的に重要であるため、肝細胞のインスリン直接作用の最も純粋な実験的情報は、インスリンで刺激されたグリコーゲン合成、インスリンで制御された転写産物、およびインスリンシグナル伝達カスケード内のリン酸化イベントである。インスリンが遺伝子転写やグリコーゲン代謝などの生理的プロセスを制御できるのは、これらのリン酸化イベントによるものだからである。
肝インスリンのシグナル伝達は、すべての細胞型と同様に、INSRのトランス自己リン酸化、活性化、足場となるシグナル伝達タンパク質の導入から始まる。肝細胞で発現している主なIRSアイソフォームはIRS1とIRS2である(196)。IRS1とIRS2の発現を様々な遺伝的要因で変化させても、どちらのアイソフォームにも明確な役割があるとは言えず、むしろ、IrS1とIrs2は肝臓において機能的に似た役割を果たしていることが示唆されている(195, 196, 662, 827, 907)。IrS1は、正常なグルコースのホメオスタシスにおいて、Irs2よりも大きな役割を果たしている可能性がある。肝臓特異的IrS1-/-マウスは、肝臓特異的Irs2-/-マウスよりも顕著な耐糖能異常を示した(195)。IrS1-/-マウスの軽度のインスリンシグナル異常は、肝臓特異的Irs2欠失を併用することでかなり悪化したが、肝臓特異的Irs2欠失のみでは、肝細胞のインスリンシグナルは維持されたまま、軽度の耐糖能異常しか生じなかった。肝特異的Irs2欠失では、インスリンによるPI3KおよびAKT活性の刺激が抑制され、著しい空腹時高血糖を伴う重度の代謝表現型が生じることが必要であった(196)。同様に、ショートヘアピンRNAを用いてIrS1またはIrs2を70-80%ノックダウンしたマウスの肝臓では、下流のPI3KおよびAKT活性に障害がなく、穏やかな表現型が得られた(827)。IrS1とIrs2を同時に欠損させたマウスでは、耐糖能が低下し、インスリンによるPI3KやAKTの活性化が阻害された(827)。肝IRS1またはIRS2に優先的な経路制御を割り当てる試みは、一貫性のない結果となった(195, 827)。しかし、IRS1とIRS2が肝のインスリンシグナル伝達において少なくとも部分的には異なる機能を果たしている可能性は依然として高い。この仮説は、1)インスリンが肝臓においてIrS1ではなくIrs2の転写を強力に制御すること、2)IRS2のユニークなKRLBモチーフがINSRのチロシンキナーゼドメインに結合し、その活性を制限する可能性があること、などから支持されている(915, 950)。肝細胞のインスリンシグナルにおける主要なPI3K触媒サブユニットはp110αであり、このアイソフォームを肝臓特異的に欠損させると、インスリン刺激によるPIP3の生成、AKTの活性化、肝臓でのグルコース産生の抑制が大きく損なわれる(790)。
肝インスリンのシグナル伝達経路の多様化は、AKTの活性化の遠因となっているようである。AKTの基質には、GSK3(グリコーゲン合成を制御)転写因子であるフォークヘッドBOXO1(FOXO1,グルコン生成遺伝子の転写を制御)mTORC1活性の複数の制御因子が含まれており、これらが脂肪生成遺伝子の発現やタンパク質合成をアップレギュレートする大規模な同化プログラムを制御している(141, 504, 604)。代謝を制御するための肝細胞のインスリン直接シグナルは、完全にAKT依存性ではないかもしれないが、代替経路はまだ説明されていない(504)。インスリンシグナルと栄養感知経路、特にmTORシグナルとの間にはかなりの機能的冗長性があるため、肝細胞におけるインスリンシグナルの代替経路の存在を証明しようとする試みは困難であった(339, 504)。このことを念頭に置いて、肝細胞インスリンシグナルの前述の生理学的分岐を順に検討する。
食後のインスリンが肝細胞に及ぼす直接的な生理機能としては、グリコーゲンの合成を促進することが重要である。ヒトでは、高血糖・低グルカゴン血症条件下での肝グリコーゲン合成速度の半減期は、門脈のインスリン濃度が20〜25μU/mlの時に起こる(697)。骨格筋と同様に、肝グリコーゲン合成はリン酸化とアロステリーの両方によって制御されており、アロステリーが非常に重要である(702)。肝細胞におけるグルコース輸送はインスリンによって制御されていないので、インスリンがグリコーゲン合成速度を完全に制御することは骨格筋に比べて少ない(440, 640)。例えば、高血糖は肝臓のグリコーゲンホスホリラーゼをグルコースアロステリーによって不活性化し、それによって正味の肝臓のグリコーゲン合成を促進するのに十分である(109, 440, 640)。高血糖はまた、グルコキナーゼを核から細胞質へと移動させ、グルコース-G6Pフラックスを可能にする(359)。しかし、正常なグリコーゲン合成には、INSRを介した肝臓のインスリンシグナルが必要である。肝臓のInsrを急性アンチセンスオリゴヌクレオチドでノックダウンしたラットでは、高血糖条件下で肝臓のグリコーゲン合成が著しく低下する(R. J. Perry and G. I. Shulman, unpublished observations)。インスリンによる肝グリコーゲン合成の促進にはいくつかのメカニズムが考えられる。肝臓ではグルコース輸送はインスリンの制御下にないが、インスリンはグリコーゲン合成酵素(GYS2)のG6Pによるアロステリーを制御している。GYS2のArg582は、G6Pによるアロステリックな活性化に必要であり、GYS2のR582A変異をヘテロに持つマウスは、絶食・再摂食実験において肝グリコーゲンの沈着が減少する(872a)。グルコキナーゼのトランスロケーションはインスリンによって促進され、Gck遺伝子もインスリンによって迅速かつ強力な正の転写制御を受ける(9, 295, 359, 360, 451, 504)。グルコキナーゼの発現増加は、GYS2でのG6Pアロステリーを増加させるだけでなく、肝のグルコース利用と貯蔵を制御するため、肝のインスリン作用には不可欠である。代謝制御解析により、グルコキナーゼの発現がグリコーゲン合成フラックスの主要な速度制御部位であることが示され(9)、また、グルコキナーゼ活性は、基質を押し出すことでデノボ脂肪生成を促進する(295)。興味深いことに、2型糖尿病のヒトでは、グルコキナーゼの発現が低下していることが報告されており、この転写抑制の程度は空腹時血糖値と相関していた(294)。AKTのリン酸化とGSK3の不活性化は、GYS2の脱リン酸化を促進するが、これもインスリンによるGYS2活性の促進に寄与していると考えられる。しかし、肝臓のAkt1およびAkt2を欠損したマウス(AKT DLKOマウス)では、断食-再摂食を行っても、逆説的にGSK3リン酸化の刺激が維持されるにもかかわらず、正味のグリコーゲン合成を促進することができなかった。このことは、AKTが必要であり、GSK3リン酸化は正味の肝グリコーゲン合成を促進するのに不十分であることを示している(504)。興味深いことに、AKT DLKOマウスではグルコキナーゼの発現は最小限であり、インスリンに反応せず、AKT DLKOマウスではグルコース-G6Pサイクリングの低下も見られた(504)。インスリンによるGYS2の活性化には、PP1活性の活性化も関与しており、GYS2の重要な制御リン酸化部位はSer7である(90, 702)。S7AおよびS644A変異のあるGYS2を過剰発現させたマウスでは、摂食時と絶食時の両方で肝グリコーゲンが増加した(703)。これらのデータを総合すると、インスリン依存性およびインスリン非依存性の両方のメカニズムによって、グルコース代謝物による肝グリコーゲン合成の正味量をアロステリックに、また基質によって制御することが重要であることを示している。
インスリンによる肝グリコーゲン代謝の制御には、グリコーゲン分解のフラックスの抑制も含まれる。そのメカニズムは上述の筋肉の場合と同様である。肝臓のホスホリラーゼは、ヒトの筋肉のホスホリラーゼと79%の配列が同一であり、その制御様式の一部は類似している。例えば、NH2-末端のSer15のリン酸化はホスホリラーゼ活性を強く活性化し、インスリンによるホスホリラーゼキナーゼの阻害とプロテインホスファターゼ-1の活性化は、インスリンによるグリコーゲン分解の抑制の主要なメカニズムである(579, 682)。インスリンによるホスホリラーゼの不活性化は、構成的に活性なAKTを発現させることで模倣可能であることから、インスリンの正統なシグナル伝達はグリコーゲン分解の抑制に寄与していると考えられる(13)。グリコーゲン分解の制御は、グリコーゲン合成の制御とも密接に関連している。PP1の肝臓型グリコーゲンターゲティングサブユニット(GL)は、活性化したホスホリラーゼに結合して阻害され、ホスホリラーゼとGYS2が同時に活性化されることを防いでいる(9, 15)。しかし、ホスホリラーゼは強力なアロステリック制御を受けており、上述のように、グリコーゲン分解を阻害するにはグルコースのアロステリック制御で十分である。肝臓のホスホリラーゼは、筋肉のホスホリラーゼとは異なり、AMPやグルコース-6-リン酸によるアロステリックな制御には比較的鈍感である(579)。むしろ、グルコースによるアロステリックな阻害は、肝臓のホスホリラーゼにとって特に重要な調節因子である(579, 682)。肝ホスホリラーゼの活性を制御する主要なアロステリック制御因子がグルコースであることは、インスリンの影響を受けずに肝細胞の細胞膜上でグルコースが迅速に平衡化されることや、肝グリコーゲン分解が血糖値の維持に重要な役割を果たしていることを考えると、非常に理にかなっている。
このようなメカニズムで、インスリンとグルコースは協調して肝臓のグリコーゲン代謝を制御しているのである。肝グリコーゲン合成フラックスの増加には高インスリン血症が必要かつ十分であり、肝グリコーゲン分解の抑制には高血糖が必要かつ十分であり、正味の肝グリコーゲン合成の促進には高インスリン血症と高血糖の両方が必要であるというモデルが生理学的データから支持されているが(640)、そのメカニズムの研究から、インスリンとグルコースがグリコーゲン分解とグリコーゲン合成の両方で役割を果たしていることが明らかになっている(78)。インスリンはグリコーゲン合成において、リン酸化による酵素活性の変化を介した許容的な役割に加えて、グルコキナーゼの転写制御を介してGSの浮遊性と基質の利用可能性を制御する。アロステリーと基質の利用可能性は、肝グリコーゲン代謝の調節の中心である(641)。インスリンがタンパク質のリン酸化、アロステリー、基質の利用可能性を通じてグリコーゲン代謝フラックスを調節する能力は、グリコーゲンの正味合成量、ひいては肝グルコース生成量の強力な調節因子となる。
インスリンが摂食状態に対応するもう1つの重要なメカニズムは、FOXO転写因子を中心としたグルコン生成遺伝子の転写抑制である。FOXO1は特によく知られたAKTの標的であり、肝細胞において重要な生理機能を持つ(103, 195, 484, 720, 857)。AKTはFOXO1の3つの残基、Thr24,Ser256,Ser319をリン酸化するが、他のキナーゼもこれらの部位を標的とすることができる(103, 857)。リン酸化されたFOXO1は核から排除され、転写因子としての活性が失われる(103)。核内で活性化したFOXO1は、転写共役者であるペルオキシソーム増殖活性化受容体γ共役者1-α(PGC1α)と結合し、グルコース-6-ホスファターゼ(G6pc)と細胞質ホスホエノールピルビン酸カルボキシキナーゼ(Pck1)の発現増加を含むグルコン原性の転写プログラムを調整する(569, 664)。活性化したFOXO1はまた、コ・リプレッサーであるSIN3Aと結合してグルコキナーゼの発現を低下させ、グルコースの輸出をさらに促進する(451)。FOXO1のグルコン生成転写プログラムの重要性は、肝臓のFOXO1制御の遺伝的欠損を持つマウスの研究によって強調されている。肝FOXO1を欠損したマウスは、空腹時低血糖を示し、HGPが低下する(526)。高脂肪食を与えたマウスでは、肝Foxo1 mRNAの発現が40%減少しただけでも、基礎的なHGPを低下させるのに十分であった(720)。Foxo1,Foxo3,Foxo4のトリプルノックアウトは特に重篤な空腹時低血糖を引き起こす(295, 296)。興味深いことに、肝臓特異的IrS1-/- Irs2-/-マウス、肝臓特異的Insr-/-マウス、肝臓特異的Akt1-/- Akt2-/-マウスなど、基礎HGPの増加とHGPのインスリン抑制機能の低下を伴う近位のインスリンシグナル障害のいくつかのモデルにおいて、Foxo1のアブレーションは、空腹時のHGPを正常化し、HGPをインスリンに対して再感作するのに十分であった(195, 504, 603, 840)。このような顕著な表現型の回復は、これらの肝インスリン抵抗性モデルにおけるFOXO1の無制限の活性化がもたらす悲惨な糖新生の結果を反映している可能性が高く、さらに重要なことは、高インスリン血症-高血糖を受けた絶食状態のげっ歯類におけるインスリンの急性の糖新生抑制には、肝インスリンの直接的な作用が必要でないことを示している。
上述のFOXO転写因子に加えて、cAMP応答要素結合タンパク質(CREB)CREB結合タンパク質(CBP)CREB-regulated transcription coactivator 2(CRTC2)を含む転写複合体が、インスリン依存的に糖新生遺伝子の発現を制御する(421)。CREB/CRTC2モジュールとFOXO1/PGC1αモジュールは重複せず、異なる制御を受けているようである。CREB/CRTC2モジュールは、断食の最初の数時間において、糖新生遺伝子の発現に重要であることが示されているが、FOXO1/PGC1αモジュールは、より長い断食においてより重要である(493)。この魅力的な現象には、CREB/CRTC2によるPGC1αのアップレギュレーションが寄与している可能性がある(421)。FOXO1がリン酸化による核外排出で制御されているように、CRTC2はインスリンに反応して塩誘導性キナーゼ2(SIK2)によりSer171でリン酸化される(188)。CRTC2がリン酸化されると、核からの脱出が促進され、ポリユビキチン化と分解が起こり、CRTC2のグルコン生成プログラムが無効になる(188)。この軸に深刻な障害があるマウスは、グルコースホメオスタシスが変化している。CRTC2のノックアウトマウスは空腹時に低血糖を示し、CRTC2の構成的活性変異を過剰発現させたマウスは高血糖を示す(321, 882)。
FOXO1/PGC1αとCREB/CRTC2の転写モジュールはよく知られており、エレガントなメカニズムである。PGC1αを薬理学的に阻害すると、肥満の高脂肪食マウスにおいて、グルコン原性遺伝子の発現、空腹時血糖値、肝インスリン感受性が低下することさえ示されている(755)。しかし、空腹時間が16時間を超えることがほとんどない現代人の日常生活において、これらの転写モジュールが肝の糖新生に及ぼす影響は、比較的小さいと提唱されている(419)。例えば、2時間のインスリン刺激でも、G6pcタンパク質レベルの検出可能な減少を引き起こすには不十分である(620)。むしろ、計算モデルによると、非転写メカニズムがグルコースの代謝フラックスを高度に制御していることが示されている(419)。このようなメカニズムには、基質の利用可能性、アロスト性、酸化還元状態、翻訳後修飾などの変化が含まれる。上述したように、インスリンがこのようなメカニズムで肝グリコーゲン代謝を制御することはよく知られている。非転写的なインスリンによる肝グルコーゲン代謝の制御も行われているが、近年ではあまり注目されていない。後述するように、インスリンによる急性の糖新生抑制には、白色脂肪細胞の脂肪分解を介した間接的な肝内糖新生の制御が重要である。しかし、インスリンは、cAMPによって誘導されるホスホフルクトキナーゼ-2/フルクトース-2,6-ビスホスファターゼ-2(PFK-2/FBPase-2)のSer36のリン酸化を打ち消すことによって、肝臓の糖新生を直接制御することもできる(691, 914)。このリン酸化の解除はFBPase-2の活性を促進し、フルクトース-2,6-ビスリン酸レベルを低下させ、それによって糖新生酵素FBPase-1を抑制する。興味深いことに、PFK-2/FBPase-2の脱リン酸化は、グルコキナーゼの核内移行を促進することにより、グルコキナーゼを阻害することもある(173)。このメカニズムは、グルカゴン/カテコールアミンの濃度が高い状態で最も有効であると考えられ、食後の正常な糖新生抑制における役割については、さらなる研究が必要である。
上述したように、HGPの急性抑制は、肝のインスリン作用を示す最も一般的な生理学的指標の一つである(33, 54, 105, 112, 115, 484, 504, 722)。この効果が直接的(すなわち、肝細胞自律的)なものか、間接的なものかという問題は、非常に注目されている(135, 136, 208, 472, 504, 620)。この点に関して、方法論的に重要なのは、グリコーゲン分解とグルコーネ生成のHGPに対する相対的な貢献度である。肝グリコーゲン含量は空腹時に指数関数的に減少し(このことは、その誘導体である肝グリコーゲン分解率も空腹時に指数関数的に減少することを意味する)ラットでは12時間後、ヒトでは48時間後にほぼ完全に枯渇する(628,704)。一方、肝グルコン生成の絶対速度は、基質(すなわち、乳酸、アラニン)の制限によりグルコン生成フラックスが減少するまで、絶食の最初の48時間は比較的一定である(図3)(628, 643, 704)。空腹時の血漿グルコース濃度はHGPを反映しているので、これらの観察結果の興味深い点は、空腹時の血漿グルコース濃度(ひいては血漿インスリン濃度)は、肝グリコーゲン量(すなわち、中枢神経系やその他のグルコースを利用しなければならない組織で利用可能な炭白色脂肪組織化物の蓄え)を反映する重要な全身性のシグナルであるということである。本章および後続の章で検討したように、直接および間接的な肝インスリン作用は、肝グリコーゲン分解およびグルコ生成の正味量に異なる影響を与えるため、これらのフラックスの相対的な寄与を理解することは、この問題に関する実験の設計および解釈に不可欠である。
図3 ヒトの空腹時における肝グルコース産生の源
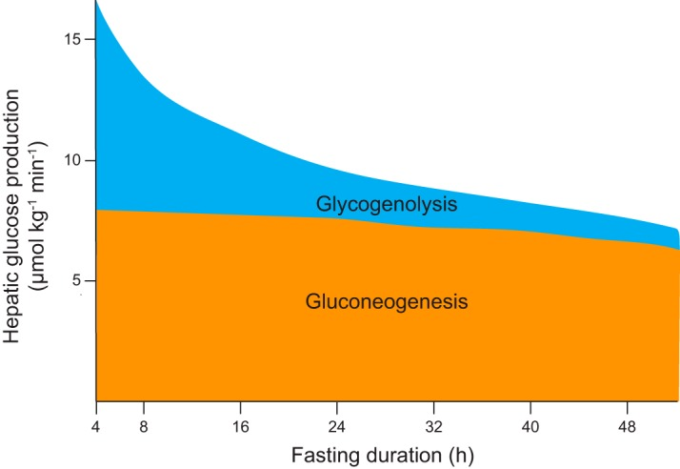
食後初期(図示せず)には、摂取したグルコースが肝グリコーゲンとして貯蔵されるため、肝臓はグルコースの正味の取り込みを行う。グルコン生成のフラックスは継続されるが、グリコーゲンの貯蔵に転用される。この期間の後、グルコン生成フラックスは肝臓でのグルコース生成に寄与し、48時間ほど比較的一定の割合で継続するが、基質の利用可能性の低下により最終的には減少する。一方、正味の肝グリコーゲン分解は、最初は肝グルコース産生の約半分を占めるが、その速度は肝グリコーゲン量に応じて指数関数的に減少する。血漿中のグルコース濃度は、断食中の肝グルコース産生速度を反映しているので、血漿中のグルコース濃度は、断食中の肝グリコーゲン含量の全身的なシグナルとなる。[データはRothmanら(704)より]。
この点を考慮すると、インスリンが肝グリコーゲン分解を直接抑制することは、インスリンによるHGP抑制の生理学的に重要なメディエーターであることは間違いない(208)。実際、ヒトでは絶食後22時間の間、肝グリコーゲン分解がHGPの約40%を占めると推定されている(704)。しかし、高インスリン血症-高血糖クランプ実験によく用いられるグリコーゲンを欠いた絶食状態のネズミでは、HGPの主な原因は肝グルコゲン生成である(620)。インスリンは数分以内に肝の糖新生を抑制するので、糖新生タンパク質レベルの変化が起こるずっと前に、上述の転写メカニズムはインスリンによる肝の糖新生の急性抑制を説明できない(218, 484, 620)。さらに、完全な肝インスリン抵抗性の複数の遺伝子モデルがインスリンに反応してHGPを正常に抑制することから、急性の糖新生抑制に関与する代替的なAKT非依存性の直接肝細胞インスリンシグナル伝達経路が存在する可能性があるが、生理学的な説明としては不十分である(105, 195, 504)。その代わり、インスリンによる肝グルコン生成フラックスの急性抑制は、主にインスリンによるWAT脂肪分解の抑制を媒介とした、大部分が間接的な効果であると考えられる(54, 136, 620, 684)。このメカニズムについては第III部で詳しく述べるが、ここでは、空腹時におけるインスリンの肝グルコース産生抑制作用を測定しても、肝におけるインスリンの直接作用を具体的に評価することはできないことを強調しておきたい。
インスリンはまた、脂質代謝に対して肝細胞に直接作用する。これらの作用の中で最も顕著なのは、de novo lipogenesis (DNL)のいくつかの遺伝子の転写アップレギュレーションであるが、トリグリセリドに富むリポ蛋白のクリアランスの増加と超低密度リポ蛋白(VLDL)の輸出の減少も報告されている(457)。全体的な効果としては、肝細胞への脂質の蓄積が促進され、他の組織による酸化のための脂肪酸の利用可能性が減少する。実際、血漿中のトリグリセリド濃度はインスリン投与後15分以内に急激に減少するが、混合食の場合は吸収されたトリグリセリドがこの効果を無効にする。
DNLは、糖新生と同様に、PI3K/AKT依存性のメカニズムにより、ゆっくりと、しかし強力に転写制御されている。しかし、グルコン生成とは異なり、DNLはインスリン刺激による脂肪生成酵素のリン酸化によって急性に制御されることもある。DNLは肝臓の脂肪生成物質の約25%を占めるにすぎないと推定されているが(これに対して、循環脂肪酸のエステル化によるものは約60%、食事性脂質によるものは約15%)インスリンによるDNLの刺激はDNLの全体的な同化作用と一致している(197)。
SREBP-1cは、アセチル-CoAカルボキシラーゼ1(Acaca)、脂肪酸合成酵素(Fasn)、グリセロール-3-リン酸アシルトランスフェラーゼ1(Gpam)など、いくつかの脂肪生成酵素の転写を促進することで、DNLを促進する(215, 442)。SREBP-1cを肝臓特異的に過剰発現させると、肝脂肪症を引き起こすのに十分である(368)。インスリンは主にSREBP-1cの転写を増加させることでSREBP-1cに作用するが、インスリンはSREBP-1cの切断と核移行も促進する:これはSREBP活性化の正統なメカニズムである(207, 330)。これらの効果は、PI3K、AKT、mTORC1の阻害により阻害されることから、これらのキナーゼがSREBP-1cの上流に存在することが示唆されている(487)。特に、肝臓特異的なAkt2-/-マウスは、レプチン欠乏のob/ob背景でも肝脂肪症を発症しないという観察結果は、インスリンによる脂質代謝の制御は、主にAKTの下流で行われていることを示唆している(458)。mTORC1の基質であるS6Kは、SREBP-1cのプロセッシングには必要であるが、その転写アップレギュレーションには必要ない(487, 604)。しかし、インスリンによるDNLプログラムの転写活性化は特に遅いことに注意する必要がある。ラットの初代肝細胞を用いたある研究では、インスリン処理後8時間経過するまで核抽出液中にSREBP-1が検出されなかった(283)。インスリンがDNLフラックスを増加させるより早い転写メカニズムは、グルコキナーゼの誘導であり(163, 295)、これにより脂肪生成基質の利用可能性が増加する。
インスリンは、DNLの転写的なアップレギュレーションに加えて、脂質生成酵素のリン酸化を制御することによっても、DNLフラックスを急性的に活性化するが、その具体的なシグナル伝達経路はまだ十分に解明されていない。例えば、ACCはインスリンに反応して急速に活性化されるが(909)、これには脱リン酸化とリン酸化の両方のイベントが関与していると考えられる(100)。インスリンは、おそらく通常これらの部位をリン酸化するAMPKを阻害することにより、ACC1の79番目のSer79とACC2の212番目のSer212の脱リン酸化を促進する(845, 908)。ACC1のSer79とACC2のSer212をともにアラニンに変異させたノックインマウスでは、肝ACCが恒常的に活性化し、その結果、肝脂肪生成が増加することから、これらの部位の生理的重要性が示されている(250)。インスリンは、他のACC残基のリン酸化を増加させる可能性もあるが、これらの修飾がACCの活性を変化させることは示されていない(305)。インスリンはまた、ATPクエン酸リアーゼ(ACLY)のリン酸化を制御する。ACLYは、トリカルボン酸サイクルの中間体であるクエン酸を脂肪生成前駆体であるアセチルCoAに変換し、グルコース代謝とDNLを結びつける。ACLYの3つのリン酸化部位はインスリン応答性である。Ser455はAKTの基質であり、Thr446とSer450はGSK3の基質である(57, 345)。ACLYのリン酸化は、クエン酸によるアロステリック阻害を阻止して酵素を活性化する(658)。しかし、インスリンがACLYの活性を高める役割を果たしていることは明らかではない(189)。例えば、ACLYのSer455はプロテインキナーゼA(PKA)によってもリン酸化されるが、PKAの活性はほとんどの場合、インスリンの作用に対抗する(658)。さらに、GSK3活性はインスリン作用によって阻害されるが、これはインスリンがACLYのリン酸化を促進してその活性を高めるというモデルとは矛盾する。したがって、インスリン刺激によるACLYリン酸化の生理的役割は不明である。リン酸化プロテオミクスの膨大なデータセットから、脂質生成経路においてインスリンによって制御されている他のリン酸化イベントが明らかになるかもしれない。
ここまで、インスリンがグリコーゲンと脂質という2つの主要な生体高分子の肝細胞での合成をどのように制御するかについて考察してきた。最後に、3つ目の生体高分子であるタンパク質について説明する。インスリンによるタンパク質合成の調節は、主に哺乳類ラパマイシン標的(mTOR)ネットワークへのシグナル伝達によって行われる。mTORは大型のプロテインキナーゼであり、結合相手によってmTORC1とmTORC2という互いに排他的な機能を持つ複合体を形成する(752)。mTORC1とmTORC2はともにインスリンシグナルカスケードと相互作用するが、mTORC1の作用の方がよく研究されている。インスリン刺激によるタンパク質合成は、肝細胞、脂肪細胞、心筋細胞など多くのインスリン応答性細胞においてmTORを介して行われるが、肝細胞のタンパク質合成速度が特に高いことと、mTORC1がインスリン刺激によるde novo lipogenesisを調節する役割を果たしていることから(前述)肝のインスリンシグナルについての議論に含めることにした。
インスリンによるmTORの活性化は、PI3K-AKT経路と双方向的に統合されている。AKTによるmTORC1の活性化は不完全に理解されているが、AKTのリン酸化と、mTORC1活性化の阻害剤である結節性硬化症複合体2(TSC2)および/またはプロリンリッチAKT基質40kDa(PRAS40)の不活性化が関与している可能性がある(349, 866b)。活性化されたmTORC1は、S6Kや真核生物翻訳開始因子結合タンパク質1および2(4EBP1/2)などの翻訳機構の構成要素をリン酸化し、5′末端オリゴピリミジン(TOP)モチーフを持つ転写産物を特徴とする広範な翻訳プログラムを誘導する(508a, 752, 839)。これらの下流効果に加えて、mTORC1シグナルは、IRS1のS6Kリン酸化と不安定化、およびアダプタータンパク質GRB10のリン酸化と安定化を促進することで、近位のインスリンシグナルにネガティブフィードバックを及ぼす(339, 944)。最後に、mTORシグナルはAKTを正に制御する。AKTのSer473に対するmTORC2のリン酸化は、おそらく細胞内インスリンシグナルの最も一般的な測定値であるが、mTORC2がインスリンによってどのように活性化されるのかは、いまだに正確にはわかっていない(453, 729)。Ser473のリン酸化は、AKTキナーゼ活性を高める活性化イベントであり、その基質特異性を変化させる可能性がある(16, 288, 362)。このように、mTORシグナルは、インスリンシグナルのすべての機能に影響を与えており、タンパク質合成のような直接的なシグナル伝達や、INSR、IRS1,AKTへのフィードバックのような間接的な調整を行っていると考えられる。重要なことは、mTORは、他の同化シグナル(例えば、アミノ酸の利用可能性)をインスリンシグナルに統合することができることである(453)。
以上の議論から明らかなように、肝細胞のインスリンシグナルは、多量栄養素の代謝のすべての分野に関連する、複雑なカスケードである(図4)。インスリンが肝臓に及ぼす主な直接作用は、グリコーゲン合成を促進することと、糖新生、de novo lipogenesis、タンパク質同化を転写的に制御することである。残念なことに、肝臓におけるインスリンの作用を実験的に調べるために使用される最も一般的な情報は、1)インスリンの肝細胞に対する部分的に間接的な作用(肝グルコース産生の抑制)2)複数の生理学的入力と間接的なインスリン制御を伴うが、優れた市販の抗体が利用可能なリン酸化事象(AKT Ser473)である。第4章では、肝インスリン抵抗性に関する知見と肝インスリン作用に関する知見を統合し、生理学的に意味のある実験戦略を提案することを試みる。
図4 肝のインスリンシグナル伝達
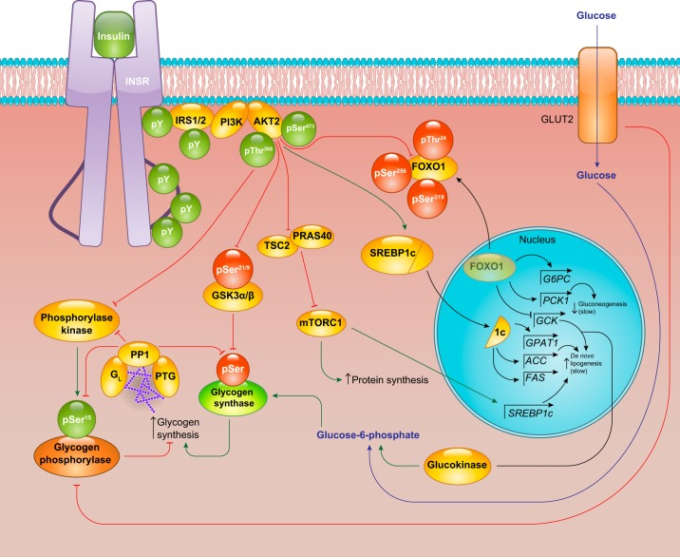
AKTシグナルは肝細胞のインスリン作用の中心である。早い作用としては、グリコーゲンやタンパク質合成装置の活性化がある。転写を介した緩やかな作用としては、グルコキナーゼのアップレギュレーション、グルコン生成能の低下、デノボ脂肪生成能の刺激などがある。緑の丸と矢印は活性化イベントを、赤の丸と矢印は抑制イベントを表す。IRS(インスリン受容体基質)GSK3(グリコーゲン合成酵素キナーゼ3)PI3K(ホスホイノシチド-3-キナーゼ)PP1(プロテインホスファターゼ1)GPAT(グリセロール-3-リン酸アシルトランスフェラーゼ)G6PC(グルコース-6-ホスファターゼ)PCK1(ホスホエノールピルビン酸カルボキシキナーゼ)SREBP1c(ステロール制御エレメント結合タンパク質1c)FAS(脂肪酸合成酵素)。
D. 白色脂肪細胞のインスリンシグナル。エフェクターと効果
白色脂肪細胞は生体内試験ではインスリンに対して非常に敏感である。血漿中の非エステル化脂肪酸(NEFA)レベルをコントロールするインスリンの能力は、高血糖を維持するのに重要であり、脂肪分解の抑制は、WAT癌におけるインスリンの重要な生理機能である(620, 684)。白色脂肪組織脂肪分解の抑制は、血漿インスリンレベルに急峻な依存性を示し、ヒトにおけるED50は約20μU/mLである(684)。糖尿病ではない健常人の血漿インスリン濃度は約5〜60μU/mLであるため(655, 684)、インスリンによる白色脂肪組織脂肪分解の生理的調節は、ED50が約60μU/mLで、200μU/mL以上の生理的に高いインスリン濃度でのみ最大レベルに達する全身のグルコース取り込みのインスリン調節と比較して、そのダイナミックレンジのはるかに大きな部分を利用することができる(694)。グルコース輸送の促進は、脂肪細胞におけるインスリンのもう一つの主要な機能であるが、WATは全身のグルコース処理のごく一部を占めるに過ぎない(430)。次に、白色脂肪細胞における脂肪分解とグルコース取り込みのインスリン制御に関与するエフェクターについて考えてみよう(図5)。
図5 白色脂肪細胞におけるインスリンシグナル伝達
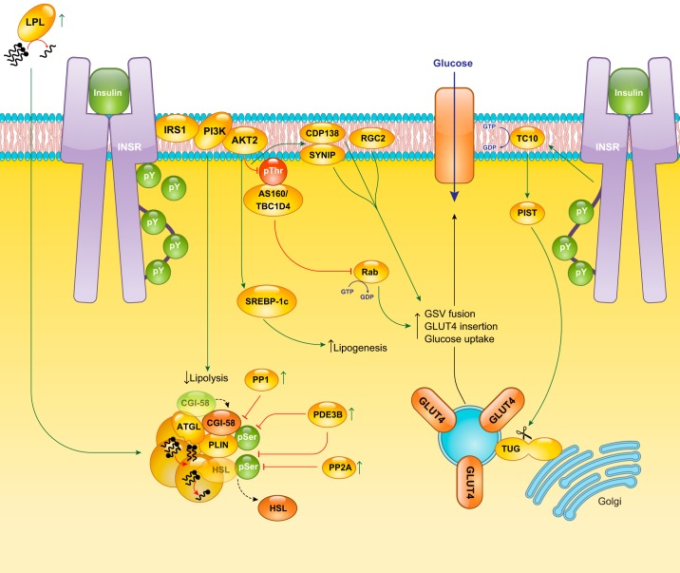
白色脂肪組織におけるインスリン作用の最も重要な生理機能は、脂肪分解の抑制とグルコースの取り込みの促進である。脂肪分解の抑制にはホスホジエステラーゼ3B(PDE3B)が必要であり、ペリリピン(PLIN)やホルモン感受性リパーゼ(HSL)のリン酸化など、cAMP刺激によるイベントの減衰が主な原因となっている。インスリンによる糖取り込みの促進は、ホスホイノシチド-3-キナーゼ(PI3K)依存性(左のインスリン受容体(INSR))およびPI3K非依存性(右のINSR)の経路で、多数のエフェクターを用いて、GLUTを含む貯蔵小胞(GSV)の移動、ドッキング、細胞膜との融合を促進する。緑の丸と矢印は活性化イベントを、赤の丸と矢印は抑制イベントを表す。IRSはインスリン受容体基質、PPはタンパク質リン酸化酵素、SREBP-1cはステロール制御エレメント結合タンパク質1c、LPLはリポタンパク質リパーゼを意味する。
インスリンによる血漿NEFAレベルの抑制は、脂肪細胞におけるトリグリセリドの脂肪分解を速やかに阻害することで起こる。インスリンは最も強力な脂肪分解ホルモンであり、迅速に作用する。ラットの血漿NEFAレベルは、食後レベルまでインスリンを上昇させてから5分以内に約90%抑制される(285, 477)。この急速な作用は、血漿中のNEFAの半減期が2〜4分と短いことによって促進される(206)。インスリンが脂肪分解を抑制するメカニズムとしては、cAMPとプロテインキナーゼA(PKA)を介したアドレナリンシグナルの減衰または逆転が最もよく理解されている(203, 367)。インスリンによる脂肪分解の制御を理解するためには、これらのcAMP/PKA依存性のメカニズムを要約することから始める。PKAは、ホルモン感受性リパーゼ(HSL)とペリリピン(PLIN)という脂肪分解に関与する2つの重要なタンパク質をリン酸化する(367, 869)。HSLはCOOH末端の3つのセリン残基(Ser563,Ser659,Ser660)でリン酸化され、細胞質から脂質滴表面へと移動する(328, 820)。白色脂肪組織脂肪分解のホルモン制御におけるHSLの重要性は、ヒトHSLのフレームシフト変異が確認されたことで明らかになった(14)。この突然変異のホモ接合患者はHSLを発現しておらず、脂肪分解の制御が著しく損なわれており、イソプロテレノールによる脂肪分解の刺激とインスリンによる脂肪分解の抑制の両方が著しく損なわれていた(14, 947)。同様に、マウスでHSLを欠失させると、アドレナリンによる脂肪分解の刺激が著しく損なわれる(293, 557, 602)。しかし、HSLは主にDAGリパーゼとして働き、脂肪トリグリセリドリパーゼ(ATGL)が最初のTAG加白色脂肪組織分解を触媒する(210, 946, 959)。ホルモンによる脂肪分解速度の完全な制御には、脂肪滴をコーティングするタンパク質であるペリリピンも必要である。ペリリピンは豊富に存在し、5つのPLINアイソフォームが組織特異的な機能を果たしている(93)。PLIN1は白色脂肪細胞で高発現しており、PKAによっていくつかのセリン残基でリン酸化される(93, 531, 946)。脂肪分解の制御におけるPLINのリン酸化の正確な機能は完全には解明されていないが、少なくとも3つの主要なメカニズムが関与していると考えられている。まず、PLINのリン酸化は、ATGLの補因子であるCGI-58に対する親和性を低下させ、CGI-58がATGLと結合してATGLの活性を約20倍に増加させる(279, 946)。第2に、PLINのリン酸化は、脂質滴表面におけるHSLの完全な活性化に重要である(546, 820)。第3に、PLINのリン酸化は、脂質マイクロベシクルの出芽を促進することにより、脂質滴表面積と体積の比を増加させることが示されている。これは、リパーゼの基質へのアクセスを増加させる可能性があるが、アドレナリン刺激に長時間さらされる必要があるため、急性の脂肪分解反応には関与していないと考えられる(521)。Plin1-/-マウスでは、基礎的な脂肪分解が亢進し、アドレナリン刺激に反応しないことから、PLINは単にリパーゼのアクセスに対する受動的な障壁ではなく、むしろ刺激された脂肪分解を積極的に制御していることが明らかになった(522, 713, 828)。PLINが脂肪分解を組織化するメカニズムを完全に理解するには、さらなる研究が必要である。PLINがリパーゼと補因子の大規模なインタラクトームを足場にして、アドレナリン刺激に対する脂肪分解反応を調整・増幅している可能性がある。
