
Contents
- Lipids and Alzheimer’s Disease
- 要旨
- 1. 背景
- 2. 脂質ラフト
- 3. 老化と脂質
- 4. 血液-脳-バリア(BBB)
- 5. 脂肪酸
- 6. 腸マイクロバイオータと腸-脳軸
- 7. ケトジェニックダイエット(KD)とケトン体
- 8. 特化されたプロ分解性脂質メディエーター(SPM)
- 9. 脂質過酸化 イソプロスタン IsoPsニューロプロスタン(NeuroPs)
- 10. グリセロ脂質
- 11. グリセロリン脂質
- 12. スフィンゴ脂質
- 13. コレステロール
- 14. アポリポタンパクE(ApoE)
- 15. スタチン
- 16. ATP結合カセット(ABC)トランスポーター
- 17. モノホスホリル脂質A
- 18. 脂溶性ビタミン ビタミンA、D、E
- 19. リピドミクス研究
- 20. 結論
- 略語
Lipids and Alzheimer’s Disease
www.ncbi.nlm.nih.gov/pmc/articles/PMC7073164/
要旨
脂質は、細胞膜の基本成分として、脳の機能だけでなく、人間の健康にも重要な役割を果たしている。脳には脂質が非常に豊富に含まれており、脂質の恒常性の乱れは神経疾患やアルツハイマー病などの神経変性疾患と関連している。
老化は脂質組成の変化と関連している。脂質ラフトのレベルでの脂肪酸の変化と脳脂質過酸化は、アルツハイマー病の初期段階で発見された。アポリポタンパク質や脂質トランスポーターの担持状態や食事脂質量などの遺伝的・環境的要因がアルツハイマー病と関連している。脂質と アルツハイマー病の間のつながりへの洞察は、この不可解な病気の代謝の側面を解明するために重要である。
最近の脂質分析法の進歩により、脂質に関する深い理解が得られるようになっていた。その結果、リピドミクスは新しい治療法の発見を目指して、病気の予知、診断、予防のためのバイオマーカーを見つけるために、アルツハイマー病の研究のホットなトピックとなっている。
キーワード
アルツハイマー病、脂質、脂肪、脂肪酸、コレステロール、リピドミクス
1. 背景
世界アルツハイマー報告書によると、2015年には世界で4,680万人の認知症患者がいて、年間約990万人が新たに発症していると推定されており[1]、認知症の世界的な負担の大きさを示している。認知症の中で最も多いアルツハイマー病は、進行性の記憶喪失、認知機能障害、行動変化を臨床的に特徴づける神経変性疾患である。アルツハイマー病の主要な神経病理学的特徴は、β-アミロイド(アミロイドβ)ペプチドの過剰産生やクリアランス障害による老人斑におけるアミロイドタンパクの蓄積であり、シナプスの損失や神経変性をもたらす神経原線維のもつれ(NFTs)の沈着である。
脳脂質は、乾燥脳重量の少なくとも50%を占めており、脂肪組織に次いで最も脂質が豊富な臓器である[2]。脂質は神経細胞膜の基本的な構造成分である。脳の脂質は、50%のリン脂質、40%以下の糖脂質、10%のコレステロール、コレステロールエステル、微量のトリグリセリドで構成されている。長鎖多価不飽和脂肪酸(LC-多価不飽和脂肪酸s)は、ドコサヘキサエン酸(DHA)およびアラキドン酸(アラキドン酸)を含む、ヒトの脳内の全脂肪酸(脂肪酸)の25~30%を占める[2]。脳脂質過酸化は、アルツハイマー病の初期イベントであることが判明した。アルツハイマー病患者の脳は、異常な脂質代謝を示唆している、グリア内のリポイド顆粒(または脂肪内包物)の高い数を表示する。ゲノムワイド関連研究(GWAS)は、アルツハイマー病とAPOE(アポリポ蛋白ECLU(クラスターチン、アポリポ蛋白Jとしても知られているSORL1(ソルチリン関連受容体1)とABCA7(ATP結合カセット、サブファミリーA、メンバー7)などの脂質恒常性に関与するいくつかの遺伝子との間の関連を発見した[3]。
液体クロマトグラフィー、ガスクロマトグラフィー、質量分析、質量分析イメージングなどの脂質分析は、組織や体液中の脂質の同定を可能にする[4,5]。脂質には、表 1 に示すように 8 つのカテゴリーがある。以下のセクションでは、各脂質群に関する研究とアルツハイマー病との関連について述べる。
表1 脂質のカテゴリー
カテゴリー/例
- 脂肪酸(脂肪酸)/アラキドン酸、ドコサヘキサエン酸(DHA)
- グリセロ脂質(GL)/モノアシルグリセロール(MAG)ジアシルグリセロール(DAG)トリアシルグリセロール(トリグリセリド、TGも含む)
- グリセロリン脂質(GP)/ホスファチジルエタノールアミン(PE)ホスファチジルコリンPCホスファチジルセリン(PS)
- スフィンゴ脂質(SP)/スフィンゴミエリン、セラミド、スルファチド、ガングリオシド
- ステロール脂質(SL)/コレステロール、ビタミンD
- プレノール脂質(PR)/カロテノイド、ビタミンE&K
- 糖脂質(SL)/脂質A
- ポリケチド(PK) /ロバスタチン
2. 脂質ラフト
2つの膜単分子膜の脂質組成は異なっている:内側のリーフレットはホスファチジルセリン、ホスファチジルエタノールアミン、ホスファチジルイノシトールに富むのに対し、外側のリーフレットはホスファチジルコリンとスフィンゴミエリンに富む[6]。
脂質ラフトは細胞膜内の動的構造であり、シグナル伝達、細胞接着、脂質/タンパク質の選別において重要な役割を果たしている。脂質ラフトは、スフィンゴ脂質、コレステロール、飽和脂肪酸、および多価不飽和脂肪酸の減少した含有量の組み合わせによって特徴づけられる。
多くのアルツハイマー病関連タンパク質は、アミロイド-βタンパク質前駆体(βAPPβ-セクレターゼ、γ-セクレターゼおよびネプリライシンなどの脂質ラフトで発見されている[7]。
脂質ラフトは、アミロイドβがApoE、およびタウと相互作用し、アミロイドβオリゴマーおよび高リン酸化タウの凝集を促進するプラットフォームとして機能する[8]。
アミロイドβ産生(アミロイド新生)は、脂質ラフト内の脂質組成に関連している[9]。
細胞障害性アミロイドβフィブリルの形成は、GM1ガングリオシド、コレステロール、スフィンゴミエリンを含む脂質ラフトによって誘発される[10]。アルツハイマー病患者の前頭前皮質からの脂質ラフトは、健康なコントロールと比較して、不飽和度および過酸化性指数の有意な低下と同様に、n-3 LC-多価不飽和脂肪酸(主にDHA)およびモノエン(主にオレイン酸)の含有量が異常に低いことを示している[11]。
脂質ラフトのレベルでのこれらの脂肪酸の変化は、アルツハイマー病の発症初期、特に前頭葉と内耳皮質で観察された[12]。
3. 老化と脂質
人間の人生の最初の20年の間に、脳脂質の量は増加し、その後50歳を過ぎると徐々に減少し始める [13]。老化は全身性遊離脂肪酸(遊離脂肪酸)レベルの上昇を伴う脂肪組織分布の変化を引き起こし、これはメタボリックシンドロームの共通の特徴である [14]。異なる脳領域では、脂質組成の加齢に伴う変化がある。
飽和脂肪酸(飽和脂肪酸)一価不飽和脂肪酸(一価不飽和脂肪酸)および多価不飽和脂肪酸は、中年男性では若い男性に比べて有意に多く、一方、DHA、アラキドン酸を含む多価不飽和脂肪酸は減少し、一価不飽和脂肪酸は加齢に伴い眼窩前頭前皮質の灰白質で増加する[15]。
同様に、加齢は炎症の増加と関連している。脂質は、多くの免疫応答をオーケストレーションするメディエーターである。いくつかの抗炎症性脂質メディエーター(SPM)は、特に加齢と関連している[16]。
4. 血液-脳-バリア(BBB)
脳血流(脳血流)と年齢との間には負の相関がある[17];さらに、血液脳関門(BBB)透過性は若年者に比べて高齢者の方が高い[18]。動的造影磁気共鳴画像法(MRI)は、BBB破壊が加齢脳の海馬で始まる初期のイベントであることを示唆している[19]。脳の低灌流とBBBの完全性の損失は、記憶と学習の障害につながる、エネルギーの利用可能性の低下と破壊されたシナプスの結果、[20]。BBBの完全性は、脳アミロイド血管症と呼ばれる脳血管の変化(違反)の結果として、アルツハイマー病の脳の実質の血漿タンパク質の検出によって証明されるように、アルツハイマー病 [21]で損なわれている。脳の灰白質と大脳皮質全体では、対照群と比較して アルツハイマー病 患者では BBB リークが有意に増加し、疾患の重症度と相関していた [22]。
Takechiらは、高脂肪食の12週間後、飽和脂肪酸食マウスのBBB機能障害は、高コレステロール食では7倍に増加したのに対し、対照群と比較して30倍に増加したことを示した。これらのマウスに脂質低下剤であるプロブコールを投与したところ、血漿中の非エステル化脂肪酸レベルが上昇し、脳血管の炎症やBBBの完全性の喪失が起こらなかったことから、プロブコールは血漿脂質を直接調節することではなく、炎症性の経路を抑制することで高脂肪食によって誘発されるBBBの障害を防ぐことが示唆された[23]。
5. 脂肪酸
脂肪酸は、より複雑な脂質の基本的なビルディングブロックである。トリグリセリド(TG)は、脂肪酸の貯蔵形態であり、ATP生産のためのエネルギーを解放しながら、β酸化を介して分解する[4]。脂肪酸の主要なカテゴリは、飽和、トランス、一価不飽和および多価不飽和脂肪酸である。脂肪酸は、二重結合の数に基づいて飽和対不飽和として分類することができる。不飽和のものは少なくとも1つ(一価不飽和、一価不飽和脂肪酸または2つ以上の(多価不飽和、多価不飽和脂肪酸)二重結合が含まれているのに対し、室温で固体である傾向がある飽和脂肪酸の二重結合はない。自然食品の脂肪酸組成は本質的に可変であり、果物や野菜は主に不飽和脂肪酸が含まれている間、肉や乳製品の高い飽和脂肪酸がある。トランス脂肪酸は、噂や腸内細菌の代謝または植物油から複数の不飽和脂肪酸の水素化のいずれかによって行われ、これらは高コレステロール血症であり、心血管疾患[24]の高いリスクと有害な結果にリンクされている。
飽和脂肪酸は、インスリン抵抗性と炎症[14]の設定で動脈硬化の開発を加速することが可能である、すべての脂肪酸の中で最も有害なものと考えられている。脳は長鎖多価不飽和脂肪酸s-DHA(22:6n-3)とアラキドン酸(20:4n-6)で非常に濃縮されている。脳では、多価不飽和脂肪酸sは、主に膜の流動性、シグナル伝達、遺伝子転写に影響を与え、神経細胞のアポトーシスと死から保護するために、神経膜のリン脂質に組み込まれている[25]。多価不飽和脂肪酸sは、炎症反応を支配する脂質メディエーターの生合成のための前駆体として機能する。N-6 脂肪酸は、プロスタグランジン、トロンボキサン、ロイコトリエン、リポキシン、レゾルビン、およびエキシンを含むエイコサノイドの前駆体である。したがって、食事のn-3/n-6 多価不飽和脂肪酸の比率は、有害な(アラキドン酸誘導体のプロ炎症性効果有益な(DHA代謝物の抗炎症、神経保護および抗酸化効果)または神経調節効果(アラキドン酸由来のエンドカンナビノイド)を有する可能性がある脂質メディエーターに代謝される膜状リン脂質の脂肪酸組成に影響を与えることができる[26]。赤血球の脂肪酸組成の変化は、認知機能障害に先立って、アルツハイマー病の初期段階でも起こる。新皮質βアミロイド負荷が低い人と比較して、βアミロイド負荷が高い人は血漿アラキドン酸が上昇し、ドコサペンタエン酸(DPA)が低かった[27]。