インスリンは、主にホスホジエステラーゼ3B(PDE3B)を介して脂肪分解を抑制する。PDE3BはcAMPを分解し、HSLやPLINに対する脂肪分解促進のPKAシグナルを弱める(147, 367, 670, 781)。Pde3bを欠損した脂肪細胞における脂肪分解の促進は、インスリンによって抑制されず、Pde3b-/-マウスでは糖負荷試験時の血漿NEFAレベルの抑制に障害がある(147)。興味深いことに、インスリンによるPDE3Bの活性化のメカニズムは不完全に定義されている。PDE3BのSer273は、インスリン刺激後に14-3-3タンパク質依存的にAKTによるリン酸化を介して活性化される(411, 483, 598, 701)。しかし、AKTはインスリンによる脂肪分解の抑制には必要ないようで、Akt2-/-マウスは摂食時には正常に脂肪分解を抑制し、インスリン負荷試験や高インスリン血症-高血糖クランプ時にもほぼ正常に脂肪分解を抑制し(423)、AKTを薬理学的に阻害しても培養脂肪細胞におけるインスリンによる脂肪分解の抑制は消失しない(192)。さらに、PDE3BのAKTリン酸化部位であるSer273は、培養脂肪細胞における脂肪分解のインスリン抑制には必要ないことがわかった(192)。PKA基質であるSer296も試験管内試験でのインスリンによる脂肪分解抑制には不要であることが示されているが(192, 483, 671)、これらのイベントの機能的重要性は不明である。PDE3Bのリン酸化を調節することでPDE3Bの活性を制御するのではなく、インスリンは主にシグナル複合体、すなわち「シグナロソーム」の形成を促進することでPDE3Bを活性化するという新しいパラダイムが提唱されている(11, 192)。
インスリンによるPDE3Bの活性化がcAMP/PKAを介した脂肪分解の抑制を媒介するという強力な証拠があるが(743)、アドレナリン入力を消失させるというこのメカニズムが、インスリンによるあらゆる生理学的条件下での渡辺の脂肪分解の抑制を説明するのに必要かつ十分であるかどうかは明らかではない。特に、このメカニズムは、アドレナリン緊張が低い状況では重要性が低いかもしれない。例えば、PKA活性が検出されなくても、インスリンはHSLの脱リン酸化を引き起こすが、これはインスリンがHSLのホスファターゼ活性を刺激していることを意味する(804)。プロテインホスファターゼ2A(PP2A)がこの効果の主な媒介者であると考えられるが、他のホスファターゼもHSLに作用するため、そのメカニズムの全容は不明である(911)。インスリンもまた、ペリリピンの脱リン酸化など、PI3K依存的ではあるが、AKT非依存的な作用を持つようである(146, 209)。HSLとは対照的に、脂肪細胞ではプロテインホスファターゼ1(PP1)が主要なペリリピンホスファターゼとして同定されており、PP1制御サブユニットのリン酸化と活性はインスリンに反応して増加する(43, 151)。
以上のことから、インスリンが脂肪分解を抑制するメカニズムはまだ完全には解明されていないが、インスリンがPDE3Bを活性化してアドレナリンキナーゼの活性を低下させると同時に、プロテインホスファターゼを活性化して脂肪分解制御タンパク質を積極的に脱リンさせるという機能モデルは、実験的な観察結果を説明するのに十分であると考えられる。ホルモンの刺激による脂質分解複合体または非脂質分解複合体の脂肪滴への集合が、脂肪分解の正味の方向性を決定するという、白色脂肪組織の脂肪分解の細胞生理学の図式ができつつある(11, 192)。現在進行中の研究によって、関連するメディエーターや相互作用がより詳細に解明されることは間違いないだろう(54, 620, 684)。
肝グリコーゲンの正味貯蔵量がグリコーゲン分解とグリコーゲン合成のバランスに依存するように、脂肪分解の正味量は、脂肪分解と遊離脂肪酸の再エステル化によるフラックスの合計である(474)。再エステル化は、脂肪細胞内または循環から生じた脂肪酸に作用する(447)。空腹時には脂肪細胞の再エステル化はほとんど起こらないが、グルコースを注入するとかなりの再エステル化が起こる(165)。さらに、1型糖尿病患者ではインスリンの補充が不十分なため、食後の脂肪酸の貯蔵が損なわれる。これらの知見は、インスリンが脂肪の脂肪酸エステル化を促進する役割を担っていることを示唆している(475)。インスリンで刺激されたグルコースの取り込みは、脂肪酸がエステル化されるグリセロール-3-リン酸の供給源となり、インスリンは脂肪組織内皮のリポタンパク質リパーゼ活性を活性化する(225, 267)。さらに、インスリンは、3T3-L1脂肪細胞における脂肪酸輸送タンパク質FATP1およびFATP4のトランスロケーションを促進する(794)。しかし、健康な成人では、インスリンは、ナイアシンによる脂肪分解の抑制に比べて、全身性の脂肪酸エステル化を促進することはなかった(17)。脂肪細胞内で遊離した脂肪酸の再エステル化率はインスリンに依存しないという観察結果(110)と合わせて考えると、脂肪細胞の脂肪酸エステル化率は、エステル化促進酵素活性に対するインスリンの急性刺激よりも、基質の利用可能性と濃度勾配に依存するという解釈が妥当である。インスリンには、脂肪細胞における他の脂肪生成促進機能があり、肝細胞におけるのと同様に、SREBP-1cとその脂肪生成転写プログラムを活性化する(398)。しかし、デノボ脂肪生成は脂肪細胞の脂肪生成のごく一部であり、前もって形成された脂肪酸のエステル化が主な脂肪生成経路である(474)。インスリンはまた、転写因子であるペルオキシソーム増殖剤活性化受容体-γ(PPARγ)を介して脂肪形成を促進する(693)。
インスリンによる細胞内のグルコース取り込みの制御は、培養した3T3-L1脂肪細胞でよく研究されている。3T3-L1脂肪細胞は線維芽細胞由来であり、真の白色脂肪細胞の特徴を完全に再現しているわけではないが、その操作のしやすさから、インスリンシグナルの分子メディエーターについて多くのことが明らかになっている(73, 282)。さらに、インスリン刺激によるグルコース取り込みの研究は、小胞輸送を研究する大規模で生産的な科学コミュニティによって促進されている。1980年にインスリンがグルコース輸送活性の細胞膜への移行を促進するという発見によって植えられた種は、実りある木へと成長し、GSVの出芽から輸送、テザーリング、ドッキング、融合までのプロセスの各ステップを分子レベルで詳細に説明する枝が伸びている(73, 174, 814)。脂肪細胞では、GLUT4のトランスロケーションには、上述した心筋細胞のグルコース取り込みに関するエフェクターと同じものが多く関与しているが、すべてではない。ここでは、脂肪細胞におけるインスリン刺激によるグルコース取り込みの分子メディエーターについて簡単に考察するが、興味のある読者には、このテーマに関するより詳細なレビューがいくつか紹介されている(47, 73, 276, 365, 413, 471)。
脂肪細胞におけるインスリン刺激によるグルコース取り込みは、筋肉と同様、IRS1-PI3K-AKT軸に決定的に依存している。IRS1のアンチセンスノックダウンは、ラット初代脂肪細胞におけるインスリン刺激によるグルコース取り込みを著しく阻害する(666)。IRS2もまた、脂肪細胞のインスリンシグナルに関与している。インスリン活性化サイクリン依存性キナーゼ4(CDK4)によるIRS2のSer388リン酸化は、脂肪細胞のインスリンシグナルを維持するポジティブフィードバック機構であると考えられる(448)。脂肪細胞のインスリン作用におけるPI3Kの活性化の重要性は、成熟脂肪細胞のPIP3リン酸化酵素PTENを誘導的に欠失させたマウスの研究で明らかになっている。3T3-L1脂肪細胞のグルコース取り込みを増加させるには、PI3KまたはAKT2を合成または光遺伝学的に活性化すれば十分であり、Tbc1d4(AS160)を欠損させたマウスでは、インスリン刺激による脂肪細胞のグルコース取り込みが完全に阻害される(118, 581, 930)。AS160関連のRab GAPであるTBC1D1は、骨格筋では重要であるが、脂肪組織では低レベルの発現であり、Tbc1d1-/-マウスではインスリン刺激によるグルコース取り込みが正常に行われる(118)。脂肪細胞におけるAS160の主な基質(すなわち、AS160が不活性なGDP結合状態を維持するRab GTPase)は、GSVのエキソサイトーシスを制御するRAB10であると考えられている(94, 726)。関連するTBC1D13-RAB35ペアもまた、他のいくつかのRabと同様に、GLUT4の細胞膜への輸送をサポートしている(179, 365)。また、GAP複合体RGC1/2とGTPase RalAが関与する別のGAP-GTPase軸も、インスリン刺激によるGSVの脂肪細胞細胞膜へのターゲティングに関与しており、RGC2はAKTの基質である(132, 133)。
AKTは、GAP-Rab相互作用による小胞輸送の制御に加えて、小胞のテザーリング、ドッキング、融合に関与する標的をリン酸化することにより、脂肪細胞におけるGLUT4のトランスロケーションを促進する。そのような基質の一つがSYNIPである。SYNIPはリン酸化されるとt-SNAREシンタシン-4から解離し、細胞膜での小胞のドッキングと融合を可能にする(544, 931)。CDP138もまた、AKTの基質としてGSVの融合に関与しているが、その正確な機能はいまだに解明されていない(926)。最後に、モータータンパク質であるミオシン5AはAKT基質であり、GLUT4が皮質のアクチンネットワークを通過して細胞膜に到達するのを助ける(939)。GSVの輸送に関与する新規のAKT基質が続々と発見されていることは、PI3K依存性のGLUT4輸送に関する現在の理解が不完全であることを示唆しているが、AKTがそのマスターコントローラーとしての役割を果たし、プロセスの全段階で多くのエフェクターに情報を提供していることは確かである。
脂肪細胞では、RhoファミリーGTPaseのRAC1は、骨格筋細胞ほどインスリン刺激によるGLUT4の輸送に重要ではないようだが、もう一つのRho GTPaseであるTC10αは、脂肪細胞ではAKTに依存しないメディエーターとして同定されている(732)。TC10αは、インスリン刺激によって活性化され、siRNAでノックダウンすると、GLUT4の移動が阻害され、インスリン刺激によるグルコース取り込みが減少する(119)。TC10は、エクソシスト複合体のEXO70と結合することで、GSVの細胞膜へのテザーリングを促進する(350)。さらに、TC10はPISTというタンパク質を介して、ゴルジ網で細胞内にGSVを隔離するタンパク質テザーであるTUGのタンパク質分解による切断を促進する(47, 73-75)。TUGの切断により、インスリン刺激によるGSVの細胞膜への動員が可能になる(943)。さらに、脂肪細胞におけるインスリン刺激によるグルコース取り込みのAKT非依存的な経路として、MUNC18CのINSRによる直接のチロシンリン酸化があり、MUNC18CはSNAREとの相互作用によってGSVの細胞膜への融合を調節する(369)。
本節で述べた脂肪細胞のグルコース取り込みを制御する経路と、上述の筋肉のグルコース取り込みを制御する経路は、かなりの部分で重複している。3T3-L1脂肪細胞では強い証拠があるが、心筋細胞では弱い証拠があるメカニズムがこのセクションの焦点となっている。前述の経路の大部分(AKT/TBC1D4シグナル、TUG切断など)は、脂肪細胞と心筋細胞の両方で機能していることが知られていることに留意する必要がある。さらに、心筋や褐色脂肪組織など、インスリン刺激によるグルコース取り込みを示す他の組織でも、これらの分子機構のサブセットが採用されている可能性が高い(342, 776, 777)。
また、次に述べるように、脂肪細胞のインスリン作用は、脂肪にとどまらず、肝の糖新生を強力に制御している。
III. 間接的なインスリン作用
A. 間接的なインスリン作用の生理的関連性
インスリン作用は、孤立した均質な細胞集団ではなく、特殊な細胞タイプを持つ複雑な多細胞生物の中で進化してきた。したがって、全身のインスリン作用の重要な要素のいくつかが細胞自律的ではなく、組織のクロストークを必要とすることは驚くべきことではない。これらの現象は培養細胞ではモデル化が困難であり、定義上、均質な孤立した細胞集団ではモデル化できないため、その分子基盤の理解はインスリンの直接作用の研究に比べて遅れている。しかし、インスリンの間接作用は、組織全体のインスリン作用に定量的に大きく寄与しており、場合によっては間接作用が直接作用よりも優位に働くこともある(6, 620, 686)。このように、インスリンの間接作用は、無傷の生物の研究において無視できない(841)。特によく研究されている例としては、脂肪組織での脂肪分解を阻害することにより、肝臓での糖新生を抑制するインスリンの作用が挙げられる(6, 54, 89, 135, 136, 620, 684, 686, 774)。他にもいくつかのインスリンの間接作用が証明されている。例えば、膵臓のα細胞におけるインスリン作用は、グルカゴンの分泌を抑制することにより、統合的なインスリン反応を強化する。また、中枢神経系のインスリン作用が末梢の代謝制御に関与していることを示す文献が数多く存在する。本レビューの範囲は、内分泌膵臓や中枢神経系ではなく、主に末梢のインスリン反応組織を対象としており、また、これらの後者のトピックに関する優れた最近のレビューが利用可能であるため(2, 201, 307, 392, 409, 444, 459, 652, 749, 863)、本セクションでは、インスリン-脂肪細胞-肝細胞軸とその肝グルコース産生の制御に焦点を当て、これらの他の重要なインスリンの間接作用については簡単にしかレビューしない。
B. 脂肪細胞-肝細胞軸:脂肪分解による肝グルコース産生の制御
脂肪酸基質の利用可能性が肝グルコース産生を制御することは以前から知られていた。灌流したグリコーゲン欠乏ラット肝を用いて、1960年代にいくつかのグループが、オレイン酸の酸化がアラニンや乳酸からのグルコン生成を増加させることを示した(315, 835, 903, 905, 906)が、これには議論があった(217)。脂肪酸による糖新生の活性化には糖新生基質が必要であり、またミトコンドリアのアセチルCoA含量の増加が観察されたことから、これらの研究者は、アセチルCoAによるピルビン酸カルボキシラーゼ(PC)のアロステリックな活性化が、脂肪酸酸化による肝糖新生の刺激効果を媒介すると提案した(7, 434, 864, 902, 903)。しかし、アセチルCoAは肝細胞内での濃度が低く、生体外では急速に分解されてしまうため、肝サンプルでの正確な測定には限界があった。さらに、単離された肝細胞を用いた研究では、インスリンがPC活性に直接作用することは証明できなかった(138)。しかし、試験管内試験の肝臓標本では、脂肪組織の脂肪分解による脂肪酸の利用可能性を制御するインスリンの効果を研究することはできない。インスリンがHGPに間接的に作用することを示す生体内試験の証拠は、1966年に膵臓摘出イヌにおいて、ニコチン酸で脂肪分解を阻害すると、血漿中のNEFAレベルとHGPの両方が低下するという報告に遡ることができる(615)。その20年後には、末梢のインスリン濃度は上昇するが、門脈のインスリン濃度は上昇せず、グルカゴンも変化しない低用量のインスリン注入が、ヒトのHGPの抑制に有効であったことが報告され、HGP抑制におけるインスリンの肝外作用の重要性が指摘された(661)。しかし、脂肪分解-アセチルCoA-PC-HGP軸の関連性を生体内試験で決定的に示すには、インスリン分泌を増加させグルカゴン分泌を減少させるNEFAの効果が混同されることや、肝アセチルCoA含量や肝ピルビン酸カルボキシラーゼのフラックス速度の測定など、方法論上の限界がある(229)。さらに、脂肪分解によりグリセロールが放出され、これが酸化還元で調節されながらも基質に大きく依存した速度でグルコースに変換される(34, 511, 620)。ソマトスタチン膵臓クランプの熟練した使用、門脈と末梢のホルモン濃度のより良い測定、脂肪酸とグリセロールの代謝を測定するトレーサー法の開発により、これらのメカニズムの生理的関連性についての洞察が得られた。その後、いくつかのグループが、インスリンのHGP抑制効果は、血漿NEFAレベルの抑制によって部分的に媒介されることを報告した(6, 476, 686, 774)。Bergmanと共同研究者(686)は、NEFAがHGPを調節する効果は、NEFAが内皮のインスリン輸送を調節することに起因するのではないかと考えた。NEFAがHGPを調節することを示す特に説得力のある証拠は、絶食イヌを用いた研究から得られた。高インスリン血症-高血糖膵臓クランプ中にNEFAを注入すると、血漿NEFAレベルとHGPの両方に対するインスリンの抑制が完全に阻止された。他の研究におけるこの効果の大きさの違いは、種の違い、門脈および末梢のインスリンとグルカゴンの制御が不完全であること、インスリンが正味の肝グリコーゲン分解を抑制する直接的な効果を持つこと、などの組み合わせによって部分的に説明される。最近、Perryと共同研究者(620, 630)は、新しい液体クロマトグラフィー・タンデム質量分析法を用いて、生体内試験での肝アセチルCoA含量と肝ピルビン酸カルボキシラーゼのフラックス速度を評価し、自由行動下のラットにおいて、インスリンが白色脂肪組織脂肪分解と肝アセチルCoA含量および肝ピルビン酸カルボキシラーゼ活性を抑制することにより、肝グルコエネシスを急性に抑制するという仮説を検証した。高インスリン血症-高血糖クランプ中、インスリンはパルミチン酸とグリセロールの代謝(すなわち脂肪分解)肝アセチルCoA含量、肝ピルビン酸カルボキシラーゼフラックス、および肝HGPを時間的に緊密に一致して急速に減少させることが示された(620)。このような肝アセチルCoAの減少を酢酸の注入によって防ぎ、同時にグリセロールを基礎代謝速度に合わせて補充すると、インスリンによる肝ピルビン酸カルボキシラーゼのフラックスとHGPの抑制効果は完全に消失した(620)。重要なことは、これらの研究は、グリコーゲンが枯渇した肝臓を持ち、実質的にHGPの100%を糖新生に頼っている絶食ラットで行われたことである。
遺伝子組み換えネズミを用いた研究でも、HGPのインスリン抑制に脂肪細胞-肝細胞軸が重要な役割を果たしていることが示唆されている。特に、肝臓特異的にAkt1,Akt2,Foxo1を欠損させたマウス(TLKOマウス)の研究は、このメカニズムを強く証明している(504,620,842)。TLKOマウスは、肝細胞のインスリン反応の重要なエフェクターを3つ欠損しているにもかかわらず、高インスリン血症-高血糖クランプ試験において、肝アセチルCoAとHGPを正常に抑制した(504)。バゴトミーもグルカゴン遮断もこの表現型を変化させなかった(842)。しかし、インスリンによる肝臓のアセチルCoA含量と肝臓へのグリセロールフラックスを減少させるWAT脂肪分解の抑制効果を、アセテートとグリセロールの注入で阻止すると、TLKOマウスも野生型マウスも、インスリンに反応してピルビン酸カルボキシラーゼフラックスと肝臓での糖新生を減少させることができなかった(620)。別の研究では、血漿中のNEFA濃度を基礎レベルに維持するためのクランプ中にイントラリピドとヘパリンを注入すると、TLKOマウスではインスリンによるHGPの抑制が阻止されたが、野生型マウスでは阻止されなかった(842)。同様に、アンチセンスオリゴヌクレオチドを用いて肝臓と肝臓上部のInsrを欠損させたラットでは、ATGL阻害剤であるatglistatinを用いて脂肪部のインスリン作用を機能的に回復させた場合にのみ、高インスリン血症-高血糖クランプ試験でHGPを抑制することができた(620)。正規の肝細胞インスリンシグナルを欠くネズミを用いたこれらの研究は、肝細胞インスリンの直接的な作用とは無関係に肝の糖新生が制御されていることを強く示唆している(620)。むしろ、これらのデータは、インスリンによる肝の糖新生の制御は、アセチルCoAのアロスタリーとグリセロール基質の利用可能性を介した白色脂肪組織脂肪分解にほとんど依存しているというモデルを支持している(620)。このパラダイムは、1)全身のInsrをノックアウトしたマウスでは、Insrの発現を肝臓でトランスジェニックに回復させても、HGPのインスリン抑制が損なわれなかったこと、2)肝臓特異的にInsrを切除しても、HGPのインスリン抑制が損なわれなかったこと、という驚くべき報告も説明できる(105, 592)。脂肪細胞レベルでの脂肪分解の変化もまた、HGPのインスリン抑制に影響を及ぼす可能性がある。脂肪組織のAtglを欠損したマウスでは、脂肪酸代謝が低下し、それに伴い肝アセチルCoAが減少し、野生型と比較してHGPの抑制効果が向上した(12, 620)。最後に、Pde3b-/-マウスのインスリン反応は、血漿NEFAレベルを抑制することができず、それに伴ってHGP抑制が損なわれるという特徴があったが、これらのマウスは肝細胞のインスリンシグナル伝達にも欠陥があった(147)。以上のことから、インスリンによる肝内グルコン生成の急性抑制には、グリセロールからグルコースへの変換とピルビン酸カルボキシラーゼのアセチルCoA活性化の両方を低下させる白色脂肪組織脂肪分解の抑制が関与しているという仮説が支持されている。
インスリンの直接作用と間接作用のHGPに対する相対的な重要性を明らかにするには、さらなる調査が必要である。特に、これらの相対的な寄与は、生理学的および病態生理学的な条件によって大きく異なる可能性があるからである。しかし、簡略化して考えると、HGPに対するグリコーゲン分解の寄与は、肝のインスリンの直接作用によって制御され、HGPに対するグルコン生成の寄与は、主に白色脂肪組織脂肪分解に対する作用を介したインスリンの間接作用によって制御されると考えることができる(図6)。したがって、グリコーゲンが十分にある(給餌した)肝臓では、HGPに対するインスリンの直接作用が優勢となり、グリコーゲンが枯渇した(絶食した)肝臓では、HGPに対するインスリンの間接作用が優勢となる。この仮説は、このテーマに関する不一致の文献を説明するものであり、種差の重要性を強調するものである。ヒトやイヌはネズミに比べて肝グリコーゲンの分解が遅いので、一晩の絶食後にはインスリンの肝グルコース代謝への直接作用が優勢になると考えられる。一方、ラットやマウスは一晩の絶食で肝グリコーゲンがほとんどなくなるため、この条件下ではインスリンの肝グルコース代謝に対する間接効果が支配的になると考えられる。この後者の効果のヒトでの定量的な意義についてはまだ検討されていないが、コントロール不良の2型糖尿病患者のHGPの増加は、正味の肝グリコーゲン分解ではなく、肝グルコン生成の増加によって完全に説明されていることは興味深いことである(513, 704)。2型糖尿病におけるNEFAレベルの上昇と高血糖の関連性は以前から認識されていたが、これらの因果関係を示すメカニズム上の証拠が増えることで、白色脂肪組織脂肪分解を標的とした治療法の革新に拍車がかかることが期待されている。
図6 脂肪細胞-肝細胞軸とインスリンによる糖新生の抑制
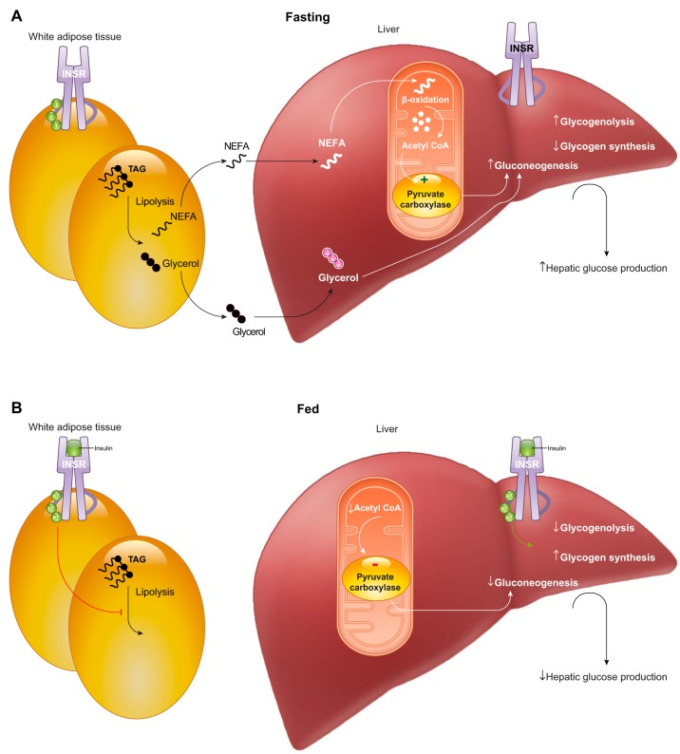
A: 断食状態では、脂肪細胞は非エステル化脂肪酸(NEFA)を循環系に放出する。肝細胞では、NEFAはミトコンドリアのアセチルCoAに酸化され、ピルビン酸カルボキシラーゼ(PC)のアロステリックアクチベーターとなる。PCはグルコン生成を促進する。これがグリコーゲン分解と相まって、空腹時の肝グルコース産生(HGP)を促進する。B: インスリン刺激時(食後など)には、インスリンの直接作用と間接作用の両方がHGPを抑制する。脂肪細胞のインスリンシグナルは脂肪分解を抑制し、血漿中のNEFA濃度、肝のミトコンドリアのアセチルCoA濃度、PC活性、およびグルコン生成フラックスを減少させる。同時に、インスリンが肝細胞に直接作用すると、グリコーゲンの純合成が促進される。この2つのプロセスにより、インスリンは迅速かつ強力に正味のHGPを抑制することができる。
C. インスリンによるグルカゴン分泌の抑制
グルカゴンの作用は正常なグルコースのホメオスタシスに重要であり(871)、特に肝臓に対する強力な抗インスリン作用を考えると、どのような代謝状態においてもインスリンまたはグルカゴンのどちらか一方の分泌が好まれ、両方の分泌は好まれないという、優雅で厳密に制御された相互パラクリン制御システムが進化してきたことは驚くべきことではない(863)。1970年、インスリン欠乏性糖尿病は、相対的なグルカゴン血症の状態であり、グルカゴン分泌促進剤であるアルギニンに対するα細胞の反応性が高いことが示され(861)、その後の研究で、インスリンがパラクリン的に作用して膵島からのグルカゴン放出を抑制することが確認された(29, 280, 523, 890)。1型糖尿病(1型糖尿病)のヒトでは、グルカゴンレベルを抑制するためには、インスリン単体でも共栄血糖および低血糖時に十分であり、非糖尿病のヒトでは空腹時のグルカゴンレベルはインスリン抵抗性と相関している(164, 223, 756)。α細胞のインスリン受容体を欠損したマウスでは、摂食時およびインスリン負荷試験時にグルカゴン濃度が上昇することから、このメカニズムの直接的な遺伝的証拠が得られた(393)。インスリンがα細胞のグルカゴン分泌を抑制するメカニズムはまだ解明されていないが、PI3K活性とホスホジエステラーゼを介したcAMPの分解が関与していることが示されている(213, 683)。全体として、インスリン作用と糖尿病におけるグルカゴンの重要性は過小評価されている(862)。グルカゴン受容体を欠損したマウスがβ細胞を破壊しても糖尿病を発症しないことは、このことを示す数多くの印象的な例の一つに過ぎない(468)。
D. 中枢神経系インスリン作用の末梢への影響
グルコース代謝の調節における中枢神経系の役割は、1850年代から認識されていた(55)が、過去20年間でこのテーマはルネッサンスを迎えている(444)。インスリンは血液脳関門を通過し(40)、神経細胞もグリア細胞もインスリン受容体を発現している(652)。インスリンの脳内での主な機能は食欲の抑制である(48, 912)。神経細胞特異的にInsrを欠失させたマウスは、食欲を抑制するためか、食事誘発性の肥満になりやすく、肝のインスリン抵抗性も併発する(104)。全身のInsr-/-マウスでInsrの発現を神経細胞特異的に救済すると、寿命が数日から数週間に延びるが、救済されたマウスは依然として重篤な糖尿病を発症する。このことは、脳のインスリン作用の有益な効果を十分に発揮するには、末梢のインスリン受容体が無傷であることが必要であることを示している(591)。実際、中枢神経系のシグナル伝達は、エネルギーバランスとは独立したメカニズムで肝臓のインスリン作用を制御することが明らかになっている。2002,ネズミの肝グルコース産生を抑制するには、1時間以上遅れて脳室内にインスリンを投与すれば十分であるという知見が得られ(588)、この分野での研究が進んだ。その後、視床下部の特定の核におけるインスリン作用が、げっ歯類において、肝グルコース産生を強力に抑制し(588, 653)、筋肉のグルコース取り込みを促進し(415)、脂肪組織の脂肪分解を抑制し(415, 737)、グルカゴンの分泌を抑制することが報告された(612)。鼻からのインスリン投与は、脳脊髄液のインスリン濃度を不均衡に増加させ、痩せたヒトでの高インスリン血糖クランプ試験において、インスリンによる肝グルコース産生の抑制効果を高めることが示されている(308)。しかし、イヌでは、脳内インスリン濃度の生理的上昇だけでは、肝グルコース産生を変化させるには不十分である(208, 675)。脳と末梢のインスリン作用を結びつけるメカニズムは不明だが、おそらく交感神経と副交感神経の流出、さらには視床下部-下垂体-副腎(HPA)軸が関与していると考えられる。例えば、ある研究では、肝迷走神経切断術がHGPに対する中枢神経系インスリンの作用を阻害することが示された(653)が、肝ムスカ性アセチルコリン受容体を欠損したマウスの正常な肝グルコース代謝(480)や、脱神経したげっ歯類の肝臓(551)や移植したヒトの肝臓(術後少なくとも2年間は脱神経したままである)(744)の正常な肝インスリン作用は、この仮説に疑問を投げかけている(673)。この分野の実験的研究の多くが生理学的に一般化できるかどうかについては、他にも重要な問題が提起されている。例えば、脳室内インスリンの投与量と投与経路が生理的でないこと、門脈-末梢間のインスリン勾配が生理的に約3:1に保たれておらず、その結果、肝外のインスリン作用が強調されていること、実験群間でグルカゴンレベルをコントロールできていないこと、げっ歯類と大型哺乳類では肝グルコース産生の源とコントロールに種差があることなどである(673)。これらの懸念に対処するための優れた試みとして、意識のあるイヌでグルカゴンを基礎的に補充し、門脈からインスリンを注入するソマトスタチン膵臓クランプを行い、PI3K阻害剤LY249002を脳室内に注入して、生理的高インスリン血症の末梢作用に対する脳のインスリン作用の寄与を評価した(674)。この実験では、急性期の脳内インスリンシグナルの阻害が、インスリンによる肝グルコース産生の抑制やインスリン刺激による全身グルコース取り込みに影響を与えないことがわかったが、生理的な意義は不明だが、肝グルコキナーゼの発現誘導にわずかな鈍化が見られた(674)。慢性的な脳のインスリン作用や脳のインスリン抵抗性が末梢のインスリン作用に大きく影響するかどうかは未解決の問題である(444)。
脳は、インスリンの直接的な作用部位であるだけでなく、末梢で分泌される様々なホルモンのシグナル統合部位でもある。中でもレプチンは、エネルギーバランスや代謝に様々な影響を及ぼす中枢性アディポカインとして、特に注目されている(537, 626)。レプチンの作用機序は数多くあり、他にもレビューされているが(166, 236, 537, 565, 626)、先に述べた脂肪細胞-肝細胞軸と直接関連する機序の一つとして、飢餓や糖尿病性ケトアシドーシスにおけるレプチンの役割が挙げられる(623, 628)。これらの状態のラットでは、血漿中のレプチン濃度が低く、HPA軸が活性化され、その結果、コルチコステロイドによる白色脂肪組織脂肪分解、ケト生成、肝グルコン生成が起こるが、レプチンを生理的に補充すると、HPA軸が抑制され、これらの影響を防ぐことができる。このように、レプチンは、これまでインスリン減少が主な原因であると考えられていた飢餓時のグルコースから脂肪酸への基質転換の制御に関与している。興味深いことに、レプチンにはホルモン作用があり、低濃度ではHPA軸主導の脂肪分解を抑制するが、高濃度ではカテコラミン主導のWAT脂肪分解を促進する(628)。1型糖尿病におけるレプチン慢性療法の抗糖尿病効果に大きく貢献している第2の重要なメカニズムは、レプチンによるグルカゴン分泌の抑制であり、これは末梢または中枢神経系へのレプチン投与によって達成される(249, 877)。慢性的なレプチン治療またはインスリンで、高グルカゴン血症、高血糖、ケトアシドーシスを改善できるという興味深い観察結果から、糖尿病の症状は、インスリン不足ではなくグルカゴン過剰の結果であるという仮説が提唱されている(862)。しかし、重要なことは、レプチンは、グルカゴン過剰状態が改善される前に、高血糖糖尿病ケトアシドーシスを回復させるということである。この急性の効果は、HPA軸に起因するWAT脂肪分解の抑制によってもたらされる(630)。
腸-脳-肝臓の軸が、げっ歯類の肝グルコース代謝を制御していることを示唆する実験もいくつかある。腸はそれ自体が内分泌器官であり、グレリン、インクレチン、FGF19,コレシストキニン(CCK)などのホルモンを産生する腸内分泌細胞がある(409)。FGF19(げっ歯類ではFGF15とも呼ばれる)は、胆汁酸の感知に応じて遠位小腸から分泌され、胆汁酸合成の腸肝制御という本来の役割に加えて、げっ歯類ではHPA軸を抑制することで耐糖能を高める中枢的な働きをする(410, 556, 622)。腸内細菌叢は、末梢のインスリン作用の中枢神経系制御にも関与している可能性がある。ラットでは、微生物が産生する酢酸が、副交感神経系を直接活性化することにより、グルコース刺激によるインスリン分泌を促進することが示された(624)。このメカニズムがヒトでも機能しているかどうかは不明である。栄養素を直接感知することは、腸が中枢神経系による末梢代謝の調節を促すもう一つのメカニズムとして提案されている。ラットでは、十二指腸脂肪酸エステル化が副交感神経の活性化を介して作用し、上部腸管脂質投与時の肝グルコース産生を抑制することが示されているが(879)、ヒトでの同様の実験(膵臓クランプ条件下で低用量の十二指腸脂質を注入)では、十二指腸脂質は血漿グルコースや肝グルコース産生に影響を与えなかった(925)。一方、CCKの分泌を介して食物摂取を抑制する上での十二指腸脂質の供給の役割については議論の余地がない(529)。げっ歯類で発見されたこれらの統合された生理学的メカニズムの複雑さは、ヒトでの慎重な確認が必要であるが、腸-脳間のシグナル伝達経路に関する研究がさらに進めば、肥満治療手術の効果のメカニズムやインクレチンの生理学など、治療に関連する分野で継続的な洞察が得られると思われる(201)。
IV. インスリン抵抗性の病態生理
A. インスリン抵抗性とは?
インスリン抵抗性という概念は、Himsworth(317)の観察にまで遡ることができる。彼は、糖尿病患者にグルコースとインスリンを同時に注射すると、2つの結果のうちの1つが生じることに注目した。糖尿病患者の中には、血糖値が安定しているか低下している患者がおり、このような患者はインスリン感受性が高いと呼ばれている。一方、血糖値の上昇が顕著な糖尿病患者は、インスリン非感受性とされた。血漿中のインスリン濃度が正常であれば、標的組織は、内因性グルコース産生の抑制、脂肪分解の抑制、利用可能な血漿中グルコースの細胞への取り込み、グリコーゲンの純合成を含む正常な協調的グルコース低下反応を起こすことができない(377, 378, 381, 595, 684)。このようなインスリン抵抗性は、それを補うためにインスリン分泌を増加させる必要があるため、空腹時の血漿インスリン濃度が上昇する(176, 380)。インスリン感受性とインスリン分泌をつなぐリアルタイムのフィードバック回路は、主な欠陥を特定するという「鶏卵(chicken-eggˮ)問題」を複雑にしているが、明らかなのは、インスリン標的組織とβ細胞の両方の欠陥が、空腹時高血糖と2型糖尿病の発症に必要であるということである(380)。慢性的な高インスリン血症と同様に、多様な生理活性因子がインスリン感受性を低下させる可能性がある(595, 724)。組織のインスリン作用におけるこれらの欠陥は、減量や低カロリー療法によって(2型糖尿病患者であっても)容易に回復可能であるが、インスリン抵抗性がある状態で栄養過多が続くと、高インスリン血症とインスリン抵抗性の悪循環が生じ、最終的にはβ細胞の機能不全に陥り、グルコースや脂質の毒性やその他の要因によって顕性2型糖尿病になる可能性が高い(380, 637)。
インスリン抵抗性の統合された生理機能は、標的細胞でのインスリン作用の欠陥によるものである。細胞内のインスリン作用の欠陥を特定する試みは、新しいエフェクターの同定によって進められてきた。INSRがインスリン作用の唯一の分子エフェクターとして知られていた1970年代から 1980年代初頭にかけて、いくつかのグループがインスリンの用量反応曲線や125I-インスリン結合試験を用いて、表面のINSR含量と生理的なインスリン作用や抵抗性との関係を明らかにした(26, 272, 378, 417, 420, 595, 787)。当時、インスリン抵抗性の原因が、「受容体の欠陥」(すなわち、細胞表面でのINSRの発現低下)にあるのか、「受容体後の欠陥」(すなわち、シグナル伝達の障害)にあるのか、というのが中心的な問題であった(図7)(417, 595)。受容体の欠陥は、肥満や糖尿病のネズミやヒト、脂肪細胞や他の細胞タイプで確認された(26, 272, 379, 418, 786, 787)。持続的な高インスリン血症に直面した際の表面INSR量の代償的なダウンレギュレーションがこの現象を部分的に説明していると思われるが、インスリン抵抗性における表面INSRの発現と調節不全を積極的に制御するメカニズムが明らかにされ始めている。例えば、ユビキチンリガーゼであるMARCH1は、INSRをユビキチン化して細胞表面の受容体の数を減らす(566)。MARCH1は、FOXO1を介して転写制御されており、肥満マウスやヒトのWATではMARCH1の発現が増加しており、FOXO1の阻害によるインスリン抵抗性と一致している(566)。細胞は予備の受容体を持っているため、細胞表面のINSR含量が正常値の5-10%以下にならない限り、最大反応の低下は見られない(378)。
図7 用量反応曲線におけるインスリン抵抗性
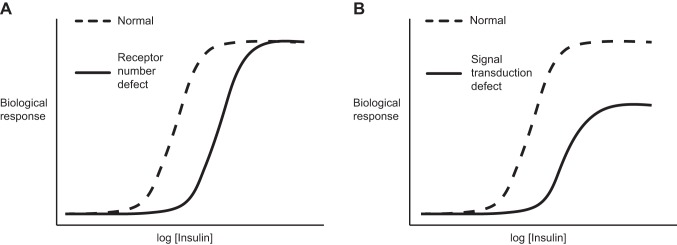
A: 表面のインスリン受容体(INSR)の含有量が減少した仮想的な細胞では、用量反応曲線は右にシフトするが、表面受容体の90%以上が失われない限り、最大生物学的反応は減少しない。B: インスリンのシグナル伝達(ポスト受容体)に欠陥がある細胞、あるいは受容体とポスト受容体の両方に欠陥がある細胞では、右シフトと最大反応の低下の両方が観察される。右のグラフは、ヒトの肥満に伴う筋肉、肝臓、脂肪組織でのインスリン抵抗性を表している。
ヒト2型糖尿病のインスリン抵抗性は、表面の受容体量が90%以下であるにもかかわらず、用量反応曲線が右にシフトし、全身のグルコース取り込みに対するインスリン反応の最大値が低下することが示されたとき、受容体と「受容体後ˮ」の両方の欠陥がインスリン抵抗性に寄与していることが推測された(417, 595)。その結果、インスリン受容体結合の低下が典型的な肥満に伴うインスリン抵抗性を説明するという仮説は、インスリンのシグナル伝達の欠陥が中心となるモデルに取って代わられて久しい(35)。さらに、典型的な肥満に伴うインスリン抵抗性のシグナル伝達(ポストレセプター)の欠陥には、ポストレセプターのエフェクターに加えてインスリン受容体自体も関与していることが長年にわたって認識されており、「受容体の欠陥」と「ポストレセプターの欠陥」の意味が混同されている(77, 247, 595, 821)。INSRがチロシンキナーゼであることが明らかになってからの30年間、この分野で行われてきたほとんどすべてのメカニズム研究は、インスリンのシグナル伝達の欠陥を明らかにすることに焦点を当ててきた。しかし、肥満に伴う典型的なインスリン抵抗性には、表面のINSR量の減少とインスリンシグナル伝達の障害の両方が関与していることは明らかである。
インスリンの「抵抗性」(インスリンEC50の増加)を、インスリンの「反応性」の低下(最大効果の低下)と区別することは有用であるが(378)、この分野ではもはや一般的な方法ではない。インスリン抵抗性と反応性を適切に区別するためには、インスリンの用量反応曲線を作成する必要があるが、これは生体内試験での研究では煩雑である。一般的に用いられている意味でのインスリン抵抗性は、したがって、このレビューの残りの部分では、最大反応の低下を伴うか否かにかかわらず、EC50が増加したインスリンの用量反応曲線として定義されている。重要なことは、インスリン抵抗性は、インスリンシグナルのスイッチオフではないということである。このため、高インスリン血症は、軽度および中等度のインスリン抵抗性において、インスリン作用を維持する効果的な代償機構である。インスリン抵抗性は組織特異的な機能的結果を示すので、次に、骨格筋、肝臓、肝臓でのインスリン抵抗性の特殊性を考察し、典型的な肥満に伴うインスリン抵抗性では、どのシグナル伝達エフェクターとどの生理機能が障害されるかに注目する。
B. 骨格筋のインスリン抵抗性の病態生理
骨格筋におけるインスリンの主な機能は、細胞内へのグルコースの取り込みを促進することであり、そのプロセスはGLUT4の移動によって制御される。インスリンで刺激された筋のグルコース取り込みは、インスリン抵抗性の影響を非常に受けやすく、実際、典型的な肥満に伴うインスリン抵抗性と2型糖尿病の主要な構成要素となっている(182, 767)。骨格筋は、インスリン刺激によるグルコース廃棄の主要な部位であるため(高インスリン血症による共食クランプ時には70〜80%が廃棄されるが、グルコースの出現部位、グルコース濃度、組織のグルコース需要がクランプ時とは異なる食後の状態では25〜30%しか廃棄されない)筋のインスリン抵抗性は全身のグルコース代謝に大きな影響を与える(182, 428)。グリコーゲン合成と解糖のインスリン刺激は、どちらも基質を供給するためにインスリン刺激によるグルコースの取り込みがそのまま必要であるため、これらの作用もインスリン作用に対して抵抗性になる(152, 768)。
筋肉のグルコース処理に対するインスリン抵抗性のメカニズムについては、様々な研究が行われてきた(図8)。初期の研究では、非酸化的グルコース代謝(すなわち、グリコーゲン合成)が筋細胞グルコースの主要な運命であることが示唆され、後に13C磁気共鳴分光法(MRS)を用いてヒトで直接確認された(58,180,768)。インスリン刺激による筋グリコーゲン合成が、2型糖尿病患者と2型糖尿病患者のインスリン抵抗性のある痩せた健康な子孫の両方で著しく(〜50%)障害されていることが明らかになったことで、筋のインスリン抵抗性が機能的に説明できるようになったが、遮断部位は特定できなかった(633, 768)。他の多くの研究では、インスリン抵抗性や2型糖尿病の筋肉でグルコース輸送が減少することが示されているが、これらの結果も同様に、グリコーゲン合成活性、ヘキソキナーゼ活性、グルコース輸送のいずれかのレベルで一次遮断を行い、最終的にグルコース輸送を減少させることでシステムを「バックアップ」した結果であると考えられる(193, 255, 767)。最終的には、細胞内のグルコースおよびG6P濃度の13Cおよび31P MRS測定により、糖尿病患者のインスリン刺激による筋グリコーゲン合成の低下の原因となっている主要な律速段階は、まさにグルコース輸送であることが明らかになった(152, 285, 633, 698)。これらの生理学的研究は、インスリンが筋細胞のGLUT4の細胞膜およびt管への移動を強力に制御し、この移動がヒトのインスリン抵抗性では欠損しているという細胞生物学的観察を補完するものであった(258, 651, 881)。これらの研究をまとめると、筋のインスリン抵抗性の問題は、インスリンとINSRの結合とGLUT4の転座をつなぐシグナル伝達カスケードに欠陥があるということになる。
図8 骨格筋のインスリン抵抗性の機能的影響
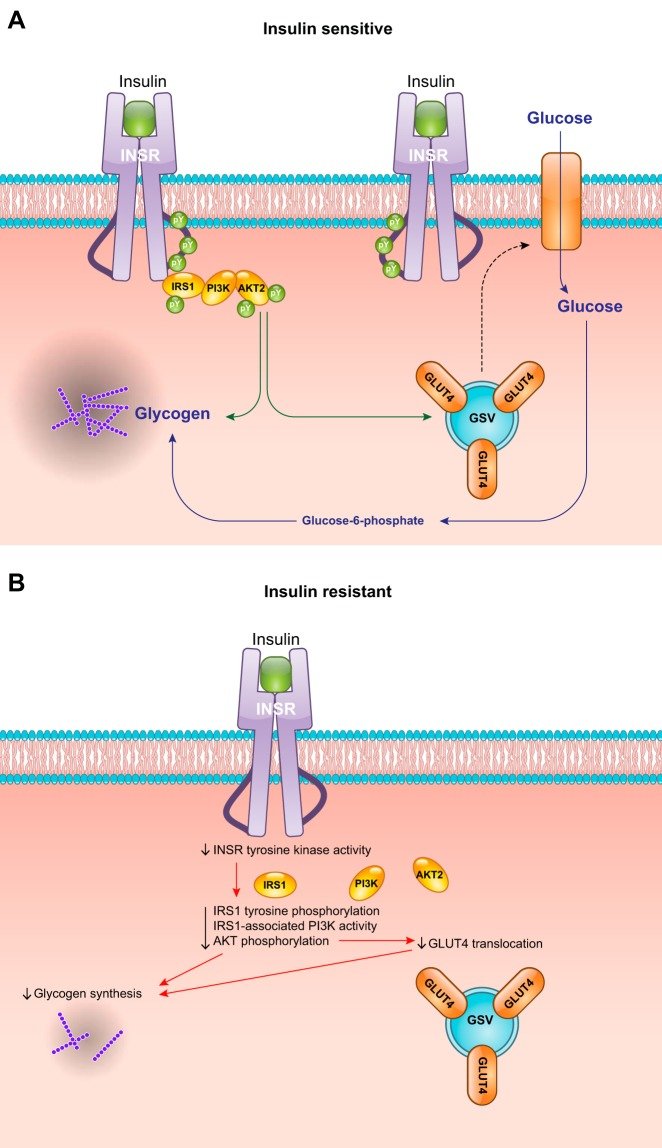
A:インスリン感受性の骨格筋細胞は、インスリンシグナルカスケードの代謝インスリン受容体基質1(IRS1)-ホスホイノシド-3-キナーゼ(PI3K)-AKTアームを活性化し、グルコースの取り込みとグリコーゲン合成を増加させる。B: インスリン抵抗性の心筋細胞では、近位のインスリンシグナルイベントが障害され、GLUT4の移動とグリコーゲン合成を促進するインスリンの能力が鈍化している。
骨格筋のインスリン抵抗性は、インスリンシグナル伝達の最も近位のレベルでの欠陥に起因する。骨格筋のインスリン抵抗性は、インスリンシグナルの最も近位のレベルであるINSR、IRS1,PI3K、AKTの活性に起因する。初期の研究では、肥満マウスの骨格筋から精製したINSRのチロシンキナーゼ活性が欠損していることが明らかになった。これにより、細胞内のインスリンシグナル伝達が損なわれていることが初めて示され、表面のインスリン受容体のダウンレギュレーションが肥満に伴うインスリン抵抗性の唯一の欠陥ではないという予測が立証された(417, 470, 595)。この観察は後に、肥満や糖尿病のヒトにも拡大され、表面のINSR量の減少と、精製した受容体からのINSRキナーゼ(IRK)活性の減少という2つのINSRの欠陥が見られた(117)。IRK活性の低下は、糖尿病ラット、妊娠性糖尿病の女性、2型糖尿病患者の若い痩せた親族の骨格筋でも観察された(66, 107, 301)。しかし、現在ではIRK活性を組織サンプルから測定することはほとんどない。代わりに、INSRの活性化は、リン酸化チロシンの免疫ブロッティングによって評価されることが多い。この実験は技術的には容易であるが、INSRのシグナル伝達を直接評価するものではない。また、INSRにはチロシンリン酸化部位が多数存在するため、これらのパラメータの関係は複雑である(83, 117, 248, 470)。IRKの活性低下は、インスリン抵抗性骨格筋で一貫して観察されるIRS1のチロシンリン酸化の減少からも示唆されている(175, 248, 285, 436, 942)。IRS1に関連するPI3K活性に対するインスリン刺激の鈍化もまた、インスリン抵抗性筋において、急性または慢性的に誘発されるかにかかわらず、再現性をもって観察される(175, 198, 285, 441, 942)。興味深いことに、肥満のヒトや2型糖尿病のヒトの骨格筋では、MAPKを介したマイトジェニックシグナルに対してインスリン抵抗性が発現しない(175)。AKTを含む他の遠位エフェクターのインスリン活性化の障害は、筋のインスリン抵抗性でしばしば見られるが、近位のインスリンシグナル伝達の欠陥が同時に存在するため、これらの遠位の欠陥が独立した起源を持つのか、あるいは近位の欠陥の単なる二次的なものなのかを判断するのは難しい。インスリン抵抗性には複数のシグナル伝達エフェクターの調節異常が関与していると考えられるが、肥満に伴うインスリン抵抗性で見られるグルコース取り込みに対するインスリン刺激の障害全体を説明するには、近位部のシグナル伝達の欠陥で十分である可能性もある。シグナル伝達経路の計算モデルが進歩すれば、インスリン刺激によるグルコース取り込みの障害という最終的な機能的欠陥に対する特定のシグナル伝達の欠陥の相対的な寄与を解明することができるだろう(81, 402, 745)。
C. 肝インスリン抵抗性の病態生理
第II章で述べたように、肝のインスリン作用はすべての多量栄養素の代謝に影響する。肝インスリンの作用には、急性と慢性、直接と間接の両方の要素がある。ここでは、インスリンによる肝臓の糖代謝と脂質代謝の調節について、肥満や2型糖尿病でどのプロセスが抵抗性になるかに注目して考える。
インスリンは、適切な基質の供給と相まって、肝臓におけるグルコースの純生産から純取り込みへの切り替えを指揮する。これには、グルコン生成とグリコーゲン分解の抑制、およびグリコーゲン合成の活性化が協調して行われる。糖新生の抑制は、急性期と慢性期に分けられ、そのメカニズムは大きく異なる。肝の糖新生の急性抑制(ラットではインスリン刺激後10分以内)は、主に肝外のインスリン作用によると思われる(6, 208, 620, 686, 774)。脂肪組織での脂肪分解を抑制すると、肝臓への脂肪酸の供給が減少し、その結果、肝臓のβ酸化フラックスが減少する。その結果、肝臓のアセチルCoA濃度が低下し、PC活性がアロステリックに低下する。脂肪分解の抑制によるグリセロール代謝の低下と合わせて、グルコン生成のフラックスが減少する(620)。このメカニズムは、インスリンによるHGPの抑制効果の低下を肝のインスリン抵抗性の代替指標として利用する際に重要となる。糖新生がHGPの大部分を占めるグリコーゲン枯渇肝では、HGPのインスリン抑制障害は、肝のインスリン抵抗性ではなく、脂肪のインスリン抵抗性を示唆しているのかもしれない。このパラダイムは、遺伝的に改変された多くの驚くべきげっ歯類モデルを調和させている。これらのモデルでは、肝のインスリンシグナルに深刻な摂動が生じても、高インスリン血症-高血糖クランプ試験中にインスリンがHGPを抑制する能力を予測できない(105, 135, 504, 592, 603, 620, 840)。この現象に関する最も印象的な研究は、従来の遺伝子ターゲティングまたはアンチセンスオリゴヌクレオチド処理のいずれかによって肝のインスリン受容体を切除したものである。肝特異的インスリン-/-(LIRKO)マウスは肝細胞のインスリンシグナルを完全に欠き、重度の耐糖能異常を示し、高インスリン血症-高血糖クランプ試験ではHGPを抑制しない(234, 539)。しかし、この欠陥は完全に回復させることができる。肝臓のINSRを置き換えるのではなく、肝臓のFoxo1を同時に切除することで、LIRKOマウスの構成的なFOXO1の活性化によって生じる無制限のグルコン生成酵素の発現(およびグルコキナーゼの抑制)を修正することができる(592,603,840)。同様に、肝臓および肝臓内のINSRをアンチセンスオリゴヌクレオチドで破壊すると、ラットのHGPのインスリン抑制が阻害されるが、この欠陥は脂肪分解阻害剤のatglistatinで肝臓内のインスリン作用をシミュレートすることで回復する(620)。肝のINSRを切除した両モデルとも、肝細胞のインスリンシグナル伝達経路が、通常のインスリンによる糖新生の抑制から切り離されていることから、HGP抑制の測定値を肝のインスリン抵抗性の指標として使用するには注意が必要であると考えられる。
2型糖尿病では、肝臓での糖新生の割合が増加し、2型糖尿病の特徴である空腹時高血糖の直接的な原因となっている(259, 346, 513)。しかし、糖新生は、様々なメカニズム(アロステリ、レドックス、基質駆動、転写、翻訳後など)によって制御されているため(647)、インスリンの肝細胞における糖新生への直接的な作用が2型糖尿病で制御されていないとは限らない。インスリンによる慢性的な糖新生の抑制は、急性インスリン刺激試験よりも絶食-再摂食試験でよく観察されるが、これはFOXO転写因子、特にFOXO1の阻害を介した肝細胞への直接的な影響である。FOXO1活性の亢進は、2型糖尿病の肝糖新生の亢進に何らかの役割を果たしているのであろうか?FOXO1のmRNAの発現と核内局在(活性の代用)は、非アルコール性脂肪性肝炎のヒトで増加しており、HOMA-IR(インスリン抵抗性の恒常性モデル評価)スコアと相関していることが報告されている(866)。さらに、上述したように、肝インスリン受容体のアブレーションは、病的な糖新生を駆動するのに必要かつ十分なFOXO1活性の奔放さと関連している(603, 840)。このような表現型は、肝のIrS1およびIrs2を切除して重度の肝インスリン抵抗性を示したマウスでも見られ、Foxo1を切除したマウスでは耐糖能が正常化した(195)。さらに、肝臓と白色脂肪組織のFOXO1をアンチセンスオリゴヌクレオチドでノックダウンすると、慢性的に脂肪を摂取したマウスの肝臓のインスリン作用が改善される(720)。ob/obやlipodystrophicマウスのような深い肝インスリン抵抗性のモデルマウスは、FOXO1活性の増加を示唆するG6pcの発現増加を示す(764)。しかし、短期間(5日間)および中期間(4週間)脂肪を摂取したラットの肝インスリン抵抗性は、グルコン生成酵素の含有量の変化を伴わないことから、遺伝的に正常なげっ歯類では、FOXO1によるグルコン生成遺伝子の転写が関与していないと考えられている(620, 629)。さらに、マウスの肝のPck1発現を90%以上低下させても、糖新生のフラックスは40%程度しか低下しなかったことから、Pck1発現の振動は糖新生を限定的に制御していることが示唆された(108)。最後に、ヒトの2型糖尿病患者を対象とした研究では、空腹時の高血糖にもかかわらず、FOXO1の標的であるG6pcとPck1の肝臓での発現は増加しなかった(719)。以上のことから、FOXO1が完全にかつ慢性的に抑制されると、げっ歯類では病的な糖新生を促進することは明らかであるが、これがヒトの典型的な肝インスリン抵抗性における空腹時高血糖の作用機序であるという強い証拠はない。肝の糖新生は多面的に制御されているので、遺伝的に正常な被験者では、転写を介した「糖新生能力ˮ」のわずかな増加では、糖新生を促進するのに十分ではないかもしれない。2型糖尿病患者の肝における糖新生の亢進に対するFOXO1の制御異常の相対的な寄与を適切に説明するためには、さらなる研究が必要である。
インスリンが肝のグリコーゲン分解を抑制し、糖新生を促進することは、第II部で述べたように、肝細胞のインスリン作用が損なわれていないことを必要とする直接的な効果である。インスリンによる肝グリコーゲン代謝の制御が糖尿病になると抵抗性になるかどうかという問題は、G6PとグルコースによってGSとホスホリラーゼが強力なアロステリック制御を受けることと、GLUT2によって肝細胞の細胞膜を横切るグルコースがインスリンに依存せずに輸送されることによって、混乱する。このためか、肝グリコーゲン代謝制御の欠陥が肝インスリン抵抗性や2型糖尿病に及ぼす影響については、意見が分かれている。例えば、ストレプトゾトシン誘発糖尿病ラットを用いた高インスリン血症-高血糖クランプ試験では、インスリン刺激による肝GS活性の低下は見られなかったが、3日間の高脂肪食ラットを用いた同様の試験では、低下が見られた(439, 721)。しかし、多くのデータは、肝インスリン抵抗性が肝グリコーゲン代謝の欠陥を伴うことを示唆している。ヒトでは、肝グリコーゲン量をMRSで測定したところ、2型糖尿病では食後4時間のグリコーゲン量が少なく、グリコーゲン合成の障害が示唆された(513)。さらに、2型糖尿病の被験者は、空腹時の肝グリコーゲン含量が低く、食後および高インスリン血症-高血糖クランプ条件下でのグリコーゲン合成が低下していることがわかった(437)。さらに、2型糖尿病患者では、グリコーゲン分解が低下し、インスリンによる抑制が不完全であることが判明した(38, 69, 513)。2型糖尿病の日常的な肝グリコーゲン代謝は、グリコーゲン合成と動員の両方に対するインスリンの制御が弱まり、正常な摂食や絶食に特徴的な大きな振動ではなく、比較的静的な肝グリコーゲン含量になるという、振幅の減少を示している。これは、グリコーゲン合成装置の翻訳後修飾、グルコキナーゼ活性の転写制御、核から細胞質への移動のいずれに対してもインスリンの作用が低下しているためと考えられる。
グルコース代謝に対する急性肝インスリン作用を理解するためには、グリコーゲン代謝に対する肝細胞の直接的な作用と、グルコン生成に対する代謝を介した大部分の間接的な作用を分離する必要があるのと同様に、肝インスリン抵抗性を理解するためには、これらの構成要素を互いに区別する必要がある(647)。真の肝インスリン抵抗性は、インスリン刺激によるグリコーゲン合成を阻害し、この機能に対するインスリンの用量反応曲線を右にシフトさせ、最大値を低下させる可能性がある。しかし、孤立した肝のインスリン抵抗性は、インスリンによる糖新生の抑制能力を阻害するとは考えられない。なぜなら、インスリンによる糖新生の抑制は、間接的な要素が大きいからである。この予測は、上述したように、肝細胞のインスリンシグナルを切除した複数のげっ歯類モデルで劇的に立証された。
D. 選択的な肝インスリン抵抗性
肝インスリンの作用は、グルコースの処理に加えて、主にSREBP-1cを介して脂質代謝を強力に制御する。インスリンが通常、肝脂肪生成を促進するのであれば、インスリン抵抗性の被験者は脂肪生成が減少すると予想される。遺伝的な完全肝インスリン抵抗性モデル(すなわち、肝インスリン受容体のアブレーション)では、血漿トリグリセリドが減少し、肝DNLが減少することから、実際にそうなっている(60, 868)。しかし、遺伝的に正常なげっ歯類やヒトでは、肝インスリン抵抗性は肝脂肪症との関連性が高く、インスリン抵抗性肝では正味の脂肪生成が一貫して上昇している。この現象は、「選択的肝インスリン抵抗性」(99)または「経路選択的インスリン抵抗性および反応性」(920)と呼ばれており、これらのモデルの支持者は、「インスリン抵抗性」の肝臓は実際にはグルコースの処理(すなわちFOXO転写プログラム)に対してのみ抵抗性であり、脂質の処理(すなわちSREBP-1c転写プログラム)に対しては抵抗性ではないと主張している(764)。肝の選択的インスリン抵抗性のメカニズムを解明しようとする試みは、インスリンシグナルのうち脂質を処理する部分はそのままで、グルコースを処理する部分が抵抗性になるというインスリンシグナルの分岐の可能性に焦点を当ててきた。初期の報告では、肝臓のIRS1はグルコースの処理に責任があり、IRS2は脂質の処理を制御していると考えられていたが、その後の研究ではそれが裏付けられていない(195, 196, 443, 662, 827)。さらに別の仮説として、単一リン酸化(pThr308)のAKTと二重リン酸化(pThr308, pSer473)のAKTの基質特異性が異なる可能性に注目している(487, 920, 921)。AKTの疎白色脂肪組織性モチーフにあるSer473のリン酸化は、いくつかのAKT基質(例えば、NH2-末端のFOXOサイト)に対する活性を可能にする一方で、他の基質(例えば、GSK3β、TSC2)に対するAKTの活性には、PDK1によるThr308のリン酸化のみが必要であると考えられている(288,362)。db/dbマウスのような肝インスリン抵抗性モデルでは、インスリンによるThr308への刺激は維持されるが、Ser473への刺激は維持されないようなので、この仮説は興味深い(921)。しかし、直接的な実験的裏付けは今のところなく、また、AKTのSer473リン酸化に対する栄養感受性の入力が複数あるため、簡単には解釈できない。肝インスリン抵抗性においてFOXO1が反応しないにもかかわらず、SREBP-1cがインスリンによって活性化され続ける最後のメカニズムは、これらの経路がインスリンに対する本質的な感受性が異なることであると考えられる。このモデルを裏付けるように、SREBP-1cの活性化に対する抵抗性を引き起こすには、FOXO1の不活性化に対する抵抗性よりも4倍高い濃度のINSR阻害剤が必要である(162)。
インスリンシグナルには多くの分岐点があり、AKTやFOXO1などのエフェクターがグルコースや脂質の処理に関与していることもあり、肝のインスリン抵抗性が経路選択的に起こるというパラドックスに対する統一的な仮説はなかなか得られていない(99, 457)。しかし、このパラドックスを解決する可能性があるのは、肝のインスリン抵抗性は選択的なものではなく、慢性的な栄養過多がインスリンに依存しないいくつかの脂肪生成の促進因子を活性化するということである。まず、脂肪のインスリン抵抗性によって促進される循環脂肪酸の基質刺激による再エステル化は、非アルコール性脂肪性肝疾患(NAFLD)における肝臓トリグリセリドの主な原因であると考えられる。NAFLDにおける脂肪酸の再エステル化の役割を示す証拠として、最近、血漿中の脂肪酸の増加が、肝のインスリンシグナルや転写作用とは無関係に、基質押し出し機構による再エステル化によって肝のトリグリセリド蓄積を促進するのに十分であることが示された(868)。注目すべきは、インスリン抵抗性のヒトでは、DNLではなく再エステル化が主要な脂肪生成フラックスであるということである(197)。次に、DNLの転写プログラムのアップレギュレーションには、インスリンは必要ない。栄養素によるDNLプログラムの誘導には、炭白色脂肪組織化物応答要素結合タンパク質(ChREBP:グルコースで刺激される)(657)とSREBP-1c(mTORC1のアミノ酸活性化で刺激される)(487)の両方が関与している。フラクトースはまた、特に強力な脂肪生成の刺激因子であり、急性には基質の押し上げ、慢性的にはChREBPとSREBP-1cの両方の活性化など、複数のメカニズムで作用する(311, 718)。インスリン抵抗性はしばしば栄養過多を伴うことから、これらの栄養感受性経路の活性化は、肝性脂肪症と肝性インスリン抵抗性の共存を説明する魅力的な要因となる。この概念では、肝インスリン抵抗性がインスリン自体のSREBP-1cへの作用を鈍らせるものの、代替的な栄養感受性経路が脂肪生成を促進するのに十分であると考えられる。このメカニズムは、NAFLDのヒトで観察されているDNLの増加を説明している(347, 449, 868)。興味深いことに、高脂肪食ラットではde novo lipogenic fluxが減少することがわかっており、この経路がインスリン感受性を維持するのではなく、インスリン抵抗性になることと一致している(184, 200, 868)。重要なことは、脂肪を摂取しても、DNLプログラムの代替的な栄養感受性ドライバー(ChREBP、mTORC1)が強く活性化されるとは考えられないことである。また、インスリン抵抗性は、単にインスリンの用量反応曲線が変化しただけであり、末梢のインスリン抵抗性に伴う門脈の高インスリン血症は、SREBP-1cを活性化する能力があり、この経路がインスリンに対して非常に敏感であることを考えると、おそらく正常なレベルまで活性化することができることを念頭に置くことも有用である(162)。インスリンに依存しない循環脂肪酸のエステル化、栄養によるDNL転写プログラムの活性化、高インスリン血症を考慮すると、インスリン抵抗性のヒトがNAFLDを発症する仕組みを理解するために、選択的肝インスリン抵抗性のパラドックスを持ち出す必要はないかもしれない(図9)。
図9 肝のインスリン抵抗性にもかかわらず、非アルコール性脂肪性肝疾患(NAFLD)が発症する機序
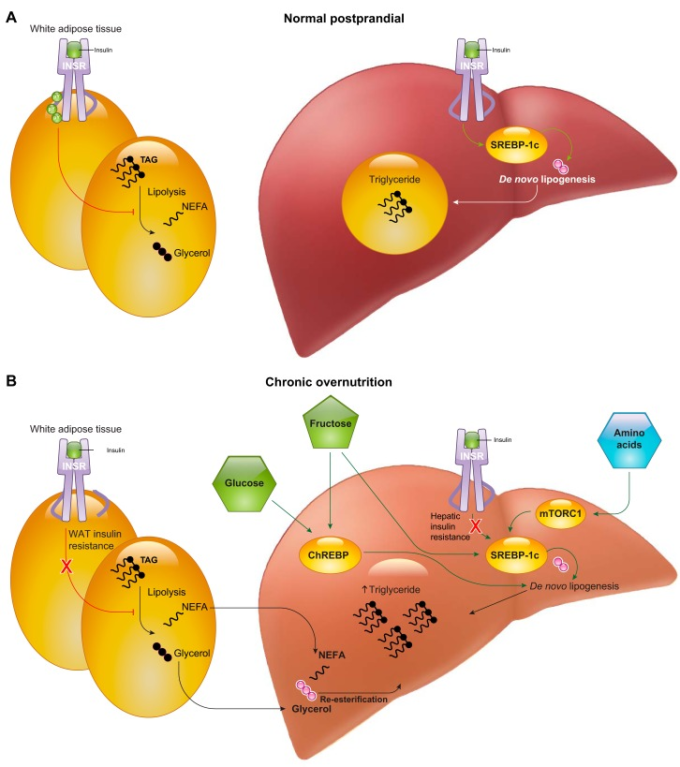
A: インスリンは通常、ステロール制御エレメント結合タンパク質1c (SREBP-1c)を介してde novo脂肪生成を活性化する。B: NAFLDと肝インスリン抵抗性の一見逆説的な共存は、選択的肝インスリン抵抗性という仮説を生み出した。これは、インスリンによる糖代謝の制御が損なわれているにもかかわらず、インスリンによる脂肪生成の活性化が維持されるというものである。しかし、肝のデノボ脂肪生成には、ChREBPやmTORC1/SREBP-1cなどの複数の入力があり、これらは慢性的な栄養過多の状態で活性化される。さらに、肝トリグリセリド合成の主な経路は、あらかじめ形成された脂肪酸の再エステル化であり、慢性的な栄養過多の状態では、食事の供給と脂肪のインスリン抵抗性の両方が原因で、脂肪酸が容易に入手可能である。インスリン受容体(INSR)によるSREBP-1cの活性化がインスリン抵抗性によって損なわれたとしても、これらの他の入力がNAFLDにつながる脂肪生成フラックスを支えることができると考えられる。NEFAは非エステル化脂肪酸、WATは白色脂肪組織を意味する。
経路選択的な肝インスリン抵抗性は、いくつかの肝インスリン抵抗性モデルにおいて近位のインスリンシグナルが障害されているという知見との整合性も難しい。1980年代後半、高脂肪食ラット(886)や2型糖尿病のヒト(116)の肝臓でIRK活性の低下が認められた。また、3日間の高脂肪食を与えたラットのインスリン抵抗性肝でも、IRK活性の低下とIRS2のチロシンリン酸化が観察された(722)。これらの証拠から、肝インスリン抵抗性の主な欠陥は、下流の「分岐点」と呼ばれるエフェクターではなく、インスリン作用を制御する最も近位で強力な場所であるインスリン受容体にあると考えられる。
肝インスリン抵抗性の最後の重要な要素は、INSRに関連した肝インスリンクリアランスの低下である(80, 222, 650)。この現象の分子的な決定要因はまだ完全には解明されていないが、インスリン抵抗性の肝細胞で観察された細胞表面のINSR含量の減少(116, 787)には、CEACAM1(660)、MARCH1(566)、CHIP(830)、p31comet(145)などの表面INSRプールの制御因子が関与している可能性がある。どのようなメカニズムであっても、肝のインスリンクリアランスが低下すると、高インスリン血症が促進される。空腹時血漿インスリン濃度は、HOMA-IRやQuantitative Insulin Sensitivity Check Index (QUICK-I)などのアルゴリズムにおいて、肝のインスリン感受性を示す一般的な指標として用いられることが多いが、β細胞の機能や末梢のインスリン感受性も血漿グルコース-インスリン回路への入力となることには注意が必要である。
要約すると、肝臓におけるインスリン抵抗性は、インスリン受容体のレベルでの欠陥に起因しており、したがって、おそらく肝細胞のインスリンシグナル伝達のすべての部分に影響を及ぼすと考えられる。肝のインスリン抵抗性がシグナル伝達系によって様々な影響を及ぼすことは、各伝達系への入力が多様で複雑であることを考えれば当然のことである。肝インスリン抵抗性を示す最も一般的な生理学的指標であるHGPや空腹時血漿インスリンの抑制は、直接的な要素と間接的な要素の両方を含んでいるため、肝細胞インスリン作用の評価としては不完全である。しかし、これらの測定値は、1)非侵襲的に測定できること、2)肝インスリン作用の直接成分と間接成分を統合した有用な画像を提供することから、特にヒトの研究においては、今後も広く使用されるべきである。肝のインスリン抵抗性を正確に測定するには、表1にまとめた複数の相補的な読み出しを用いるのが最適である。研究者が肝細胞のインスリン作用そのものについて主張しようとする場合には、間接成分を含まない読み出しを使用することが望ましい。これらの読み出しは、肝のインスリン抵抗性の程度と性質を判断するのに使用できるが、原因となる因子については限られた知見しか得られない。実験上の注意点はIVFの項で述べている。
表1 肝性インスリン抵抗性の測定値
| 読み出す | インスリン抵抗性の変化 | 直接/間接 | 急性/慢性 | ノート |
|---|---|---|---|---|
| 正味の肝グリコーゲン合成の活性化 | ↓ | 直接 | 急性 | 高インスリン血症と高血糖症の両方が必要です。 |
| 肝臓の糖新生の抑制 | ↓ | 間接 | 急性 | NEFAとグリセロールの代謝回転および肝臓のアセチルCoA含有量の減少につながるWAT脂肪分解の抑制に関連しています。肝内脂肪分解の抑制によるインスリンの小さな直接効果もあるかもしれません。 |
| 肝臓のブドウ糖生産の抑制 | ↓ | 直接的および間接的 | 急性 | 種と絶食期間に応じて、糖新生とグリコーゲン分解のさまざまな寄与。 |
| INSRTyrリン酸化とIRK活性 | ↓ | 直接 | 急性 | |
| IRSTyrリン酸化 | ↓ | 直接 | 急性 | |
| AKT Ser / Thrリン酸化 | ↓ | 直接 | 急性 | Ser473リン酸化への複数の入力。 |
| 糖新生遺伝子発現 | ↑ | 直接 | 慢性 | 特にG6pc、Pck1。 |
| デノボ脂質生成 | ↓ | 直接 | 慢性 | mTORC1、ChREBPを含む複数の入力。 |
| 空腹時血漿インスリン | ↑ | 直接的および間接的 | 慢性 | 大規模な疫学研究に役立つ、肝臓のインスリン抵抗性の粗い代理。 |
INSRはインスリン受容体、IRKはインスリン受容体キナーゼ、IRSはインスリン受容体基質、WATは白色脂肪組織、NEFAは非エステル化脂肪酸。
E. 脂肪細胞のインスリン抵抗性の病態生理
脂肪細胞のインスリン抵抗性への関心は、栄養吸収源であると同時に内分泌器官でもある脂肪組織の驚くべき複雑さを解明する研究者たちによって、近年復活してきた(735)。筋肉や肝臓と同様に、2型糖尿病患者では、脂肪のインスリン受容体チロシンキナーゼ活性が低下している(246, 247)。この欠損は、細胞膜のインスリン受容体量の減少と相まって(378, 379, 418, 595, 786)、シグナル伝達の観点から、脂肪のインスリン抵抗性を説明するのに十分であると考えられる。実際、体重を減らすと、脂肪のインスリン抵抗性と脂肪細胞のIRK活性の欠陥の両方が修正される(247)。しかし、残念なことに、このような脂肪細胞のインスリン受容体の欠損のメカニズムは、ほとんど解明されていない。
脂肪細胞はインスリン刺激によるグルコースの取り込みを行うが、骨格筋と同様に、この機能は肥満や2型糖尿病では抵抗性になる(377)。しかし、脂肪細胞はインスリン刺激によるグルコース処分の定量的に重要な部位ではなく、ヒトでは経口グルコース負荷の5%未満を占める(363, 428)。にもかかわらず、マウスでGLUT4を脂肪に特異的に欠失させると、脂肪率や体重に変化はなく、肝臓や骨格筋のインスリン抵抗性が生じる(1)。このことは、インスリン抵抗性が脂肪細胞のグルコース取り込みに間接的かつ生理的に重要な影響を与えることを示唆している(725)。脂肪細胞のグルコース取り込みによるグローバルなインスリン感作作用は、グルコースによるChREBPの活性化を介して部分的に行われる可能性がある。ChREBPの活性化は、脂肪生成遺伝子の発現を促進し、脂肪組織が栄養吸収源として機能することを可能にし、その結果、肝臓や筋肉への基質供給が減少する(310)。また、脂肪細胞のグルコース取り込みは、脂肪酸エステル化のためのグリセロール-3-リン酸を生成し、脂肪組織の異所性脂質の貯蔵を促進し、肝臓や筋肉への脂質の供給を減少させる。また、脂肪のGLUT4発現と全身のインスリン感受性との関連は、レチノール結合タンパク質4(RBP4)などのアディポカインによるもの(935)や、ニコチンアミドN-メチル基転移酵素の発現の変化によるもの(432)とも言われている。