遊離脂肪酸、特に遊離脂肪酸の皮質上昇は、試験管内試験でアミロイドとタウのフィラメントの集合を誘導する。より大きな刺激は、通常、不飽和脂肪酸に関連付けられているが、すべての長鎖脂肪酸は、ある程度[28]にアセンブリを強化した。リノール酸、アラキドン酸、α-リノレン酸、DHA、エイコサペンタエン酸(EPA)とオレイン酸を含む6つの不飽和脂肪酸をテストする研究では、これらすべての不飽和脂肪酸は正に神経斑やNFTの負担と負の認知パフォーマンスと相関して関連付けられていたことを示した。アルツハイマー病の病理学-中前頭前野と下側頭回に脆弱な脳領域では、リノール酸、αリノレン酸、およびアラキドン酸の減少とDHA [29]の増加があった。これらのすべての6つの不飽和脂肪酸は、直接アミロイドβ40およびアミロイドβ42ペプチドと相互作用し、アミロイド線維形成、特にオレイン酸とDHAを防止することにより、優れた抗凝集特性を表示することができる[30]。しかし、神経保護α-セクレターゼ切断可溶性APP(sAPPα)分泌および細胞膜流動性の調節における異なる不飽和脂肪酸の役割を調査すると、4個以上の二重結合を有するアラキドン酸、EPAおよびDHAのみが、膜流動性およびsAPPα分泌を増加させることができるのに対し、ステアリン酸(SA、18:0リノール酸、αリノレン酸およびオレイン酸はできない[31]。
5.1. N-3脂肪酸である。ドコサヘキサエン酸(DHA
DHAはn-3 脂肪酸であり、すべての脳領域で最も豊富な多価不飽和脂肪酸である[32]。DHAはα-リノレン酸に由来し、その過程でEPAを形成する。出生前に、脂肪酸の濃度はプラトーに達するが、DHAは、シナプス形成の直前に急速な蓄積で増加し続けている例外である[33]。ヒトのDHAの供給源には、魚などの食事源だけでなく、肝臓でのDHA生産が含まれている[34]。肝臓は、人間の脳[35]の要件よりも短い鎖n-3 脂肪酸前駆体であるαリノレン酸とEPAからDHAの1.8〜36倍を合成する。脳内のDHAレベルは、欧米の食生活[34]などのn-3 脂肪酸の低食生活消費量の条件で、食事由来のn-3 脂肪酸の肝臓の代謝能力に依存する。
年齢の増加に伴い、DHAレベルの漸進的な低下がある。正常な老化は、全体的な脳の萎縮につながるが、低いDHAレベルは、増加した海馬の萎縮[36]に関連付けられている。アルツハイマー病患者は、疾患抵抗性領域を含む脳全体でDHAレベルが低下しているが、最も顕著な減少は海馬にある。ミニ精神状態検査(MMSE)によると、DHAの含有量は認知症[37]と正の相関関係を持っている。アルツハイマー病患者からの肝臓はまた、テトラコサヘキサエン酸(THA)を含む短鎖n-3前駆体の高いレベルのDHAの低レベルが含まれているが、DHA合成の最後のステップであるペルオキシソームβ酸化を介してDHAにTHAの生体変換の欠陥を示唆している[38]。しかし、いくつかの研究では、赤血球やアルツハイマー病とコントロール個体の脳組織間のDHA含有量に有意な差がないことが示されている[39,40]。
フラミンガム研究では、血漿DHAレベルの上位4分の1の被験者は認知症のリスクが47%低かった[41]。軽度認知障害(MCI)患者では、プラセボで治療した群と比較して、n-3 脂肪酸処理群では認知機能低下率の低下と認知機能の改善が示されたが、アルツハイマー病患者では効果はなかった[42]。マウスでは、8週間のDHAを濃縮した食事療法は、いずれかの認知機能の低下を遅らせる[43]または7ヶ月間より長く処理されたマウスの学習能力を向上させた[44]。DHAの複数の効果は、プレセニリン1(PS1)とc-ジュンN末端キナーゼ(c-JNK)を阻害することにより、アミロイドβ沈着とタウリン酸化を減少させ、脳血流を増加させるなど、アルツハイマー病の病因に拮抗することが示されているα-セクレターゼによるAPPの切断を強化しながら、βとγ-セクレターゼの活性を低下させ、樹状突起棘密度を増加させ、海馬でシナプス機能を回復させる[45,46,47]。n-3 脂肪酸はSPMのための前駆体であるので、MCIを持つ患者のn-3 脂肪酸の補給は、SPM resolvin D1 [48]の生産を増加させ、アミロイドβ[49]の貪食を支持して中間的なM1-M2表現型に非常にproinflammatory M1からマクロファージをシフトした。多くの研究では、DHAの有益な認知効果をサポートしているが、2つの二重盲検無作為化比較試験(RCT)を含む他の研究では、効果は報告されていない[42]。さらに、多価不飽和脂肪酸sは一般的に神経保護とみなされているが、過酸化損傷はその二重結合に影響を与える傾向がある[50]。
4つのn-3およびn-6 脂肪酸のうち、DHAは、EPA、アラキドン酸およびリノール酸に続いて、最大の程度に酸化された。したがって、DHAの長期投与は慎重に検討する必要がある。最近、重水素強化多価不飽和脂肪酸s(D-多価不飽和脂肪酸s)は、通常の水添多価不飽和脂肪酸s(H-多価不飽和脂肪酸s)よりも脂質過酸化の活性酸素誘発連鎖反応に強く、H-多価不飽和脂肪酸sと比較して大脳皮質と海馬のプロスタグランジンF2αとF2-イソプロスタンが明らかに減少していることが明らかになった[51]。D-多価不飽和脂肪酸を与えられたAPP/PS1 ADマウスは、H-多価不飽和脂肪酸を与えられたものと比較して、海馬の脂質過酸化生成物とアミロイドβ40/アミロイドβ38産生が低下したが、学習と記憶障害の変化はなかった[52]。
5.2. N-6脂肪酸 リノール酸(リノール酸)とアラキドン酸(アラキドン酸
アラキドン酸とその前駆体リノール酸は、n-6 脂肪酸である。MCIとアルツハイマー病の患者は、アラキドン酸のレベルが上昇していたが、リノール酸のレベルが低下し、リノール酸のレベルは、1つの研究[53]で健康なコントロールからMCIにアルツハイマー病患者にMCIに進行的に減少した健康なコントロールと比較していた。アラキドン酸カスケードの活性化は、アミロイドβの増加につながり、インターロイキン-1βによって誘導されるワーキングメモリの障害を引き起こす[54]。2%のアラキドン酸を含む食事を21週間与えたマウスでは、アミロイドβの産生および沈着が増加した[55]。アラキドン酸は、酵素5-リポキシゲナーゼ(5-LO)によってロイコトリエンに、シクロオキシゲナーゼ(COX)によってプロスタグランジンおよびトロンボキサンに変換され代謝されるが、これらはすべて炎症性作用と関連している[56]。5-LO酵素経路はアルツハイマー病においてアップレギュレートされている。5-LOの過剰発現はアミロイドβレベルの上昇をもたらすのに対し、5-LOの阻害はアミロイドβおよびγ-セクレターゼの減少をもたらす[57]。
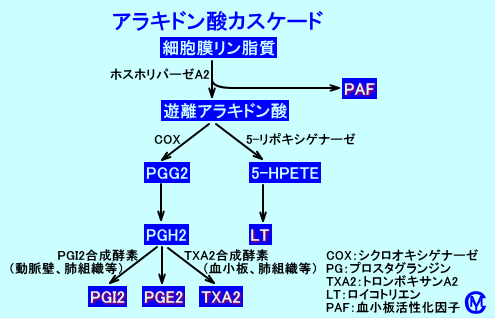
hobab.fc2web.com/sub2-arakidonnsannkasuke-do.htm
5.3. N-9 脂肪酸 オレイン酸
オレイン酸は、n-9 脂肪酸と最も豊富な食事脂肪酸は、ほとんどの研究でアルツハイマー病に対する保護とみなされる。オレイン酸はアルツハイマー病脳の前頭皮質と海馬で減少していた[58]。オレイン酸が豊富なオリーブオイルなどの地中海料理の利点が強調され、アルツハイマー病の発症や加齢に伴う認知機能の低下に対して保護されている[59]。オレイン酸の補給は、アルツハイマー病の細胞モデルや動物モデルにおいて、分泌されるアミロイドβを減少させ、アミロイド形成を改善した[60]。アルツハイマー病患者は健忘症に関連したプロリルエンドペプチダーゼ(PEP)活性が増加しており、オレイン酸は不飽和脂肪酸の中で最も高いPEP阻害活性を示した[61]。さらに、ある研究では、オレイン酸 はγ-セクレターゼ活性の増加をもたらし、トランスフェクションされた細胞では PS1 と アミロイドβが増加することが示されている[62]。
5.4. n-6 脂肪酸と n-9 脂肪酸の組み合わせ
Amtulらは、試験管内試験でのアミロイドβ産生に対するn-6,n-9 脂肪酸またはその組み合わせの効果を研究した。リノール酸添加は、C-99トランスフェクションしたCOS-7細胞のアミロイドβ40レベルを有意に増加させ、アラキドン酸添加はアミロイドβ40およびアミロイドβ42を含む全アミロイドβレベルを増加させた;一方、オレイン酸添加細胞はアミロイドβ40,アミロイドβ42および全アミロイドβレベルの減少をもたらした。したがって、n-9 脂肪酸(オレイン酸)に富む食事の消費量の増加とn-6 脂肪酸(リノール酸およびアラキドン酸)の摂取量の減少は、アルツハイマー病の低いリスクにリンクされている可能性がある。リノール酸-オレイン酸-アルブミンまたはリノール酸-オレイン酸-アラキドン酸-アラキドン酸-アルブミンの混合物とAPP695トランスフェクトされたCOS-7細胞のインキュベーションは、強く濃度依存的な方法でほぼ2倍の総アミロイドβレベルを強化し、脂質の混合物は、個々の脂質の効果よりも強力であると思われるアミロイドβ産生に相加的な効果を持っていることを明らかにした。この実験は、アミロイドーシスへの影響について、単一の脂質ではなく、脂質の複合体を研究することの重要性を強調している[63]。
5.5. 食事性脂肪酸
脂肪酸のすべての異なる種のうち、ほとんどの研究では、アルツハイマー病と認知機能の低下のリスクの増加は、食事摂取量の高い飽和脂肪酸と相関していることをサポートしている[64]。シカゴ健康と老化プロジェクト(CHAP)によると、飽和脂肪摂取量の上位5分位の人のアルツハイマー病のリスクは、リスクがトランス脂肪のために2.5倍であったのに対し、最低5分位の人と比較して2.2倍であった[65]。大豆油ベースの食事と比較して、40%飽和脂肪酸を含む欧米化食を4ヶ月間与えたトランスジェニックマウスでは脳アミロイドβ値が増加したが、DHAを補った食事を与えた人では大豆油ベースの食事を与えた人よりもアミロイドβ値が低かった[66]。一価不飽和脂肪酸を豊富に含む食事は、しばしば地中海式食事法として知られており、加齢に伴う認知機能の低下を遅らせ、アルツハイマー病のリスクを軽減することと関連していることから提唱されている[64]。しかし、一価不飽和脂肪酸の食事摂取量は、食事摂取量の中で飽和脂肪酸やトランス脂肪酸と一致することが多かった。現実世界における食事性脂質とアルツハイマー病の効果を調査する研究は、有益な脂肪酸と有害な脂肪酸の両方を構成する脂肪酸の組成が異なることでしばしば混同され、否定的な所見につながっている[64]。
6. 腸マイクロバイオータと腸-脳軸
短鎖脂肪酸(SCFA)は、主に食物繊維や未消化の複合炭水化物の嫌気性発酵中に大腸菌によって産生される炭素原子数1~6個の鎖長を持つ脂肪酸である[67]。酢酸塩、酪酸塩、およびプロピオン酸塩などの短鎖脂肪酸は、BBBを透過するか、または腸-脳軸を介して脳にその効果を発揮することができる[68]。酪酸塩は、Gタンパク質共役型受容体(GPCR)シグナル経路や抗炎症シグナル伝達を介してヒストン脱アセチル化酵素阻害剤として神経保護効果を有する[69]。海馬のヒストンアセチル化を改善し、学習過程に関連する遺伝子発現をアップレギュレートすることができる[70]。ADマウスモデルでは、酪酸ナトリウムの治療は、学習と記憶機能を促進し、アミロイドプラーク沈着を減少させ、海馬ニューロンの樹状突起棘密度を復元[70,71]。酢酸は、ミクログリアに影響を与え、BBB透過性[72]を減少させることができ、アルツハイマー病ショウジョウバエ[73]でダウンレギュレートされることが判明した