生理学的には、白色脂肪組織インスリン作用の極めて重要な機能は、脂肪分解の抑制である。セクションIIで述べたように、脂肪分解はインスリンに対して非常に敏感である。この抑制効果はげっ歯類やヒトでは迅速(〜10分)である(620)。血漿中のNEFAの主な供給源は脂肪組織であり、脂肪分解性基質の放出は肝の糖新生を分単位で制御する重要な因子であり、また、糖新生の亢進は2型糖尿病を規定する空腹時高血糖の主要な要因であることから、脂肪細胞におけるインスリンの脂肪分解抑制作用に対する抵抗性は、病態生理学的に非常に重要である。
もし、脂肪細胞のインスリン抵抗性が脂肪分解の抑制を阻害するのであれば、2型糖尿病では血漿中のNEFA濃度が上昇することが予想される。コントロール不良の2型糖尿病患者では、血漿中のNEFA濃度が、痩せた糖尿病でない対照群と比較して、有意に上昇していることが複数の研究で確認されている(134, 245, 266, 685, 816)。2型糖尿病患者の痩せた健康な親族では、食後の血漿NEFA濃度の抑制が損なわれている(31)。さらに、血漿NEFA濃度の上昇は、2型糖尿病の発症を予測する(121, 611)。血漿中のNEFA濃度の上昇は、十分にコントロールされた2型糖尿病患者では、はるかに緩やかで、時には存在しないこともある(245, 286, 685, 816)。しかし、これらの濃度の測定値は、より生理学的に重要なパラメータであるフラックスのはるかに大きな違いを示している。高インスリン血症-高血糖クランプ条件下では、2型糖尿病患者では血漿NEFA濃度よりも血漿NEFAターンオーバーの方が相対的にはるかに上昇していた(286)。また、2型糖尿病では、空腹時のグリセロール回転率もグリセロールからのグルコン生成速度と同様に上昇している(586, 663)。さらに、肥満手術を受けた患者では、基礎的な脂肪分解の減少がHOMA-IRの改善と相関している(265)。重要なことは、ヒトでは肥満そのものと血漿NEFA濃度に相関がないことであり、2型糖尿病の血漿NEFAの増加は、単純な質量効果ではなく、機能的欠陥(すなわち、脂肪細胞のインスリン抵抗性)が介在していることを示している(388)。脂肪細胞のインスリン抵抗性に関するこのような解釈と一致するように、グリセロール回転で測定される脂肪分解フラックスのインスリン抑制は、インスリン抵抗性の肥満のヒト青年、糖尿病のヒト、および2型糖尿病のヒトの一親等の親族で障害されている(263, 309, 370, 696)。300人以上のコホートにおいて、空腹時血漿インスリン濃度と空腹時血漿NEFA濃度の積で評価した脂肪のインスリン抵抗性は、耐糖能正常から耐糖能異常、2型糖尿病へと進行する過程で連続的に増加し、脂肪のインスリン抵抗性が2型糖尿病の病態に寄与していることが示唆された(261)。空腹時血漿インスリン濃度と血漿NEFA濃度の積を脂肪のインスリン抵抗性の実用的な指標として用いることは、最近、multistep pancreatic clampに対して検証された(789)。
脂肪分解の抑制に対するインスリン抵抗性が、主に脂肪分解の亢進、脂肪酸の再エステル化の低下、あるいはその両方に関与しているかどうかは定かではない。14C]パルミチン酸を用いて高インスリン血症-高血糖クランプ中の脂肪酸動態を追跡しモデル化した刺激的な研究では、正味の脂肪分解は痩せた2型糖尿病患者と肥満の2型糖尿病患者で差がなかったが、肥満の2型糖尿病患者では組織への取り込みから脂肪酸が逃げる割合が高く、この後者の所見が糖尿病患者の血漿NEFAレベルのインスリン抑制の欠陥を説明すると結論づけられた(692)。同様に、肥満の非糖尿病患者と2型糖尿病患者を対象とした研究では、経口グルコース負荷試験中の血漿グリセロールとNEFAレベルは両群間で同等であり、ex vivoでのイソプロテレノール刺激による脂肪分解の抑制も両群間で同等であったが、2型糖尿病患者では脂肪酸トランスポーターFABP4の発現が低下しており、脂肪分解ではなく脂質の貯蔵がインスリン抵抗性の原因であると結論づけている(618)。
インスリン抵抗性の肝臓におけるグリコーゲン代謝が、空腹時と摂食時の両方で正味の振幅が減少するのと同様に、インスリン抵抗性の脂肪組織では、空腹時に脂肪分解が最大限に促進されず、摂食時に脂質の蓄積が最大限に促進されないという証拠がある。健康な痩せた男性と腹部肥満の男性を対象とした複数回の食事摂取に関する研究では、この現象が浮き彫りになった。脂肪組織の毛細血管床を流れる正味の脂肪酸フラックスの方向は、空腹時には放出に有利で、食後には貯蔵に有利であり、いずれの場合も肥満の男性ではその大きさが小さかった(536)。その結果、肥満の男性では、摂取した脂肪のうち適切に脂肪組織に蓄えられる割合が大幅に減少し、脂肪組織以外の組織への脂質の蓄積が促進されたと考えられる。
興味深いことに、骨格筋や肝臓とは対照的に、脂肪細胞のインスリン抵抗性を媒介する特定の分子欠陥はほとんど明らかにされていない。むしろ、脂肪細胞のインスリン抵抗性の病因の理解は、インスリンシグナルを損なう可能性のあるオートクラインまたはパラクラインの炎症性サイトカインに焦点を当てている(27, 289)。これらのメカニズムについては、セクションVIIで詳しく検討している。しかし、インスリンによる脂肪分解の抑制に対する抵抗性は、高脂肪食をラットに3日間与えただけで明らかになる(722)。これは、炎症が顕著になる前のことであり、脂肪細胞のインスリン抵抗性の初期の欠陥には、非炎症性のメカニズムが関与している可能性が高いことを示唆している。
脂肪細胞のインスリン抵抗性における脂肪分解に関与するタンパク質(ATGL、HSL、PLIN、FSP27)の具体的な役割については不明である。しかし、正味の脂肪分解を抑制することができないのは、NEFAのエステル化がうまくいかないことに起因する可能性もある。実際、脂肪分解を抑制するインスリン抵抗性のメカニズムとして、NEFAのエステル化が関与している可能性があり、インスリン抵抗性のグルコース取り込みとのメカニズム上の関連性が指摘されている。インスリンで刺激されたグルコースの取り込みは、解糖を促進し、グリセロール-3-リン酸を得ることができる3つの炭素の前駆体を生成する。グリセロール-3-リン酸は、脂肪酸のエステル化とインスリン刺激による脂質の適切な貯蔵に必要である(474)。インスリンのグルコース取り込みに対する抵抗性は、このメカニズムによって、インスリン刺激時に脂肪細胞が脂質の純輸出から純貯蔵に切り替わるのを防ぐことができるかもしれない。
このように、白色脂肪細胞のインスリンに対する優れた感受性は、諸刃の剣であるかもしれない。この感度の高さは、食後の栄養貯蔵を迅速かつ適切に行うことを可能にする。しかし、栄養過多に対する最初の応答者としての白色脂肪細胞は、栄養ストレスに起因するインスリン抵抗性に対して脆弱である。適度な脂肪細胞のインスリン抵抗性であっても、脂肪分解を抑制するインスリンの用量反応曲線が急峻であるため、肝臓や骨格筋への脂肪酸の供給が増加し、これらの組織のインスリン抵抗性が促進される(セクションV参照)。全身のグルコースホメオスタシスにとってWATインスリン作用が絶対的に重要であることは、脂肪ジストロフィーの極端な、しかし可逆的なインスリン抵抗性によって強力に証明されている(79, 406, 642, 714, 785)。白色脂肪組織分は不活性な貯蔵庫ではなく、実際には全身のインスリン感受性と抵抗性の要となっているのかもしれない。
F. インスリン抵抗性の統合生理学と実験的考察
インスリン作用と抵抗性は、このような統合された生理学的背景の中で最もよく理解されるので、インスリン作用を評価する実験方法には慎重な設計と解釈が必要である(18)。前述のHimsworthの「グルコース-インスリン耐性テスト」(317)は、インスリン作用の実験的テストにおける重要な考慮点を示している。インスリン抵抗性を直接測定するためには、研究者はインスリン作用とインスリン分泌を注意深く区別する必要がある。この区別は、標準的なグルコース負荷試験(GTT)で簡単に説明できる(23, 33, 534)。図10は、普通の餌を与えたマウス、高脂肪食(HFD)を8週間与えたマウス、自己免疫性のインスリン炎のモデルである非肥満糖尿病(NOD)マウスを用いて腹腔内GTTを行った場合の仮想的な結果を示したもので、インスリン炎の発症後、顕在化した1型糖尿病が発症する前に調査したものである(358,361)。NODマウスとHFDマウスでは血糖値が上昇しているが、血糖値とインスリンの関係を見ると、NODマウスの耐糖能異常はインスリンの作用異常ではなく、β細胞の機能異常であることがわかる。逆に、HFD給餌マウスでは、高血糖に伴うインスリン濃度の上昇が見られ、耐糖能低下の主な原因がインスリン抵抗性であることを示している。重要なことは、耐糖能の改善は、同様にインスリン分泌の亢進またはインスリン作用に対する過敏性のいずれかの結果であるということである。前者はスルホニルウレア剤などのインスリン分泌促進剤による治療(688)、後者はFGF21などのインスリン感受性増強剤による治療(112, 401)でモデル化できる。Himsworthは古典的な研究(317)の中で、先見の明をもって、上記のような理由から、外因性インスリンなしのグルコースチャレンジでは、2つのタイプの糖尿病患者を区別するには不十分であると指摘した。現代では、Andresが開発した高インスリン血症-黄血球クランプ法を採用することで、この曖昧さを実験的に回避することができる(33, 534)。この方法では、被験者のインスリン濃度を一致させることで、インスリン分泌の違いをほぼ否定することができる。インスリン濃度を一定にして上昇させた状態で、共役血糖を維持するために必要なグルコースの注入速度は、全身のインスリン感受性を反映している(181)。高インスリン血症-高血糖クランプは,全身のグルコース代謝,内因性グルコース産生(EGP),組織特異的グルコース取り込みを追跡するための同位体標識グルコースおよびグルコースアナログの注入と組み合わせることにより,標的組織におけるインスリン作用を生体内試験で実験的に評価する最も強力な方法であり,ヒトおよび動物モデルの両方で広く使用されている(33, 534, 760)。
図10 糖負荷試験の解釈には、血漿インスリン濃度の測定が必要である
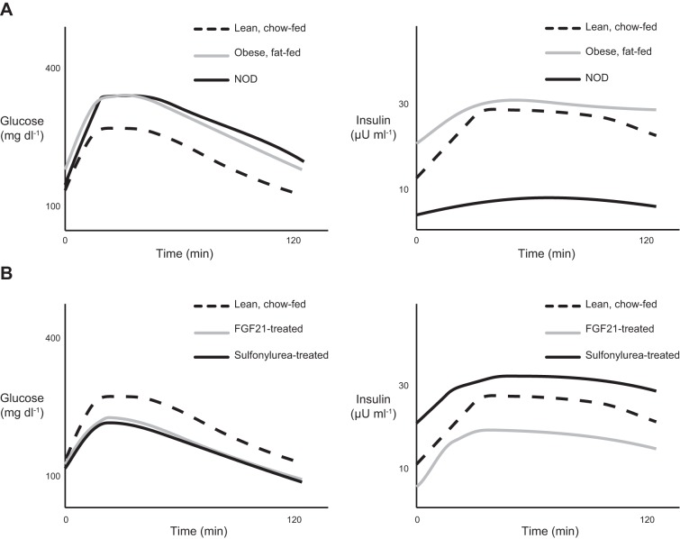
A: 食事誘発性肥満マウスと糖尿病予備力の非肥満糖尿病(NOD)マウスは、どちらも痩せた餌を与えたコントロールマウスに比べて耐糖能異常を示すが、その原因は異なる。食事誘発性肥満マウスでは、インスリン分泌反応は正常あるいは亢進しているが、インスリン抵抗性により高血糖となっている。糖尿病予備力であるNODマウスは、インスリン分泌不全により耐糖能が低下している。
B: 耐糖能の改善は、同様にインスリン感受性の増加(線維芽細胞成長因子21(FGF21)処理マウス)またはインスリン分泌の増加(スルフォニルウレア処理マウス)に起因する。
高インスリン血症-高血糖クランプ試験におけるEGPの測定は正確で有用であるが、限界がある。例えば、EGPはHGPと同じであると考えられているが、EGPはすべての供給源から血漿中にグルコースが出現する速度を反映しており、空腹時には腎性グルコース生成によるわずかな寄与も含まれる。さらに、インスリンによるEGPの抑制を測定しても、肝グリコーゲン合成に対するインスリンの直接的な作用と、肝糖新生に対するインスリンの間接的な作用を区別することはできない。肝臓でのグリコーゲン合成速度は、おそらくEGP抑制よりも直接的な肝臓のインスリン作用を読み取るのに適しているが、第II節で述べたように、肝臓のグリコーゲン代謝はグルコースの有無に非常に敏感であるため、生体内試験での評価はより困難である。インスリンによるグリコーゲン合成促進作用を実験群間で正確に比較するためには、被験者間で厳密に一致させた高血糖が必要である(640)。インスリン刺激後の肝グリコーゲン量を簡単に測定するには、基礎グリコーゲン量を最小にするために長期間の絶食が必要である。このような長期間の絶食は正常な生理状態を表さないため、げっ歯類では好ましくないことが多い(32, 33, 534)。基礎肝グリコーゲン量が大きく変動するという問題を回避する方法として、高インスリン血症・高血糖クランプ中に均一に標識した13C-グルコースの肝グリコーゲンへの取り込みを測定する方法が成功している。肝グリコーゲンと血漿グルコースのm+6モル分率、および肝グリコーゲンの総濃度を知ることにより、輸液中の直接経路によるグリコーゲン沈着の絶対速度を計算することができる(153, 645)。血漿グルコースから肝臓のUDP-グルコースへのm+6モル分率の希釈を測定することにより、グリコーゲン合成に対する直接経路と間接経路の相対的な寄与度が明らかになる(645)。直接経路のグリコーゲン合成速度の絶対値と、全グリコーゲン合成フラックスに対する直接経路の寄与率の両方を知っていれば、高インスリン血症・高血糖クランプ中の全グリコーゲン合成速度を計算することができる。ヒトでは、非侵襲的なMRS法を用いてグリコーゲン合成およびグリコーゲン分解の両フラックスを測定している(640, 641, 704, 832)。しかし、インスリンは肝グリコーゲン合成を調節する上で、単に許容的な役割を果たしているだけであり、肝心なのは血漿(門脈)グルコース濃度であることを強調しておかなければならない。すべての被験者の門脈グルコース濃度を注意深く照合しないと、インスリン自体が肝グリコーゲン代謝に及ぼす影響を評価することは困難である。
インスリンによる脂質代謝の制御を生体内試験で評価するための有効な方法もある。インスリンによる脂肪分解の抑制は、標識したパルミチン酸とグリセロールを用いた高インスリン血症-高血糖クランプ試験で追跡することができる(620, 910)。肝デノボ脂肪生成のインスリン制御は、転写を介した作用であるため、急性注入試験では評価できないが、重白色脂肪組織素白色脂肪組織の補給などの効果的なトレーサー法により、数日間にわたってDNLを測定することができ、インスリン抵抗性モデルではDNLが低下することが明らかになっている(868, 910)。インスリンによる脂肪生成転写プログラムのアップレギュレーションを測定するために、一般的に採用されているプロトコルは、絶食(例:24時間)-再摂食(例:6時間)である。栄養素によるmTORの活性化は、他のホルモンの変化と同様に重要な問題である(452, 764)。
上述および図11に示した様々な方法に共通しているのは、代謝経路のフラックスを計算するために同位体トレーサーを使用していることである。トレーサーを用いた研究は、一般的に代謝物濃度の研究よりもリソースを必要とするが、インスリンの作用をより適切に評価することができると考えられている。メタボロミクスの進歩により、何百もの代謝物を迅速かつ正確に自動測定することで仮説の立案が容易になったが、インスリンは代謝フラックスを変化させることで作用する。このようなフラックスの変化は、特定の代謝物の蓄積や減少を促進する可能性があるが、濃度の違いは常にフラックスの違いの結果として生じる。これまでも、そしてこれからも、代謝の統合的な生理現象の最も重要なメカニズムを解明することができるのは、究極的にはフラックスの測定なのである。
図11生体内試験でのインスリン抵抗性を評価するための同位体トレーサー法
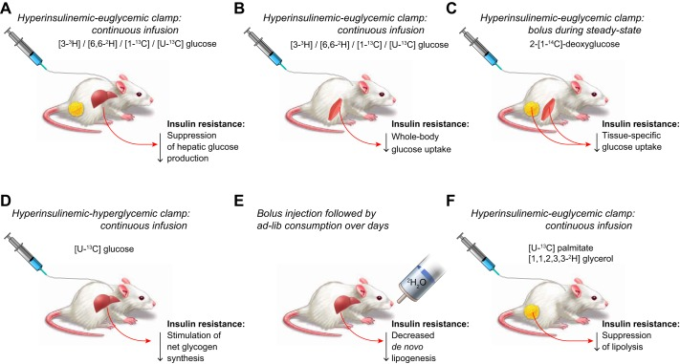
6つの方法が例示されている。実験プロトコルとトレーサーの投与方法は斜体で、トレーサーは太字で、インスリン抵抗性の影響は右下にある。A:数種類のグルコース同位体を用いて全身のグルコース代謝を追跡することができる,Rd. 高インスリン血症-高血糖クランプ中、Rdとグルコース注入速度Fがともに既知である場合、内因性グルコース生成量はRdからFを引くことで算出できる。インスリン抵抗性の被験者では、インスリンによる内因性グルコース産生の抑制効果が低下する。B:高インスリン血症-血糖値クランプ条件下では、Rdの70〜80%が骨格筋のグルコース取り込みによって占められるため、骨格筋のインスリン抵抗性はRdの低下を伴うことが多い。C:代謝されないグルコースアナログである2-デオキシグルコース(2-DG)は、グルコース-6-ホスファターゼを持たない組織(骨格筋や脂肪組織などで、肝臓は含まれない)ではリン酸化されて体内に取り込まれる。組織の2-DG-6-リン酸濃度は、インスリン抵抗性で低下するインスリン刺激によるグルコース取り込み量の推定に使用できる。D: m+6グルコースは自然界にはほとんど存在しないため、[U-13C]グルコースは肝グリコーゲン合成の有用なトレーサーとなるが、肝インスリン抵抗性では減少する。肝グリコーゲン合成を促進するためには、高インスリン血症と高血糖の両方が必要である。E: 重白色脂肪組織素化またはトリチウム化した白色脂肪組織を肝パルミチン酸に取り込ませることで、肝デノボ脂肪生成(DNL)を測定することができるが、同位体の定常状態に達するには数日間の投与が必要である。DNLは、高脂肪食のネズミなど、インスリン抵抗性のモデルでは減少する。インスリンによるDNLの制御は、転写を介したゆっくりとしたプロセスであるため、この方法は研究対象の生理学に適合している。F: [U-13C]パルミテートや[1,1,2,3,3-2H]グリセロールなど、いくつかのパルミテートやグリセロールのトレーサーを脂肪分解の追跡に使用することができる。インスリンによる白色脂肪組織からの脂肪分解の抑制は、脂肪のインスリン抵抗性で損なわれている。注目すべきは、空腹時のRaグリセロールは、Raパルミチン酸よりも正確な脂肪分解の指標となる可能性が高いことである。なぜなら、パルミチン酸は脂肪細胞内で再エステル化される可能性があるからである。
G. インスリン抵抗性 「何が」と「なぜ」か
セクションIVでは、インスリンシグナルのエフェクターとインスリン抵抗性になったインスリンの直接的な代謝作用に焦点を当てて、骨格筋、肝臓、肝臓、肝臓におけるインスリン抵抗性の中心的な特徴を説明しようとした。インスリン抵抗性組織の病態生理学的特性は、ここで説明した以外にも確実に存在する。インスリンシグナルは多様な細胞機能と接点を持ち、インスリン抵抗性では無数のシグナル伝達経路が変化していることが確認されている。さらに、20種類以上ある肥満と2型糖尿病の特徴的な動物モデルは、それぞれ代謝疾患の明確な病態生理を示しており、ヒトの肥満に伴う2型糖尿病と重なる部分もあるが、モデルにしようとしているヒトの疾患のすべての側面を再現しているわけではない(412)。
インスリン抵抗性のように様々な症状が現れるプロセスを研究する研究者にとっての課題は、原因と結果、一次的な欠陥と二次的な結果を区別することである。第V-VII章では、本章で述べたインスリン作用の障害の原因となる最初の傷害を突き止めるための努力を振り返る。インスリン抵抗性の状態で活性やレベルが変化する遺伝子や代謝物の候補を同定するだけでは十分ではない。因果関係を推論するためには、候補となるメディエーターがインスリン抵抗性に必要であるか、十分であるか、あるいは必要かつ十分であるかを評価する妨害が必要である。また、候補となる経路が、典型的な肥満に伴うヒトのインスリン抵抗性に作用しているかどうか、したがって、実行可能な治療標的となりうるかどうかを検討するためには、ヒトでの研究が必要である。げっ歯類の研究では、栄養過多に対するインスリン作用の最も初期の欠陥を同定しようとするアプローチが、インスリン抵抗性のメカニズムの基礎を解明する可能性が最も高い(548, 721, 854)。インスリン抵抗性の二次的な結果である欠陥が、インスリン抵抗性をさらに悪化させる可能性があり、それゆえに有効な治療ターゲットとなることを認識することが重要である。しかし、ここでは、インスリン抵抗性の一次障害を明らかにすることに主眼を置いている。基本的な仮定は、これらの一次障害を特定して改善することができれば、すべての二次障害を防ぐことができるということである。まず、インスリン抵抗性の最も古く、最も広く研究されている仮説、すなわち、脂質代謝フラックスの変化の直接の結果としてのインスリン作用の欠陥について概説する。
V. 脂質によって誘発されるインスリン抵抗性
A. 脂質誘導性インスリン抵抗性の紹介
脂質によるインスリン抵抗性の現象は、おそらく1941年にウサギに脂質を静脈内投与したところ、インスリンによる低血糖が起こらなくなったという報告が最初であろう(940)。1960年代初頭には、NEFAやケトン体濃度の上昇がインスリン刺激によるグルコース取り込みの障害につながるという報告が相次いだ(257,677,765,904)。これらの知見を総合して、Randleら(677-679)は、骨格筋や心筋の酸化的基質選択を制御するグルコース-脂肪酸サイクルを提唱した。グルコース-脂肪酸サイクル仮説では、酸化基質の選択は基質の供給によって制御される相互関係にあると考えた。すなわち、脂肪酸の供給があれば脂肪の酸化が促進され、グルコースの酸化が抑制され、その逆もまた同様である(676)。脂肪の酸化が進むと、ミトコンドリアのアセチルCoAとNADHが蓄積され、PDHが阻害されて、ピルビン酸がミトコンドリアに入って酸化されるのが制限されるのである。また、細胞質内のクエン酸が増加すると、ホスホフルクトキナーゼ-1のアロステリックフィードバック阻害により解糖系フラックスが低下する。その結果、グルコース-6-リン酸が増加すると、今度はヘキソキナーゼがアロステリックに阻害され、細胞内グルコース濃度が上昇することになる。
ランドルのグルコース-脂肪酸サイクルは、脂質によるインスリン抵抗性の最もわかりやすく人気のある実験モデルである、急性脂質注入法を用いて広く研究されている。このような研究では、リポタンパク質リパーゼを活性化し、血漿NEFA濃度をさらに上昇させるために、ヘパリンを添加した20%トリグリセリド乳剤を使用するのが一般的である。このような輸液は、筋肉のグルコース取り込みに対するインスリン抵抗性を効果的に誘発するが、それは輸液を開始してから数時間後のことであり、実験者は脂質に触れる時間と範囲を完全にコントロールすることができる(68, 198, 229, 285, 464, 698, 836)。この方法は、2meq/l以上の超生理的な血漿NEFA濃度を得るために用いることができるが,0.75meq/lのような高い生理的濃度を目標とした輸液でも、インスリン刺激によるグルコースの取り込みを阻害することができる(68)。急性脂質輸液は、脂質によるインスリン抵抗性のメカニズムを研究する上で魅力的な実験モデルである。その理由の一つは、慢性的な食事性脂質によるインスリン抵抗性のモデルに見られる、基礎的な高インスリン血症、炎症、身体組成の変化など、混同される生理学的な代償を回避できるからである。
急性脂質注入時の骨格筋のグルコース代謝をMRSで調べることにより、Randle仮説を生体内試験で検証することができた(図12)。具体的には、細胞内のグルコース-6-リン酸(G6P)およびグルコース濃度、ならびに解糖系およびグリコーゲン合成速度をMRSで測定し、脂質の酸化が解糖系フラックスを減少させ、G6P濃度を増加させるというRandleの予測を直接検証した。これらの研究により、Randle仮説が予測した増加とは逆に、脂質誘発性の急性インスリン抵抗性ではG6Pおよびグルコース濃度が実際に減少することが明らかになった(198, 698)。解糖とグリコーゲン合成も減少したが、これはランドル仮説ではなく、グルコース輸送の障害によるものであった(198, 698)。これらの知見は、インスリン受容体の活性化とGLUT4の転座を結びつける分子メカニズムの解明や、インスリン抵抗性や2型糖尿病においてGLUT4の転座やグルコース輸送が障害されるという観察結果と一致していた(152, 258, 633, 847)。今後、脂質による筋のインスリン抵抗性のメカニズムを研究する際には、脂質への曝露とインスリンシグナルの阻害を関連付ける必要があることが示唆された。
図12 Randle仮説と脂質による骨格筋のインスリン抵抗性
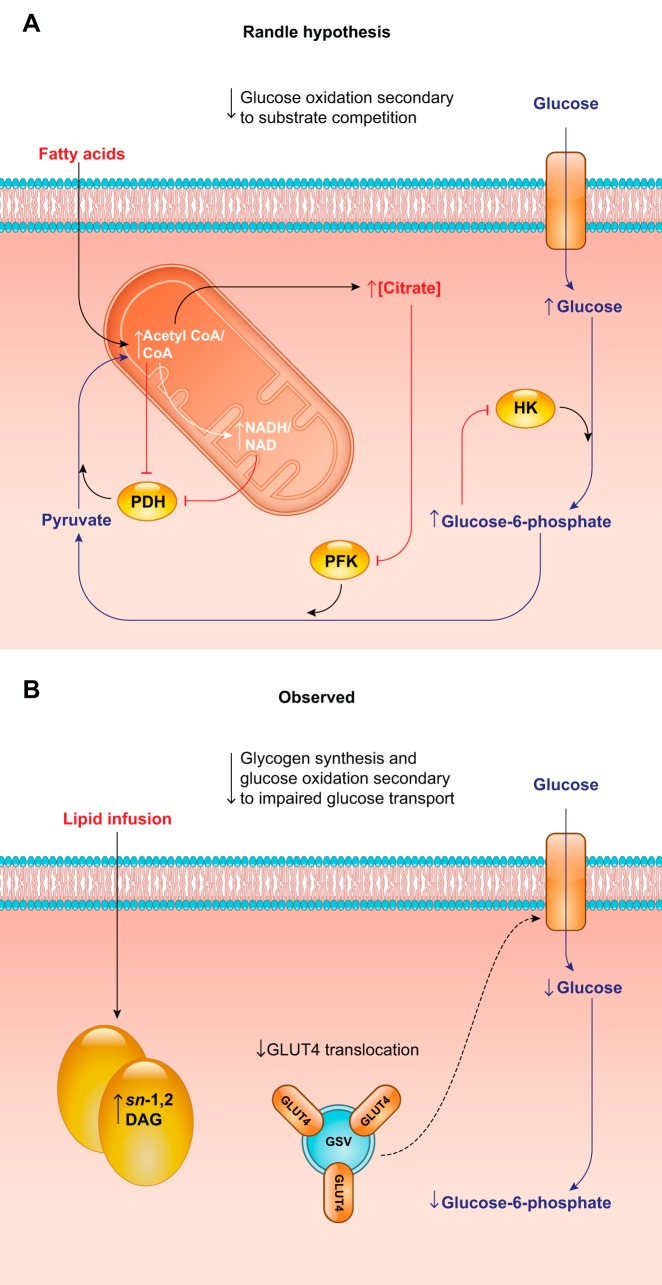
A: Randleと共同研究者は、脂質によって誘発される筋肉のグルコース酸化の障害は、脂肪酸との基質競合による二次的なものであると提案した。脂肪酸の酸化が増加すると、ミトコンドリアのアセチルCoA/CoAとNADH/NAD+の比率が上昇する。これにより、ピルビン酸デヒドロゲナーゼ(PDH)とホスホフルクトキナーゼ(PFK)がアロステリックに阻害され、解糖系フラックスが減少する。続いてグルコース-6-リン酸濃度が上昇すると、ヘキソキナーゼ(HK)がアロステリックに阻害され、細胞内グルコース濃度が上昇することになる。B: 急性脂質注入時に細胞内グルコースとグルコース-6-リン酸を測定したところ、この環境下では、これらの代謝物の濃度は増加するのではなく、減少することが明らかになった。筋のインスリン抵抗性とGLUT4の転座障害を関連付ける研究と合わせて、これらのデータは、脂質によって誘発される筋のインスリン抵抗性の主な欠陥としてグルコース輸送障害を示唆している。
脂質によるインスリン抵抗性にグルコース-脂肪酸サイクルが関係しているかどうかは、他の生体内試験モデルで検証されている。例えば、Pdk2とPdk4を欠損したマウス(Pdk2/4 DKO)では、骨格筋のPDHが構成的に脱リン酸化され、活性化しているため、グルコースを優先的に酸化してしまう(669)。Pdk2/4 DKOマウスでは、グルコース-脂肪酸サイクルが機能していない。しかし、Pdk2/4 DKOマウスは、細胞内脂質の増加に伴って重篤な筋インスリン抵抗性を発症することから、脂質による筋インスリン抵抗性にはグルコース-脂肪酸サイクルは必要ないことが示唆される(669)。
全体として、グルコース-脂肪酸サイクルは、脂質を数時間注入した後に生じるインスリン刺激による筋肉のグルコース取り込みの顕著な障害(68, 71, 698)にも、ヒト肥満/2型糖尿病のインスリン抵抗性にも関与していないことが分かっている。しかし、グルコース-脂肪酸サイクルは、酸化的な基質選択を制御するための生理学的に重要なプロセスであることは間違いない(244)。例えば、深いインスリン抵抗性が生じる前の、急性脂質注入の最初の3時間では、細胞内のG6P濃度が上昇し、解糖系フラックスが減少し、Randle仮説の予測と一致する(372, 373)。この現象を説明するには、Randleが提唱したアロステリックメカニズムで十分であると考えられる。マロニルCoAがカルニチンパルミトイルトランスフェラーゼ-1を阻害することで、グルコースによる脂肪酸酸化が抑制されるという相互制御機構は、この基質選択回路の統合的な性質を際立たせている(533)。このように、グルコース-脂肪酸サイクルは、基質の利用可能性を全身で制御するための細胞の自律的な反応と考えることができ、摂食状態ではグルコースを、絶食状態では脂肪酸を効率的に酸化することができる。
血漿中のNEFAターンオーバーの増加とインスリン抵抗性との関連性が確立されていることから、脂質注入モデルが筋肉のインスリン抵抗性に広く関連していることが明らかになった(134, 245, 266, 632, 684, 685, 816)。しかし、ヒトのインスリン抵抗性で観察される血漿中のNEFAターンオーバーや濃度の増加はわずかである(632)。NEFA濃度が生理的に高くなることが多いことに加えて、急性静脈内脂質曝露は肥満に伴うインスリン抵抗性を正確にモデル化することができない。すなわち、脂質曝露は慢性的ではなく急性であり、脂質の供給は経腸や肝臓/脂肪組織からではなく静脈内であり、血漿脂肪酸濃度は動的ではなく静的である。その結果、急性の脂質曝露は、通常、脂質によるインスリン抵抗性の直接的なメカニズムの研究にのみ用いられる。ある実験的擾乱が組織のインスリン感受性に及ぼす影響を調べる研究のほとんどは、慢性的な介入を適切に行う。
典型的な肥満に伴う脂質誘導性インスリン抵抗性の最も一般的なモデルは、高脂肪食を与えたネズミである。マウスやラットの系統は、食事によって誘発される肥満やインスリン抵抗性に対する感受性が大きく異なるが、C57BL/6JマウスやSprague-Dawleyラットのような感受性の高い系統は、扱いやすいモデルシステムとなっている(239, 548, 898)。しかし、これらのげっ歯類は、肥満になりやすいという点ではヒトによく似ているが、脂肪を与えたげっ歯類の研究を解釈する際には、いくつかの重要な生理学的な違いを念頭に置く必要がある(428)。例えば、げっ歯類の短期間の脂肪摂取プロトコルでは、骨格筋のインスリン抵抗性を伴わない著しい肝脂質の蓄積とその結果としての肝インスリン抵抗性が生じるが(721,722,854)メタボリックシンドロームのヒトでは、骨格筋のインスリン抵抗性が肝インスリン抵抗性よりも先に生じると考えられている(182,639,725)。高脂肪食を与えると肥満と耐糖能異常を起こすマウスがある一方で、痩せ型で代謝的に健康なマウスもあるという驚くべき傾向は、エネルギー消費、膵臓機能、組織のインスリン感受性のすべてが表現型の生成に重要な役割を果たすという、グルコースホメオスタシスの統合的な性質を強調している(428)。一般的に使用されている食事誘発性肥満のげっ歯類モデルには、ob/obマウス、db/dbマウス、Zucker fa/faラットなどがあり、レプチン満腹軸の自然発生的な変異を利用して、著しい摂食亢進とその結果としての肥満を実現している。これらのモデルは有用であるが、レプチンは単なる満腹感のスイッチではなく(341, 801)変異の結果は多岐にわたるということを念頭に置いて解釈する必要がある。さらに、高栄養状態のネズミのモデルは、通常、インスリン抵抗性が発現してからかなり時間が経過した病的な肥満の成人で研究される。このような表現型は、インスリン抵抗性の初期症状を明らかにしようとする研究には不向きである。このような研究には、実験を開始するまで正常な体組成と代謝表現型を維持できる高脂肪食の齧歯類が有用である。しかし、高脂肪食のネズミと遺伝子的に高脂肪食のネズミの両方には、肝臓と骨格筋における異所性脂質の増加という、ヒトの肥満に伴うインスリン抵抗性と共通する表現型がある。肝臓と筋肉のインスリン抵抗性の異所性脂質仮説については、以下と図13を参照してほしい。
図13 肝臓と筋肉のインスリン抵抗性の異所性脂質仮説
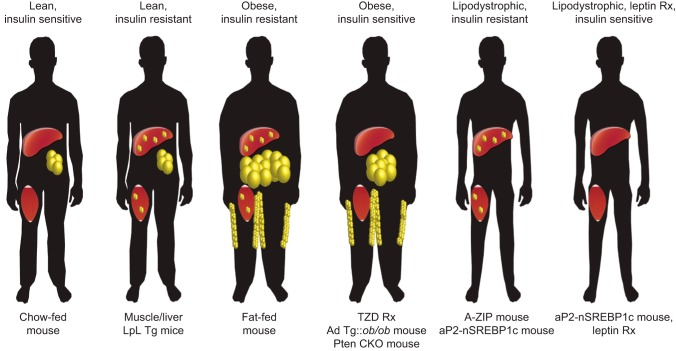
中心となる組織原理は、白色脂肪組織、特に皮下デポにおける脂質の蓄積は生理的に適切であるが、肝臓や骨格筋における脂質の蓄積は不適切であり、代謝的に有害であるというものである。このような「異所性」の部位に脂質が蓄積すると、インスリン抵抗性が生じる。ヒトの表現型はシルエットの上に、対応するマウスモデルは本文の下に記載している。
ヒトのインスリン抵抗性における最も再現性の高い関連性の一つは、肝内トリグリセリド(IHTG)との関連である(96, 627)。アルコールの過剰摂取を伴わないIHTGの増加と定義されるNAFLDは、先進国で最も一般的な肝疾患である(870)。肥満者の約3分の2,そして2型糖尿病を持つ肥満者のほぼ全員がNAFLDに罹患している(45, 772, 843, 883)。同様に、IHTGは、インスリン抵抗性の例外的に強い予測因子である(106, 422, 520, 728)。肝のインスリン抵抗性におけるIHTGの役割を示す因果関係の証拠は、ヒトとげっ歯類の両方の研究から得られた。糖尿病のヒトでは、適度な減量によりIHTG含量を減少させると、末梢に大きな影響を与えることなく、インスリンによる肝グルコース産生の抑制と空腹時血糖値が正常化する(637)。内臓脂肪は、肝インスリン抵抗性に寄与すると提案されているが、IHTGの優れたバイオマーカーである可能性が高いである(260)。IHTGは、肝インスリン抵抗性のはるかに強い予測因子であり、IHTGの減少を伴わない卵巣脂肪の外科的除去は、肝インスリン抵抗性を是正しない(220, 221)。さらに、脂肪異栄養症の人は、内臓脂肪がなくても、IHTGが大幅に増加し、重度の肝インスリン抵抗性と関連している(536)。また、内臓脂肪は、全身の脂肪酸代謝にはほとんど寄与していない(388)。2型糖尿病ではないがNAFLDのある人では、体重の変化を考慮してもNAFLDを改善することで2型糖尿病のリスクが減少する(933)。ヒトでは、1回の経口脂肪ボーラスでも、IHTGを増加させ、肝のインスリン感受性を低下させるのに十分である(313)。
脂肪組織量ではなくIHTGがインスリン抵抗性を予測するという発見の副次的な命題は、肝臓から肝臓への脂肪の再配分がインスリン抵抗性を修正するはずであるということである。実際、多くのモデルがこの命題を支持している。重度のインスリン抵抗性を有するリポジストロフィーA-ZIP/F-1マウスでは、脂肪組織を移植することにより、IHTGと細胞内脂質の顕著な減少に伴い、肝臓のインスリン作用が正常化する(262, 406)。重度のNAFLDと肝インスリン抵抗性を有するリポディストロフィーのヒトやリポディストロフィーのマウスは、レプチンを投与することでIHTGの大幅な減少と肝インスリン作用の正常化を達成している(79, 642, 763)。この「再分配仮説」は、リポディストロフィーのモデルを超えて広がっている。Ptenを欠失させて脂肪細胞のインスリン感受性を高めたマウスは、高脂肪食を与えると肝臓ではなく脂肪組織に脂肪が蓄積され、食事誘発性の肝インスリン抵抗性から完全に守られる(555)。Pck1(PEPCK)をマウスの脂肪組織に特異的に過剰発現させると、脂肪細胞内でのグリセロン生成と脂肪酸の再エステル化が促進され、血漿中のNEFAレベルが低下する。その結果、これらのマウスは肥満になるが、肝臓と骨格筋ではインスリン感受性を示す(242)。また、PPARγを活性化することで脂肪形成を促進するチアゾリジン系の糖尿病治療薬のインスリン感受性を高めるために、脂肪の再分布が関与していると考えられている(723)。おそらく最も印象的なのは、レプチン欠損ob/obマウス(Ad Tg ob/ob)でアディポネクチンを過剰発現させると、脂肪が大量に再分布し、Ad Tg ob/obマウスの体重は肥満のob/ob対照マウスの2倍にもなるが、IHTGは減少することである(408)。驚くべきことに、これらの巨大なマウスは、空腹時のグルコースとインスリンのレベルが餌を与えられた痩せたマウスと同等であり、インスリン感受性も正常である(408)。逆の再分配の劇的な例として、db/dbマウスの脂肪組織特異的なレプチン受容体の再発現は、肥満を防ぐが、IHTGの蓄積、高インスリン血症、4週齢での糖尿病の発症をもたらし、db/db対照群の14週齢と比較している(878)。
IHTGが肝のインスリン抵抗性と密接に関係しているという仮説は、様々な介入モデルによって裏付けられている。マロニルCoAデカルボキシラーゼを肝臓で異所的に発現させたマウスは、高脂肪食を与えても肝脂肪の蓄積から保護され、肝のインスリン感受性が維持される(24)。同様の表現型は、エストロゲンを投与した卵巣摘出雌マウス(111)やFGF21を投与したマウス(112)など、様々なモデルマウスで観察されている。2つの独立した研究では、トリアシルグリセロールヒドロラーゼCES2をdb/dbマウスまたは高脂肪食マウスで過剰発現させると、耐糖能の改善に伴ってIHTGが減少することが報告されている(495, 705)。さらに、SREBP-1cを肝臓特異的に過剰発現させたマウスは、肝臓特異的なトリグリセリドの蓄積と肝臓特異的なインスリン抵抗性を示する(368)。IHTGと肝インスリン作用の関係を生体内試験で最も直接的に調べるには、ミトコンドリアのプロトンフォアである2,4-ジニトロフェノール(DNP)を用いてIHTG貯蔵量を急速に減少させる方法がある。短期間の脂肪摂取により肝インスリン抵抗性が生じたラットでは、DNPの投与によりIHTGと肝インスリン作用の両方が正常化した(721)。後者の結果は、薬理学的に異なる形のDNPを用いて拡張され、ストレプトゾトシン誘発糖尿病ラット、Zucker糖尿病脂肪ラット、脂肪異栄養症マウス(3)、非アルコール性脂肪性肝炎を誘発するメチオニン/コリン欠乏食を与えたラットなど、より極端な形態の肝インスリン抵抗性において、IHTGと肝インスリン作用の両方が劇的に改善されることが示された(621, 629)。