7. ケトジェニックダイエット(KD)とケトン体
高脂肪低炭水化物食はKDと呼ばれている。代謝経路では、β-ヒドロキシ酪酸(β-HB)とアセト酢酸の2種類の主なケトン体が生成される。第三のタイプのケトンであるアセトンは、アセト酢酸の酵素的脱炭酸によって生成され、主に呼気中に排泄される[74]。ケトン体は、解糖をバイパスしてトリカルボキシサイクルに入るアセチル-CoAの産生のための基質として作用することができる。ケトン体は、脳の代替燃料として低血糖症(例えば、絶食、より高いレベルの運動、糖尿病または神経病理学的状態)に対する正常な応答を表す[75]。KDの2〜3日後、ケトジェネシスは、一般に、肝細胞のミトコンドリアマトリックスで行われる。ケトン体はモノカルボン酸トランスポーター1(MCT1)を介してBBBを通過し、その後、脳内のニューロンに入ることができる[76]。食事エネルギーの60〜80%は、古典的なKDからのLC脂肪酸(炭素数14〜22)によって提供される。中鎖トリグリセリド(MCT)を含む代替KDが開発され、エネルギーの約45%のみが中鎖脂肪から来ているため、より大きな炭水化物成分とより良い患者の耐性を可能にする。MCTベースのKDは、約60%のオクタン酸(8-炭素脂肪酸)と40%のデカン酸(C10 脂肪酸)で構成されており、MC脂肪酸(6-12炭素)に代謝されることができる[77]。
MCT-KDは2つの理由で古典的なKDよりも効果的である。LC脂肪酸sの再エステル化がリンパ系を介して吸収され、肝臓に到達する前に末梢循環を通過するカイロミクロンに組み込むのに対し、
(1)門脈への腸からの直接吸収、
(2)ミトコンドリアにアクセスする前に補酵素Aにカルニチンによる活性化を必要とするLC脂肪酸sと比較して、β酸化のためのカルニチン依存性の活性化の必要性はない。オクタン酸およびデカン酸もまた、ケトン体産生とは無関係な認知促進作用を有する[78]。
ブドウ糖代謝の低下は、発症の数十年前にアルツハイマー病患者の脳で観察された。対照的に、ケトン体の代謝は、少なくともアルツハイマー病の初期段階では変化しない[79]。したがって、ケトン体は低血糖症の間、アルツハイマー病患者の脳内の代替燃料であるように思われる。
アルツハイマー病トランスジェニックマウスにおけるKDは、対照と比較して脳内の総アミロイドβレベルの低下をもたらした[80]。ラット海馬の培養細胞へのβ-HBまたはアミロイドβまたはその組み合わせの添加は、アミロイドβのみに曝露された細胞と比較して、β-HBが生存細胞数を2倍にし、細胞サイズと神経突起の伸長を増加させることで、アミロイドβの毒性を逆転させたことを示しており、ケトン体が神経成長因子としても作用することを示唆している[81]。ケトン体sのもう一つの可能性のある神経保護メカニズムは、脳内の酸化性リン酸化とATP生成を改善するミトコンドリア生合成[82]のアップレギュレーションのためのそれらの機能と関連付けられている。ケトン体は、ミトコンドリアのグルタチオンレベル[83]とグルタチオンペルオキシダーゼ活性[84]を増加させることにより抗酸化作用を有する。KDを給与した動物は、ミトコンドリアの呼吸鎖複合体の効率を改善することでフリーラジカルの減少を示した[85]。また、KDは、脳の血管機能を改善し、有益な腸内細菌叢(Akkermansia muciniphilaと乳酸菌)の成長を増加させ、代謝プロファイルを強化し、完全にアルツハイマー病 [86]の疾患プロセスを減衰させることができる。
ヒトでは、軽度および中等度のアルツハイマー病を持つ被験者にMCT-KDを投与する無作為化二重盲検試験は、改善された記憶と認知機能[87,88]を示したが、有益な効果は、APOEのε4キャリア[87]を除外した。MCT-KDを3ヶ月間使用した後、1ヶ月のウォッシュアウトを行った;食事療法中、アルツハイマー病AS認知サブスケールの平均スコアは改善し、ウォッシュアウト後はベースラインに戻った[89]。したがって、記憶を維持するためにアルツハイマー病患者に長期KDが必要かどうかは、さらなる調査が必要である。しかし、神経変性疾患を持つ人々には栄養不良のリスクがあり、KDは食欲低下を引き起こし、消化器系の副作用を伴う可能性があるため、高齢者へのKDの適用には懸念がある[90]。
8. 特化されたプロ分解性脂質メディエーター(SPM)
前述したように、加齢は炎症と関連したプロセスである。アルツハイマー病は炎症が亢進した疾患であり、炎症の解決の障害はアルツハイマー病患者に見られる[16]。炎症の解決は、炎症の解決と恒常性を回復するための抗炎症特性を有する、特殊化プロ解決メディエーター(SPM)と呼ばれる脂質メディエーターのファミリーによって制御されている。SPMの主要なタイプは、レゾルビン、リポキシン、プロテキンおよびマレシンである。SPMは、多価不飽和脂肪酸s-アラキドン酸(リポキシン前駆体EPA(Eシリーズのレゾルビン前駆体DHA(Dシリーズのレゾルビン、プロテキン、マレシン前駆体)によって産生される[91]。炎症から解決段階への移行は、最初にアラキドン酸由来のリポキシンの増加とプロ炎症性プロスタグランジンおよびロイコトリエンの減少によって特徴づけられ、その後、n-3 脂肪酸由来のSPMの増加につながる[92]。
アルツハイマー病患者の海馬ではSPMの低レベルがある。アルツハイマー病患者の死後検査では、非アルツハイマー病患者と比較して脳脊髄液と海馬でリポキシンA4(LXA4)が減少していることが示された。脳脊髄液中のLXA4およびレゾルビンD1(RvD1)レベルはMMSEスコアと相関していた [93]。内耳皮質では、年齢をマッチさせた対照群と比較して、アルツハイマー病患者ではMaresin 1(MaR1Protein D1,resolvin D5の発現が減少した。MaR1とRvD1は、ヒトのミクログリアにおけるアミロイドβ42誘導性炎症をダウンレギュレートし、アミロイドβの取り込みを伴うミクログリアの貪食を増強した[48,94]。複数の家族性アルツハイマー病変異を持つAPPとPS1を発現する5x家族性ADマウスは、野生型に比べて海馬のSPMが低かった。SPMs-レゾルビンE1(RvE1)またはLXA4を単独または併用して処置した後、5x家族性ADマウスはSPMsのレベルを回復し、炎症過程を反転させ、アミロイドβ病理学の減少に伴う神経炎症を減少させた[95]。アスピリンをトリガーとしたLXA4治療は、NF-κB活性化、プロ炎症性ケモカインおよびサイトカインの減少、ならびに抗炎症性IL-10およびトランスフォーミング成長因子-βのレベル上昇をもたらした。さらに、ミクログリアは、アミロイドβクリアランスと認知機能を促進するファゴサイトーシスが改善された表現型にシフトした[96]。
9. 脂質過酸化 イソプロスタン IsoPsニューロプロスタン(NeuroPs)
脂質の過酸化は、反応性酸素種の攻撃に敏感な多価不飽和脂肪酸sの含有量が多いため、中枢神経系における酸化ストレスの主な症状である[97]。脳脂質の過酸化副産物には、F2-イソプロスタンス(F2-IsoPs)およびイソフラン(IsoFF4-ニューロプロスタンス(F4-NeuroP)およびニューロフラン(NeuroF)が含まれる。F2-ジホモ-イソプロスタンス(F2-ジホモ-IsoP)およびジホモイソフラン(ジホモ-IsoF4-ヒドロキシ-トランス-2-ノネナール(4-HNE4-ヒドロキシ-2-ヘキセナール(4-HHEアクロレイン、およびマロンジアルデヒド(MDA)[98]。IsoPsまたはイソプロスタノイドはプロスタグランジン様化合物であり、その中でもF2-IsoPsはアラキドン酸の過酸化によって産生される[99]。F2-IsoPsの増加がアルツハイマー病患者の脳と脳脊髄液で認められた[100]。脳脊髄液中のF2-IsoPsレベルは疾患の進行と相関しており、その増加は100%の精度で正常なコントロールと区別することができる[101]。したがって、F2-IsoPsの上昇は、アミロイドβが沈着する前であっても、アルツハイマー病患者における脂質過酸化の初期バイオマーカーとして機能することができる[102]。脳のF2-IsoPsレベルを低下させると、βAPP/PS1マウスにおけるアミロイドβ沈着とプラーク形成が有意に減少した[103]。さらに、マウスの食事でアルミニウムを補充すると、脳のF2-IsoPs形成が増加し、加速されたアルツハイマー病表現型につながった[102]。
F4-NeuroPsはDHAの酸化に由来するものであり、その発現はアルツハイマー病患者の脳脊髄液と脳で高い[104]。MCIやアルツハイマー病患者の脳では、F4-NeuroP、IsoP 8,12-iso-iPF2α-VI、HNE、MDA、アクロレインのレベルも上昇していた[11,105]。したがって、酸化的損傷と脂質過酸化はアルツハイマー病の初期イベントである[106]。尿サンプル中の17種類の脂質過酸化バイオマーカーの同時測定に超高性能液体クロマトグラフィー質量分析法(UPLC-MS/MS)を用いたところ、17(RS)-10-epi-SC-Δ15-11-dihomo-IsoF、PGE2,NeuroP、IsoP、IsoFは軽度アルツハイマー病患者と対照群で差が認められ、脳脊髄液と比較して入手が容易であることから、初期のアルツハイマー病バイオマーカーとして有望視されている[107]。しかし、先の研究では血漿中のF2-IsoPsと尿中のF4-NeuroPsのレベルがアルツハイマー病患者の中枢神経系のレベルを正確に反映していないことが示されているため、さらなる調査が必要である[104]。
10. グリセロ脂質
10.1. グリセロ脂質 トリグリセリド(TG)
TGは最も優勢なグリセロ脂質である。脂質低下剤gemfibrozilの4〜6ヶ月で治療した高脂血症肥満被験者は、より良い脳灌流と認知パフォーマンスを持っていた[108]。また、動物実験では、海馬のN-メチル-d-アスパラギン酸(NMDA)成分を介して長期的な電位の維持が損なわれることによって作用する可能性のあるTGと認知機能障害との間の因果関係が確認されており、その効果はゲムフィブロジルによっても逆転される可能性がある[109]。しかし、非標的リピドミクス解析を用いて、Proitsiらは、低鎖および極低鎖トリグリセリド(LCTGs/VLCTGs)とアルツハイマー病との間に関連性を見出したが、対照群とアルツハイマー病患者との間に血清TGに差はなかった[110]。また、MCI被験者においてもTGは変化しなかった[111]。
10.2. グリセロ脂質 モノアシルグリセロール(MAG)とジアシルグリセロール(DAG)
MAGとDAGは、MCI状態であるアルツハイマー病の経過の初期に前頭皮質と血漿中で上昇している[111,112]。DAGの上昇は大脳皮質だけでなく、MCI個体の白質にも認められ、ホスファチジルエタノールアミン(PE)のホスホリパーゼ分解と関連している可能性が示唆された[113]。アミロイドβ1-42ペプチドは、SH-SY5Y神経芽腫細胞におけるホスホリパーゼD(PLD)を介したDAGの産生を増強する可能性がある。PLD産生されたDAGは、アミロイドβを介した可溶性アミロイド前駆体蛋白質α(sAPPα)分泌の減少に関与していることが示唆された。
モノアシルグリセロールリパーゼ(MAGLとして知られている)は、MAGの遊離脂肪酸およびグリセロールへの加水分解を触媒し、脳内のエンドカンナビノイド2-アラキドノイルグリセロール(2-AG)を代謝する主要な酵素である[115]。
en.wikipedia.org/wiki/Monoacylglycerol_lipase
MAGLの不活性化はβ-セクレターゼ1(BACE1)の発現を抑制し、アルツハイマー病のマウスモデルにおいてアミロイドβの産生と蓄積を減少させた。また、MAGLを阻害することにより、抗炎症作用や神経保護反応が発現し、アルツハイマー病動物モデルにおいてシナプス機能や認知能力が改善された[115]。MAGL 阻害薬 JZL184 は、APdE9 トランスジェニックマウスモデルにおいて、ミクログリアの炎症反応を低下させ、総 アミロイドβ負荷を減少させた[116]。ダウン症マウスモデルであるTs65DnマウスにJZL184を投与したところ、海馬の長期増強が増加し、アミロイドβ40とアミロイドβ42のレベルが低下したことも明らかになった。また、選択的MAGL阻害薬であるURB602は、シクロオキシゲナーゼ-2(COX-2)上昇やERK1/2,NF-κBリン酸化を抑制するとともに、IκB-α分解を減少させることで、ホモシステイン介在性障害に対する神経保護効果を示した[117]。
11. グリセロリン脂質
11.1. グリセロリン脂質(ホスホグリセリド)
グリセロリン脂質は、細胞膜を規定する脂質の主要なタイプである。ヒトの脳の主要なリン脂質はエタノールアミンホスホグリセリド(35.6%)[118,119]であり、その優勢な形態はエタノールアミンプラスミン(PlsEtns)であり、ホスファチジルエタノールアミン(PE)は残りの量を構成している。ホスファチジルコリン(PC)は、ヒトの脳におけるコリンホスホグリセリドの主要な形態であり、32.8%を占めている[118]。筋肉を除いて、エタノールアミン形質体はコリン形質体の10倍以上である。
アルツハイマー病患者の脳では、PCとPEが有意に減少し、リン脂質脱アセチル化産物であるグリセロホスホコリンが前頭皮質、一次聴覚野、頭頂皮質で増加していた[120]。PE(P-16:0/20:40)およびPE(P-16:0/22:6)の減少は、アルツハイマー病重症度と相関していた。形質生物はγ-セクレターゼ活性を低下させることができ、それらの枯渇の結果、アクレースされたアミロイドβが得られる[121]。PI(16:0/20:4PI(16:0/22:6)およびPI(18:0/22:6)の欠乏は、アミロイドβ42生合成の促進に関与していた。アルツハイマー病では、エタノールアミン含有プラズマローゲンおよび血小板活性化因子(PAF)前駆体の加水分解の亢進は、PlsEtnsおよびPAF代謝産物[すなわち、PC(O-16:0/2:0)およびPE(P-16:0/0:0)]の蓄積をもたらし、これはタウの病理を加速し、小胞放出を亢進し、神経細胞の損失をシグナルする[122]。PAFは免疫細胞の活性化に重要なエーテル-グリセロリン脂質である[123]。
11.2. グリセロリン脂質 エタノールアミンプラスミン(PlsEtn)欠乏とペルオキシソーム機能不全
PlsEtnは、ニューロンにおける全エタノールアミンホスホグリセリドの50%以上、ミエリンにおける含有量の85%以上を占めている[124]。PEと比較して、PlsEtnの選択的欠損がアルツハイマー病患者の脳で決定され、脂質欠損は特に側頭皮質で解剖学的特異性を示した[125]。ヒトの脳では、アルツハイマー病の非常に初期の段階では、前頭前野、頭頂部、側頭部の白質にプラズマローゲンの最大40%の減少があったが、進行した段階では灰白質の30%の減少が発生し、この減少は、疾患の重症度と相関していた[126]。PlsEtnの欠乏は認知症の臨床症状に何年も先行し、その減少の程度は疾患の重症度と相関している[127]。しかし、別の研究では、認知症の若年者と老年者の灰白質ではPlsEtnとPEが減少しているが、MCI群では認知機能が損なわれていない被験者と比較して変化していないことが示されている[113]。プラズマローゲンはその生合成に無傷のペルオキシソームを必要とするため、組織内での減少はペルオキシソーム機能障害と一致している[4]。アルツハイマー病患者におけるペルオキシソーム機能不全の結果としてのPlsEtnとPCの減少は、I-II期のものと比較して、V-VI期の病理学的ステージを持つ皮質領域におけるペルオキシソームβ酸化の基質であるC22:0とVLC脂肪酸s(C24:0とC26:0)の蓄積を伴っていた[128]。ペルオキシソームの喪失は、共焦点レーザー顕微鏡によって、異常なタウリン酸化を伴う神経細胞の過程で実証されている[128]。フーリエ変換赤外顕微鏡では、老人斑とその周辺は酸化脂質の存在として特徴づけられている[129]。したがって、酸化ストレスは、アルツハイマー病の病因と進行の原因である可能性があり、PlsEtnの欠乏は、アルツハイマー病における酸化ストレス仮説をサポートしていることを示している。脳におけるPlsEtnの抗アポトーシス作用は、海馬の神経細胞死の抑制によって示され、それはカスパーゼ-3およびカスパーゼ-9切断の抑制、およびAKTおよびERKシグナル伝達のリン酸化の亢進と関連していた[130]。C57/6 Jマウスにプラズマローゲンとリポポリサッカライド(LPS)を7日間腹腔内注射すると、神経炎症が抑制され、海馬におけるアミロイドβの蓄積が廃止された [131]。60~85歳のMCIおよび軽度アルツハイマー病患者を対象とした多施設無作為化二重盲検プラセボ対照試験では、ホタテ貝から精製したPlsEtnを1mg/日投与し、24週間投与した。治療群では、軽度アルツハイマー病患者はプラセボ群と比較して血漿PlsEtnの減少が小さく、認知機能が改善したことが明らかになった[132]。
11.3. グリセロリン脂質 ホスファチジルコリン(PC
アルツハイマー病では3つのPCが有意に減少していることが判明した。PC(16:0/20:5)PC(16:0/22:6)PC(18:0/22:6)[133]。Mapstoneらは、健康な高齢者患者を対象に5年間の観察研究を行い、MCIまたはアルツハイマー病の血漿中に減少した7つのPC、1つのリゾファチジルコリン、2つのアシルカルニチンからなる10の代謝物を同定し、その減少は、平均して2~3年以内にMCIまたはアルツハイマー病に転化する認知的に正常な個体を(90%以上の精度で)特定できることを明らかにした[134]。コリンプラズマローゲンの73%の減少は、非アルツハイマー病対照と比較してアルツハイマー病患者の死後前頭前野(ブロドマン領域9)で発見された[135]。シチジン-5-二リン酸-コリン(CDP-コリン)は、遊離コリンをホスファチジルコリンとコリンプラズマローゲンに取り込むリン脂質代謝経路に参加している。若年性アルツハイマー病患者にCDP-コリンを1ヶ月間投与すると、精神パフォーマンスの有意な改善がみられた[136]。ほとんどの研究でアルツハイマー病におけるPCレベルの低下が報告されているが、矛盾する所見も報告されている。Proitsiらは、アルツハイマー病と最も強く関連する脂質はPC 40:4とPC 36:3であり、どちらもアルツハイマー病で増加していることを発見した[110]。Kennedyらは遺伝子発現プロファイリングとゲノムワイドスクリーンを組み合わせ、PC(O-16:0/2:0)がアルツハイマー病で上昇することを発見した[137]。コントロール脳と比較して、脳脊髄液 PCの増加がアルツハイマー病で観察された。正常な老化の間に、リゾホスファチジルコリン、コリンプラズマローゲン、およびリゾPAFの血漿レベルが有意に増加する;これらのコリン含有リン脂質の同様の、しかしより顕著な変化は、アルツハイマー病患者で観察された[138]。
11.4. ホスホリパーゼ
ホスホリパーゼは、細胞膜から多価不飽和脂肪酸を解放するためにリン脂質の加水分解を触媒し、切断部位に基づいて4つの異なるクラスに分けることができる。アラキドン酸を放出するホスホリパーゼA2(PLA2)は、アルツハイマー病に関与している[139]。プロ炎症性酵素ホスホリパーゼA2は、低密度リポタンパク質(LDL)および高密度リポタンパク質(HDL)とともに複合体として活性型で血漿中を循環している。血漿中のリポタンパク質関連ホスホリパーゼA2のレベルは、アルツハイマー病患者では高かった[140]。脳脊髄液中の分泌ホスホリパーゼA2活性もアルツハイマー病患者では有意に高かった[141]。
ホスホイノシチド(PI)シグナル伝達経路は、細胞増殖、細胞周期制御、アポトーシス、膜輸送、細胞骨格制御、ホルモン分泌、神経伝達物質シグナル伝達、イオンチャネル活性、細胞および組織の極性、神経組織におけるCa2+制御などの様々な細胞機能の制御に関与している。PI特異的ホスホリパーゼC(PLC)は、神経伝達に関与する重要な酵素の一つであり[142]、アルツハイマー病を含むいくつかの脳障害にリンクしている[143]。PLCは6つのアイソザイム(β、γ、δ、ε、ζ、η)を有する[144]。以前の研究では、アルツハイマー病ではPI特異的なPLC活性は変化しないことが示されていたが[145]、PLCアイソザイムの1つであるPLCδに対する免疫染色により、この酵素がNFTに蓄積していることが示された。クロマトフォーカシングプロファイリングは、コントロールと比較して、アルツハイマー病の脳でPLCγ1の有意な減少とホスホリパーゼCδ1活性の付随的な増加を示した、アルツハイマー病におけるPLCアイソザイムの関与が異なることを示唆している[146]。また、PLCは細胞内Ca2+の調節にも重要な役割を果たしている。PLCηは認知と記憶に関連する脳領域に非常に豊富に存在し、Ca2+信号の変調と増幅に関与している[147]。
ホスホリパーゼD(PLD)は、PCの加水分解をホスファチジン酸(PA)とコリンに触媒する酵素であり、後者はアセチルコリン合成前駆体として作用する。アルツハイマー病 の初期の病理学的変化は、コリン作動性ニューロンから始まる、神経細胞の損失を伴うコリン作動性機能障害によって特徴付けられる。PLD経路は、アミロイド形成において重要な役割を果たすことが実証されている[148]。マウスP19胚性癌細胞におけるAPPの過剰発現はPLD活性を増加させた[149]。アミロイドβは培養神経細胞においてPLD活性を刺激し、PLD活性化がアミロイドβ誘導神経毒性とアルツハイマー病に関与していることを示唆している[150]。PLDは3つのアイソフォーム-PLD1,PLD2,およびPLD3-を有する。PLD1は、おそらくPS1活性を媒介することによって、APP処理のダウンレギュレーションを調節した[148]。アルツハイマー病患者の側頭皮質と海馬からのシナプトソームは、PLD1の発現の増加を示した。PLD1の阻害は、初期段階(アミロイドβ駆動)と後期段階(アミロイドβとタウ駆動)の3xTg-ADマウスの海馬におけるシナプス機能障害をブロックする[151]。PLD3の発現は、海馬や大脳皮質を含むアルツハイマー病に脆弱な脳領域で上昇している[152]。PLD3コーディングバリアントのキャリアは、アルツハイマー病後期発症リスクが2倍に増加する [152,153]。PLD3の過剰発現は細胞内APP、細胞外アミロイドβ40およびアミロイドβ42を有意にダウンレギュレーションするが、PLD3のノックダウンは細胞外アミロイドβ40およびアミロイドβ42を増加させる[152]。しかし、いくつかのフォローアップゲノム研究では、アルツハイマー病におけるPLD3リスクバリアントの影響を再現することができなかった[154,155]。アルツハイマー病発症におけるPLD3の関連性を明らかにするためには、さらなる検証研究が必要である。