リポタンパク質リパーゼ(LpL)は、標的組織の局所で作用し、循環しているトリグリセリドから脂肪酸を放出して組織に取り込ませる。LpLの活性を調節することで、肝脂質の蓄積が肝のインスリン抵抗性と関連しているという仮説を検証することができる。LpLは肝臓では内因性に発現していないが、LpLの肝臓での過剰発現はIHTGを増加させ、肝臓特異的なインスリン抵抗性を引き起こすのに十分である(404)。げっ歯類では、脂肪由来のLpL阻害剤であるアンジオポエチン様8(Angptl8)のアンチセンスオリゴヌクレオチドによる阻害や、LpL阻害剤であるアンジオポエチン様4(Angptl4)の脂肪組織特異的な欠失により、肝臓への脂質の取り込みが促進され、IHTGの蓄積や肝臓のインスリン抵抗性が予防される(28, 867)。マウスのLpL活性化因子ApoA5をアンチセンスオリゴヌクレオチドで阻害すると、血漿トリグリセリドは対照群に比べて3倍に増加するが、IHTGは減少し、高脂肪食を与えても肝インスリン作用は維持されるという興味深い表現型が得られた(113)。南アジアの痩せた男性では、LpL阻害剤でもあるアポリポタンパク質C3(APOC3)をコードする遺伝子に共通の変異があるため、血漿中のApoC3濃度が高く、高トリグリセリド血症になり、IHTGが増加し、比較的低い体格指数でも肝性インスリン抵抗性になる傾向がある(638)。同様に、アポリポタンパク質C3を過剰発現させたマウス(ApoC3 Tgマウス)は、体組成は野生型のコントロールとほぼ同じであるが、肝トリグリセリドの増加と肝インスリン抵抗性を示する(462)。
このように、IHTGと肝インスリン抵抗性の間の因果関係を支持するデータが、ヒトとげっ歯類の両方の研究から多数得られているにもかかわらず、いくつかの興味深い研究では、この2つを分離している(224)。ヒトでは、一般的なPNPLA3 I148M変異アリルは、IHTGと強く関連しているが、この多型がインスリン抵抗性と関連しているかどうかについては、研究によって意見が分かれている(386, 607, 700, 875)。I148Mアリルをホモ化したマウスは、高ショ糖食でIHTGが増加したにもかかわらず、正常な耐糖能を示したが、肝のインスリン作用は直接評価されなかった(778)。興味深いことに、Pnpla3I148Mノックインマウスは、野生型と比較して、高脂肪食を与えても肝脂肪症が悪化せず、したがって、血糖値、空腹時インスリン、グルコース(イン)トレランスも同様であった(778)。他のげっ歯類モデルでも、IHTGと肝臓のインスリン抵抗性との関連性が問われている。インスリンシグナル伝達に肝臓特異的な障害を持つ様々なマウスは、定義上、インスリン抵抗性を示するが、脂肪肝は発症しない。肝の脂肪酸酸化に欠陥のある他のマウスでは、IHTGが蓄積しているにもかかわらず、空腹時に低血糖を示し、インスリン感受性と耐糖能が高い。これらのモデルは、代謝表現型を混乱させる糖新生をサポートする脂肪酸酸化の役割が確立されていることを強調している(216, 399, 587)。このようなモデルは、脂肪酸酸化が糖新生をサポートする役割を果たしていることを強調しているが、これは代謝の表現型を混乱させるものである(216, 399, 587)。IHTGと肝のインスリン抵抗性を分離した、特に興味深いマウスモデルは、肝臓特異的なScap-/-マウスである(550)。SCAPタンパク質は、SREBPの処理に必要であり、Scapを欠損させるとSREBPの脂肪生成プログラムが阻害される(550)。ob/obマウスでScapを欠損させると、IHTGは正常化したが、空腹時血糖値とインスリン濃度はわずかに改善しただけであった(550)。Scapを肝臓で誘導的に欠失させた後、16週間の高脂肪食を与えると、HFDの間、IHTGは増加せず、空腹時のグルコースとインスリン濃度は、野生型脂肪食を与えた対照群と同様になった(550)。肝のインスリン作用は直接評価していないが、これらのモデルは刺激的であり、慢性的な栄養過多の状態では、肝の脂質蓄積の回復や防止だけでは、他の多くの代謝異常-特に脂肪のインスリン抵抗性や過剰な脂肪分解-を克服するのに十分ではない可能性があることを示している。
骨格筋の脂質蓄積とインスリン抵抗性の関係は、肝臓よりも複雑で、議論の余地がある。1990年代後半、先駆的な1H-MRS研究により、正常体重の非糖尿病患者において、細胞内脂質(IMCL)は、高インスリン血症による高血糖クランプ中のインスリン感受性を極めて強く予測する因子であり、血漿NEFA濃度よりも強いことが明らかになった(438)。非糖尿病のピマインディアンでは、肥満度ではなく筋トリグリセリドが全身のグルコース摂取量と負の相関を示した(610)。さらに、脂肪異栄養症のA-ZIP/F-1マウスにWATを移植したり、脂肪異栄養症のヒトにレプチンを投与したりして、筋肉のインスリン抵抗性を改善すると、筋肉のトリグリセリドが著しく減少する(406, 642)。さらに、LpLを骨格筋で選択的に欠損させたマウス(したがって、血漿トリグリセリドを燃料として使用できない)は、筋肉のインスリン抵抗性から保護される(876)。逆に、LpLを骨格筋に選択的に過剰発現させたマウスでは、IMCLが増加し、骨格筋のインスリン抵抗性が発現する(404)。しかし、IMCLが筋のインスリン抵抗性に関与しているかどうかを判断するには、長年にわたって評価されてきた「アスリートのパラドックス」が複雑に絡み合っている。高度に訓練されたインスリン感受性の高い持久系アスリートは、2型糖尿病患者で観察されるレベル以上のIMCLレベルを示す(275, 488, 560)。2型糖尿病の人と持久系アスリートがIMCLの貯蔵量を増加させる経路は大きく異なるが、アスリートのパラドックスは、筋のインスリン抵抗性の病因における貯蔵トリグリセリドの因果関係を否定する証拠となる(866e)。
非脂肪組織への脂質の蓄積が、その組織のインスリン抵抗性と因果関係があるという命題は、インスリン抵抗性の異所性脂質仮説として知られている。後述するように、異所性脂質仮説は、非脂肪組織における組織や細胞の自律的なプロセスを示唆しているが、異所性脂質によるインスリン抵抗性には脂肪組織も中心的な役割を果たしている。もし、過剰なエネルギーがすべて脂肪組織に蓄えられるのであれば、異所性脂質によるインスリン抵抗性は発症しないと考えられる。上述したアディポネクチン過剰発現のトランスジェニックob/obマウスは、体重が肥満のob/obマウスの2倍にもなるにもかかわらず、正常な耐糖能を維持しており、この原理の劇的な例である(408)。異所性脂質仮説の間接的なヒトの遺伝的証拠としては、末梢脂肪の貯蔵能力を制限するゲノム変異が、空腹時インスリン、血糖値クランプデータ、糖負荷試験などの複数のパラメータで評価されるインスリン抵抗性と強く関連していることが挙げられる(501)。
蓄積されたトリグリセリド自体が細胞内のインスリン作用を直接損なう可能性は低いことを認識していた研究者たちは、長い間、脂質の蓄積とインスリン抵抗性をメカニズム的に結びつける可能性のある脂質部分を特定しようとしてきた(766)。ここでは、最も注目されているジアシルグリセロール、セラミド、アシルカルニチンの3つの脂質クラスについて検討する。
B. DAG-ノーブルプロテインキナーゼC軸とインスリン抵抗性
DAGとインスリン抵抗性を関連付ける初期の研究は、細胞内脂質量とインスリン抵抗性を関連付けるMRS法の開発に先立って行われた。むしろ、これらの初期の研究は、sn-1,2-DAGの腫瘍類似体であるホルボールエステルが試験管内試験および生体内試験でインスリン抵抗性を引き起こすという観察から始まった(821, 822, 866c)。この観察結果を受けて、筋肉、心臓、肝臓のDAGを測定したところ、肥満や糖尿病のラットではすべての組織でDAGが増加していた(593, 852)。トリアシルグリセロール(TAG)合成の最終中間体であるDAGの量は、脂質の処理が正常に行われているほとんどすべてのげっ歯類およびヒトのモデルの筋肉および肝臓におけるTAGの量と一致している(112, 113, 445, 621, 627, 629, 669, 724, 767)。その結果、上述したIMCL/筋のインスリン抵抗性とIHTG/肝のインスリン抵抗性の関連性は、DAGとインスリン抵抗性の関連性でもある。現在までに、5つのヒトの研究で、肝DAGと肝のインスリン抵抗性との間に有意な正の相関関係が認められている。この相関関係は、HOMA-IR(445,506,705)または高インスリン血症-高血糖クランプ試験中のHGPの抑制(512,834)によって測定されている。
DAGは、プロテインキナーゼC(PKC)の活性化に必要な生理活性シグナル脂質であることが知られていたため、すぐにインスリン抵抗性におけるPKC活性の役割の可能性に注目が集まった。しかし、この分野の初期の研究では、哺乳類細胞に存在するいくつかのクラスとアイソフォームのPKCについての知識が不十分であったことと、非常に非特異的な薬理学的阻害剤が障害となった。その結果、1980年代から 1990年代初頭にかけてのこの分野の文献には、インスリン作用に対するPKCの刺激作用と抑制作用の両方を記述した矛盾した報告が含まれていた(160, 353-355, 387, 558, 866c)。
分子生物学の進歩と入念な生化学的特性解析により、PKCは徐々に3つの主要なクラスに分けられるようになった。従来型PKC(cPKC、アイソフォームα、β、γ)は、完全な活性化にCa2+とDAGの両方を必要とし、新規PKC(nPKC、アイソフォームδ、ε、θ、η)はDAGのみを必要とし、非定型PKC(aPKC、アイソフォームζ、λ、ι)はCa2+もDAGも必要としない(799)。従来のPKCアイソフォームは迅速に活性化され、ホスホリパーゼCを介したシグナル伝達に大きく関与し、cPKCの完全な活性化に必要なCa2+とDAGの平行なスパイクを作り出す(580, 799)。これとは対照的に、nPKCはDAGによってゆっくりと持続的に活性化される。これは、nPKCのDAG結合C1ドメインがcPKCに比べてW/Yのアミノ酸を1つずつ置換しているためで、nPKCのDAGに対する親和性は2倍になっている(199)。この特性により、nPKCは、慢性的な細胞内脂質蓄積によるインスリン抵抗性を媒介するのに最も適したPKCアイソフォームであると先験的に考えられている。
実際、インスリン抵抗性の骨格筋や肝臓では、nPKCの活性化が一貫して観察されている。高脂肪食ラットの骨格筋では、PKCαやPKCζではなく、PKCεやPKCθのトランスロケーションが見られた。この研究では、筋肉のTAGおよびDAG含量とPKCθトランスロケーションとの間に正の線形関係があることも注目された(739)。その後、いくつかのグループにより、肥満、インスリン抵抗性、および/または糖尿病ラットの筋肉における骨格筋PKCθおよびPKCεトランスロケーションが測定され、すべてのモデルではなく、いくつかのモデル(285, 348, 356, 456, 667)で増加していることが判明した(357)。ヒトの研究では、PKCθ(819)とPKCε(619)の転座が、痩せ型の対照群と比較して2型糖尿病の筋肉で増加していることが判明している。肝のインスリン抵抗性に関与するPKCアイソフォームを同定し、その特徴を明らかにしようとした結果、筋肉とはやや異なる状況が明らかになった。PKCθは肝細胞ではあまり発現していないので、他のアイソフォームを調べた。肥満の2型糖尿病患者やZucker糖尿病ラットを用いた初期の研究では、膜に結合したPKCεとPKCζが、痩せた対照群に比べて増加していた(161)。ラットでは、脂質-ヘパリンを注入すると、肝臓でのPKCδのトランスロケーションが増加することが報告されているが、他のPKCアイソフォームのトランスロケーションは報告されていない(450)。2004,Samuelらは、3日間の高脂肪食ラットを急性肝インスリン抵抗性モデルとして用い、肝臓に高発現しているすべてのPKCアイソフォーム(α、β、δ、ε、ζ)のトランスロケーションを測定し、このモデルではPKCεのトランスロケーションのみが増加していることを明らかにした(721)。その後、数十種類の高脂肪食を与えたげっ歯類モデルにおいても、肝臓のPKCεトランスロケーションが観察された(646)。孤立したPKCε活性化の発見は、その後、肥満のヒトの肝生検でも再現された(445)。興味深いことに、PKCεの転座は、肝細胞のDAG含量とHOMA-IRで評価したインスリン抵抗性の両方と強い相関があり、この関係を示した6つのPKCアイソフォームのうち唯一のものであった(445)。しかし、ヒトの肥満肝臓では、PKCεとPKCδの両方のmRNA発現が増加していることも報告されている(59)。
nPKCが組織のインスリン抵抗性を引き起こすかどうかを判断する1つの方法として、ある介入を行うとインスリン感受性の変化がnPKCのトランスロケーションの変化に追随することを示す、相関関係に基づいた方法がある。脂肪を与えたラットにチアゾリジン系のインスリン感受性増強剤であるロシグリタゾンを投与すると、筋肉のDAG含量が減少し、それと並行してnPKCの転座も減少した(742)。同様に、慢性的に高脂肪食を与えたラットに低脂肪食を1回与えると、筋肉中のPKCθ(PKCεではない)のトランスロケーションが減少し、インスリン刺激による筋肉のグルコース取り込みが正常化した(46)。糖尿病になりやすいNZOマウスで断食を行うと、アドリブ食のコントロールと比較して、骨格筋DAGの減少、肝DAGの減少、PKCεの活性化の減少と関連して、高血糖の発症を防ぐことができた(39)。脂質/ヘパリン注入による急性筋のインスリン抵抗性は、この研究のための生産的なモデルシステムでもある。脂質によって誘発される急性筋インスリン抵抗性の特徴は、その効果が現れるまでに3〜5時間の時間差があることであり、再現性が高い(71, 942)。ある研究では、急性脂質注入は、PKCεではなく、PKCθの顕著なトランスロケーションと時間的に並行して、筋肉におけるインスリンシグナルとグルコース取り込みを障害した(285)。別の研究では、インスリン抵抗性を誘発するのに必要な4-5時間の脂質注入は、細胞内の長鎖アシルCoA、DAG含量、およびPKCθ活性化のピークと相関したが、セラミドやトリグリセリド含量とは相関しなかった(942)。
nPKCのトランスロケーションとインスリン感受性の間の動的な相互関係を調べる研究は、ヒトでも行われている。急性期に脂質を経口または非経口的に投与すると、インスリン刺激による筋肉のグルコース取り込みが減少し、それに伴って筋肉内のPKCθトランスロケーションが増加した(585, 819)。げっ歯類で観察された急性脂質注入時の筋細胞DAGの遅れた一過性の上昇は、ヒトでも観察されている(819)。
これらの相関性のある研究は、げっ歯類の遺伝子操作を用いてDAG/nPKC仮説をさらに検証するための強力な推進力となった。この目的のために、多くのモデルが使用されてきたが、結果はまちまちであった。これらは大きく分けて、脂質によるインスリン抵抗性にDAGが必要かどうか、あるいは十分かどうかを検証するモデルと、脂質によるインスリン抵抗性にnPKCが必要かどうか、あるいは十分かどうかを検証するモデルに分かれる。ここでは、この2つの大きなカテゴリーについて順次検討していく。
肝臓や筋肉におけるトリアシルグリセロール合成の主要経路であるケネディ経路では、脂肪性アシル-CoA部位がsn-グリセロール-3-リン酸に順次付加され、グリセロール-3-リン酸アシルトランスフェラーゼ(GPAT)を介してまずリゾホスファチジン酸が生成され、次にアシルグリセロールリン酸アシルトランスフェラーゼ(AGPAT)を介してホスファチジン酸が生成される。3位のリン酸はホスファチジン酸ホスファターゼ(PAP、別名リピン)によって除去され、sn-1,2-ジアシルグリセロール(DAG)が生成され、最後にジアシルグリセロールアシルトランスフェラーゼ(DGAT)によってアシル化されてトリアシルグリセロール(TAG)が生成される。この経路は、細胞性インスリン抵抗性のDAG仮説を遺伝子操作によって検証するための論理的な出発点となった。肝のGPATであるミトコンドリアGPAT(mtGPAT)を欠損したマウスは、高脂肪食を摂取すると、予測通り、脂肪アシル-CoA濃度が上昇し、肝のDAGおよびTAG濃度が低下した(575)。肝臓のDAGの減少は、PKCεの転位の減少を伴っていた(575)。DAG/nPKC仮説と一致して、mtGPAT-/-マウスはHFD誘発の肝インスリン抵抗性から保護され、高インスリン血症-高血糖クランプ試験中のHGP抑制の増強と、肝インスリンシグナルの増加の両方で証明された。IRS2に関連したPI3K活性やAKT活性が増加することが明らかになった(575)。同様に、mtGPATをアデノウイルスで肝に過剰発現させたマウスでは、肝のDAGが増加し、PKCεのトランスロケーションが増加し、肝のインスリン抵抗性が見られた(567)。AGPAT2またはlipin-1の全身欠損は、マウスおよびヒトにおいて重篤なリポディストロフィーを引き起こし、これらのモデルを用いてDAG/nPKC仮説を検証しようとする試みを混乱させる(170, 634)。主要な肝リピンであるリピン-2を生殖細胞系列で欠失させたマウスの解析も、代償としてのリピン-1のアップレギュレーションによって混乱をきたす(205)。しかし、shRNAによるリピン-1またはリピン-2の急性ノックダウンは、HFD給餌マウスの耐糖能を改善し、肝臓のDAGおよびTAG含量を減少させる(707, 708)。同様に、アデノウイルスによるlipin-2の肝臓での急性過剰発現は、耐糖能を低下させるのに十分であり、肝臓のDAGおよびTAGの増加を伴う(708)。骨格筋にlipin-1を過剰発現させたマウスは、深刻な肥満とインスリン抵抗性を示すが、このモデルにおける原因となる細胞メカニズムは完全には解明されていない(649)。最後に、DGATの遺伝子を改変したマウスでは、やや矛盾した結果が得られている。骨格筋にDGAT1を過剰発現させたマウスでは、予想通り、心筋細胞のTAGが増加し、心筋細胞のDAGが減少し、HFD誘発のインスリン抵抗性から保護された(491)。Dgat1-/-マウスは、予想に反してHFD誘発性の肥満とインスリン抵抗性から保護されたが、これはエネルギー消費の増加によるものと思われる。Dgat1-/-マウスの孤立した脂質処理ヒラメ筋は、インスリン刺激による2-デオキシグルコースの取り込みが鈍化し、DAG/nPKC仮説と一致した(491, 782)。DGAT2の阻害もまた、複雑な表現型をもたらす。筋特異的にDGAT2を過剰発現させたマウスでは、DAGが減少し(ただし、比較的インスリン感受性の低い解糖系筋線維でのみ)驚くべきことに、セラミド含量の増加と関連して緩やかな耐糖能異常を示す(473)。一方、肝臓のDGAT2を過剰発現させたマウスでは、TAGと、意外にもDAGの含有量が増加していた(371, 547)。これらのマウスでは、肝PKCεの転位が増加し、重度の肝インスリン抵抗性が生じていることが、高インスリン血症-高血糖クランプ試験でのHGP抑制が著しく損なわれていることからも明らかである(371)。肝臓特異的DGAT2過剰発現マウスを用いて行われた2つの研究のうち1つは、トランスジェニックマウスと野生型コントロールとの間でクランプEGPに差が見られなかったが、コントロールとトランスジェニックマウスの両方が肝インスリン抵抗性と一致するクランプEGP率を示したので、これらの実験の解釈は難しい(547)。これと一致して、DGAT2をアンチセンスオリゴヌクレオチドを介して肝臓でノックダウンした高脂肪食ラットでは、肝臓のDAGとPKCεの転位が減少し、肝臓のインスリン感受性が改善された(144)。ケネディ経路とは異なるが、モノアシルグリセロールアシルトランスフェラーゼ(MGAT)も、DAGのもう一つの脂肪生成源である。アデノウイルスのshRNAやアンチセンスオリゴヌクレオチドを用いてMgat1をノックダウンすると、肝脂肪症が減少し、HFD給餌マウスの耐糖能が改善された(466, 791)。しかし、MGAT1 ASOを用いた2つ目の研究では、耐糖能の改善はHFD肥育マウスやob/obマウスのIHTGの減少を伴わず、予想外にDAGの増加を伴うことが分かった(298)。この研究では、PKCεの膜/シトゾール比は正式には決定されなかったが、MGAT1 ASOにより膜のPKCε含量が減少したことから、肝のDAG総量は増加したものの、PKCεの活性化に利用できるDAGプールは増加していないことが示唆された(298)。興味深いことに、MGAT1 ASOを投与したマウスでは、DAG量が増加したにもかかわらず、de novo lipogenesisの割合が減少したことから、インスリン抵抗性を媒介する上で、定常状態のDAG量よりもlipogenic fluxの方が重要である可能性が示唆された(791)。以上のことから、脂質生成経路を阻害するマウスモデル(図14)は、予期せぬ生理的代償によって複雑化することが多いが、それでも脂質によって誘発されるインスリン抵抗性のDAG/nPKC仮説と概ね一致している。
図14 肝トリグリセリド合成が阻害されたげっ歯類モデル
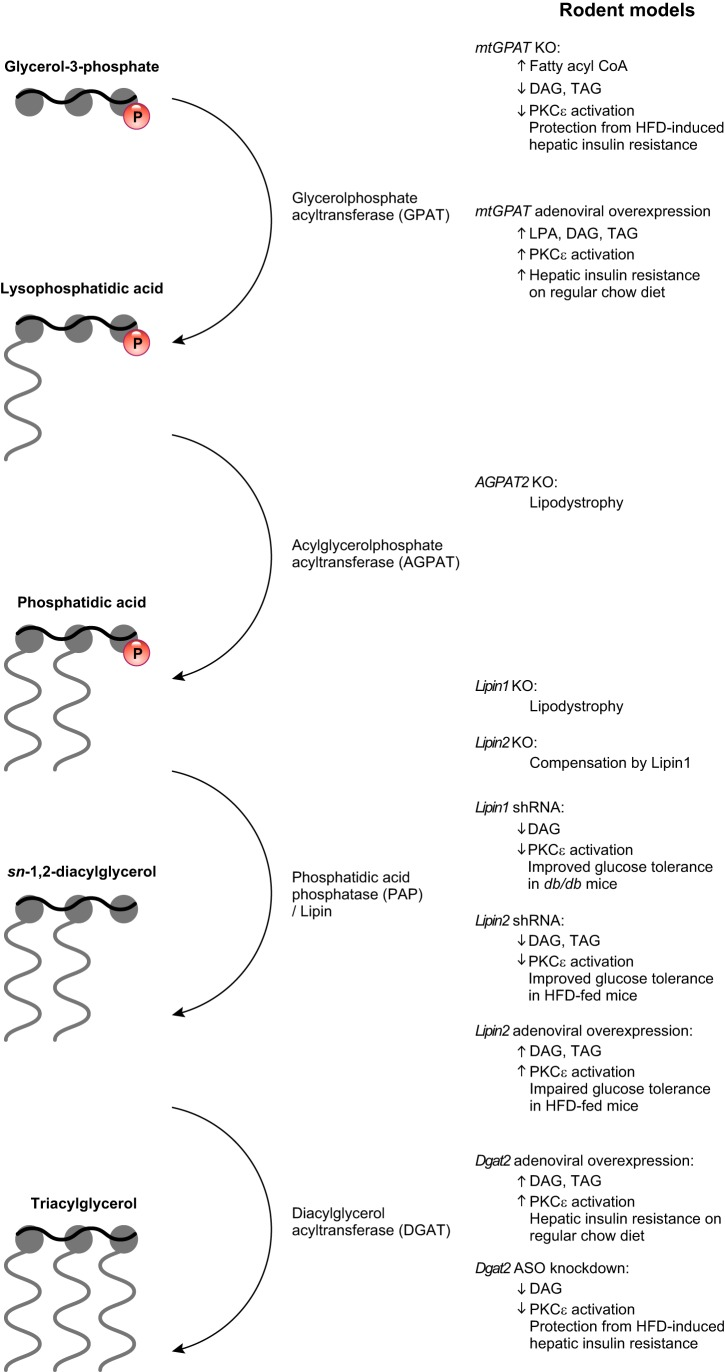
脂質誘発性の肝インスリン抵抗性におけるジアシルグリセロール(DAG)の役割を調べるために、様々な機能喪失型および機能獲得型の遺伝的なげっ歯類モデルが作成されている。これらのモデルは、脂質による肝のインスリン抵抗性についてのDAG/プロテインキナーゼC(PKC)ε仮説とほぼ一致している。TAG, トリアシルグリセロール, HFD, 高脂肪食, KO, ノックアウト. 詳細および参考文献は本文をご参照ほしい。
遺伝的に脂質代謝が変化した他のモデルでは、DAG/nPKC仮説が取り上げられている。脂肪酸伸長酵素Elovl6を欠損したマウスは、高脂肪食を与えると肝不全になりやすかったが、肝臓のDAGやPKCεの転位は増加せず、高脂肪食による肝臓のインスリン抵抗性から守られた(527)。DAGをPAに変換するジアシルグリセロールキナーゼ(DAGK)の摂動も研究されている(リピンの逆反応)。哺乳類のDAGKには10種類のアイソフォームがあり、基質特異性、組織での発現プロファイル、細胞内での局在などが異なっており、脂質代謝やインスリン抵抗性における役割に関するデータはまだ出始めたばかりである(50, 517, 518, 571, 573)。DAGKδは、2型糖尿病患者で減少しており、Dgkd+/-マウスでは、筋細胞のDAGが増加し、筋のインスリンシグナルやインスリン刺激によるグルコース取り込みが低下していた(137)。さらに、DAGKζをノックダウンすると、3T3-L1脂肪細胞におけるインスリン刺激によるGLUT4トランスロケーションが損なわれる(492)。不思議なことに、DAGKζノックアウトマウスは、筋肉のDAGが上昇しているにもかかわらず、HFDで筋肉のインスリン感受性が改善されたが、これらのマウスは、成長障害、脂肪の減少、燃料選択の変化も見られ、その表現型の解釈は複雑であった(50)。DAGKεの阻害による生理的な影響も不明であり、あるグループはHFD誘発の耐糖能異常からの保護を報告し、別のグループは耐糖能異常に対する感受性の増加を報告している(518, 571)。
トリグリセリドの脂肪分解を遺伝子的に阻害したマウスからは、多様で魅力的な表現型が得られている。タグ加白色脂肪組織分解酵素であるカルボキシルエステラーゼ2(CES2)を過剰発現させると、高脂肪食マウスの肝臓のDAGが減少し、耐糖能が改善された(705)。脂肪細胞特異的なAtgl-/-マウスは、脂肪細胞の脂肪酸酸化が抑制され、肝細胞のDAGが減少し、高インスリン血症による高血糖クランプ時のインスリンによる肝グルコース産生抑制が改善されるという、DAG仮説に合致したモデルの一例である(12)。同様に、HFD飼料を与えたマウスの肝臓にAtglをアデノウイルスで過剰発現させると、肝DAGが減少し、肝インスリンシグナルが改善された(855)。肝臓特異的なAtgl-/-マウスは、重篤な肝脂肪症を発症するが、グルコースおよびインスリン耐性は正常であり、IHTG自体は肝臓のインスリン抵抗性を誘発しないという仮説と一致する(916)。同様に、Atglを標的としたshRNAをアデノウイルスで12週間投与したマウスでは、肝のTAGとDAGが増加したが、耐糖能は正常に維持され、空腹時血糖値はコントロールよりも低かった(597)。これらの表現型は、肝内脂肪分解がグルコン生成をサポートするために重要であることを反映していると考えられる(例えば、グリセロールがグルコン生成に入り、アセチルCoAがPCを活性化するなど)。
遺伝学的または薬理学的な介入によって肝のインスリン作用を変化させた少なくとも2ダースのげっ歯類の研究では、肝のDAG含量と肝のインスリン感受性との間に逆の関係が観察されている(646)。しかし、上述した以外にも(298, 547, 597)いくつかの遺伝子改変マウスモデルでは、DAGの蓄積と肝のインスリン抵抗性との間に関連性がないような表現型が得られており、脂質によるインスリン抵抗性のDAG-PKC仮説には懐疑的な見方がされている(20, 231)。ChREBPを過剰発現させたマウスでは、いくつかの脂肪生成遺伝子の発現が増加し、肝DAGが増加して肝脂肪症になることが予想された(49)。しかし、ChREBPを過剰発現させたマウスは、普通の餌を食べても正常な耐糖能とインスリン抵抗性を示し、HFDによる耐糖能と肝インスリンシグナルの障害からも保護された(49)。この研究では、PKCεの活性化は測定されていないが、脂肪原性DAGの蓄積が肝のインスリン抵抗性に十分であるかどうかは疑問であり、脂質を迅速に貯蔵することの利点を指摘している(496)。肝インスリン抵抗性のDAG仮説と矛盾するもう一つの重要なマウスモデルは、肝臓特異的なミクロソームトリグリセリド転送タンパク質(Mttp)ノックアウトマウスである(543)。このマウスは、超低密度リポタンパク質(VLDL)の分泌に障害があり、体重や脂肪組織量が正常であるにもかかわらず、DAG、セラミド、TAGなどの肝脂質が蓄積している(543)。肝脂質の蓄積にもかかわらず、肝臓特異的なMttp-/-マウスは、高インスリン血症-高血糖クランプ試験において正常な肝インスリン感受性を示した(543)。これらのマウスではPKCεの活性化は評価されておらず、脂肪が正常であることから、間接的な肝インスリン作用(脂肪分解の抑制を介する)は損なわれていないと考えられ、このことがクランプ中に観察された肝グルコース産生の正常な抑制を説明しているのかもしれない(543)。もう一つのモデルは、肝臓特異的なHdac3-/-マウスである。Hdac3-/-マウスは、非常に小さな脂肪滴とアシルCoAプールの減少を特徴とする独特の肝脂肪症を発症し、空腹時インスリンの減少、耐糖能の改善、普通の餌でのインスリン耐性の改善を示す(812)。Hdac3-/-マウスでは、肝臓のDAGが数倍に増加したにもかかわらず、PKCεの活性化は観察されず、肝臓の脂質と肝臓のインスリン作用との間の不一致が、肝臓のPKCεの活性化と肝臓のインスリン作用との間の一致を伴っているもう一つの例である(812)。
DAG/nPKC仮説に対する重要な挑戦は、ATGLコアクチベーターCGI-58をアンチセンスオリゴヌクレオチドでノックダウンしたマウスから始まった。これらのマウスは、重篤な肝脂肪症を呈し、肝DAG含量が増加しているにもかかわらず、肝インスリンシグナルは正常に維持され、高インスリン血性共役血糖クランプ中の肝グルコース産生を正常に抑制する(98, 115)。これらのデータは、DAGの増加が肝のインスリン抵抗性を誘発するのに不十分であるという説明もあるが、より微妙な解釈も可能である。例えば、PKCεは、CGI-58 ASO処理マウスでは、脂質誘発性インスリン抵抗性で一般的に観察されるような膜結合画分ではなく、脂質滴に移行することが観察された(115)。脂質滴のDAG/nPKC活性は、分泌装置や細胞膜などのインスリン受容体の輸送やシグナル伝達に関与するコンパートメントからPKCεを隔離する役割を果たしている可能性がある。ペリリピン5をアデノウイルスで肝に過剰発現させたマウスでは、肝のインスリン感受性が維持されたまま肝のTAGとDAGが増加するという同様の表現型が見られたが、これも同様のメカニズムによるものと考えられる(848)。DAGの局在化の重要性が認識されるようになった結果、近年では複数の細胞内コンパートメントのDAG量を測定することが標準的になってきた。
細胞内のDAGを細分化するもう一つの方法は、アシル鎖の組成(長さと飽和度)である。In vitroでは、異なるDAG種がPKCアイソフォームを適度に異なる力で活性化する可能性があるが、このような違いの明確な分子基盤は特定されていない(510)。生体内では、アシル基の飽和度とインスリン抵抗性との間に、正の相関、負の相関、あるいは無相関が観察されている(21, 53, 819, 866d)。DAGの組成は、主に循環脂肪酸の利用可能性の関数であり、この問題を解決するために、急性脂質注入や合成げっ歯類HFDを使用して行われた研究の有用性は制限されるかもしれない。また、多くの脂質種について相関係数を算出した研究では、多重比較を慎重に補正することが不可欠である。
最後に重要なことは、DAGの立体異性体に関することである。DAGは、炭素数3のグリセロール骨格に沿って2つの脂肪アシル鎖が配置されることにより、sn-1,2,sn-2,3,sn-1,3の3つの立体異性体のいずれかとして存在する。PKCアイソフォームを活性化できるのはsn-1,2-DAGのみである(680)。この立体特異性は生理学的に重要な意味を持つ。脂肪分解酵素であるATGLは、PKCを活性化するsn-1,2-DAGではなく、sn-1,3-DAGを優先的に生成することから、細胞内の脂肪分解がnPKCを介した細胞のインスリン抵抗性を促進する要因ではないと考えられている(210)。脂肪分解におけるDAG立体異性体の重要性は、HSLノックアウトマウスの研究でも明らかになっている。HSLノックアウトマウスでは、筋肉中のsn-1,3-DAG含量が増加したにもかかわらず、インスリン刺激によるグルコース摂取量が増加した。DAG/nPKCを介したインスリン抵抗性は、脂肪分解性DAGではなく、脂肪生成性DAGのフラックスによってもたらされると考えられる。興味深いことに、Dgat2のmRNAの発現は、AtglまたはCGI-58のいずれかを肝臓特異的に欠失させたマウスのインスリン感受性脂肪肝、およびCGI-58 ASO処理マウスで減少しており、脂肪生成抑制補償を示唆している(98, 290, 916)。DAGの立体異性体を特定した測定法はあまり報告されておらず、自然なアシル移動によりサンプル調製中にsn-1,2-からsn-1,3-DAGに変換される傾向がある。しかし、最近の研究では、スポーツ選手、痩せ型コントロール、肥満被験者、2型糖尿病被験者を含むヒトコホートにおいて、立体異性体、コンパートメント、種によって細分化された筋DAG濃度が報告されている(619)。これらの研究者は、2型糖尿病患者では、痩せた対照群と比較して、全細胞ライセートとサルコレム膜画分でsn-1,2-DAGの総量が増加していることを発見したが、ミトコンドリア/ER、核、細胞質画分では増加していなかった(619)。関連する実験として、Pestaらは、インスリン感受性の高いスポーツ選手は、肥満のインスリン抵抗性被験者と比較して、筋肉中のPKCθトランスロケーションが減少していることを発見した。このことは、筋肉中の総TAGおよびDAG含量が同様に増加しているにもかかわらず、DAGのコンパートメントおよび/または立体異性体の分布の違いが、これらの集団間の筋肉のインスリン感受性の違いを説明している可能性を示唆している(D. Pesta, D. Zhang, G. Shulman, M. Roden, 未発表結果)。この分野では、方法論の改良と標準化、および複数の集団にわたるデータ収集の両面で、さらなる研究が必要となるであろう。
また、すべてのモデルで測定されたDAG/nPKC軸の変化は、慎重に解釈しなければならないことも重要である。というのも、DAGレベルとnPKC活性化を定量化するために現在利用できる実用的な方法は、比較的粗雑で、細胞内の空間的分解能や時間的分解能に欠けているからである。例えば、DAGのコンパートメントやPKCのトランスロケーションを調べるために一般的に使用される「膜」または「微粒子」の画分には、細胞膜、ミトコンドリア膜、小胞体、ゴルジ膜、その他の膜端が含まれる(115)。研究者は通常、あるマウスモデルにおけるDAG/nPKC摂動のパターンが、典型的なヒトのインスリン抵抗性で観察されるパターンと一致することを確信することはできない。また、DAG/PKC軸の活性化に関連するコンパートメントが細胞膜であることは、広く想定されていることではあるが、確実ではない。DAG-PKCεの相互作用は、主にゴルジ体で起こることが3T3線維芽細胞で示されており、ゴルジ体にアンカーするタンパク質RACK2が局在している(469, 648)。nPKCのDAG結合C1ドメインにある1つのトリプトファン残基もまた、ゴルジ体へのターゲティングを促進するようである(199)。ゴルジ膜は、DAG/PKC軸を調べるために典型的に用いられる「膜ˮ」画分の構成要素であるが、細胞性インスリン抵抗性におけるゴルジでのDAG/nPKC軸の活性化を特に調べた研究はない。sn-1,2-DAGの生成は、他のオルガネラで最もよく研究されている。すなわち、細胞膜ではホスホリパーゼCによるホスファチジルイノシトール4,5-ビスホフエートの切断(PKCα、β1,β2,γなどの古典的なPKCアイソフォームの活性化の主要なメカニズム)小胞体ではホスファチジン酸ホスファターゼによるKennedy経路またはMGAT活性によるものである(211)。しかし、sn-1,2-DAGはゴルジ体にも存在し、そこで膜タンパク質の輸送にシグナルを与える(36, 730)。さらに、RACK2は、小胞輸送を媒介するコートマー複合体の構成要素であり、これらの構造間の広範なクロストークを強調している(648)。全体として、異なるDAG部位がどのようにインスリン抵抗性に関与しているかについて分かっていることを考慮すると、DAGがnPKCアイソフォームを活性化するという最初の仮説を合理的に改良するには、脂肪生成sn-1,2-DAGがゴルジ体で作用してnPKCアイソフォームを活性化するということになるかもしれない(図15)。
図15 ジアシルグリセロール(DAG)の細胞内局在と立体異性体
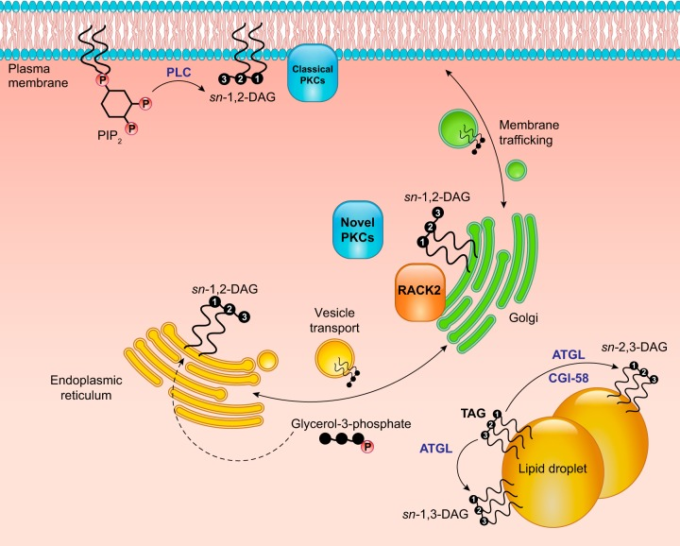
DAGは細胞内の複数の部位で様々な酵素の作用により産生されるが、ここではその一部を紹介する。細胞膜では、ホスホリパーゼC(PLC)がホスファチジルイノシトール-4,5-二リン酸(PIP2)からsn-1,2-DAGを生成する。このDAGは、古典的なプロテインキナーゼC(PKC)アイソフォームの活性化に特に重要であり、PLC活性の下流で放出されるCa2+も必要とする。ケネディ経路の脂肪生成酵素は小胞体に局在し、sn-1,2-DAGを生成する。新規のPKCアイソフォームは、ゴルジ体に顕著に局在する。特にPKCεでは、アダプタータンパク質であるRACK2がゴルジ体に局在していることがその一因となっている。RACK2は小胞体とゴルジ体の間の小胞輸送にも関与しており、ゴルジ体のDAGはタンパク質の細胞膜への輸送を制御している。注目すべきは、脂肪滴に存在する脂肪分解酵素(ATGL)によって生成されるDAGはsn-1,3立体異性体であり、PKCアイソフォームを活性化するとは考えられないことである。