11.5. グリセロリン脂質 カーディオリピン
カーディオリピンは、主にミトコンドリア内膜に存在するリン脂質であり、ミトコンドリア電子輸送鎖酵素の流動性と活性の維持に関与している[156]。正常な老化脳は、カルジオリピンの減少とカルジオリピンの過酸化の増加を示し、その結果、ミトコンドリア電子輸送鎖の複合体Iの活性が低下する[157]。シナプスのミトコンドリア膜におけるカルジオリピンの減少は、アルツハイマー病患者の脳で報告されている[158]。リポソームを含むカルジオリピンは神経成長因子のBBBを越える能力を促進し、アミロイドβ1-42の神経毒性を減少させることで神経細胞の生存率を高める。しかし、臨床応用を確認するためには、その後の研究が必要である[159]。
12. スフィンゴ脂質
スフィンゴ脂質には、極頭群に応じて、ホスフィンゴ脂質とグリコスフィンゴ脂質の2つの主要なクラスがある。前者にはスフィンゴミエリン(SM)が含まれ、後者にはセレブロシドとガングリオシドが含まれる[160]。スフィンゴ脂質は、アポトーシス、カルシウムホメオスタシス、タウリン酸化、アセチルコリン生合成、アミロイド生成経路を介してAPPの代謝を促進する[161]。
12.1. スフィンゴ脂質 スフィンゴミエリン(SM)とスフィンゴミエリンナーゼ(スフィンゴミエリン酵素)
SM は脳内で最も豊富なスフィンゴ脂質であり、ミエリン鞘に豊富に存在する[162]。31P核磁気共鳴研究を用いて、SMの増加がアルツハイマー病被験者の脳組織で発見された。アルツハイマー病脳におけるメタボロームアッセイでは、アシル残基を持つ3つのSM(SM C16:0,SM C16:1,SM C18:1)とアシル残基を持つ1つのヒドロキシスフィンゴミエリン(SM (OH) C14:1)を含むSMの高濃度は、アルツハイマー病病理の重症度と認知異常のリスクの増加と関連していることが判明した[161]。しかし、アルツハイマー病におけるSMの変化は研究間で一貫性がなかった。SMは脂質ラフトの重要な構成要素であり、γセクレターゼ阻害剤として作用し、アミロイドβ40およびアミロイドβ42ペプチドの合成を減少させる[163]。
スフィンゴミエリン酵素(SMase)は、スフィンゴミエリンを加水分解してセラミドに触媒する酵素である。スフィンゴミエリン酵素はpHに基づいて、酸性スフィンゴミエリン酵素(ASM)中性スフィンゴミエリン酵素(NSM)およびアルカリ性スフィンゴミエリン酵素(alkSM)の3つの形態に分類される[164]。マウスモデルの研究によると、アミロイドβの脳内注射はスフィンゴミエリン酵素とセラミドレベルを促進した[165]。アミロイドβはNSMを活性化したが、ASMは活性化しなかった。NSMの阻害は、3-O-メチルスフィンゴミエリンまたはアンチセンスオリゴヌクレオチドを用いた遺伝子ノックダウンによるアミロイドβ誘発オリゴデンドロサイトの死を減衰させた[166]。遺伝子発現レベルに応じて、ASMとNSM2はアルツハイマー病においてアップレギュレーションされていた[167]。ASMはリソソソーム糖タンパク質である。アルツハイマー病患者やマウスのデータによると、ASMの活性は血漿、線維芽細胞、脳で上昇しており、これはリソソソームの枯渇によるオートファジー分解不全に寄与していると考えられている[167]。また、家族性アルツハイマー病のマウスモデル(APP/PS1)では、ASMを遺伝子的に部分的に阻害することで、リソソソームの生合成が回復し、オートファゴサイトの欠陥が改善され、その結果、アミロイドβの沈着が減少し、記憶障害が改善された。同様の効果は、APP/PS1マウスにおいて、ASMを薬理学的に正常範囲に回復させた後にも認められた[168]。
en.wikipedia.org/wiki/Acid_sphingomyelinase
12.2. スフィンゴ脂質 セラミドとエクソソソーム
セラミドはスフィンゴ脂質の代謝において、また脂質の第二のメッセンジャーとして重要な役割を果たしている。セラミドは、SMの加水分解から生成されるか、小胞体でde novo合成される。それらは炎症および神経細胞のアポトーシス、特にMAPキナーゼ(MAPK)およびAKT経路と関連している[169]。
リピドミクス研究は、アルツハイマー病脳[167,169,170]、特にセラミドCer16,Cer18,Cer20,およびCer24で増加したセラミドレベルを発見した[167]。老人斑では飽和セラミドCer(d18:1/18:0)とCer(d18:1/20:0)が多く見られた[171]。セラミドのレベルの上昇もまた、アルツハイマー病患者の脳脊髄液 [172]および血清中に認められた。Cer 16:0およびCer 24:0の高ベースライン血漿レベルは、高齢女性におけるアルツハイマー病リスクの増加と一致しており[173]、Cer 22:0およびCer 24:0レベルの上昇は、海馬体積の喪失および認知機能の低下を示唆している。Hanらは、アルツハイマー病の初期段階ではセラミドの増強を観察したが、その濃度は疾患の重症度に伴って減少した[174]。セラミドは、APPの処理を調節するβ-セクレターゼ酵素BACE1の安定化によってアミロイドβ形成を増強する。アミロイドβ形成は、正のフィードバックループとしてのスフィンゴミエリン酵素によるセラミドへのSMの分解を触媒することにより、セラミドのレベルの増加を誘導する[169]。
エクソソソームは、40~150nmの範囲の直径を持つナノ粒子であり、多胞体(MVB)の内側への出芽によって生成され、MVBが細胞膜と融合する際に細胞から分泌される。細胞外小胞(EV)は、セラミドや他のガングリオシドと同様に濃縮されたエクソソソームである[175]。最近の研究では、脳内のエクソソーム分泌の増加が、エクソソームがアミロイドβ凝集を加速することができるため、アルツハイマー病の進行に積極的な役割を果たすことができることがわかった[176]。セラミドはエキソソソームの多包性エキソソームへの出芽を誘発するが、神経細胞におけるエキソソーム分泌はNSM-2を阻害することで有意に減少させることができる[177,178]。NSM2 欠損 5X家族性アルツハイマー病 マウスにおけるエクソソソーム分泌の減少は、5X家族性アルツハイマー病 アルツハイマー病 モデルマウスと比較して、グリア細胞活性化、タウリン酸化と総 アミロイドβプラーク沈着を減少させ、認知機能を改善し、アルツハイマー病 病因を改善した [179]。
12.3. スフィンゴ脂質 スフィンゴシン1-リン酸塩(S1P)
S1Pは、スフィンゴシンキナーゼ(SphKs)によるセラミドとスフィンゴシンの加水分解により生成され、スフィンゴシンキナーゼ1(SphK1)とスフィンゴシンキナーゼ2(SphK2)を含む2つのアイソザイムを有するスフィンゴシンキナーゼ(SphKs)によって生成される。スフィンゴシン1-リン酸リアーゼ(SPL)はS1Pを分解することができる。アルツハイマー病脳ではS1Pのレベルが低下していた [180,181]。S1Pを蓄積したSPLの欠損は、リソソーム中のAPPやアミロイド原性C末端断片の分解を阻害するとともに、γ-セクレターゼの活性を低下させ、アミロイド生成を減少させる[182]。Sphk1はプロサバイバルシグナル伝達メディエーターである。グルコースリロードストレス/グルコース欠乏は海馬ニューロンにおける Sphk1 の発現と活性をアップレギュレートした [183]。SphK1の発現低下とSPLの発現増加は、プロサバイバルS1Pの損失とともに、アルツハイマー病脳の内葉皮質におけるアミロイドβの蓄積に関連していた[184]。逆に、Sphk2はアルツハイマー病の脳でアップレギュレーションされた。A S1Pトランスポーター スピンスターホモログ2(Spns2)は、生体内試験および試験管内試験で活性化されたミクログリアのプロ炎症反応を増強した。マウスのSpns2ノックアウトは、アミロイドβ42誘発性ワーキングメモリの障害を改善した[185]。S1P受容体の刺激は、いくつかの試験管内試験および生体内試験のADモデルにおいて有望と思われる[186]。これらの研究は、スフィンゴ脂質代謝がアルツハイマー病の病態において中心的な役割を果たしていることを示唆している。
12.4. スフィンゴ脂質 スルファチド
サルファチドは、ほぼすべてのオリゴデンドロサイトで合成されるミエリンの必須成分であり、オリゴデンドロサイト膜の安定化に関与している。サルファチドの急激な減少はアルツハイマー病の初期に観察されたが、進行期までは濃度の変化はほとんど見られなかった[187]。PIは一定であるが、脳脊髄液中の硫酸化物もアルツハイマー病の初期には減少する。硫化物.PI比がアルツハイマー病初期のマーカーではないかと提案された。PI比がアルツハイマー病の早期診断のマーカーとなる可能性が示唆されている[188]。ApoEはAPPトランスジェニックマウスにおける硫化物の代謝/移動/恒常性を媒介し、硫化物レベルを調節する[189]。ApoE4の過剰発現は、トランスジェニックマウスの脳内スルファチド濃度の60%の低下を引き起こした。サルファチドは、アミロイドβのApoE関連粒子への結合を増強し、アミロイドβの取り込みを促進し、これはリソソームにおけるアミロイドβの蓄積につながる[189]。
12.5. スフィンゴ脂質 ガングリオシド
ガングリオシドと呼ばれる酸性のグリコスフィンゴ糖脂質は、その糖質部分の内側のガラクトシル残基に1つ以上のシアル酸が結合している。ガングリオシドは中枢神経系に多く存在し、脂質ラフトや外側の血漿膜に好んで集積している。ガングリオシドは、シアル酸残基の数により,0(またはアシアロ)a、b、cの系列に分類される。
アルツハイマー病脳の基底側脳と前頭皮質と側頭皮質でガングリオシド系列(GM1,GD1a、GD1bおよびGT1b)の有意な減少があったが、これはおそらく皮質ニューロンの変性と相関している[190]。ガングリオシドのサブタイプである “a”-ガングリオシド(GM1とGD1a)と “b”-ガングリオシド(GD1bとGT1b)の分析は、”b”-ガングリオシドがアルツハイマー病患者に優先的に影響を与え、異なる研究間で一貫して減少を示したことを示した[190]。主要なガングリオシドと比較して、GD3,GM2,GM3,GM4 のような単純なガングリオシドは、アルツハイマー病 脳の前頭部と頭頂皮質で増加しており、これは神経細胞死の間に加速されたアストログリオシスおよび/またはガングリオシドのリソソソーム分解と相関している可能性がある。
可溶性アミロイドβは、生理的条件下でガングリオシドを含む脂質ラフトに結合するために高い親和性を発現する[191]。ガングリオシドが介在するアミロイドβの構造変化に起因する独特のガングリオシド結合型のアミロイドβ(Gアミロイドβ)がアルツハイマー病脳で発見された[192,193]。ガングリオシドはアミロイドβ凝集と細胞毒性を調節することができるが、GM1ガングリオシドに結合したアミロイドβは最も強いアミロイドβ播種能を示した[192,194]。D-およびL-トレオ-1-フェニル-2-デカノイルアミノ-3-モルホリノ-1-プロパノールを用いたグリコスフィンゴ脂質の枯渇は、内因性APPおよびアミロイドβの分泌を著しく抑制した。逆に、外因性脳ガングリオシドの添加はこれらの効果を逆転させた[195]。