インスリン抵抗性におけるDAGの役割を調べるために作られたいくつかのモデルを検討した後、今度は、脂質によって誘発されるインスリン抵抗性におけるnPKCの活性化の役割を遺伝学的に検証するために作られたモデルに注目してみよう。このようなモデルとして最初に報告されたのは、骨格筋に特異的にドミナントネガティブでキナーゼを死滅させたPKCθを異所性に発現させたマウスであった(753)。意外なことに、このマウスは加齢に伴う肥満を発症し、耐糖能異常と筋肉のインスリンシグナルの障害を伴っていた(753)。同様に、全身のPKCθノックアウトマウスは、身体活動とエネルギー消費の低下により、HFDによる肥満とインスリン抵抗性に陥りやすかった(256)。しかし、PKCθノックアウトマウスは、脂質によるインスリン抵抗性の最も単純なテストである急性脂質注入を行うと、骨格筋のインスリン抵抗性から完全に保護された(405)。PKCθの過剰発現もまた、培養心筋細胞のインスリン抵抗性を引き起こすのに十分である(292)。これらの異なる結果は、PKCθが脂質によるインスリン抵抗性の病態生理学的役割を担っている可能性に加えて、正常な骨格筋のインスリン作用やエネルギーバランスにおいても、まだ決定されていない生理学的役割を担っている可能性を示唆している。また、PKCθは脂質によるインスリン抵抗性に関与している可能性が高いが、肥満に伴う筋のインスリン抵抗性に完全に必要なわけではないことも示された。
脂質によるインスリン抵抗性におけるPKCεの役割は、遺伝学的にも検証されている。PKCεのアンチセンスオリゴヌクレオチドを肝臓と白色脂肪組織に特異的にノックダウンすると、ラットに3日間のHFDを与えたとき、肝臓のインスリン抵抗性を防ぐことができる(722)。さらに、全身のPKCεノックアウトマウスは、肝臓のTAGおよびDAG含量が増加するにもかかわらず、1週間のHFDを与えても耐糖能異常から完全に保護される(668)。PKCεノックアウトマウスで高脂肪食を与える期間が長くなると、肝臓の表現型の評価がβ細胞機能の改善と混同されるが、耐糖能の改善は持続する(741)。このように、遺伝的モデルは、脂質による肝のインスリン抵抗性の病態生理にPKCεが関与していることを示している。
PKCδの活性化が脂質によるインスリン抵抗性にどのように寄与しているかについては、まだ十分に解明されていない。PKCδの移動は、脂質を投与したラットの肝臓で観察されており(450)、マウスモデルでも、このnPKCが肝臓のインスリン作用に有害な役割を果たしていることが支持されている。PKCδをノックアウトしたマウスは、全身または肝臓に特異的に存在し、糖新生や脂肪生成の転写プログラムが減少し、肝臓のインスリンシグナルが改善される一方、PKCδを肝臓にアデノウイルスで過剰発現させたマウスは耐糖能が低下する(59)。しかし、NAFLDのヒトや3日間高脂肪食を与えたラットでは、肝内のPKCδの転位が増加しておらず、これらの観察結果の生理学的な妥当性が疑問視されている(445, 721)。PKCδノックアウトマウスの脂質生成能の低下は、DAG/PKCε軸の活性化を低下させ、肝のインスリン作用の改善に直結すると考えられる。骨格筋のインスリン抵抗性におけるPKCδの役割は複雑である。In vitroの研究では、PKCδの活性化が実際に筋細胞のインスリン作用を増強する可能性が示唆されている(91, 92)。しかし、この仮説は生体内試験のデータでは支持されていない。若いマウスの骨格筋では、PKCδの発現は低く、興味深いことに、脂肪を摂取すると減少する(482)。そのため、筋肉特異的にPKCδを欠失させたマウス(M-PKCδKOマウス)では、若いマウスのHFDによる筋肉のインスリン抵抗性を防ぐことができない(482)。しかし、骨格筋のPKCδの発現は加齢とともに増加し、老齢のM-PKCδKOマウスは加齢に伴う耐糖能異常から保護される(482)。脂質によるインスリン抵抗性にPKCδがどのように関与しているかについては、今後の研究が必要である。
これらの遺伝子モデルは、脂質誘導インスリン抵抗性における様々なnPKCの組織特異的な役割について重要な手がかりを与えてくれたが、DAG/nPKC軸の病態生理学的な意義を確立するには、活性化されたnPKCがインスリン作用を損なう分子メカニズムを解明する必要がある。もし、DAG/nPKC軸が脂質によるインスリン抵抗性を媒介するのであれば、インスリンシグナル伝達物質のセリン/スレオニンリン酸化が最も直接的なメカニズムとなる。この仮説は長い間追求されてきたが、特定のセリン/スレオニンリン酸化部位と脂質誘導インスリン抵抗性を結びつける決定的な証拠は、最近になってようやく現れ始めた。
PKCによるインスリンシグナルの阻害を調べる初期の研究では、ホルボールエステル、特にホルボール-12-ミリスチン酸-13-アセテート(PMA)(12-O-テトラデカノイルホルボール-13-アセテート(TPA)としても知られる)が用いられた。フォルボールエステルがINSRのチロシンリン酸化とインスリン刺激によるグリコーゲン合成を低下させることが初めて確認されたのは1984年のことである(822)。この阻害作用は、INSRのセリンおよびスレオニンのリン酸化と関連しており、アルカリホスファターゼ処理によって回復した(821, 822)。これらの証拠に加えて、1986年には、精製されたPKCが(Ca2+依存性の活性はあるものの)精製されたINSRをリン酸化できることが示され(77)PKCがIRKの活性を直接阻害できることが示唆された。INSRのセリンおよびスレオニンリン酸化の特異的な部位を探したところ、いくつかの候補が見つかったが、そのほとんどが比較的構造化されていないCOOH-末端尾部にあった。これらの部位には、Thr1348 (478)、Ser1327 (156)、Ser1305/Ser1306 (479)、Ser1006 (802)、Ser1035/Ser1037 (489, 802)が含まれていた。しかし、ホルボールエステルは、複数のPKCアイソフォームの非特異的な活性化剤であり、上記の研究のほとんどは、ホルボールエステルまたはPKCαやPKCβIIなどのcPKCアイソフォームのいずれかを使用していた。慎重な研究にもかかわらず、これらの候補部位は結局、生体内試験でのインスリン抵抗性に関与していない(82, 394, 564)。このパラダイムの例外として考えられるのは、PKCα、β、ζおよびTANK結合キナーゼ1の試験管内試験基質であるSer994であり、肥満に伴うインスリン抵抗性のいくつかのげっ歯類モデルでは、基礎状態で肝INSRのSer994リン酸化が増加することが示されている(155, 559, 802)。しかし、INSRのキナーゼ活性におけるSer994リン酸化の役割については、そのメカニズムが明らかにされていない。
脂質による骨格筋のインスリン抵抗性にはPKCθの活性化が関与し、脂質による肝のインスリン抵抗性にはPKCεが関与していることが明らかになったことから、これらの特異的なアイソフォームに着目して機構解明が進められた。2004,試験管内試験のキナーゼアッセイと細胞実験により、心筋細胞においてIRS1のSer1101がPKCθの基質であることが確認された(494)。IRS1 Ser1101は、インスリン刺激後15分以内にリン酸化され、IRS1のチロシンリン酸化を阻害することから、インスリン作用を弱める急性の負のフィードバック回路として作用する可能性が示唆された(494)。筋肉におけるIRS1のSer1101リン酸化の増加は、急激な脂質投与を受けたヒトで報告されており、筋肉のインスリン抵抗性の発現と関連している(819)。しかし、PMA、腫瘍壊死因子(TNF)-α、アラキドン酸、オレイン酸などの他の刺激でもIRS1 Ser1101のリン酸化が増加することが確認されている(494)。さらに、アミノ酸活性化キナーゼであるS6キナーゼ1(S6K1)は、栄養状態に応じてIRS1のSer1101をリン酸化することが明らかになっており、この機能的に重要なリン酸化部位が複雑かつ統合的に制御されていることが示唆されている(846)。IRS1は50以上のセリン・スレオニン部位でリン酸化されるが、そのほとんどがインスリンなどの代謝刺激によって動的に制御されている。あるセリン・スレオニンのリン酸化イベントがIRS1のチロシンリン酸化を阻害する構造的な根拠はほとんどわかっていない(169, 300)。インスリン感受性や抵抗性における特定のIRSリン酸化部位の因果関係を明らかにすることの難しさは、長い間、c-Jun NH2-terminal kinase (JNK; VII節参照)を介して炎症により誘発されるインスリン抵抗性のマーカーと考えられてきたIRS1のSer307の場合に典型的に示されている(465)。IRS1のSer307リン酸化がインスリン抵抗性を媒介することを示唆する多くの証拠があるにもかかわらず、Ser307Ala変異をホモ接合で導入したマウスは、驚くべきことに、食事によって誘発されるインスリン抵抗性の影響を受けやすくなるどころか、受けなくなった(167)。同様に、よく研究されているIRS1のリン酸化部位であるSer302のアラニンノックインマウスでは、筋肉のインスリン作用に欠陥は見られなかった(168)。IRS1のセリン/スレオニンリン酸化は非常に複雑であるため、単一のIRSリン酸化部位がIRSシグナルの強度をオン/オフ制御できるという仮説には疑問があるようだ。PKCθによるIRS1のSer1101のリン酸化の重要性は不明であるが、PKCθによる筋のインスリン抵抗性の他のメディエーターの可能性についても報告されている。PKCθは、パルミチン酸処理した筋管において、ホスホイノシド依存性キナーゼ1(PDK1)のSer504およびSer532をリン酸化し、これらのリン酸化事象は、PDK1を介したAkt Thr308のリン酸化の低下と関連していることが報告されている(873)。PKCθの最後の興味深い標的は、グアニン交換因子であるGIV/ガーディンである。GIVは、INSR、IRS1,PI3Kと相互作用し、インスリン刺激によるグルコース取り込みに必要であるが(514)、PKCθはSer1689のリン酸化によってGIVを阻害し、PI3K-AKTシグナルを減少させる(500)。GIVは、L6筋管におけるグルコース取り込みに対するパルミチン酸誘導のインスリン抵抗性に必要であること、リン酸化Ser1689Asp GIV変異の発現は、L6筋管におけるインスリン刺激によるグルコース取り込みを消失させるのに十分であること、多嚢胞性卵巣症候群の女性においてGIVのSer1689リン酸化はピオグリタゾン治療によって大きく減少することが示された(514)。しかし、このリン酸化イベントの生理的意義を完全に理解するには、今後の研究が必要であり、近位のインスリンシグナルカスケード内の他のPKCθ基質がまだ発見されていない可能性がある。
PKCεと肝インスリン抵抗性の間のメカニズムを理解するための努力も、興味深い結果をもたらした。肝インスリン抵抗性のシグナル伝達障害は、近位のIRK活性にまで遡ることができるため(116, 155, 161, 722)PKCεがIRK活性を直接阻害するという仮説が立てられた。PKCεは肝臓においてINSRβと共沈することが示されており、直接的な相互作用が示唆されている(722)。また、PKCεを標的としたアンチセンスオリゴヌクレオチド(ASO)を投与したラットから免疫沈降した肝臓のINSRは、HFDによるIRK活性の低下から完全に保護されていた(722)。しかし、PKCεがIRKの活性を阻害するメカニズムは、最近まで不明であった。試験管内試験のPKCε/IRKキナーゼアッセイのリン酸化ペプチド質量分析法により、IRK活性化ループ内にThr1160という新たなリン酸化部位があることが明らかになった(645)。このスレオニンは、ショウジョウバエのような遠縁の後生動物でも保存されており、そのリン酸化は、立体障害と静電的な反発によって活性化されたIRKの正常な配置を崩すと予測されている(645)。実際、リン酸化されたThr1160Glu変異は、ほぼキナーゼ死に近い状態のINSRを作り出す(645)。逆に、Thr1160Ala変異は、試験管内試験でのPKCεによるIRKの阻害を消失させた(645)。InsrT1150Aノックインマウスは、HFDによる肝インスリン抵抗性から保護されたことから、Thr1160リン酸化は脂質による肝インスリン抵抗性の生理学的メカニズムであることが示された(645)。これらの研究は、DAG/PKCε軸の活性化が、INSR Thr1160の直接的な抑制的リン酸化を促進するという、脂質による肝インスリン抵抗性モデルを支持するものである(図16)。重要なことは、この欠陥はインスリン受容体に局在するが、受容体数の減少ではないため、ポスト受容体の欠陥として分類するのが最適であり、インスリンの用量反応曲線を右方向および下方向にシフトさせる。しかし、このシフトしたインスリン用量反応曲線のかなりの部分では、DAG/PKCε/INSRを介した肝のインスリン抵抗性は、軽度から中等度のインスリン抵抗性にしばしば伴う門脈高インスリン血症によって克服することができる。この概念は、セクションIVで議論したように、インスリン刺激による肝グリコーゲン合成とインスリン刺激によるde novo lipogenesisの両方に適用されるはずである。このモデルは、肝のインスリン抵抗性で典型的に観察されるシグナル伝達パターン(例えば、PKCεの活性化、IRKの阻害、およびすべての下流のエフェクター)を説明するのに十分であるが、それにもかかわらず、脂質によって誘発される肝のインスリン抵抗性のための追加のPKCε依存性または非依存性のメカニズムが作用している可能性が高いと思われる。
図16 脂質誘発性肝インスリン抵抗性におけるジアシルグリセロール(DAG)/プロテインキナーゼC(PKC)ε軸
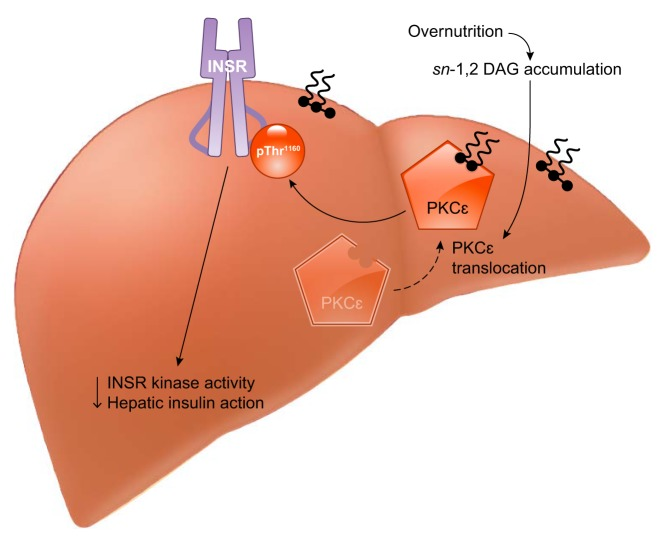
慢性的な過栄養状態が続くと、肝内の脂質が蓄積される。この脂質は主に比較的不活性なトリグリセリドとして蓄積されるが、トリグリセリド合成の最終中間体である生理活性脂質DAGの量も増加する。DAGは、PKCの細胞膜への移動を促進することでPKCアイソフォームを活性化し、特にPKCεの移動は、脂質によって誘発される肝インスリン抵抗性の設定において慢性的かつ再現性よく増加する。PKCεは、インスリン受容体(INSR)のチロシンキナーゼドメインの活性化ループにあるThr1160を直接リン酸化して阻害することで、インスリンの作用を低下させる。
これらの研究は、DAG/PKC仮説に光を当てると同時に疑問を投げかけ、より焦点を絞った特異的な仮説を可能にしている。遺伝学的および薬理学的な操作により、脂質によるインスリン抵抗性においてDAG/nPKC軸の活性化が因果関係を持つことを示す強力な証拠が得られており、「野生型」のげっ歯類やヒトを用いた相関研究では生理学的な関連性が示唆されている。遺伝子組み換えマウスの時代になって、この仮説は強化され、また挑戦されたが、生理的でない代償という注意点が常に存在している。ジアシルグリセロールがいつ、どこで、どのようにしてnPKCを活性化し、インスリンシグナルを損なうのか、理解を深めるためには、高度な細胞生物学と分析化学を、特に脂質処理経路が損なわれていない細胞や組織に適用することが必要である。
C. セラミドとインスリン抵抗性
スフィンゴ糖脂質は、セリンと主にパルミトイルCoAが結合してできたもので、何百もの異なる脂質種を含んでいる(325)。スフィンゴ糖脂質がインスリン作用と相互作用することが最初に示唆されたのは、スフィンガニンとスフィンゴシンが3T3-L1線維芽細胞におけるインスリン刺激による2-デオキシグルコースの取り込みを阻害したという試験管内試験の報告からである(574)。また、スフィンゴシンは、培養脂肪細胞におけるインスリン刺激による最大グルコース取り込みと脂肪生成を阻害することが示された(695, 779)。スフィンゴシンに脂肪性アシル基を共有結合で付加して作られるセラミドの初期の研究では、短鎖(C2,C6)の細胞伝染性セラミドが使用され、多くの場合、超生理学的な濃度で使用された。これらの研究では、培養液にセラミドを添加するとインスリン作用が阻害されるという点でほぼ一致しているが、阻害される部位が異なることが報告されている。セラミドによってINSRチロシンキナーゼ活性が阻害されると報告したグループもあれば(385, 617)近位のインスリンシグナルには障害がないとしたグループもあった(574, 740, 809, 874, 956)。1998年に発表された、C2セラミドがインスリン作用を阻害するのは、INSR-IRS-PI3Kの活性化とは別の、AKTのレベルであるという報告は、セラミドによるインスリン抵抗性のメカニズムのコンセンサスを得るのに、特に大きな影響を与えた(809, 956)。C2セラミドがAKTを阻害するメカニズムとしては、PP2A活性の上昇(715, 748, 806, 961)と、非定型PKCζの活性化によるインスリン刺激によるAKTのトランスロケーションの欠損(805)の両方が関係しているとされている(67, 241, 659, 806)。その後のセラミドによるインスリン抵抗性の研究では、主なメカニズムとしてAKTの阻害が注目されている。
短鎖セラミドは初期の試験管内試験研究では便利な実験ツールであったが、最も一般的な内因性セラミドはパルミチン酸や他の長鎖飽和脂肪酸(SFA)を含んでいる。そのため、最近のセラミドによるインスリン抵抗性の研究では、SFAの影響に注目している。パルミチン酸は、培養心筋細胞や肝細胞において強力にインスリン抵抗性を誘導するが、特にAKTのSer473リン酸化をインスリン抵抗性の主要な指標とした場合、その効果は顕著である(126, 740)。しかし、インスリン刺激によるグリコーゲン合成、グルコース取り込み、HGP抑制などのインスリン抵抗性の機能的指標を用いた場合、パルミチン酸はオレイン酸やリノール酸などの不飽和脂肪酸(USFA)と同程度にインスリン抵抗性を誘導する(323, 740)。USFAはセラミドレベルを増加させないので、セラミドが脂質によるインスリン抵抗性の唯一のメディエーターではないことになる(124)。
SFAによる筋肉のインスリン抵抗性におけるセラミドの役割については、セラミドの合成を阻害してインスリンの作用を評価する試験管内試験および生体内試験の研究で検討されている。これらの研究の多くは、セラミド生合成の第一段階であるセリンパルミトイルトランスフェラーゼ-1の活性を阻害する天然真菌製品ミリオシンを用いている(127, 545)。例えば、パルミチン酸を多く含むラード油を注入すると、ラットでは急性筋のインスリン抵抗性が生じ、それに伴って筋細胞のDAGとセラミドが増加する。ミリオシンを投与すると、このインスリン抵抗性が部分的に予防され、DAGではなくセラミドの蓄積が抑制された(322, 323)。さらに、C2C12筋管にミリオシンを前処理すると、パルミチン酸誘発のインスリン抵抗性は完全に阻止されたが、リノール酸誘発のインスリン抵抗性は阻止されなかった(323)。しかし、別の研究では、筋セラミド含量が正常化したにもかかわらず、ミリオシンを投与してもSFA給餌マウスの耐糖能異常は回復しなかった(243)。セラミドによるインスリン抵抗性に関する多くの研究では、セラミド生合成を特異的に変化させるように設計された研究であっても、DAGのようなインスリン抵抗性の他の推定されるメディエーターの変化を含むリピドーム全体の変化を併発することが重要な交絡因子となる(101, 102)。例えば、セラミド研究の「主力」と言われるミリオシン(127)は、複数の肥満モデルにおいて、エネルギーバランス、体重増加、異所性脂質の蓄積を変化させることが示されている(934)。ミリオシンを投与したネズミの表現型全体に対する、これらの既知の筋インスリン感受性調節因子のそれぞれの相対的な寄与を評価することは困難である。
セラミドは、脂質による肝のインスリン抵抗性に関与している可能性についても研究されている。ジヒドロセラミド合成酵素1を1コピー欠損したマウス(DeS1+/-)は、インスリン負荷試験においてインスリン感受性が改善された(ただし、全身のセラミドレベルは野生型の同胞と同等であり、耐糖能は変化しなかった)(323)。超長鎖(C22/C24)セラミドを産生するセラミド合成酵素2のハプロ不全マウス(CerS2+/-)では、肝内の総セラミド量は変化しなかった(672)。しかし、CerS2+/-マウスでは肝セラミドのアシル鎖組成が変化してC16:0(パルミトイル)セラミドが増加し、肝脂肪症とインスリン抵抗性の悪化が認められた(672)。セラミド生合成を阻害する多くの実験モデルにおいて肝脂肪症が共存していること(61,323)は、原因となるメディエーターを特定する努力を複雑にしている。遺伝的に脂質代謝を阻害したモデルでも、肝臓のトリグリセリドが上昇したげっ歯類やヒトでは、一般にDAGも上昇している(115, 144, 371, 547)。さらに、最近のヒトのリピドミクス研究では、肝セラミドとHOMA-IRスコアとの間に正の関連があることが指摘されているが、肝DAGとの間にも同様の関連が観察されている(506)。しかし、セラミド分解酵素である酸セラミダーゼを組織特異的に過剰発現させたマウスを用いた最近の研究は、このパラダイムに疑問を投げかけている。酸性セラミダーゼを肝臓に特異的に過剰発現させたマウス(Alb-ACマウス)は、肝臓のセラミド量を減少させ、肝臓のDAG量が増加したにもかかわらず、HFDによる肝臓のインスリン抵抗性を防ぐことができた(922)。しかし、興味深いことに、Alb-ACマウスをHFDで飼育すると、肝脂肪症が顕著に予防された(肝TAG量が約3倍減少)(922)。このような肝臓のDAGとTAGの異常な解離のメカニズムは、酸セラミダーゼを脂肪組織特異的に過剰発現させたマウスでも観察されたが(922)、その理由は明らかではない。興味深いことに、この研究では、酸セラミダーゼの誘導による肝のインスリン抵抗性の逆転は、肝の脂肪症の逆転と平行していた(922)。セラミド含有量は、組織特異的な酸セラミダーゼの過剰発現によって変化した数多くの生理学的パラメーターの一つに過ぎないが、この研究は、肝のインスリン抵抗性におけるセラミドの役割を示唆するものであることは確かである。
結局のところ、セラミドが細胞のインスリン作用をどのように損なうかという問題は、インスリン抵抗性の状態でセラミドの生合成とセラミドレベルがどの程度変化するかによって、その意義が決まる。この後者の問題については、多くの研究が行われており、その結論は様々である。最も古い報告では、肥満のZucker fa/faラットでは肝臓と骨格筋のセラミド含量が増加していることが判明した(852)。また、Zucker diabetic fatty (ZDF)ラット(323)、ラードオイルを注入したラットの肝臓と筋肉(323)、ob/obマウスの肝臓(8)、肥満のヒトの筋肉(4, 21, 803)の肝臓と筋肉でもセラミドが増加していた。しかし、これらの一般的なインスリン抵抗性モデルは、いずれも組織内の脂質量が全般的に増加することを特徴としているため、インスリン抵抗性におけるセラミドの特異的な役割を評価することはできない。実際、脂質によってインスリン抵抗性が誘発されるモデルの多くは、筋肉と肝臓の両方において、セラミドレベルの上昇とは無縁である。例えば、C57BL/6Jマウスでは、8週間の高脂肪食を与えても、筋肉のセラミド量は変化せず、肝のセラミド量は減少した(548)。Liposyn IIの急性注入は、筋肉のセラミドを変化させることなく、筋肉のインスリン抵抗性を強力に誘導した(942)。ピルビン酸脱白色脂肪組織素酵素キナーゼ2および4を欠損したマウス(Pdk2/4-/-)は、筋肉中のグルコースを構成的に酸化し、その結果、脂質による筋インスリン抵抗性を発現したが、筋セラミドレベルは増加しなかった(669)。ラットでは、肝のインスリン抵抗性にもかかわらず、SFAリッチ、USFAリッチの3日間のHFDでも肝のセラミド量は増加しなかった(253)。肥満の非糖尿病患者を対象とした4つの独立した研究では、3つの研究で肝セラミド含量はインスリン抵抗性と相関しなかったが、1つの研究では正の相関が観察された(445, 506, 512, 834)。ApoA5のノックダウンにより組織内の脂質供給量が減少したマウスでは、筋肉および肝臓のインスリン感受性の改善は、組織内のセラミド含量の違いとは関連しなかった(113)。野生型のフルクトースを与えた対照と比較してde novo lipogenesisが低下した肝臓特異的なXbp1-/-マウスでは、肝臓のインスリン感受性は上昇したが、肝臓のセラミドレベルは上昇した(375)。さらに、卵巣摘出雌マウスにおけるエストラジオールのインスリン感受性向上作用は、DAG/nPKC軸の活性化の補正と関連していたが、肝臓や筋肉のセラミドには変化が見られなかった(111)。これらのモデルは、肝臓や筋肉のセラミドレベルの上昇は、脂質によるインスリン抵抗性には必要ないことを示唆している。
ジアシルグリセロールを介したインスリン抵抗性の研究では、アシル鎖の長さ、立体異性体、細胞内局在によって測定値が細分化されてきたように、いくつかのセラミド研究では、特定のセラミド種とその細胞内局在に焦点が当てられている。特に筋肉のC18:0セラミドは、3つのヒトの研究(52, 619, 844)において、高インスリン血症-高血糖クランプ時のインスリン感受性と逆相関することが確認されているが、この関係はすべての研究で観察されたわけではない(21, 149, 819)。興味深いことに、細胞内分画を用いた最近の研究では、このC18:0セラミドの関係は、サルコレンマ、ミトコンドリア/ER、および核の各コンパートメントで同様に観察された(619)。しかし、筋生検をサルコレム膜下画分と筋原線維内画分に分けて行った別の分画研究では、C16:0とC18:1セラミドのみにHOMA-IRとの強い相関関係が認められた(149)。C18:0セラミドはアシル鎖が飽和していることから、上述のPP2A/AKT機構に加えて、膜の流動性を低下させることでインスリンの作用を阻害するという仮説が立てられているが、これらの仮説はまだ直接検証されていない(52)。しかし、興味深いことに、C18:0セラミドは、マウスの高脂肪食摂取時に初めて増加する筋肉のセラミド種である(854)。
脂肪細胞のインスリン抵抗性におけるセラミドの役割は不明だが、この分野の研究は加速している。ヒトを対象とした研究では、脂肪のセラミド含有量とHOMA-IRとの相関関係(64)や、肥満の糖尿病患者では、肥満の非糖尿病患者に比べて脂肪のセラミドが増加することが報告されている(122)。さらに、脂肪組織特異的なセラミド分解の誘導に伴う全身のインスリン感作は、直接的なものであれ、組織のクロストークを介したものであれ、インスリン作用における脂肪セラミドの役割を示唆している(922)。実際、C16:0セラミドは、肥満患者の脂肪組織で増加しており、関連する合成酵素CerS6のmRNAの発現が増加していることが報告されている(686)。また、高脂肪食を与えると、マウスの脂肪組織のセラミドが増加することも報告されている(856)。白色脂肪組織分特異的セリンパルミトイルトランスフェラーゼ(Sptlc2)を欠損させたマウスでは、脂肪細胞のセラミド生合成を阻害することで、HFDによる体重増加や高血糖からマウスを守ることができたが、驚くべきことに、この遺伝的摂動はほとんどの白色脂肪組織分セラミド種の存在量に変化を与えなかった(122)。脂質誘発性肝インスリン抵抗性におけるセラミドの役割を研究している多くのげっ歯類モデルと同様に、エネルギーの全体的な変化は、これらのマウスに見られる代謝の改善を一つのメカニズムに帰する努力を困難にしている。
セラミドによるインスリン抵抗性の関連性を評価するためのもう一つの重要な問題は、インスリン抵抗性におけるシグナル伝達の遮断部位に関するものである。もしセラミドが典型的な肥満に伴うインスリン抵抗性の原因であり、セラミドがAKTの阻害を通じてインスリン抵抗性を誘導するのであれば、近位のインスリンシグナルは無傷であり、検出可能な欠陥はすべてAKTの下流にあると予想される。AKTのSer473リン酸化の欠陥は、筋肉と肝臓の両方のインスリン抵抗性で確かに検出されるが(113, 115, 669, 921)近位のインスリンシグナル伝達の欠陥も顕著である(117, 470, 722)。興味深いことに、GM3ガングリオシドのようなグルコシルセラミドは、INSRの膜マイクロドメインの局在が変化することで近位のインスリンシグナルが損なわれることが示唆されており(125, 376)、グルコシルセラミド合成酵素を特異的に阻害すると、肥満のネズミのインスリン感受性が改善される(8)が、インスリン抵抗性における特定のグルコシルセラミドの役割を理解するには、さらなる研究が必要である。しかし、典型的なインスリン抵抗性において認められる近位のインスリンシグナルの障害は、セラミドによるAKTの阻害がインスリン抵抗性の中心的な欠陥であるという仮説にとって問題となる。AKTのレベルでの主要な欠陥は、実際にはGRB10の安定化のようなAKTが制御するネガティブフィードバック機構の喪失を通じて近位のインスリンシグナルを増強すると予測される(339, 944)。さらに、セラミドによるインスリン抵抗性のメカニズムの一つとして提案されているPP2Aを急性に阻害してAKTのリン酸化を増加させても、脂肪食ラットのインスリン感受性を回復させるには不十分であり、予想外に筋肉のインスリン抵抗性を悪化させた(254)。セラミドが近位のインスリンシグナルを現在のところ未確認のメカニズムで阻害するのでなければ、セラミドのAKTへのシグナル伝達が典型的な肥満に伴うインスリン抵抗性を完全に説明できるとは考えられない。
以上を総合すると、セラミドがインスリン抵抗性を引き起こすというメカニズムの証拠は、骨格筋で最も強く、肝臓とWAT癌ではより曖昧なデータとなっている(644, 810)。典型的な肥満に伴う組織のインスリン抵抗性におけるセラミド誘導性インスリン抵抗性の相対的な重要性は不明である。セラミドシグナルはいくつかのモデルにおいてインスリン抵抗性を促進するのに十分であるかもしれないが、脂質誘導性インスリン抵抗性には必要でないようである。
D. アシルカルニチン、代謝の柔軟性、インスリン抵抗性
骨格筋のインスリン抵抗性のモデルであるDAG/nPKCモデルとセラミド/AKTモデルは、いずれも過剰な脂質を生理活性物質に移行させる不適切な同化作用を示唆している。しかし、もう一つの仮説は、トリカルボン酸(TCA)サイクルのフラックスと一致しない過剰な脂質の不適切な異化作用が、骨格筋におけるインスリン作用を低下させるというものである。この仮説を後押しした重要な観察結果は、ex vivoの骨格筋ホモジネートにおいて、高脂肪食の摂取により脂肪酸酸化(FAO)の割合が増加したことであり、これは、心筋細胞における過剰脂質の運命が単に異所性の貯蔵ではないことを示唆している(427)。しかし、このFAOの増加は、CO2産生量の増加(すなわち、完全な酸化)を伴わず、代わりに筋細胞のアシルカルニチン(脂肪酸がカルニチンと結合してミトコンドリアへの侵入を可能にしたもの)の増加を伴っており、これは不完全なFAOのマーカーである(427)。深刻なインスリン抵抗性を持つザッカー糖尿病脂肪ラットの筋アシルカルニチンプロファイリングでは、長鎖アシルカルニチン、特に鎖長10炭素以上のアシルカルニチンが顕著に増加していた(427)。これらの増加は、TCAサイクル代謝物の減少と相まって、TCAフラックスに対するFAOの不適合とその結果としてのミトコンドリアストレスを反映していると解釈された(427)。この結果は、運動によって活性化される転写因子であるPGC-1αが、脂質の完全酸化を促進し、高脂肪食によって減少するという以前の観察結果を補完するものであった(426)。しかし、筋肉特異的にPGC-1αを過剰発現させると、FAOの割合が増加するにもかかわらず、予想外に筋肉のインスリン抵抗性が生じることが後に示された。これらのマウスは、筋肉のDAG含量の増加とPKCθの活性化も示した(143)。さらに、培養液にカルニチンを加えると、パルミチン酸によるL6筋管のインスリン抵抗性誘導が促進されることが報告されており、脂質によるインスリン抵抗性にFAOが関与していることが示唆されている(427)。これらの結果は興味深いものではあるが、カルニチンを補給することで高脂肪食を与えたネズミの筋肉のインスリン感受性が高まるという他の報告(583, 866a)との整合性をとるのはやや難しい。
血漿中のアシルカルニチンを測定することで、先天性FAOエラーを非侵襲的に調べることができる(746)。血漿中にアシルカルニチンが出現するメカニズムは完全には解明されていないが、血漿中のアシルカルニチン濃度は細胞内のレベルを反映していると考えられている(654, 746)。それでは、血漿中のアシルカルニチン濃度は、筋肉中の不完全なFAOのバイオマーカーとして利用できるのだろうか?この仮説の大きな問題点は、血漿アシルカルニチン濃度は骨格筋だけでなく、すべての組織のFAO率の関数であることである。特に肝臓は脂質を優先的に酸化するので、血漿中のアシルカルニチン濃度に大きな影響を与えていると考えられる(19, 746)。興味深いことに、Pdk2/4-/-マウスでは、骨格筋においてグルコースの酸化が恒常的に行われており、筋細胞の中鎖および長鎖アシルカルニチンは野生型のコントロールと比較して著しく減少していたが、血漿レベルは変化しなかった(669)。このように筋と血漿のアシルカルニチンのプロファイルが異なることは、ヒトでも報告されている(784)。このことから、血漿アシルカルニチンを骨格筋の不完全FAOの検査に用いることはできないと考えられるが、いくつかのグループが試みている(5, 541)。
不完全なFAO、特にアシルカルニチンは、どのようにしてインスリン抵抗性を媒介するのだろうか?一つの可能性は、アシルカルニチンがインスリンシグナルを直接阻害することである。C2C12筋管を生理学的に適切な濃度(5〜25μM)のC4:0,C14:0,C16:0アシルカルニチンで処理すると、インスリン刺激によるAKTのSer473リン酸化が阻害されるが、その効果はわずかで(20〜30%の減少)用量依存性はなかった(10)。近位のインスリンシグナル伝達エフェクターは評価されなかった。不完全なFAOは、活性酸素種(ROS)の産生を増加させることにより、筋肉のインスリン抵抗性を誘導することも提案されている。パルミチン酸を投与すると、インスリン刺激によるグルコース取り込みの低下に伴い、活性酸素の産生が増加するが、ミルドン酸によるFAOの薬理学的阻害により、両方の効果が阻止される(10)。しかし、アシルカルニチン自体は、直接、活性酸素産生を増加させることは示されていない。むしろ、アシルカルニチン濃度は、FAOが細胞内のATP需要をどの程度上回っているかを反映していると考えられ、その場合には活性酸素の産生が増大すると考えられる(235)。活性酸素によるインスリン抵抗性のメカニズムについては、第VI章で述べている。
アシルカルニチンによるインスリン抵抗性の生理的根拠は不明である。インスリン感受性の高いラットでは、筋肉のアシルカルニチン濃度は栄養状態によって制御されており、HFDによる筋肉のインスリン抵抗性は、この栄養制御の喪失を伴うが、アシルカルニチン濃度の絶対的な増加は、絶食・給餌状態のネズミのレベルと比較して生じない(427)。このことは、もしアシルカルニチンが脂肪を与えられたネズミの筋肉でインスリン作用を損なうならば、インスリン感受性のネズミの絶食状態でもそうなることが予想されることを示しているが、これはネズミの骨格筋における絶食のインスリン感作効果が知られていることと矛盾する(32)。