アルツハイマー病被験者の内耳皮質では、SM、ガングリオシドであるGM3,リゾビスホスファチジン酸、コレステロールエステルの豊富な量が認められ、病態生理は内因性の内分泌障害と関連していることが示唆された。GM3とコレステロールエステルの増強は、家族性アルツハイマー病トランスジェニックマウスモデルで再現された。ホスホリパーゼD2の遺伝的アブレーションはGM3レベルを完全に正常化し、シナプスおよび行動障害を回復させた。この研究は、ガングリオシド、ホスファチジン酸とホスホリパーゼD2の生成物の代謝との間のクロストークを示唆している、アルツハイマー病病因におけるガングリオシド異常の重要な役割を示す[112]。
GM1は老化マウスにおいて、D-ガラクトース損傷から海馬の神経新生を保護することができる[196]。Bシリーズガングリオシド、特にGD3は、試験管内試験で神経幹細胞の自己再生能力の維持を制御している[197]。LIGA20,LIGA4およびPKS3のような半合成GM1は、親の天然化合物よりも活性が高く、より速く、より長く作用し、グルタミン酸誘発性神経細胞死の強力で効果的なアンタゴニストである。AlaらはGM1の筋肉内注射を12週間使用したのに対し、Flickerらはアルツハイマー病患者を治療するために6週間同じレジメンを使用した。安全ではあるが、治療は軽度から中等度のアルツハイマー病患者に認知的利益を提供しなかった[198]。
Svennerholmらは、早期発症のアルツハイマー病患者にGM1を1年間脳室内投与することでアルツハイマー病の進行を止め、運動機能や読解力、言語感覚などの認知機能を改善したと報告している[199]が、GM1の長期投与はアルツハイマー病患者に有用である可能性を示唆している。松岡らは、PS/APPマウスにGM1を2日おきに2週間末梢投与すると、若齢マウスの脳内アミロイドβが減少したが、重度のアミロイドβ負荷を持つ6-7ヶ月齢マウスの脳内アミロイドβは減少しなかったことを報告しており、GM1の早期投与が脳アミロイドーシスの軽減や予防につながる可能性を示唆している[200]。Yangらは、アミロイドβ1-40注射によるADモデルラットの海馬歯状回へのGM1のマイクロインジェクションが、海馬におけるマロンジアルデヒド(MDA)とHNEレベルの同時減少による脂質過酸化と酸化ストレスの減少を介して学習と記憶障害を改善することを実証した[201]。
13. コレステロール
ヒトや動物の主要なステロール脂質はコレステロールである。脳内コレステロールの主な供給源は、BBBが血漿リポタンパク質が効率的に脳内に入るのを妨げるため、de novo生合成に由来する[202]。神経細胞へのコレステロール供給が不足すると、シナプス可塑性や神経伝達が損なわれ、タウ病理や神経変性を誘発する[203]。
コレステロールとコレステロールエステルはアミロイド形成において重要な役割を果たしている[202,204]。コレステリルエステルや遊離コレステロールの低発現は、アミロイドβ産生の増加と相関し、神経細胞膜コレステロールの損失はアミロイド発生を引き起こした[205]。老化の間、神経細胞からのコレステロールの枯渇もまた、神経伝達の障害、シナプスの喪失、タウ病理の亢進、神経細胞の死と関連している[206]。
逆に、コレステロールは動物モデルにおいて脳アミロイドーシスを誘発または増悪させた [66,207]。コレステロールの上昇はアミロイドβ形成に関与しており、アルツハイマー病患者の初期段階で観察された[208]。アミロイドβの産生は主に脂質ラフト中のβ-セクレターゼ1(BACE1)レベルによって決定され、βAPPを切断してアミロイドβを生成する酵素である[204]。高コレステロールは、α-、β-、およびγ-セクレターゼのすべてのタイプのAPPタンパク質分解性セクレターゼを調節することを含む、様々な経路でAPPの処理に影響を与える。コレステロールはまた、その線維化、輸送、分解、およびクリアランスプロセスを含む多くの側面でアミロイドβ代謝を媒介する[209]。家族性高コレステロール血症のマウスモデルでは、アミロイドβ誘導神経毒性に対する感受性を高めるために、BBB透過性と酸化ストレスが増加している[210]。高コレステロール血症食は動物モデルのアルツハイマー病病理を加速させ、有意に増加したアミロイドβ負荷を引き起こした[207]。1%のコレステロールを7ヶ月間摂取させたウサギでは、神経細胞内のコレステロールが増加し、BACE1レベルの上昇と海馬におけるアミロイドβ42とリン酸化タウの蓄積を伴っていた[211]。コレステロール低下薬であるスタチンは脳内のアミロイドβの蓄積を減少させた[212]。別の研究では、コレステロール保持はBACE1の活性に直接影響を与えないが、BACE1を提示する脂質ラフトでβAPPのクラスタリングと転位を誘導し、βAPPの切断が起こるエンドソームに急速に内包され、アミロイドβ産生の増強につながることが示された[213]。
MCIおよび認知症のない参加者は、認知症のある参加者に比べて初診時の総コレステロールが低かった[214]。家族性高コレステロール血症患者ではMCIの発生率が高く、最終的に大多数の症例でアルツハイマー病に進行した[215]。アルツハイマー病患者の死後脳サンプルでは、HDLのレベルが有意に低く、LDLコレステロールのレベルが高いことが示された[216]。ある集団ベースの研究では、中年期に高血圧と組み合わせたコレステロールが、後年のアルツハイマー病のリスクを有意に上昇させることが明らかになった[217]。ほとんどの研究とは対照的に、Framingham Heart Studyでは、高コレステロール血症は認知機能の改善と関連していた。アルツハイマー病、MCI、対照者の血漿サンプルのLC/MSを用いても、コレステロールとアルツハイマー病との関連は認められず[218,219]、スタチンを処方されたアルツハイマー病患者と対照者の頻度に差はなかった[110]。
コレステロールとは異なり、24S-ヒドロキシコレステロール(24S-OHC)や27-ヒドロキシコレステロール(27-OHC)のようなオキシステロールとして知られている酸化コレステロール代謝物はBBBを通過することができ、アルツハイマー病において重要な役割を持つことがますます認識されている[219]。脳内コレステロールの恒常性は、コレステロールの生合成と血液と脳の間のオキステロールの拡散によって決定される[220]。コレステロールのレベルが生理的基準を超えると、24S-OHCに変換され、神経毒性のために神経細胞から積極的に排除される[221]。アルツハイマー病初期では、より高い24S-OHCレベルが血漿中に認められる[222]。しかし、アルツハイマー病の慢性期および進行期では血清中の24S-OHCレベルが低下しており、血清総コレステロールが認知症リスクに及ぼす影響は中年期には生じるが、晩年には生じないことを示す臨床観察と一致している。アルツハイマー病の後期では24S-OHの減少があるが、27-OHCや25-ヒドロキシコレステロールなどの他のオキシステロールは有意に増加している[223]。27-OHCの高レベルは、散発性アルツハイマー病と同様に若年性アルツハイマー病の脳と脳脊髄液で発見されている[224]。27-OHC治療は試験管内試験でタウリン酸化とアミロイドβ産生を増加させ[225,226]、レチノイドX受容体γ(RxRγ)[227]を介して試験管内試験および生体内試験で樹状突起棘の損失を引き起こしたが、シナプス機能および可塑性に対する27-OHCの作用はまだ不明である。マウスのCYP27A1遺伝子ノックアウトは、27-OHCを枯渇させ、高コレステロール食によって誘発される記憶障害を改善することができ、これは27-OHCが食事性コレステロールによって引き起こされる記憶障害の主な原因であることを示している[228]。コレステロール値とアミロイド生成またはアルツハイマー病リスクとの間のこれらの相反する結果は、脳コレステロールのホメオスタシスが厳密に制御されており、低値でも高値でもアルツハイマー病につながる可能性があることを示唆している。
en.wikipedia.org/wiki/27-Hydroxycholesterol
www.ncbi.nlm.nih.gov/pmc/articles/PMC6653998/
pubmed.ncbi.nlm.nih.gov/25453744/
14. アポリポタンパクE(ApoE)
ApoEはリポタンパク質の主成分であり、脳内のコレステロールおよびリン脂質の輸送を媒介する[209]。E2,E3およびE4はApoEの重要なアイソフォームであり、それぞれが異なる対立遺伝子(ε2,3および4)によってコードされている。ApoE3はヒトでは主要なアイソフォーム(77-78%)であり、ApoE4とApoE2はそれぞれ14-15%、7-8%を占めている[229]。ApoE4は大型でTGが豊富な超低密度リポタンパク質(VLDL)に結合し、ApoE2とApoE3は小型でリン脂質が豊富な高密度リポタンパク質(HDL)に優先的に結合する。ApoE2は総コレステロール値を低下させるのに対し、ApoE4は総コレステロール値を上昇させる。
19番染色体上のAPOE ε4は、散発性アルツハイマー病の約50%に寄与するアルツハイマー病の最も有病率の高い遺伝的危険因子として知られている[209]。ApoE2はアルツハイマー病のリスクを減少させるが、ApoE4対立遺伝子はアルツハイマー病のリスクの増加と発症年齢の低下をもたらす。1つのε4対立遺伝子はアルツハイマー病のリスクを3倍に増加させるのに対し、2つの対立遺伝子は12倍の増加を与える [231]。APOE ε4対立遺伝子のホモ接合体を持つ個体は、血漿中のコレステロールレベルが高く、脳脊髄液中の24S-OHCレベルが上昇している [232]。リン脂質の血漿中および脳内アラキドン酸/DHA比が高いことが、APOE ε4キャリアを発現する家族性ADマウスモデルでは、APOEアイソフォームを発現する個体と比較して観察された[233]。
さらに、ApoE4キャリアは、食事中のn-3 脂肪酸の欠乏によって顕著に影響を受ける。n-3 脂肪酸欠乏の食事を摂取した場合、ApoE4を保有するマウスは、同じ食事を摂取した他のAPOEマウスと比較して、組織や臓器のn-3 脂肪酸レベルの低下が大きかった。できるだけ早くDHAを補給することで、病気の進行を抑制し、ApoE4マウスの神経学的・行動学的欠損を逆転させることができる可能性がある[234]。
ApoEはLDL受容体関連タンパク質に結合することでアミロイドβの内部化を媒介し、アミロイドβクリアランスに影響を与え、アミロイドβ凝集を促進する[235]。ApoEを欠損すると、脂質ラフト中のアミロイドβ量が減少し、アミロイドβフィブリルの形成に失敗した[236]。ApoEは、イソホルムおよび用量依存的な方法で老人性プラーク負荷に影響を与える(ApoE4 > ApoE3 > ApoE2)[237,238]。ApoE4はアミロイドβの凝集および沈着を促進し、脳内のアミロイドβクリアランスを損なう[238,239,240]。