インスリン抵抗性モデルで観察される筋アシルカルニチンの増加についての別の説明は、それらが単に相対的な酸化的基質選択を反映しているというものである。上述した空腹時およびFFD給餌ラットで観察された筋アシルカルニチンレベルのパターンは、空腹時および高脂肪給餌ラットのヒラメ筋における酸化的基質選択(VPDH/VTCA)の同位体測定と完全に一致しており、FAOの相対的な割合は、空腹時の空腹ラットと高脂肪給餌ラットでは、基礎的にもインスリン刺激後にも同様である(T. Alves, R. Perry, Y. Rahimi, and G. Shulman, unpublished data)。同様の結果は、インスリン抵抗性のヒト骨格筋の痩せ型および肥満型でも報告されている(395)。これらのデータを簡単に解釈すると、インスリン抵抗性骨格筋が摂食状態、つまりインスリン刺激状態に移行する際に相対的なグルコース利用率を高めることができないという、代謝の柔軟性のなさを反映していると考えられる。筋のインスリン抵抗性における代謝不全の概念は、Randleのグルコース-脂肪酸サイクルに端を発しているが、脂質によって誘発されるインスリン抵抗性の最新モデルを取り入れている(397, 561)。脂質誘発性インスリン抵抗性における代謝の柔軟性喪失のメカニズムは、まだ完全には解明されていないが、入手可能なメカニズムデータと一致する説明としては、DAGやセラミドなどの生理活性脂質部分がインスリンシグナルを阻害し、インスリン刺激によるGLUT4の移動を減少させることで、インスリンを介したグルコースの酸化的代謝への利用可能性の増加が阻害されるというものである。この現象は、脂質の利用可能性の増加と並行して起こるため、脂肪を与えられたインスリン抵抗性の筋肉では、FAOの相対的な割合が増加することになる。このように考えると、代謝の柔軟性のなさは、脂質によるインスリン抵抗性の原因ではなく、結果として生じるものと考えられる。アシルカルニチン濃度が主に相対的なFAOフラックスを反映しているとすれば、筋肉のインスリン抵抗性との関連も、脂質によるインスリンシグナルの障害という主な欠陥の二次的なものかもしれない。
脂質によるインスリン抵抗性のメカニズムを解明することは、これまで困難な課題であった。実際、異所性脂質の蓄積が肝臓や筋肉でのインスリン作用を損なうという核心的な前提でさえ、意見が分かれている。特定の生理活性脂質が肝細胞のインスリンシグナルに障害を与えるという、完全に定義されたメカニズムの枠組み(DAG/PKCε/INSR軸)は1つしか記述されていないが、脂質によって誘発される骨格筋のインスリン抵抗性は、より多くのメカニズムが注目されている。しかし、部分的には多くのメカニズムが解明されているものの、それぞれに弱点があり、ヒトの疾患におけるそれぞれの役割を明確にするにはさらなる研究が必要である。しかし、現在のところ、このセクションで取り上げた脂質メディエーターであるDAG、セラミド、アシルカルニチンが、骨格筋のインスリンシグナルに障害を与えるメカニズムとして提案されているのは、図17にまとめたとおりである。
図17 脂質による骨格筋のインスリン抵抗性の分子メカニズムの提案
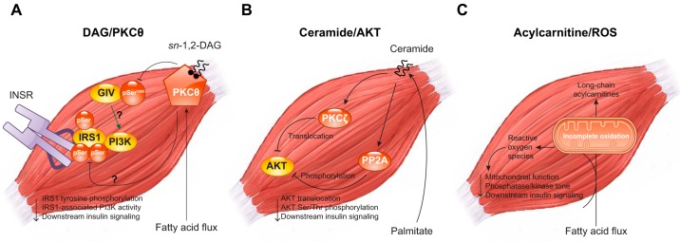
A:ジアシルグリセロール(DAG)は、プロテインキナーゼC-θ(PKCθ)を活性化することで、筋のインスリン抵抗性を引き起こすと提唱されている。インスリンシグナルカスケードにおけるPKCθの標的は不明確であるが、インスリン受容体基質1(IRS1)やGIVなどが考えられる。PI3Kは、ホスホイノシチド-3-キナーゼである。B: セラミドは、少なくとも2つのメカニズムでAKT活性を低下させることにより、骨格筋のインスリン抵抗性を媒介することが提案されている。PP2A、プロテインホスファターゼ2A。C: 不完全なミトコンドリア脂肪酸酸化は、結果として生じるアシルカルニチン種の直接的な影響、または様々な細胞プロセスを調節する活性酸素種の生成を介して、骨格筋のインスリン抵抗性を媒介することが提案されている。
VI. 細胞内栄養ストレスとインスリン抵抗性
A. 小胞体ストレスとインスリン抵抗性
肥満に伴うインスリン抵抗性の根本的な原因は、栄養の過剰供給である。第5章で述べた脂質によるインスリン抵抗性のメカニズムは、主要な生体高分子の一つである脂肪の慢性的な増加に対する細胞および分子の反応を概説したものである。しかし、インスリン抵抗性の発症には、栄養過多に対する他の細胞自律的な反応も関与していると考えられる。その1つが、小胞体ストレスによって活性化されるアンフォールドタンパク質応答(UPR)である。
UPRは、細胞がタンパク質合成の需要と能力を一致させるための、複雑でエレガントな、よく理解されているメカニズムである(191)。UPRの3つの枝は、3つの小胞体膜タンパク質によって制御されている。PKR-like eukaryotic initiation factor 2α kinase (PERK)、inositol-requiring enzyme 1 (IRE1)、activating transcription factor 6 (ATF6)である(331)。ストレスのない正常な小胞体では、これら3つのタンパク質は、シャペロンBiP/GRP78がPERK、IRE1,ATF6に結合することで不活性に保たれている。ミスフォールドしたタンパク質が蓄積すると、BiPが解離し、UPRが活性化される(331)。小胞体ストレス応答の下流には、NF-κBやc-Jun NH2-terminal kinase (JNK)などが存在し、代謝調節への関与が示唆されている。
2004年には、小胞体ストレスが生体内試験での肥満やインスリン抵抗性に直接関係するという初めての報告がなされた(605)。このブレイクスルー研究では、高脂肪食を与えたマウスと肥満/肥満マウスの両方で、リン酸化eIF2α、PERK、JNKなどの小胞体ストレスのマーカーが肝臓で強く増加していることが最初に指摘された(605)。小胞体ストレスを化学的に誘導すると、IRS1のJNK依存的なセリンリン酸化に関連して、IRS1レベルでのインスリンシグナルが損なわれた(465, 605)。これらの研究は、肥満のインスリン抵抗性マウスでJNK活性が上昇しているという同グループの以前の報告(318)と合わせて、小胞体ストレスがJNKによるIRS1のリン酸化を抑制することでインスリン抵抗性を誘導するという仮説を推し進めた。不思議なことに、最初の研究(318)では、全身のJnkを欠失させると、肥満マウスの肝臓におけるINSRシグナルが改善されたが、後の研究(605)では、JNKの活性化はINSRシグナルの障害とは関連しなかった。肝のインスリン抵抗性におけるINSRの直接的な欠損の役割が知られていることから(116, 722)、JNKがIRS1のレベルでインスリンシグナルを阻害するという仮説は、INSRの阻害を説明するには他の欠損が存在しなければならないことを意味する。さらに、肝臓特異的なJnk-/-マウスは、肝脂肪症と肝インスリン抵抗性になりやすいが、なりにくいわけではないことが判明し、肝細胞のインスリン抵抗性におけるJNK活性化の役割が疑問視されている(709)。
肥満においてUPRが活性化されているという最初の観察結果は、マウスの肝臓(572, 792)、ヒトの肝臓(284, 445)および脂肪組織(70, 284, 757)で繰り返し確認されている。小胞体ストレスとインスリン抵抗性の関係を説明するメカニズム研究は、主に、栄養過多時に小胞体ストレスが発生する論理的な候補組織である肝臓で行われてきた。
肝インスリン抵抗性における小胞体ストレスの役割を研究するための主要なモデルシステムは、X-box binding protein 1 (XBP1)に欠陥のあるマウスである。XBP1のmRNAスプライス変異であるXBP1sは、転写因子であり、UPRの重要なエフェクターである(237, 938)。全身のXbp1+/-マウスでは、小胞体ストレス(PERKやJNKの活性化など)が見られ、糖不耐性やインスリン作用の低下と関連しており、その影響はINSRの近位部にまで及んでいることが明らかになっている(605)。しかし、Xbp1+/-マウスは、野生型の対照マウスよりも体重が増加するため、異所性脂質によるインスリン抵抗性など、他の肥満関連メカニズムが表現型に寄与している可能性がある(605)。実際、XBP1がde novo lipogenic transcriptional programを活性化するという2008年の報告から、小胞体ストレスとインスリン抵抗性の関係を示す別の仮説が浮上した(460)。肝臓特異的にXbp1を欠失させたマウスでは、脂肪生成を促す高フラクトース食を摂取すると、特に顕著なDNLの欠損が見られた(460)。しかし、Xbp1を肝臓特異的に欠失させると、予想に反してXbp1の上流制御因子であるIRE1のレベルが上昇し、JNKの活性化が促進された(375)。このモデルでは、BiP/GRP78レベルやeIF2αのリン酸化など、その他の小胞体ストレスの兆候も観察された(375)。これらの観察結果を総合すると、肝ERストレスが亢進し、肝脂質が低下するという新しい表現型のマウスが生まれた。高インスリン血症-高血糖クランプ試験を行ったところ、フルクトースを与えた肝臓特異的なXbp1-/-マウスは、肝のインスリン感受性が上昇した(375)。これらのデータは、UPR活性化とJNK活性では肝のインスリン抵抗性を引き起こすには不十分であることを示しており、また、小胞体ストレス関連のインスリン抵抗性の多くのモデルにおいて、XBP1が介在する脂質生成が一因である可能性を示唆しており、小胞体ストレスと脂質誘発性インスリン抵抗性のDAG/nPKCモデルを一体化させている(図18A)。興味深いことに、高脂肪食マウスにXbp1をアデノウイルスで過剰発現させると、肝脂肪症、PKCεの活性化、肝インスリン抵抗性が抑制されたことから、XBP1と脂肪生成の関係は、これまで考えられていたよりも複雑である可能性が示された(314)。同様に、XBP1をリン酸化して活性化するIKKβを活性化すると、複数のマウスモデルにおいて、食事によって誘発された肝脂肪症と肝インスリン抵抗性が回復した(490)。XBP1の阻害と活性化の両方が肝のインスリン作用を改善するという知見は、簡単には説明できない。
図18 栄養素ストレスとインスリン抵抗性
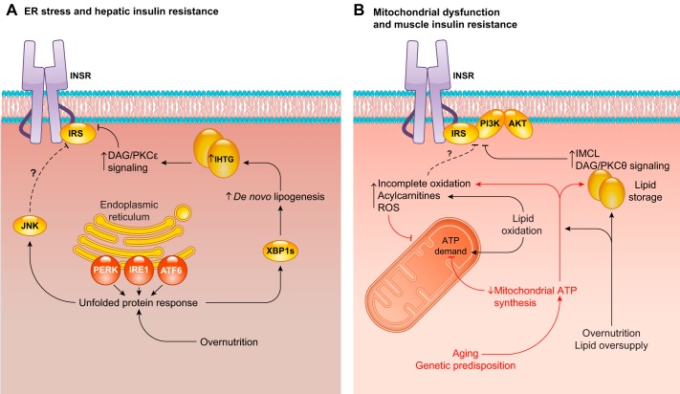
A: 小胞体(ER)ストレスは、アンフォールドタンパク質反応(UPR)として表れ、肝臓のインスリン抵抗性の複数のモデルで活性化される。小胞体の内腔にミスフォールドしたタンパク質が蓄積すると、UPRが始まり、PKR-like eukaryotic initiation factor 2α kinase (PERK)、inositol-requiring enzyme 1 (IRE1)、activating transcription factor 6 (ATF6)などが活性化される。UPRの主要なエフェクターには、c-Jun NH2-terminal kinase (JNK)とX-box binding protein 1 (XBP1s)のmRNAスプライス変異がある。JNKは、近位のインスリンシグナルを直接阻害する可能性があるが、これについては議論の余地がある。XBP1sは、de novo脂質生成プログラムを転写的に活性化し、肝脂肪症を促進し、その結果、ジアシルグリセロール(DAG)/プロテインキナーゼC(PKC)εシグナルを介して、肝インスリン抵抗性を促進すると考えられる。B: 筋のインスリン抵抗性はミトコンドリアの機能障害と関連しているが、原因と結果の関係は明確に定義されていない。高齢者や2型糖尿病(2型糖尿病)の両親から生まれたインスリン抵抗性の若い子孫は、骨格筋のミトコンドリアのATP合成量が減少している。これは、安静時のミトコンドリアのATP合成速度が最大値以下であるため、ATP需要が減少していることを反映していると考えられる。このような基質酸化の低下は、脂質の蓄積を促進し、細胞内脂質(IMCL)を増加させる。IMCLの蓄積は、DAG/PKCθ軸の活性化を介して近位のインスリンシグナルを阻害し、これらの人々に観察されるインスリン刺激によるグルコース取り込みの低下の原因となっていると考えられる。あるいは、慢性的な栄養過多や脂質の過剰供給により、ミトコンドリアの脂肪酸酸化能力が増大する。しかし、ATPの需要は比較的柔軟性に欠けるため、この増加量は供給量には及ばず、IMCLが蓄積し、不完全な脂肪酸酸化の割合が増加することになる。不完全な脂肪酸酸化は、アシルカルニチン種と活性酸素種(ROS)を生成する。アシルカルニチンは定義されていないメカニズムでインスリンシグナルを障害すると考えられており、活性酸素はミトコンドリア機能の障害を含む幅広い細胞作用を有する。活性酸素によるミトコンドリア障害は、これらの影響を悪化させ、IMCLの蓄積と活性酸素の生成をさらに促進すると考えられる。
全体として、小胞体ストレスそのものが、ヒト肝のインスリン抵抗性における重要な主要な欠陥であるかどうかを解明することは困難であった。ERストレスのいくつかのマーカー(eIF2αリン酸化、CHOP誘導)は、ヒト肝のインスリン抵抗性と関連しているが、ERストレスの最も機構的に重要な指標(XBP1スプライシング、JNKリン酸化)は関連していない(445)。さらに、小胞体ストレスと脂質代謝の相互作用は、小胞体ストレスの多くのモデルが異所性脂質の蓄積のモデルでもあることを意味する。異所性脂質はUPRの活性化によってインスリン抵抗性を誘導するのか、それとも小胞体ストレスは異所性脂質を介した一次障害の二次的な結果に過ぎないのか?脂質の過剰蓄積、特に飽和脂肪酸の過剰供給が小胞体ストレスを誘発するという証拠があるが(251)、肝特異的Xbp1-/-マウスにおける肝インスリン感受性の増加、肝脂質蓄積の減少、小胞体ストレスの増加という三要素は、結局のところ、小胞体ストレスが肝インスリン抵抗性を誘発するには異所性脂質の蓄積が必要であることを示している。
しかし、小胞体ストレスは肝性インスリン抵抗性の主要な欠陥ではないかもしれないが、主要な脂肪毒性メカニズムを悪化させる可能性は十分にある。この仮説を裏付けるように、BiP/GRP78の過剰発現によってob/obマウスのERストレスを改善すると、肝脂肪症が減少し、肝のインスリン感受性が増加する(382)。さらに、小胞体ストレスはlipin-2(PAからDAGの合成を触媒する)の発現を増加させることが示されている。lipin-2の過剰発現は、DAG/PKCε軸を活性化することで肝インスリンシグナルを損なうが、高脂肪食マウスでlipin-2をノックダウンすると、肝インスリンシグナルが改善される(708)。
白色脂肪組織分の小胞体ストレスは、肝の小胞体ストレスに比べてあまり研究されていないが、白色脂肪組織分の小胞体ストレスと脂肪分解の亢進との関連を示した文献は少ないが、興味をそそられる。小胞体関連トリグリセリド合成酵素DGAT1は、遊離脂肪酸をトリグリセリドに再エステル化する役割を果たしており、脂肪組織特異的Dgat1-/-マウスでは、寒冷曝露や絶食時にWAT癌の小胞体ストレスが生じる(139)。このようなUPRの活性化は、脂肪組織のマクロファージの浸潤と関連しており、小胞体ストレスと白色脂肪組織における炎症との関連が指摘されている(139, 331)。小胞体ストレスによる脂肪分解促進効果は、PKAの活性化とそれに伴うペリリピンのリン酸化によって媒介される可能性がある(76, 187, 958)。脂肪細胞のインスリン作用における脂肪分解の重要性を考えると、小胞体ストレスが全体的な脂肪分解のトーンにどのように寄与するかを理解することは大きな意味を持つ。たとえ近位のインスリンシグナルが無傷であっても、インスリンがcAMP-媒介性脂肪分解を抑制する能力は、小胞体ストレスによるcAMP-媒介性脂肪分解の活性化によって阻害され、機能的なインスリン抵抗性となる可能性がある。
筋のインスリン抵抗性における小胞体ストレスの役割はまだ不明である。2004年に行われた肝臓の小胞体ストレスとインスリン抵抗性に関するブレイクスルー研究では、高脂肪食マウスの骨格筋では小胞体ストレスマーカーが誘導されないことが報告された(605)。しかし、他の研究者たちは、ヒトの筋肉のインスリン抵抗性において、小胞体ストレスのすべてのマーカー(IRE1αやJNKの活性化)ではなく、いくつかのマーカー(BiPやCHOPの発現)の軽度の増加を観察した(416)。また、ヒトで6週間の高脂肪食を与えると、細胞内脂質の増加を伴う耐糖能異常が引き起こされるにもかかわらず、骨格筋ではいずれのERストレスマーカー(BiP、CHOP、IRE1α、PERKの発現)も誘導されないという報告もある(183)。さらに、構成的に活性なJNK変異を骨格筋に特異的に発現させても、JNKを骨格筋特異的にアブレーションしても、筋のインスリン作用やグルコースホメオスタシスは変化しなかった(606a)。
B. ミトコンドリアのエネルギー、酸化ストレス、インスリン抵抗性
慢性的なカロリー過多で栄養ストレスを受けるのは、小胞体だけではない。ここでは、栄養素、特に脂質の過剰供給に伴うミトコンドリアの酸化ストレスの発生とその意義について考えてみる。大まかに言うと、心筋細胞に運ばれた脂肪酸の主な運命は、酸化と貯蔵の2つである。過剰な脂質の貯蔵が心筋細胞のインスリン抵抗性に関係しているという証拠(第5章参照)により、コインの反対側である脂質の酸化についての研究が盛んに行われるようになった(561)。この分野の初期の仮説では、IMCLの蓄積とその結果としてのインスリン抵抗性は、脂質の酸化が不適切に低下することによって引き起こされるのではないかと考えられていた。実際、高齢者、2型糖尿病の両親を持つ痩せた若い子供、糖尿病予備力など、インスリン抵抗性のヒト集団において、ミトコンドリア活性の低下(すなわち、31P-MRSで測定されるATP合成量)を示す明確な証拠があり、2型糖尿病の根底にある筋細胞のエネルギー状態を知る上での潜在的な病因となっている(219, 503, 635, 636)。また、インスリン抵抗性のヒトでは、骨格筋ミトコンドリアの構造的損傷と総含有量の減少が見られるという強い証拠がある(148, 396, 552, 614)。このことは、必ずしもミトコンドリアの機能不全を意味するものではない。安静時のミトコンドリアは、通常、最大の酸化能力を発揮していない(699)。さらに、ミトコンドリアの活動は、基質の利用可能性の変化ではなく、主に細胞のATP需要によって決定される(326)。しかし、そのメカニズムにかかわらず、インスリン抵抗性骨格筋のミトコンドリア活性の低下は、全身のカロリー需要を大きく変化させ、細胞内の栄養分の過剰供給や脂質によるインスリン抵抗性を促進する可能性がある。
近年、ヒトの遺伝子研究により、脂質の貯蔵と利用というパラダイムに合致した形で、2型糖尿病とミトコンドリアのエネルギーを結びつけるメカニズムを持つ代謝リスク対立遺伝子が同定された。そのような遺伝子の一つであるSLC16A11は、小胞体と細胞膜に局在するモノカルボン酸(ピルビン酸、乳酸)トランスポーターであり、特に肝臓で高発現している(706,872a)。SLC16A11のリスクハプロタイプは、メキシコ系の人々に特に多く見られ(対立遺伝子の頻度は約30%)メキシコ系の人々の2型糖尿病リスク増加の約20%を占めると推定されている(872a)。SLC16A11の2型糖尿病リスクハプロタイプに含まれるいくつかの変異は、SLC16A11の発現を低下させ、細胞膜への局在性を損なう。siRNAでSLC16A11をノックダウンした肝細胞では、アシルカルニチン、DAG、TAGの蓄積が見られ、FAOの低下が示唆された。これらの研究から、2型糖尿病リスクハプロタイプのSLC16A11変異は、ミトコンドリアの脂肪酸酸化障害に起因する細胞内脂質の蓄積を促進することで、糖尿病リスクを高めることが示唆された。もう一つの興味深いリスク対立遺伝子は、N-アセチルトランスフェラーゼ2(NAT2)遺伝子にあり、インスリン抵抗性に関連する遺伝子を探索するゲノムワイド関連解析で明らかになった(414)。マウスのオーソログであるNat1を全身で欠失させたマウスは、基礎代謝量の低下、エネルギー消費量の減少、白色脂肪組織、褐色脂肪組織、肝臓、心臓、骨格筋など様々な組織でのミトコンドリア機能障害を示した(114, 130)。その結果、これらのマウスは、HFDを摂取すると、肝臓ではDAGによるPKCεの活性化、筋肉ではPKCθの活性化を伴う、脂質による肝臓および筋肉のインスリン抵抗性を引き起こす素地があった(114)。
しかし、ミトコンドリア活性とインスリン作用の関係は、インスリン抵抗性の心筋細胞が脂質を酸化するのではなく貯蔵するという、上記の単純なモデルよりも複雑である。興味深いことに、高脂肪食を数週間与えると、げっ歯類では、ex vivoでの脂肪酸酸化能力の増加と並行して、骨格筋のインスリン抵抗性が発現する(427, 548, 853)。これは、利用可能な脂質の増加を十分に補えず、IMCLの蓄積とインスリン抵抗性につながる適応反応を反映していると考えられる(549)。実際、薬理学的なAMPK活性化やアセチル-CoAカルボキシラーゼ2欠失によってFAOを強力に活性化しても、HFD誘発性の筋肉インスリン抵抗性からマウスを保護するには不十分であり、ミトコンドリアエネルギーにおいて基質供給よりもATP要求が重要であることが強調されている(320, 596)。また、脂質の酸化障害が筋のインスリン抵抗性を引き起こすという当初の仮説とは逆に、相対的なβ酸化フラックスの増加自体が骨格筋のインスリン抵抗性を促進するという証拠もある。この仮説は、IMCLが蓄積しているにもかかわらず、β酸化フラックスをブロックすると筋のインスリン感受性が向上するといういくつかのモデルによって支持されている(230, 287, 427)。しかし、FAOの増加は、構成的にグルコースを酸化するPdk2/4-/-マウスの筋インスリン抵抗性で示されるように、脂質誘導性の筋インスリン抵抗性に必要でないことは確かである(669)。
どのようにしてβ酸化フラックスが筋インスリン作用を損なうのか?もう一つの可能性は、活性酸素である。活性酸素は、基質の酸化がATP需要を上回ったとき(すなわち、栄養過多の状態)に「電子放出弁」の役割を果たす(37, 235)。すべての活性酸素はスーパーオキシド(O2–)に由来するが、主要な生理活性酸素は過酸化白色脂肪組織素(H2O2)であると考えられており、ミトコンドリアの酸化還元状態を伝えるセカンドメッセンジャーとして機能している(235)。高脂肪食を与えると、3日以内にミトコンドリアのH2O2産生量が増加し、その効果は慢性脂肪食マウスでも持続し、この酸化された酸化還元状態は、還元型グルタチオンと酸化型グルタチオンの比率(GSH/GSSG比)の低下として、細胞全体で検出可能である(22)。さらに、肥満のヒトの骨格筋では、ミトコンドリアのH2O2生成量が増加し、GSH/GSSG比が低下している(22)。多くのプロテインキナーゼやホスファターゼの活性は、システインチオールの酸化還元状態によって制御されていることから、酸化された細胞環境は、インスリン作用の正常なネガティブフィードバックを特徴づけるセリン/スレオニンリン酸化現象を促進するのではないかと提案されている(235)。例えば、JNKのようなストレスキナーゼが活性化されることが観察されている(570)。重要なことは、細胞の酸化還元状態は非常に動的であるということである。H2O2産生の増加とインスリン作用の低下を関連付けるモデルでは、慢性的な栄養過多の状態では、食後に細胞環境を酸化させる傾向が、相対的な絶食時に細胞環境を低下させる傾向を上回り、通常は自己修正機能を持つこのシステムがホメオスタシスを回復できなくなるとしている(235)。例えば、インスリン受容体が活性化されると、NADPHオキシダーゼ4(NOX4)による局所的なH2O2の産生が増加し、局所のホスファターゼ(PTP1B、PTENなど)が不活性化され、近位のインスリンシグナルが増幅される(37, 515, 920)。このことは、活性酸素消去物質であるグルタチオンペルオキシダーゼ1を欠損したマウスの表現型が、HFDによるインスリン抵抗性から保護されたことを説明していると思われる(497)。
ミトコンドリアの活性酸素生成を阻害することで、筋肉のインスリン抵抗性を防ぐことができるという仮説を裏付ける実験的証拠がある。ミトコンドリアのH2O2を消去することは、薬理学的な手段、またはミトコンドリアにカタラーゼを遺伝子導入したマウス(MCATマウス)によって、HFD誘発性の筋肉インスリン抵抗性から保護される(22, 338, 747)。注目すべきは、MCATマウスは加齢による筋肉インスリン抵抗性からも保護され、加齢した野生型マウスと比較して、ミトコンドリア活性が維持され、DAG/PKCθ軸の活性化が減少したことである(463)。しかし、酸化ストレスを標的とした治療の可能性については、依然として議論がある。抗酸化物質の補給に関する研究では、相反する結果が得られており(177,549)ヒトの抗酸化物質補給に関する78件の無作為化臨床試験のシステマティックレビューでは、死亡率に対する効果は認められなかった(63)。
これらの研究結果を総合すると、慢性的な脂質の過剰供給に直面した心筋細胞は、代謝的に柔軟性を失い、相対的な脂質利用率を高めることで代償しようとするという統一モデルが示唆される。しかし、細胞内のATP需要は基質供給に見合うだけ増加しないため、脂質利用率は制限され、IMCLの沈着を防ぐには不十分である。これと並行して、相対的なFAOの増加は、ミトコンドリアにダメージを与える活性酸素の生成を促し、インスリン抵抗性筋のミトコンドリア活性の低下を誘発するのである。このようなミトコンドリア活性の低下は、正のエネルギーバランスへの傾向を悪化させ、IMCLの沈着と筋肉のインスリン抵抗性をさらに助長する(図18B)。興味深いことに、いくつかのトランスジェニックマウスモデルでは、重度のミトコンドリア機能障害を誘発しながらも、筋グルコース取り込みの改善が認められ、このモデルに疑問を投げかけている(327)。これらのモデルには、ミトコンドリアの転写因子Tfamを欠損させたマウス、ミトコンドリアのアポトーシス誘導因子Aifを欠損させたマウス、転写共役因子PGC-1αとPGC-1βを欠損させたマウスなどがある(656, 913, 945)。このようなモデルでは、ミトコンドリア機能の低下が細胞内脂質の蓄積を招き、その結果、筋のインスリン抵抗性を引き起こすという仮説を再評価しなければならない。一方、前述のトランスジェニックモデルに見られるような重度のミトコンドリア機能障害では、嫌気性解糖の増加、脂肪酸化の減少、ADP:ATP比の増加によるAMPK依存性グルコース輸送の活性化などが起こり、明らかにインスリン感受性が増加すると考えられる。ミトコンドリア機能として評価されるパラメータが、生体内試験 ATP合成機能、総酸化能、ミトコンドリア密度、またはその他の読み出しであるかどうかの違いも、導き出される結論に影響を与える可能性がある(42)。
VII. インスリン抵抗性の統合された生理学的メカニズム
A. インスリン抵抗性におけるマクロファージと脂肪細胞の相互作用と炎症性シグナル伝達
ヒトの肥満は、脂肪組織の膨張を特徴とする。この効果には、脂肪細胞の過形成と肥大化の両方が寄与している。しかし、この膨張は恒常的なストレスであり、脂肪細胞の細胞死の増加と関連している(562)。ストレスを受けた脂肪細胞からの走化性シグナルは、細胞の先駆者である骨髄由来のマクロファージをリクルートする(892, 928)。これらの脂肪組織マクロファージ(ATM)は、死んだ脂肪細胞の周りに「王冠のような構造」を作り、オートクライン、パラクライン、エンドクライン効果を持つサイトカインを分泌する(562, 594)。肥満で刺激されたATMは、細菌感染などで活性化されるマクロファージとは異なる、特徴的な活性化パターンを示す(86, 431)。重要なことは、急性炎症能力が低下したマウスを用いた研究により、脂肪組織の急性炎症は、適切な組織のリモデリングとWAT癌の栄養貯蔵に不可欠であり、この能力が失われると、異所性脂質の貯蔵が増加し、食事誘導性のインスリン抵抗性が悪化することが明らかになったことである(896)。メタボリックに活性化したATMは、死んだ脂肪細胞を除去するためにリソソームを外に出し、過剰な脂肪酸を吸収するためにPPARγ駆動の転写プログラムを用いている(154, 431)。つまり、インスリン抵抗性に関連する不適応な状態は、慢性炎症なのである。ヒトの肥満は、代謝のホメオスタシスを変化させる慢性的な炎症状態であることが、現在では十分に確立されているが(424, 467)、炎症がさまざまな組織でインスリン抵抗性を引き起こすメカニズムや、インスリン抵抗性や2型糖尿病の発症におけるこれらのプロセスの重要性については、依然として活発な研究が行われている(446, 717)。ここでは、炎症とインスリン抵抗性との間に提案されているメカニズムのうち、主にWAT癌に焦点を当てる。
炎症によるインスリン抵抗性の基本的なモデルは、マクロファージの活性化に続いて、マクロファージがパラクリンや内分泌因子を産生し、脂肪細胞や肝細胞などの標的細胞にインスリン抵抗性を誘導するという「2ヒット」モデルである(594)。この2つのプロセスはよく研究されており、複雑である。
炎症を起こしたWAT癌では、WAT癌の常在マクロファージと単球由来のマクロファージの両方が活性化される。しかし、他の複数の免疫細胞タイプが肥満脂肪組織の炎症環境に寄与している。実際、高脂肪食摂取時に最初に脂肪組織に浸潤する炎症細胞はマクロファージではなく、好中球である(212, 823)。好中球は、高脂肪食摂取時にATMをリクルートして活性化するのに重要な役割を果たしていると考えられる(783)。脂肪B2リンパ球もまた、肥満の脂肪組織に蓄積され、その減少は、ATMの活性化を阻害することによってHFD誘発のインスリン抵抗性を軽減する(937)。CCR2やMCP-1を遺伝的または薬理学的に阻害すると、肥満に伴うATMの浸潤が減少し、インスリン感受性が改善することが知られているが(383, 891)、Ccr2-/-マウスを用いた別の研究では、ATMの浸潤は変化せず、耐糖能が悪化することが予想されている(351)。メタボリックに活性化されたATMは、従来のM1-炎症性/M2-抗炎症性のパラダイムでは容易に分類できず、むしろパルミチン酸の曝露によって試験管内試験で再現できる第3の極性状態を採用しているようである(431)。また、ATMの活性化は、内臓脂肪層に存在するナチュラルキラー(NK)細胞が脂肪細胞のストレスを感知し、インターフェロン-γ(IFN-γ)やTNF-αを介してマクロファージの活性化を促進することによっても引き起こされる可能性がある(461, 895)。脂肪細胞とNK細胞の相互作用を遮断したり、NK細胞を枯渇させたり、IFN-γシグナルを遮断したりすることで、HFDによるATMの活性化や耐糖能異常からマウスを守ることができた(461, 601, 895)。上述したように、代謝的に活性化されたATMが純粋に有害であるという特徴は単純化しすぎであり(927)、脂質の貯蔵や死んだ脂肪細胞の除去といった代謝的に有益なATM機能も肥満では亢進している(154, 929)。
結局のところ、ATMの活性化(”ファーストヒット”)がインスリン抵抗性を促進するメカニズムは、炎症性サイトカインの生成という “セカンドヒット “を必要とすると考えられている(594)。インスリン抵抗性に最もよく関与しているサイトカインはTNF-αとインターロイキン(IL)-1βであるが、ロイコトリエンB4やガレクチン-3を含む他のサイトカインも最近の研究対象となっている(332, 336, 364, 485, 486, 894)。TNF-αは、げっ歯類およびヒトの肥満脂肪組織で増加していることが明らかになった最初の炎症性サイトカインである(332, 336)。TNF-αを中和すると、肥満したfa/faラットの全身のインスリン刺激によるグルコース取り込みが改善されたことから、肥満に伴うインスリン抵抗性にTNF-αが関与していることが示唆された(336)。さらに、Tnfa-/-マウスはHFD誘発のインスリン抵抗性から保護され、TNF-α受容体の除去は肥満/obマウスをインスリン抵抗性から部分的に保護した(865)。しかし、TNF-αの作用を局所的に阻害すると耐糖能が低下するという最近の結果から、TNF-αとインスリン抵抗性との関係は、当初考えられていたよりも複雑であることがわかった(896)。
TNF-αなどのサイトカインとインスリン抵抗性を結びつける有力な仮説は、インスリン標的細胞におけるサイトカイン受容体の活性化が、インスリン作用を直接的または間接的に阻害するシグナル伝達経路を活性化するというものであった。In vitroでは、TNF-αを含むいくつかのサイトカインは、IRK活性を直接阻害することによってインスリン抵抗性を誘導するが(333-335, 485)、阻害効果を得るために必要な用量は、インスリン抵抗性の被験者の血漿中で測定された用量よりもしばしば桁違いに高い(332, 334, 335)。パラクライン効果(マクロファージから脂肪細胞、クッパー細胞から肝細胞)が働いている可能性は高いが、生体内でパラクラインシグナルを定量化することは困難である。
サイトカイン受容体の活性化とインスリンシグナルの障害とを結びつけるメカニズム回路を解明しようとする努力は、サイトカインシグナルと小胞体ストレスの両方に作用する多面的なエフェクターにほぼ集約されている。JNKである。JNKは、複雑な炎症性転写プログラムを誘導するだけでなく、IRS1を直接リン酸化する。JNK活性は、肥満したインスリン抵抗性の肝臓、白色脂肪組織、骨格筋で上昇し(318)、肝臓では、HFD開始後3日目にはJNK活性化が見られる(721)。Jnk1-/-マウスは、HFDによって誘発された肥満とインスリン抵抗性から保護されていたが、エネルギー消費量が増加していたため、中核体温が上昇していたことから、インスリン感受性の表現型がJNKによるインスリンシグナル伝達経路への直接的な影響によるものではないと考えられた(318)。Jnkの阻害とエネルギー消費量の増加との関連性は、ドミナントネガティブJNKを用いて脂肪細胞やマクロファージにおけるJNK活性を阻害した実験(849)や、JNK特異的アンチセンスオリゴヌクレオチドを用いた実験(775)でも裏付けられている。
しかし、組織特異的なJnkノックアウトマウスは、JNKの生物学についてより微妙な洞察を与え、Jnk欠失によるインスリン感作作用が、場合によっては体組成への影響とは切り離されることを示唆している(711)。脂肪細胞特異的にJnkを欠失させると、HFDを与えたマウスの体重増加を防ぐことはできなかったが、肝のインスリン感受性は維持され、肝の脂質蓄積からの保護に関連していた(710)。マクロファージにおけるJnk1/Jnk2の欠損(ΦKOマウス)が、高脂肪食マウスにおけるATMの浸潤と全身のインスリン抵抗性を防ぐという観察結果は、JNKがマクロファージにおける炎症誘発状態の確立に重要な役割を果たしていることを示唆しており、HFD誘発のインスリン抵抗性におけるマクロファージの活性化の重要性をさらに立証した(302)。しかし、ΦKOマウスは、通常の餌を与えても全身のインスリン感受性が同様に改善した(302)。組織のJNK活性の上昇とATMの浸潤は高脂肪食の特徴であるため(302, 318)、ΦKOマウスの代謝表現型が通常のチャウ食でも高脂肪食でも同じように顕著であった理由は不明である。しかし、これらのデータの一つの解釈として、脂肪細胞やマクロファージにおけるJNKの活性化が、肝臓などの遠方の組織におけるHFD誘発インスリン抵抗性に寄与しているというものがある。
骨格筋では、JNKの活性化は肥満に伴うインスリン抵抗性には十分ではなく(606a)、骨格筋特異的なJnkの欠失が肥満に伴うインスリン抵抗性から保護するかどうかについては報告が分かれている(606a, 712)。筋特異的Jnk欠失でHFD誘発筋インスリン抵抗性からの保護が観察された1つの研究では、筋LpLの発現も低下していたことから、インスリン感受性表現型は、JNKが近位インスリンシグナルを阻害する直接的な作用ではなく、脂肪毒性からの保護に起因する可能性があると考えられている(712)。さらに、肝臓特異的なJnkの欠失は、マウスを肝性インスリン抵抗性から保護するのではなく、肝性脂肪症と耐糖能異常を促進する(709)。さらに、JNKの活性化が細胞内のインスリンシグナルを障害すると考えられている正統的なメカニズムであるIRS1のSer307リン酸化は、生体内のインスリン抵抗性を媒介するとは考えられない。IRS1 Ser307Alaをノックインしたマウスでは、インスリン作用が維持されるのではなく、悪化する(167)。リン酸化型IRS1 Ser307Asp変異でも、インスリンシグナルは損なわれない(888)。JNKによるインスリン抵抗性の代替メカニズムについては、転写メカニズムを含めて、さらに研究が必要である。以上のことから、JNKとインスリン抵抗性との関連は、筋肉や肝臓などの標的組織における直接的なインスリンシグナルの遮断ではなく、おそらくマクロファージ-脂肪細胞-肝細胞軸が関与する間接的な影響であると考えられる。