ApoEはまた、脳リン脂質のホメオスタシスの重要な決定因子であり、ApoE4アイソフォームはこのプロセスにおいて効果的ではない。Zhuらは、ApoE4キャリアの死後のヒトの脳組織は、アルツハイマー病の初期段階でApoE3対応するものと比較して、ホスホイノシトール二リン酸(PIP2)の低いレベルを持っていたことを発見し、同様の結果はまた、ApoE4対立遺伝子を発現する初代ニューロンとApoE4ノックインマウスの脳で発見された。ApoE4キャリアでは、PIP2分解酵素であるホスホイノシトールホスファターゼであるシナプトジャニン1(Synj1)を遺伝子的にノックダウンすると、脳内のPIP2の恒常性が回復し、認知障害が回復した[241]。Synj1の発現増加に伴う二次的なPIP2の変化は、以前の研究で言及されている[242]。
15. スタチン
スタチンはコレステロール生合成律速段階の調節因子である3-ヒドロキシ-3-メチル-グルタリル-CoA(HMG-CoA)還元酵素を阻害する。国民健康保険のデータセットから、スタチンを早期に使用したアルツハイマー病患者では、スタチンを使用していない患者よりもアルツハイマー病の進行が統計的に減少した[243]。サチンはアルツハイマー病のリスクを67~73%に低下させた[244]。ApoE4を有するアルツハイマー病患者にスタチン治療を行った場合、治療を行わなかった場合と比較して、10年間の追跡調査で認知機能障害が少ないことが示された[245]。Canadian Study of Health and Agingでは、80歳未満では認知症の発症率を低下させるのにスタチンの効果が認められたが、80歳以上では認められなかった[246]。これは、晩年のコレステロール低下が認知症リスクに影響を与えないことを示した以前の研究と一致しているようである。スタチンの有益な効果を示した研究の中でも、アルツハイマー病リスクの低下は、スタチンの種類、性別、人種/民族によって異なる。シンバスタチンとアトルバスタチンは、プラバスタチンとロスバスタチンと比較して、異なる人種や性別でより一貫した効果を示しているようである[247]。2つのRCT-Heart Protection study (HPS) [248]とPROSPER study [249]は、スタチンによるコレステロール低下が認知症を予防するという概念に強く反論した。軽度から中等度のアルツハイマー病患者を対象とした2つのRCT(アトルバスタチンを72週間使用し、その後8週間の休薬を行ったLEアルツハイマー病e試験[250]と、シンバスタチンを24ヶ月間使用した別の試験[251])では、疾患の進行に対する効果は示されなかった。
スタチンはタウリン酸化を抑制し[252]、BACE1およびAPP産生を減少させ[253]、アミロイドβと直接相互作用してアミロイドーシスを減衰させることができる[254]。シンバスタチンとアトルバスタチンは、アストロサイト上のネプリライシン(NEP)の細胞外アミロイドβ分解を、ERK介在性経路を誘導することで増加させた[255]。
アミロイドーシスに対するスタチンの効果は、コレステロール生合成とは独立した経路が関与している可能性がある。メバロネート経路としても知られるHMG-CoA還元酵素経路は、コレステロールに加えてイソプレノイド、ゲラニルゲラニルピロリン酸(GGPPおよびイソプレノイド中間体であるファルネシルピロリン酸(FPP)を合成する[256]。FPPとGGPPの高い皮質レベルは有意にタウリン酸化、NFT密度と若年性アルツハイマー病と相関している。皮質のhFPPSとhGGPPS mRNA発現は正にアルツハイマー病個人のHMG-CoA還元酵素のレベルと相関しているが、組織コレステロールのレベルではなく[257]。ADマウスモデルでは、アトルバスタチンはFPPを減少させることで抗炎症効果を発揮する[258]。また、シンバスタチンはNF-κBを減少させることで神経炎症反応を改善し、酸化的損傷を救済し、海馬細胞のアポトーシスを減衰させた[253,259]。
16. ATP結合カセット(ABC)トランスポーター
ABCトランスポーターは、細胞の恒常性を維持するために様々な分子の積極的な輸送を媒介する細胞内小器官の膜と同様に、細胞膜にも存在している[260]。ABCトランスポーターは、脳内のコレステロールやリン脂質の輸出を媒介することで、脂質の恒常性の重要な調節因子となっている。
ABCサブファミリーA(ABCA)のうち、脳内には6つのトランスポーター(ABCA1,ABCA2,ABCA3,ABCA5,ABCA7,ABCA8)が発現している[261]。現在、ABCAファミリーの4つのメンバーがアルツハイマー病と関連していることが報告されている(ABCA1,2,5,7)。ABCA1はApoEを介してアミロイドβ形成を調節する。ABCA2は、APPのβ-またはγ-セクレターゼ切断を促進する。ABCA5はアミロイドβの分泌を阻害し、ABCA7はアミロイドβの取り込みとクリアランスを媒介する[7]。
ABCA1はコレステロールトランスポーターである。ABCA1とリポ蛋白結合は脂質の排出に重要である。ABCA1欠損マウスは脳脊髄液中のコレステロール量が減少していた[262]。ABCA1の機能喪失変異は、より高いアルツハイマー病リスクと強く関連している[263]。ABCA1の低発現はアミロイドβのクリアランス障害をもたらすが[262,264]、高発現はマウスADモデルにおけるアミロイドの沈着を阻害する[265]。ABCA1欠損はAPP/PS1トランスジェニック背景のApoE4マウスではアミロイドβ凝集を増加させたが、ApoE3マウスでは増加しなかったことから、アミロイドβクリアランスに対するABCA1の効果はApoEアイソフォームに依存することが示された[266]。
ABCA2は低密度リポ蛋白質受容体を介して脂質代謝を調節している[267]。前頭前皮質組織と血液からのマイクロアレイ遺伝子発現データセットは、対照群と比較してアルツハイマー病ではABCA2の過剰発現が見られることを示した。ABCA2のmRNA発現とメチル化はアルツハイマー病のリスクと関連している[268]。ABCA2はアミロイドβとコロケーションしていた[269]。ヒト胚性腎細胞およびN2a神経芽腫細胞におけるABCA2の試験管内試験での過剰発現は、転写の増加を介したAPP遺伝子の発現の増加と関連し、BACE1によるAPPの切断を促進した[269]。ABCA2のノックダウンはAPPのγセクレターゼ処理を変化させ、試験管内試験および生体内試験でのアミロイドβ産生を減少させた[270]。
ABCA5はアミロイドβのクリアランスではなく、アミロイドβ産生に作用する。ABCA5はAPPのmRNAおよびタンパク質レベルを変化させることなく、アミロイドβ40およびアミロイドβ42の生成を減少させることから、アミロイドβレベルの減少はAPPの処理の変調によるものであることが示唆されている[271]。
GWASでは、ABCA7が遅発性アルツハイマー病の遺伝的危険因子であることが明らかになっている[272]。ABCA7はコリンリンリン脂質を輸出し、lysoPCはその主要な脂質基質の一つである[273]。ABCA7発現はマクロファージやミクログリアを含むヒト食細胞で上昇しており[261]、ERKシグナル伝達を介してアポトーシス細胞のクリアランスを媒介している[274]。したがって、ABCA7欠失はアミロイドβの食細胞クリアランスを減少させた[275]。ABCA7の過剰発現は、ADマウスのアミロイドβ沈着を減少させ、認知行動を改善した。一方、ABCA7の過剰発現は小胞体ストレスを減少させ、細胞生存率を促進することでアミロイドβ神経毒性を緩和した[276]。ABCA7はアルツハイマー病に対する保護作用があるように思われるが、逆にアルツハイマー病個体ではABCA7の発現が増加している。アルツハイマー病で観察されるABCA7の増加は、不十分な代償的変化を反映していると提案された[277]。ABCB1のレベルは低下し[278]、BBBでの活性はアルツハイマー病患者では有意に低下する[279]。ABCB1は、アミロイドβ40およびアミロイドβ42と直接相互作用することにより、脳毛細血管内皮細胞の先端膜を横切るアミロイドβ輸送を積極的に媒介する[280]。
ABCG2はアミロイドβと直接相互作用し[281]、BBBを介したアミロイドβ40およびアミロイドβ42の排出を促進し、それによって脳内のアミロイド沈着を減少させる。ABCG2は活性酸素種(ROS)の発生を防ぎ、ROS-応答性NF-κB経路の活性化を促進し、結果として炎症性遺伝子の発現を減少させる。ABCG2の過剰発現はアミロイドβ産生を減少させ、これはアミロイドβPP処理酵素の活性に対する活性酸素の正の調節効果の阻害と関連している可能性がある[282]。反対に、ABCG2のmRNAとタンパク質発現は、アルツハイマー病脳で強くアップレギュレーションされていることが判明した[281]。アルツハイマー病におけるABCG2のアップレギュレーションは、NF-κBシグナル伝達経路とそれに伴う炎症性反応を抑制するための酸化ストレス時の代償メカニズムである可能性がある[282]。
17. モノホスホリル脂質A
モノホスホリル脂質A(MPL)は、リポ多糖類(LPS)由来のToll様受容体4(TLR4)アゴニストであり、LPSに似た免疫応答を促進することができるが、より強力ではない [283]。APPを過剰発現するトランスジェニックマウスでは、MPLをアジュバントしたアミロイドβ42を免疫化すると、脳内アミロイドβの蓄積が60%減少した[284]。非ヒト霊長類にアミロイドβ42とMPLをアジュバントした免疫化を行うと、アミロイドβの大きさがより小さい種にシフトし、脳からの毒性アミロイドβの除去が容易になる可能性がある[285]。
試験管内試験と生体内試験の両方の研究で、MPLがミクログリアによるアミロイドβの取り込みを刺激することが示されている。APP/PS1マウスにMPLを繰り返し腹腔内注射したところ、脳内でのアミロイドβ産生が減少し、認知障害が回復した[283]。MPLの神経保護性は、強い炎症性反応を誘発することなくアミロイドβのミクログリアの貪食を刺激する能力によるものと考えられる[286]。マウスへのLPSの経口投与は腹膜マクロファージを活性化し[287]、TLR4経路を介して一次ミクログリアによるアミロイドβ1-42の貪食活性を高める可能性がある[288]。アミロイドβ1-42を投与したラットにMPLまたはTLR2アゴニスト-Pam3Cysを低用量で脳室内投与した研究では、記憶機能が改善された;アミロイドβによって誘導される障害された長期増強が回復した;TNF-αとアミロイドβの沈着が減少した;ミクログリアマーカーであるアルギナーゼ1の発現が増強された;海馬ミクログリアの分極が増加して抗炎症表現型になった;などの結果が報告されている[289]。
18. 脂溶性ビタミン ビタミンA、D、E
18.1. ビタミンA
ビタミンA、プロビタミンAカロテノイド、ビタミンA誘導体レチノイドは抗酸化物質と考えられている。アルツハイマー病患者では、血清中のβ-カロテンとビタミンAのレベルが対照と比較して有意に低下した[290]。ビタミンA欠乏はアミロイドβ蓄積を促進し[291]、認知機能低下の割合は高齢者におけるビタミンAの血清レベルと負の相関があった[292]。トランスジェニックADマウスをビタミンAで8週間腹腔内投与したところ、脳タウリン酸化とアミロイドβの沈着が減少し、ミクログリアとアストロサイトの活性化が減少し、神経変性が減衰し、空間記憶と学習が改善された[293]。動物モデルでのいくつかの有望な結果にもかかわらず、アルツハイマー病治療におけるビタミンAまたはカロテノイドのためのヒト臨床試験が不足している。
18.2. ビタミンD
ビタミンDはステロールのカテゴリーに属するセコステロイドに属するが、議論の便宜上、ここではプレノール脂質に分類される他の2つのビタミンと一緒に配置されている。ヒトの研究では、低循環25-ヒドロキシビタミンD(25-OHD)レベルと認知症との間に相関関係があることが明らかにされている[294]。アルツハイマー病海馬CA1細胞では、ビタミンDホルモン受容体(VDR)mRNAがダウンレギュレートされている[295]。VDRは脳の可溶性・不溶性アミロイドβを減少させる役割を持っている[296]。ADラットモデルでは、ビタミンD3を濃縮した食事は、アミロイドβペプチドとアミロイドプラークの減少、炎症の減少、脳内の神経成長因子の増加をもたらした。また、ビタミンDは加齢に伴う炎症状態の増加を抑制した。その結果、ビタミンDのサプリメントは、老化とADモデルの両方で学習と記憶のパフォーマンスを向上させた[297]。
18.3. ビタミンE
ビタミンEは4つのトコフェロールと4つのトコトリエノールで構成され、抗酸化作用を持つ。すべてのビタミンE異性体の中で、RRR-α-トコフェロールは生体内での生理活性が最も高く、ヒトに必須の唯一のアイソフォームである[298]。ビタミンEは、DHAやアラキドン酸などの不飽和脂肪酸が豊富で、脳や網膜に高濃度で存在する。細胞膜内に存在し、膜の安定化、DHAの酸化損傷からの保護、膜修復の促進などの機能を持つ[298,299]。2018年に実施されたメタアナリシスでは、いくつかのケースコントロール研究やゲノムワイドな関連研究では関連性は認められなかったが、アルツハイマー病患者では認知的に正常な被験者に比べてビタミンEのレベルが低いことが明らかになった[300]。動物実験では、アミロイドβ負荷を減少させることで認知機能の低下を緩和するビタミンEサプリメントの効果が示されている[303]。また、ビタミンE治療は中等度の重度のアルツハイマー病患者では病気の進行を遅らせる。しかし、4つの二重盲検無作為化試験と同年に発表された別の1つの試験を含む2017年のコクランレビューでは、ビタミンEが認知症への進行を予防するというエビデンスはなく、MCI、認知症、アルツハイマー病患者の認知力を改善することもなかった[304,305]が、Dyskenらが行った1つの研究を除いては、ビタミンE投与群では1年あたり19%の臨床的進行の遅延があり、機能低下が遅くなることが示されている[306]。最近の研究では、患者のベースラインのビタミンEレベル、治療に使用されるビタミンEのアイソフォームまたは供給源、遺伝的変異など、ビタミンEサプリメントのバイオアベイラビリティと有効性に影響を及ぼす可能性のある因子が指摘されている[307,308]。
19. リピドミクス研究
リピドミクス解析では、アルツハイマー病患者は健康なコントロールと比較して、エーテルステロール、PC、SM、リン脂質が減少していることが特徴である[309]。Kimらによると、アルツハイマー病の血漿中に14の有意に上昇した脂質があり、PE、DAG、TG、セラミドを含むLDL/VLDLの2倍以上の増加を示した。3種の脂質(TG 50:1,DAG 18:1_18:1,PE 36:2)は脳萎縮の程度と高い相関を示し、MMSEと併用した場合、MCIの初期段階の鑑別の候補として利用することができた[275]。LC/MSベースの非標的メタボロミクスアプローチを用いて、Trushinaらは、認知的に正常な人と比較した場合、スフィンゴ脂質とコレステロール輸送がアルツハイマー病患者の血漿と脳脊髄液の両方で変化していることを発見した[310]。
20. 結論
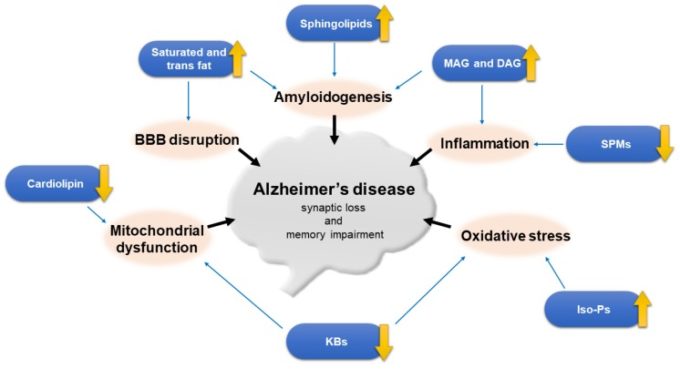
脂質のホメオスタシスの異常は加齢と関連しており、アルツハイマー病の病態に大きく寄与している。脂質異常とアルツハイマー病との関連メカニズムは、腸内細菌叢や腸-脳軸の変化、神経細胞のシグナル伝達経路、BBBの障害、ミトコンドリア機能不全、酸化ストレス、炎症などで構成されており、これらが一緒になってシナプス機能の低下、最終的には記憶障害につながると考えられている。いくつかの脂質は、アルツハイマー病の発症に不可欠な役割を果たし、バイオマーカーとして提案されている(図1)。アルツハイマー病脳内の脂質の障害は、次のカテゴリに分類される。
図1 アルツハイマー病の病因における脂質の役割
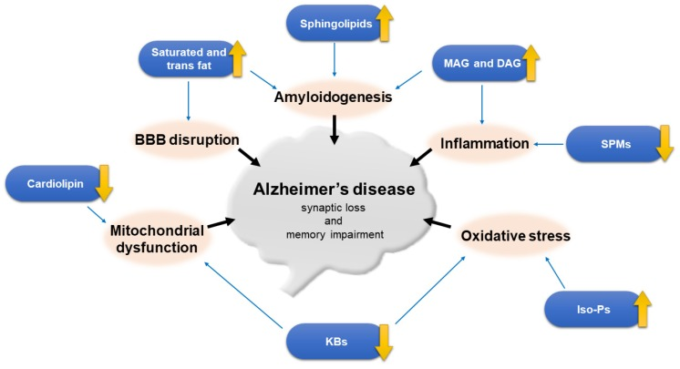
- 飽和脂肪とトランス脂肪は、BBB機能障害とアミロイドβ凝集につながるだろうし、強くアルツハイマー病のリスクに関連付けられていたので、可能であれば、食事から排除し、不飽和脂肪で置きかえる必要がある。
- アラキドン酸(アラキドン酸)などのn-6 脂肪酸は認知機能の面で脳に有害であるが、多価不飽和脂肪酸のうち、n-3とn-9 脂肪酸は有益である。DHAのレベルは、アルツハイマー病の脳で減少しており、そのサプリメントは、早期に与えられた場合は特に治療的に有望であるが、いくつかの無作為化比較試験は認知的な利点を示していなかった。特異的炎症収束性脂質メディエーター(SPMs)と呼ばれる多価不飽和脂肪酸sの脂質メディエーターは、抗炎症反応に重要な役割を果たし、アルツハイマー病の脳で減少している。F2-イソプロスタンス(F2-IsoPs)やNeuro-Psなどのイソプロスタンスは、アルツハイマー病患者における酸化ストレスや脂質過酸化の増加の結果として増加している。
- トリグリセリド(TG)やジアシルグリセロール(DAG)などの糖脂質の上昇は、アルツハイマー病との相関が報告されているが、それらの役割はあまり定義されていない。モノアシルグリセロールリパーゼ(MAGL)阻害剤は、アルツハイマー病の病態を改善する可能性があると考えられている。
- グリセロリン脂質は細胞膜の主要な脂質成分である。エタノールアミン形質転換体はアルツハイマー病脳では早期に減少する。コリン形質転換体の変化はあまり一貫性がないが、リン脂質脱アセチル化産物、例えばグリセロホスホコリンが増加している。ホスホリパーゼA2(PLA2)とD1(PLD1)はアルツハイマー病で増加する。
- スフィンゴ脂質のうち、スフィンゴミエリンとセラミドのレベルの増加は、いくつかの研究ではスフィンゴミエリンの減少または変化がないことが示されたが、アルツハイマー病の脳で見られる。スフィンゴミエリン酵素、特に酸性スフィンゴミエリン酵素と神経スフィンゴミエリン酵素2は、アルツハイマー病でアップレギュレートされている。スフィンゴシン1-リン酸(S1P)とスルファチドはアルツハイマー病の初期段階から枯渇する。ガングリオシリーズガングリオシドが減少している一方、GM2,GM3,GD3,GM4 などの単純ガングリオシドは、アルツハイマー病 の脳で上昇している。
- アルツハイマー病上のコレステロールの影響は、動物とヒトの両方で疫学的およびリピドミクス研究の間で議論の余地がある。それは、中年期の血漿コレステロールの増加レベルが上昇した アルツハイマー病 リスクと関連していると思われるし、スタチンの使用は、晩年のコレステロール値が アルツハイマー病 リスクと関連していないのに対し、この年齢で導入されたときに有益と思われる、この年齢でのスタチン治療も認知症のリスクに影響を与えない。27-ヒドロキシコレステロール(27-OHC)や24S-ヒドロキシコレステロール(24S-OHC)などのオキステロールはアルツハイマー病患者の脳脊髄液や脳内で上昇するが、24S-OHCのレベルは疾患経過の後期に低下する。したがって、脳内コレステロールのホメオスタシスは適切な範囲内で調節されるべきであり、低値でも高値でも全く健康的ではない。ApoE4対立遺伝子の保有状況は、対立遺伝子に依存した方法で発症年齢の低下とアルツハイマー病のリスクの増加を示唆している。ABCトランスポーターはコレステロール輸送に関連しており、アミロイドβ排出とアミロイド生成に重要な役割を果たしている。
- 脂溶性ビタミンA、DおよびEは、潜在的な利点を持つ抗酸化物質とみなされ、これらのビタミンの血清レベルは、認知的に正常な人と比較してアルツハイマー病患者では低下する。しかし、無作為化比較試験では、ビタミンE投与群の認知機能の低下が遅いことを示した1つの研究を除いて、それらの日常的な使用を支持することはできない。
- Toll様受容体4アゴニストであるモノホスホリル脂質A(MPL)は、ミクログリアによるアミロイドβの取り込みを刺激することができ、その治療への応用が研究されている。
- ケトジェニックダイエットは、治療中の認知機能低下を改善する可能性があると考えられているが、この不自然な食事が長期的な使用に適しているかどうかについては、さらなる調査が必要である。
研究では、アルツハイマー病の診断と進行に対する脂肪酸の効果は単純ではないことが示されており、正確な洞察を得るために複数の脂質を説明する必要がある。理想的な治療法がまだ見つかっていないアルツハイマー病有病率が上昇している時代には、食事の脂質量を変えるなどの生活習慣の改善が、現実的で自然な形で病気と向き合う方法のようである。しかし、人間は雑食動物であり、脂質だけでなく、他のエネルギー源や微量元素も摂取するため、食事によるアルツハイマー病への影響の研究は難しく、食生活の改善は理想的とは言えない。しかし、脳内には脂質が豊富に存在することから、脂質がアルツハイマー病の病態に及ぼす影響を知ることは、疾患修飾のための貴重な情報となる。リピドームを含めたメタボロームの継続的な探索が、この分野のさらなる発展につながることを期待している。
略語
- NFTs 神経原線維のもつれ
- DHA ドコサヘキサエン酸
- アポE アポリポタンパクE
- CLU クラスタイン
- SORL1 ソルチリン関連受容体1
- ABCA7 ATP結合カセット、サブファミリーA、メンバー7
- GL グリセロ脂質
- MAG モノアシルグリセロール
- DAG ジアシルグリセロール
- TG トリアシルグリセロール
- GP グリセロリン脂質
- PE ホスファチジルエタノールアミン
- PC ホスファチジルコリン
- PS ホスファチジルセリン
- SP スフィンゴ脂質
- SL ステロール脂質
- PR プレノール脂質
- SL 糖脂質
- PK ポリケチド
- APP アミロイドタンパク質前駆体
- SPMs 特異的炎症収束性脂質メディエーター
- EPA エイコサペンタエン酸
- OL オレイン酸
- SA ステアリン酸
- THA テトラコサヘキサエン酸
- PS1 プレセニリン1
- c-JNK c-Jun N末端キナーゼ
- LO リポキシゲナーゼ
- COX シクロオキシゲナーゼ
- PEP プロリルエンドペプチダーゼ
- IsoPs イソプロスタン
- NeuroPs ニューロプロスタン
- KD ケトジェニックダイエット
- MCT1 モノカルボン酸トランスポーター1
- NMDA N-メチル-D-アスパラギン酸
- PlsEtns エタノールアミンプラスモン
- PI ホスホイノシチド
- PLD ホスホリパーゼD
- SMs スフィンゴミエリン
- S1P スフィンゴシン1リン酸塩
- BACE1 β-セクレターゼ1
- RxRγ レチノイドX受容体γ
- MPL モノホスホリル脂質A
- LDL 低密度リポ蛋白質
- HDL 高密度リポ蛋白質