実際、サイトカインによる脂肪分解が炎症とインスリン抵抗性の関係を媒介するという新しいパラダイムが提唱されている。HFDを与えたΦKOマウスは、HFDを与えた野生型と比較して、高インスリン血症-高血糖クランプ中のパルミチン酸とグリセロールの代謝が低下し(すなわち、脂肪分解の低下)肝臓でのグルコース産生の抑制が改善された(620)。TNF-αは、脂肪細胞の脂質滴へのリパーゼのアクセスを制御すると考えられている脂質滴タンパク質であるペリリピンおよび/または脂肪特異的タンパク質27(FSP27)の発現を低下させることにより、脂肪分解を促進する可能性がある(455, 681)。さらに、肥満/obマウスの血漿中のNEFA濃度の上昇は、TNF-αの喪失によって痩せたマウスのレベルまで回復した(865)。しかし、重要なことは、慢性肥満における白色脂肪組織脂肪酸代謝の増加は、サイトカインを介した脂肪分解だけでなく、死んだ脂肪細胞からの脂肪酸流出にも起因していると考えられることである。この後者のメカニズムの重要性は、代謝的に活性化されたATMを機能的に欠く、全身および骨髄特異的なNox2-/-マウスの研究によって強調された(154)。これらのマウスは死んだ脂肪細胞を蓄積し、深刻な肝脂肪症とインスリン抵抗性を発症した(154)。全体として、脂肪分解は、栄養ストレスによるものであれ、炎症によるものであれ、骨格筋や肝臓への脂質の供給を増加させ、脂質によるインスリン抵抗性の経路(例えば、DAG/nPKC軸)を活性化し、また、ピルビン酸カルボキシラーゼのアセチルCoA活性化を介して肝の糖新生を促進することにより、インスリン抵抗性を引き起こす可能性がある(620)。この仮説は、脂質誘導性インスリン抵抗性と炎症誘導性インスリン抵抗性というパラダイムを統合し、脂質誘導性インスリン抵抗性の細胞自律的な欠陥を制御する統合的な生理学的メカニズムを指摘している。
炎症は2型糖尿病の一次障害なのか、それともむしろ増悪因子なのか?いくつかの証拠から、脂肪組織の炎症は脂肪のインスリン抵抗性には必要ないことが示唆されている。例えば、脂肪組織のインスリン抵抗性は、げっ歯類では高脂肪食を1週間与えただけで検出されるが(115, 722)、脂肪細胞の死とATMの浸潤は、HFDを4週間与えても最小限で、HFDを12週間与えないと顕著にはならない(582, 807)。ゲノム解析による証拠も、2型糖尿病を炎症性疾患として分類することを否定している。疾患に関連する一塩基多型(SNP)を細胞型特異的なエピジェネティック制御活性と関連付けて大規模に解析すると、調べたすべての既知の自己免疫疾患および炎症性疾患が強固にクラスタリングされることが明らかになった(226)。2型糖尿病のSNPは、リンパ系細胞や骨髄系細胞では明らかにこのような濃縮は見られず、2型糖尿病リスクの強い遺伝性(599)は、マクロファージなどの炎症細胞の遺伝的影響を介していない可能性が高いことが示された(226)。脂肪組織の炎症もまた、インスリン抵抗性には必要ではない。部分的および完全な脂肪ジストロフィーの複数のモデルでは、ATMが有意に活性化されていなくても、重度のインスリン抵抗性が認められる(406, 642, 957)。この現象で特に興味深いのは、CIDEC(Cell Death-inducing DFFA-like effector c)としても知られるFSP27を欠損したマウスである(825, 957)。FSP27-/-マウスは、エネルギー貯蔵ストレス(高脂肪食、レプチン欠乏、褐色脂肪組織の欠如)にさらされても、脂肪層の拡大やATMの著しい浸潤は見られなかったが、それにもかかわらず、重篤な肝脂肪症を伴う重度の肝インスリン抵抗性を発症した(825, 957)。さらに、mTORC2の構成要素であるRictorを脂肪に特異的に欠失させて脂肪のインスリン抵抗性を誘導すると、MCP-1の発現が増加し、マクロファージが浸潤したことから、脂肪組織の炎症を引き起こすには脂肪のインスリン抵抗性が十分であることが示唆された(762)。これらの証拠を総合すると、脂肪組織の炎症は肥満に伴うインスリン抵抗性を伴ったり、悪化させたりすることはあっても、それが主要な欠陥ではない可能性が高い。
臨床で使用されているいくつかの抗炎症剤は、2型糖尿病に対する有効性が評価されている(271)。げっ歯類とヒトの炎症とインスリン作用の統合には種差があるため、マウスとヒトで得られたいくつかの抗炎症剤の結果が大きく異なっているのかもしれない。例えば、TNF-αアンタゴニストのエタネルセプトとインフリキシマブは、マウスでは良好な効果を示したものの、ヒトでは一貫したインスリン感受性向上効果を示していない(25, 56, 467, 795, 885)。サリチル酸塩のプロドラッグであるサルサレートは、無作為化されたヒト臨床試験において、中程度の安定した血糖降下作用を示しているが、サリチル酸塩がAMPKを直接活性化し、ミトコンドリアのアンカップリングを誘導する能力を持つことから、その抗高血糖作用には抗炎症メカニズムは不要であると考えられている(268-270, 303, 780)。肥満制御キナーゼIKKεとTBK1を阻害する抗炎症剤amlexanoxは、エネルギー消費量を増加させることにより、マウスの脂肪組織の炎症と肥満を回復させるが、これは、AMPKとエネルギーバランスの負の制御因子としてのTBK1の役割と一致する(687, 955)。2型糖尿病患者を対象とした小規模な無作為化二重盲検臨床試験では、12週間のアムレキサノックスの投与により、糖化ヘモグロビンがわずかに(0.5%未満)だが統計的に有意に減少した(600)。興味深いことに、アムレキサノックスに対する反応は、IHTGの低下と強く関連していた(600)。アムレキサノックスで認められた血糖値の改善に対する脂肪組織の炎症の抑制とエネルギー消費量の増加の相対的な寄与は不明であるが、アムレキサノックス治療に対する反応が良好であった被験者では、ベースラインでのC反応性タンパク質レベルが高い傾向にあった(600)。しかし、血清IL-6レベルも反応した人では上昇しており、このサイトカインが(一般的ではないが)頻繁に代謝を悪化させることとは相反する所見である(403)。この例は、炎症の代謝的に有益な効果と代謝的に有害な効果が複雑に絡み合っていることを示しており、他の抗炎症剤の抗糖尿病効果を検証する現在の取り組みを複雑にしている可能性がある(271, 467, 716)。
B. 循環分枝鎖アミノ酸とインスリン抵抗性
1969年、ヒトの血漿中の全アミノ酸を測定した結果、3種類の分岐鎖アミノ酸(BCAA:バリン、ロイシン、イソロイシン)の濃度は、痩せた対照群に比べて肥満群で上昇し、空腹時のインスリンと正の相関があることが明らかになった(228)。血漿中のアミノ酸とグルコースのホメオスタシスとの関係は複雑で、インスリン分泌、肝グルコース産生、末梢グルコース処分への影響がすべて確立されている(72, 238, 435)。21世紀に入ってからは、メタボロミクスの手法により、HOMA-IRスコアと循環中のBCAA濃度との間に強い関連性があることが確認され、BCAAとインスリン抵抗性との関連性への関心が再び高まっている(129, 516, 577, 918)。さらに、血漿中のBCAA濃度は、将来の2型糖尿病リスクを予測し(608, 880, 919)、BCAA濃度を増加させるゲノム変異は、メンデリアンランダム化研究において2型糖尿病と関連していた(502)。脂肪組織のBCAA分解酵素の発現は、げっ歯類やヒトの肥満モデルで一貫して減少しており、肥満に伴う循環BCAAの増加の原因と考えられている(312, 508, 751, 759)。
これらの関連性が確立されると、BCAAがインスリン感受性を積極的に調節しているのか、それとも受動的に反映しているのかが重要な問題となる。これは、現在、活発に研究されている分野であり、論争の的となっている(508, 576)。BCAAの補給だけでは、普通の飼料を与えたラットのインスリン抵抗性を誘導するには不十分であるが、高脂肪飼料を与えたラットのインスリン抵抗性には寄与する(577)。BCAAがインスリン作用を低下させるメカニズムとしては、主に2つが提案されている。1つは、ロイシン(733)を感知するmTORをBCAAが慢性的に活性化することで、IRS1レベルでのインスリンシグナル伝達に負のフィードバックを与えるものである(577)。mTORC1で活性化されたリボソームS6K1は、肥満や糖尿病のげっ歯類やヒトで慢性的に活性化されており(846)、IRSシグナルのネガティブレギュレーターとして知られている(339, 846, 859, 860, 948)。さらに、高脂肪食/BCAAを補給したラットのインスリン抵抗性は、mTORC1阻害剤であるラパマイシンによる治療で回復した(577)。しかし、BCAA/mtORC1軸の活性化は、インスリン感受性の改善とも関連している(例えば、運動やBCAAの補給)(508, 509, 954)。さらに、ミトコンドリアBCAAトランスアミナーゼ(BCATm)を欠損させたマウスでは、血漿中のBCAA濃度が大幅に上昇するが、HFD誘発の肥満やインスリン抵抗性からは保護される(758)。
BCAAによるインスリン抵抗性のもう一つのメカニズムとして提案されているのは、BCAAそのものが原因ではないということである。むしろ,生理活性部位はBCAAの異化産物(プロピオニルCoA,スクシニルCoA,分岐鎖ケト酸など)であると提案されている。ある仮説では、BCAAの代謝が増加すると、毒性のあるミトコンドリアのBCAA異化産物が生成され、その結果、ミトコンドリアの酸化代謝が損なわれて(508, 576)、ミトコンドリアストレスが誘発され、インスリン抵抗性との関連が指摘されているJNKなどのストレスキナーゼが活性化されると提案されている(576, 711)。この仮説を裏付ける証拠、特にヒトのインスリン抵抗性においてBCAAの異化作用がどのように変化するかについての正確な知識は、現在のところ限られている。最近、BCAA異化産物である3-ヒドロキシイソ酪酸(3-HIB、バリンの異化産物)が、心筋細胞の脂肪酸取り込みのパラクライン陽性制御因子として同定された(366)。3-HIBは心筋細胞から分泌され、内皮性脂肪酸輸送と心筋細胞への脂質取り込みを促進することが示された。3-HIBを添加した飲料白色脂肪組織を与えたマウスは、骨格筋のDAG/PKCθ軸の活性化と耐糖能異常を示した(366)。また、db/dbマウスや2型糖尿病のヒトの筋肉では3-HIBの濃度が上昇しており、生理的な関連性が示唆されている(366)。この興味深いメカニズムは、BCAAによるインスリン抵抗性と脂質によるインスリン抵抗性を結びつけるものである。
もう一つの仮説は、血漿中のBCAA濃度の上昇は、インスリン抵抗性の結果であるというものである。この仮説は、インスリン抵抗性のリスク対立遺伝子が血漿BCAA濃度の上昇と関連していたが、BCAA濃度の上昇のリスク対立遺伝子はHOMA-IRスコアの上昇とは関連していなかったというヒトの遺伝学的研究によって支持されている(516)。メカニズム的には、タンパク質合成に対するインスリンの同化作用に抵抗することで、循環BCAAレベルが上昇するという些細な説明が可能であるが、ロイシンターンオーバーに対するインスリンの抑制作用は、2型糖尿病患者と対照群で同等であることが報告されている(299, 507)。また、BCAAの異化作用が酸化ストレスを促進するのではなく、脂質の利用可能性や利用方法によって酸化ストレスが誘発され、BCAAの異化酵素が阻害されるのではないかと提案されている(508)。また、肥満に伴う血漿中BCAA濃度の上昇を促すBCAA異化酵素の脂肪組織での発現低下は、インスリン抵抗性による二次的なものである可能性もある(508)。さらに、BCAA異化作用は脂肪生成の基質となるアセチルCoAを供給するため(分化した3T3-L1脂肪細胞における脂肪生成アセチルCoAの30%はBCAA異化作用に由来すると推定されている)(281)、肥満による脂肪のBCAA異化酵素発現の低下は、脂肪組織における適切な栄養貯蔵を阻害し、肥満でBCAA異化能力が維持されている骨格筋などの組織における異所性の脂質沈着を促進する可能性がある。後者のメカニズムは、BCAAの代謝シグネチャーを、確立されたインスリン抵抗性の脂肪毒性メカニズムと結びつけるという点で魅力的ではあるが、直接的な実験的裏付けには乏しい。現在、多くの重要な問題が解決されていないため、この分野は今後の調査が必要である。
C. インスリン抵抗性におけるアディポカインとヘパトカイン
過去20年の間に新たに同定されたペプチドホルモンの爆発的な増加は、20世紀初頭の内分泌学の研究者がインスリン、性ステロイド、サイロキシン、その他の基本的なホルモンの発見で報われた時代を思い起こさせる(65)。今も昔も、新しいホルモンが発見されると、大きな興奮と治療への期待が寄せられる。しかし、多くの新しいホルモンは、病態生理的な意義や治療の可能性がまだ不明である。ここでは、最も研究が進んでいる新規ホルモンのうち、ヒトのインスリン抵抗性における役割を示す証拠に焦点を当てて、簡単に紹介する。
この総説では、基質の貯蔵と運搬を司る白色脂肪組織と肝臓の役割を強調してきた。しかし、これらの臓器は内分泌器官でもあり、いくつかのアディポカインやヘパトカインがヒトのインスリン抵抗性に関与していることがわかっている。ここでは、ヒトのインスリン抵抗性との関連性が特に強く示唆されている4つの循環メディエーター、レチノール結合タンパク質4(RBP4)アディポネクチン、フェツインA(FetA)FGF21に限定して議論する。
RBP4は 2005年に脂肪GLUT4過剰発現マウスと脂肪GLUT4非発現マウスで相互に転写調節を行う遺伝子として同定された(935)。RBP4は肝臓と白色脂肪組織両方で産生される分泌タンパク質であるが、マウスの循環するRBP4のほぼ全ては肝臓から産生されている(838)。マウスでは、Rbp4をトランスジェニックで過剰発現させたり、RBP4を慢性的に投与すると、全身のインスリン抵抗性が誘導され、Rbp4を欠失させるとインスリン作用が改善される(935)。また、3つの異なるヒトコホートにおいて、血清中のRBP4レベルは、肥満度(BMI)空腹時血漿インスリン、高インスリン血糖クランプ試験時のインスリン刺激による末梢グルコース取り込み障害と相関していた(278)。さらに、運動によるインスリン感作効果は、血清RBP4濃度の低下の程度と高い相関関係があった(278, 425)。ヒトの肥満における脂肪のRBP4発現の増加は、血清RBP4濃度の約2倍と関連している(277, 278)。RBP4が末梢のインスリン作用を低下させるメカニズムは明らかではない。提案されているメカニズムは、肝細胞の脂肪生成プログラムの直接的な活性化(924)と脂肪組織マクロファージ(ATM)の活性化(553, 584)である。後者のメカニズムは、RBP4で処理した樹状細胞を野生型マウスに移植し、ATMの浸潤を増加させ、耐糖能を悪化させた研究で裏付けられている(553)。このパラダイムは、RBP4と脂肪分解による末梢および肝臓のインスリン抵抗性を結びつけるものである。
アディポネクチンは、1995年に脂肪細胞特異的な分泌タンパク質として発見され、高分子量オリゴマーとして5-10μg/mlの濃度で循環している(51, 736)。この濃度は内分泌ホルモンとしては非常に高いが、アディポネクチンのオリゴマーの四分一構造から、そのモル濃度はわずか10〜30nMである(51)。肥満の人ではアディポネクチンの濃度は一貫して低いが、一般的には5〜10μg/mlの範囲に収まっている(337, 850)。血漿中のアディポネクチン濃度が100μg/ml以上に急性的に上昇すると、肝でのグルコース産生が減少するが、この効果の生理的意義は不明である(51)。メタボリックディジーズのヒトでは循環アディポネクチン濃度が適度に低下していることは再現性のある知見であり、脂肪組織機能障害のバイオマーカーとして広く用いられているが、これらの小さな変化がどの程度、全身のメタボリックディジーズに寄与しているかは不明である。例えば、2型糖尿病の両親の痩せた子孫は、インスリン抵抗性であるが、血漿アディポネクチンレベルは変化しない(636)。メカニズム的には、アディポネクチンのグルコース低下作用をAMPKの活性化によるものとする試み(932)は、AMPKの活性化がHGPの抑制には不十分であること(511)や、肝臓のAMPK活性化因子であるLKB1を欠損したマウスではアディポネクチンがHGPを抑制するという観察結果により、疑問視されている(324, 542)。
2011年には、アディポネクチン受容体AdipoR1およびAdipoR2が活性化されると、セラミダーゼ(すなわちセラミド分解)活性が誘導されるという報告がなされ、アディポネクチンとインスリン抵抗性の他のパラダイムとの間に興味深い関連性があることが明らかになった(324)。アディポネクチンによる急性期のHGP抑制を説明するには十分ではないと思われるが、慢性期に肝臓特異的なセラミダーゼを活性化すると、肝脂肪症やHFDによる肝インスリン抵抗性を防ぐのに十分である(922)。これらの知見は、肥満のob/ob対照マウスの2倍の体重にもかかわらず、約3倍の循環アディポネクチンの増加と正常なインスリン感受性を示すob/obアディポネクチントランスジェニックマウスのような、アディポネクチンとインスリン感受性を結びつける多くのモデルのメカニズムの一つの可能性を提供する(408)。驚くべきことに、アディポネクチンの急性誘導性欠失は、肝の脂質取り込みの増加および肝のセラミドの増加と同時に、肝のインスリン抵抗性を迅速に(2週間以内に)誘導するのに十分である(923)。これらの結果を総合すると、AdipoR1/2アゴニストはインスリン抵抗性を治療するための有効な戦略となる可能性があり、実際、そのようなアゴニストであるAdipoRonは、高脂肪食およびdb/dbマウスにおいて有益な効果を示すことが報告されている(590)。これらのモデルでは、AdipoRonは体重増加に影響を与えなかったが、耐糖能とインスリン感受性を改善し、肝臓と筋肉のトリグリセリド含量を減少させた(セラミドレベルは特に報告されていない)(590)。アディポネクチンの多面的な作用は実に驚異的であり(851, 936)、インスリン抵抗性との関係では、異所性の脂質量、セラミドレベル、脂肪組織の炎症の調節などが考えられる。アディポネクチンの作用の分子メカニズムはまだ完全には解明されておらず、アディポネクチンレベルの低下がヒトの疾患にどのように関与しているかは不明であるが、それでもアディポネクチンのアゴニズムは治療法の革新のための有望な領域であると考えられる。
以前、第V章でSFAをセラミド前駆体として考察した。その中で、SFAはセラミド生合成の基質となり、インスリン抵抗性を促進すると提案されている(323)。しかし、SFAによるインスリン抵抗性の別のメカニズムとして、Toll-like receptor 4 (TLR4)シグナルの活性化が提案されている(761)。メカニズム的には、SFA-TLR4シグナルとインスリン抵抗性の関係には、セラミド生合成酵素の転写アップレギュレーションが関与していると提唱されている(62)。SFAはTLR4のリガンドではないが(734)、脂肪酸結合糖タンパク質であるFetAがそのようなリガンドであることが後に示され、SFAとTLR4の活性化を結びつけることになった(606)。FetAは肝臓で産生されるヘパトカインであり、脂肪細胞におけるSFA-TLR4シグナルを活性化し、これが炎症性サイトカインの産生を促進すると考えられている(606)。この仮説を裏付けるように、FetAを欠損したマウスは、食事によって引き起こされるインスリン抵抗性から保護される(525)。ヒトでは、血清中のFetAとNEFAの濃度には統計的な相互作用があり、FetAとNEFAのどちらか一方の濃度が高い場合、もう一方の濃度が高い場合は耐糖能異常が予測される(798)。つまり、FetAがIL-6やTNF-αなどのアディポカインを誘導し、そのアディポカインが肝細胞のシグナル伝達経路を調節するという、脂肪細胞-肝細胞間のクロストークが双方向に行われているのである(797)。興味をそそるパラダイムではあるが、SFA-TLR4軸の全体的な病態生理的意義はまだ不明である。ある研究では、Tlr4-/-マウスの雌はHFDによるインスリン抵抗性から保護されたが、雄はそうではなかった(761)。別の報告では、Tlr4-/-マウスも、TLR4やアダプタータンパク質MyD88をノックダウンしたマウスも、SFAによるセラミド蓄積、DAG/PKCε軸の活性化、肝のインスリン抵抗性から保護されなかった(253)。さらに、FetAはINSRチロシンキナーゼ活性を直接損なうことが示されており(実際、この文脈で発見された)(30, 524, 793)、細胞性インスリン作用に対するFetAの効果を説明するのに、TLR4シグナルを持ち出す必要はないかもしれない。
最後に、ヘパトカインであるFGF21は、FAOとエネルギー消費を増加させ、血漿脂質を減少させるという点で非常に注目されている(738, 797)。薬理学的用量のFGF21は、痩せた餌を与えたマウスでもインスリン感受性を向上させ、脂肪を与えたマウスではインスリン抵抗性を回復させるが、これは異所性脂質の蓄積を減少させることが一因である(112)。しかし、種特異的、組織特異的、用量依存的に、一見矛盾した生理学的プロセスに関与しているため、FGF21の生理機能を完全に理解することはまだできない(233)。例えば、げっ歯類の研究では、空腹時の生理的反応のメディエーターとしてFGF21に主眼が置かれているが、ヒトの血漿中FGF21濃度は、空腹時に10日経過するまで上昇しない(227, 233)。さらに、FGF21濃度は肥満やインスリン抵抗性のヒトでは逆説的に上昇することから、肥満はFGF21抵抗性の状態である可能性が示唆されている(123, 953)。実際、シグナル伝達反応で測定したFGF21抵抗性は、肥満マウスで明らかになっている(232)。脂肪酸制御転写因子PPARαがFGF21の発現を正に制御していることから、肥満の肝脂肪症が血中FGF21の増加を促し、それが肥満のFGF21抵抗性を促進すると提唱されている(233)。しかし、インスリンの薬理学的投与が2型糖尿病患者のインスリン抵抗性にもかかわらず血糖コントロールを達成するように、FGF21およびFGF21アナログは、FGF21抵抗性にもかかわらず、ヒトの肝脂肪症や非アルコール性脂肪性肝炎(NASH)の治療に効果を発揮する可能性がある(252, 264, 824)。残念ながら、骨減少症はオンターゲットの主要な副作用であり、FGF21またはその類似体の臨床的有用性を制限する可能性がある(824, 893)。
VIII. まとめ
生体システムが非常に複雑であることから、統一された簡潔で分かりやすいモデルでインスリン抵抗性を理解しようとする努力は愚の骨頂かもしれない。確かに、正常なインスリン作用は、異なる細胞型間で重要なエフェクターを共有しているにもかかわらず、特にカプセル化することができない無数の機能を果たしている。特に、脂質と糖質の両方の代謝を制御するインスリンの複雑な関係を理解することは、何世代にもわたって研究者にとって価値ある課題となってきた(532)。しかし、これまでのセクションで述べてきたインスリン抵抗性のいくつかの推定されるメディエーターを考えると、統一される可能性のある領域を指摘したり、目的論的な憶測に陥ったりしたくなる。
インスリン抵抗性のメディエーターと考えられるものをすべて結びつける基本的な要素は、栄養素の過剰供給との関係である。この総説で取り上げた各メカニズムは、栄養由来の毒性代謝物(DAG、セラミド、アシルカルニチン、BCAA)の増加、栄養利用プロセスの過負荷(小胞体ストレス、酸化ストレス)栄養ストレスを介した細胞毒性への反応(炎症)のいずれかによって、インスリン抵抗性を引き起こすと提案されている。さらに、細胞内ストレス経路や炎症によって引き起こされるインスリン抵抗性の病態生理は、生理活性脂質によって引き起こされるインスリン抵抗性と共通している。小胞体ストレスはde novo lipogenesisを促進する。老化したインスリン抵抗性のヒトのミトコンドリア機能障害は、正のエネルギーバランスと異所性の脂質貯蔵を促進する。脂肪組織の炎症は脂肪分解を促進し、非脂肪組織への基質供給を増加させる。そこで我々は、インスリン抵抗性の統合モデルを提案する。このモデルでは、栄養過多に対するいくつかの同時反応が収束して衝突し、骨格筋と肝臓における異所性脂質の蓄積とその結果としてのインスリン抵抗性を促進する(図19)。
図19 組織のインスリン抵抗性に関する統合生理学的視点
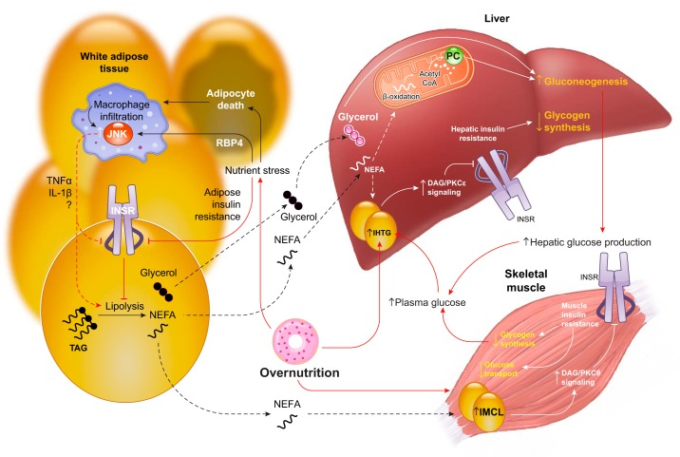
慢性的な栄養過多は、全身のインスリン抵抗性の究極の原因であり、組織自律的なメカニズムとクロストーク依存的なメカニズムの両方によってインスリン抵抗性を促進する。慢性的な栄養過多は、骨格筋や肝臓への脂質の蓄積を促進し、それらの組織でインスリン抵抗性を引き起こす。さらに、慢性的な栄養過多は、脂肪細胞に栄養ストレスを与え、脂肪細胞のインスリン抵抗性と脂肪細胞の死を引き起こす。アディポカインであるRBP4の増加やその他の炎症性シグナルにより、マクロファージが白色脂肪組織に動員される。マクロファージでは、c-Jun NH2-terminal kinase (JNK)の活性化などの炎症性シグナルにより、腫瘍壊死因子-α (TNF-α)、インターロイキン-1β (IL-1β)などのパラクリンメディエーターが産生される。これらの炎症性サイトカインは、脂肪細胞の脂肪分解を直接的に、またはインスリンシグナルを損なうことで間接的に増加させる可能性がある。炎症による脂肪細胞の脂肪分解の亢進は、非エステル化脂肪酸(NEFA)とグリセロールの回転を増加させる。これは、糖新生に対する直接的(グリセロールのグルコースへの変換)および間接的(NEFA由来のアセチルCoAによるピルビン酸カルボキシラーゼ(PC)の活性化)な刺激作用を有するとともに、肝内トリグリセリド(IHTG)の蓄積を促進し、その結果、脂質による肝インスリン抵抗性が生じ、インスリンによる肝グリコーゲンの純合成の刺激が損なわれることになる。これらの影響により、肝でのグルコース産生が増加する。また、脂肪分解が慢性的に亢進すると、細胞内脂質(IMCL)の蓄積が促進され、その結果、脂質による筋のインスリン抵抗性が生じる。筋のインスリン抵抗性によるグルコース処理の低下は、肝臓でのグルコース利用率を高め、IHTGの蓄積を促進し、肝のインスリン抵抗性を悪化させる。
栄養過多がこれらすべての代謝異常の中心的な原因であるならば、最も明白な治療法はカロリー制限である。カロリー制限の細胞への影響は複雑で不完全に理解されているが、肥満のインスリン抵抗性被験者に低カロリー食を適用した場合の生理学的効果は、この総説で紹介した仮説を検証するのに有効である。最近、Perryら(625)は、インスリン抵抗性2型糖尿病のラットモデル(高脂肪食または西洋食を4週間与えた後、低用量ストレプトゾトシン/ニコチンアミドを投与して空腹時高血糖を達成)において、3日間の超低カロリー食(VLCD:通常の摂取カロリーの25%)がもたらす代謝上の影響をまとめた。VLCDは、体重を有意に減少させることなく、血糖値とインスリン値をほぼ正常化させた。これは、IHTG、肝アセチルCoA、肝膜関連DAG、および肝PKCε活性化の低下と関連していた。変化しなかったパラメータは、肝セラミド、血漿グルカゴン、炎症性サイトカインのパネル、血漿FGF21,血漿BCAA、および肝ERストレスマーカーであった(625)。高インスリン血症-高血糖クランプ試験では、VLCDはAKTの活性化を増加させ、HGPをインスリンで抑制した(625)。興味深いことに、肝のインスリン作用の直接的および間接的な成分の両方がVLCDによって改善された。VLCDによって見られたHGPの改善は、酢酸の注入(VLCDによる肝アセチルCoAの減少を防ぐため)によって無効になったり、グリコーゲンホスホリラーゼ阻害剤(VLCDによるインスリン刺激による肝グリコーゲン合成の改善を再現するため)によって再現された(625)。このような迅速な介入の有用性は、高血糖を引き起こすパラメータと、二次的な結果や増悪要因となるパラメータを区別するのに役立つことである。この結果は、肝のDAG-PKCε軸の活性化とメタボリックに起因する糖新生を否定するものである。
しかし、すべての研究には限界があり、上記の研究の大きな限界は、この総説で引用されている多くの研究に共通するものである。インスリン抵抗性の研究に最も関係するげっ歯類とヒトの違いの一つは、栄養過多になると組織がインスリン抵抗性を発現する順序である。げっ歯類では、高脂肪食を数日与えただけで、肝脂肪症と肝のインスリン抵抗性が生じるが、骨格筋のインスリン抵抗性が生じるには数週間を要する(429)。一方、その数週間の間に、肝臓は膨張し、最終的には炎症を起こし、脂肪分解を促進し、さらに肝糖新生を促す(620)。2型糖尿病患者の若くて健康な痩せた子供は、骨格筋のインスリン抵抗性を示すが、IHTGは正常で、肝のインスリン作用も正常である(639)。しかし、筋肉のインスリン抵抗性は肝の脂肪生成を促進し、最終的にはNAFLDと肝のインスリン抵抗性が発症する。ヒトにおいて、脂肪組織のインスリン抵抗性がこのパラダイムにどのように当てはまるのかは、比較的不明である。
実際、脂肪組織のインスリン抵抗性は、特に活発に研究されているテーマである(176)。脂肪組織は過剰なエネルギーを貯蔵するように適応されており、代謝異常を引き起こすことなく過剰なエネルギーを貯蔵することができる[病的な肥満にもかかわらず通常のインスリン感受性を維持しているアディポネクチントランスジェニックob/obマウスが最も顕著に証明している(408)]。その結果、脂質によるインスリン抵抗性のように、骨格筋や肝臓でよく知られている栄養ストレスのパラダイムは、白色脂肪細胞には明らかに当てはまらないのである。質量比で少なくとも50%の脂質を持つ細胞タイプ(528)において、DAGなどの組織脂質を総体的に測定しても、意味のある情報は得られないと思われる。しかし、慢性的な栄養過多は脂肪細胞にとって明らかにストレスであり、肥満した白色脂肪組織に見られる王冠のような構造は脂肪細胞の墓石である。代謝フラックスの時空間的なコンパートメント化を研究する新しい技術が開発されれば、脂質の貯蔵に特化した細胞が脂質毒性にも脆弱であるという一見矛盾した仮説を検討することができるかもしれない。炎症は、脂肪のインスリン抵抗性の有力なメディエーターであるが、HFDは、検出可能な脂肪の炎症が発生する前に、脂肪のインスリン抵抗性を引き起こすようである。小胞体ストレスは肥満した白色脂肪組織に存在し、脂肪分解に関係していると思われるが、この潜在的な関連性についてはさらなる研究が必要である。インスリン作用の統合された生理学における脂肪組織の重要な役割は、21世紀の代謝研究の主要なテーマとなっている(735)が、脂肪組織のインスリン抵抗性の裏付けに関するより深いメカニズムの理解が急務である。
生理系はホメオスタシスを求める(424)ので、インスリン抵抗性のテレオロジーはホメオスタシスのレンズを通して見るべきである。しかし、進化は恒常的なカロリー過多の環境下で起こったわけではないので、インスリン抵抗性の生理学的結果は、よくて一時的、悪くても不意打ち的なものである可能性もあるだろう。メカニズム的には、インスリン抵抗性を引き起こすリン酸化現象やその他のシグナル伝達現象は、適切に活性化されたネガティブフィードバック機構であると考えられる。さらに悪いことには、インスリン抵抗性を引き起こすリン酸化現象は、通常はインスリン反応に必須ではないが、正常なネガティブフィードバックを媒介するキナーゼ(S6K1など)と進化的に類似しており、適切な状況下で類似した基質をリン酸化することができる、病的に活性化されたキナーゼ(nPKCなど)によって、正常なネガティブフィードバック機構が併合された結果である可能性がある。このようなことが起こるのは、永久的な栄養過多の状態が進化の歴史の中で十分に稀であり、生理的な負のフィードバック機構を病的に利用することに対して大きな選択圧が働かなかった場合に限られる。このようなシナリオはあり得るが、それでも、インスリン抵抗性を適切で偶発的ではない恒常性維持機構として扱う有力な仮説がいくつかある。
考えられるのは、インスリン抵抗性が、栄養過多に対する細胞の自律的な防御反応として進化したということである。ミトコンドリアの酸化ストレスや小胞体のストレスといった細胞の栄養ストレス応答は、栄養の過剰供給が細胞の生存を脅かすことを示している。小胞体ストレスがシグナルカスケードを誘導し、膜の合成を促進し、新しいタンパク質の合成を減少させることでホメオスタシスを回復させるのと同様に、インスリン抵抗性は、グルコースの利用、高分子の合成、その他の同化プロセスを制限する栄養過多に対する適切な反応であると考えられる。栄養不足の細胞では、同化作用を続けることは、メリットよりもコスト(酸化的ストレスや小胞体ストレスなど)の方が大きいかもしれない。
もう一つの可能性は、インスリン抵抗性が多臓器の生理的反応であり、栄養が豊富な時に白色脂肪組織にカロリーを蓄えることを促進することで、生体に利益をもたらしているということである。インスリンで刺激された筋肉や肝臓の同化作用を停止させれば、WAT癌のような別の目的地に基質が移動するはずである。この考え方の最大の欠点は、肝臓や骨格筋とはおそらく異なるメカニズムではあるが、栄養過多になると白色脂肪組織もインスリン抵抗性になることである。
脂質によって誘発されるインスリン抵抗性に適用される最後の興味深い理論は、インスリン抵抗性を断食への有益な適応として扱うものである。空腹時には脂肪分解が促進され、相対的に脂質の利用率が高まる。その結果、空腹時には肝臓や骨格筋にトリグリセリドの蓄積が誘導される(306, 796)。ヒトでは空腹時に骨格筋のインスリン抵抗性が強くなることが示されているが(519, 887)、マウスではそのようなことはない(306)。しかし、マウスの研究は、一晩の空腹でも体組成が大きく変化する(約10%の体重減少)ため、複雑である。肝臓や骨格筋における脂質によるインスリン抵抗性は、断食中に糖新生を促進し、グリコーゲンの貯蔵を最小限に抑え、中枢神経系のためにグルコースを保存するという有用な目的を果たすことができる。この仮説と一致するように、飢餓状態のラットの肝臓では、DAGの蓄積、PKCの活性化、IRK活性の阻害が観察されている(387, 628)。最近では、メキシコのテトラフィッシュの川に住む個体群と洞窟に住む個体群を対象とした注目すべき研究により、インスリン抵抗性の飢餓適応仮説が進化的に支持された(690)。栄養不足の洞窟環境に適応した魚は、多くの場合、インスリン受容体の部分的機能喪失変異により、顕著なインスリン抵抗性を示した(690)。カブトガニはより大きな脂肪を蓄え、飢餓状態でも体重を維持できるようになった(690)。カブトガニには、代謝率の低下や概日周期の変化など、他にも独立した代謝適応が見られるが、これらの観察結果は、栄養が制限された環境では、インスリン抵抗性が進化的に選択されることを示唆している。しかし、マウスとヒトでは絶食時の反応が著しく異なるため、この仮説を裏付ける他の証拠は不足している。さらに、空腹時は低インスリン血症の状態であり、インスリン抵抗性の生理的な関連性は、再摂食時の反応を促進する役割以上には明らかではない。
インスリン抵抗性は、その生理学的な起源にかかわらず、慢性的な栄養過多の状況下では不適応となる。インスリンの作用と抵抗性をより完全に理解することは、既存の抗糖尿病治療を賢く利用することを可能にし、新しい治療法の開発を可能にし、そしておそらく最も重要なことは、2型糖尿病の流れを止めるための予防戦略に役立つだろう。