
Contents
Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications
www.ncbi.nlm.nih.gov/pmc/articles/PMC6139360/
要旨
加齢に伴い、脳の様々な部位に鉄が沈着することで、正常な認知機能や行動が損なわれる可能性がある。鉄代謝異常は、フェントン反応によりヒドロキシルラジカルを発生させ、酸化ストレス反応を誘発し、細胞脂質、タンパク質、DNAの構造と機能を損傷させ、最終的には細胞死に至る。
アルツハイマー病では、鉄の恒常性のバランスが崩れている。過剰な鉄分はβアミロイドの沈着や神経原線維のもつれの形成に寄与し、アルツハイマー病の発症を促進すると考えられている。そのため、鉄を標的とした治療戦略が新たな方向性を示している。
鉄キレート剤としては、デスフェロキサミン、デフェリプロン、デフェラシロックス、クリオキノールなどが注目されており、科学実験や一部の臨床試験で良好な結果が得られている。
従来の鉄キレート剤の長期適用の限界と副作用を考えると、α-リポ酸とラクトフェリンは、自己合成された天然の低分子として、アミロイドβ凝集、タウパチー、神経細胞障害をブロックするという非常に興味をそそる生物学的活性を示している。
臨床的な有用性についてのエビデンスは乏しいものの、鉄イオンを中心としたキレーション治療は、アルツハイマー病治療のための貴重なアプローチであるとの推測は、依然として広く普及している。
キーワード
アルツハイマー病、鉄、キレーション、α-リポ酸、ラクトフェリン
序論
アルツハイマー病は、高齢者に発症する神経変性疾患である。多くの患者さんは初期に記憶力の低下を示し、病状が悪化すると言語障害、方向性の喪失、不安行動なども見られるようになる(Cheng er al)。 後期患者については、認知、感情、行動などの精神活動に異常が生じ、身体機能が徐々に失われていく(Ikononomovic er al)。 社会の発展や人間環境の変化に伴い、アルツハイマー病の発症率は年々増加している。
米国アルツハイマー協会が2017年に行った疫学調査では、米国のアルツハイマー病患者数は550万人を超え、65歳以上の人は2倍の確率でアルツハイマー病を発症していることが判明している。しかし、アルツハイマー病の発症機序は依然として不明であり、アルツハイマー病を完全に治癒させたり、症状を和らげたりする薬はまだ開発されていない(Blennow et al 2006;Huang and Mucke 2012;Mullard 2012;Reese et al 2012;Selkoe 2012)。
アルツハイマー病の病原性仮説
脳におけるアルツハイマー病の2つの主要な病理組織学的特徴は、細胞外βアミロイド蛋白質(アミロイドβ)の沈着によって形成される老人斑(SP)と、神経細胞の微小管に関連するタウ蛋白質の過リン酸化によって形成される神経原線維絡み(NFTs)である(Colvez et al 2002)。このような明らかな病理学的特徴に基づいて、アルツハイマー病の発生メカニズムについては、アミロイドカスケード仮説とNFTs仮説の2つの仮説が提唱されていた。研究の深化に伴い、最近では炎症仮説や金属イオン仮説も提唱され、アルツハイマー病の発症メカニズムを継続的に充実させていた。
アミロイドカスケード仮説は、β-アミロイド前駆体タンパク質(APP)のアミロイド分解経路に由来する。この仮説は、APPのアミロイド分解経路によって産生されるアミロイドβ1-42は、有意な神経毒性を有し、タウタンパク質の凝集と過リン酸化を誘導し、NFTsを形成し、神経細胞の損傷を引き起こし、最終的には認知症に至るというものである(Klein et al 2001)。NFTs仮説は、アルツハイマー病患者の神経細胞において、高リン酸化タウタンパク質の凝集によって形成された多数の繊維のもつれが存在することに由来する。この仮説は、高リン酸化タウタンパク質が正常なタウタンパク質と競合してチューブリンと結合し、微小管の集合と解離のダイナミックなバランスを崩し(Cardenas-Aguayo Mdel et al 2014年)軸索輸送の障害と細胞内老廃物の蓄積をもたらすことを示唆している。神経細胞は徐々に退化し、認知症を引き起こす。研究者らは、アミロイド斑やNFTに加えて、TNF-α、IL-1β、IL-6およびその他の炎症性因子の発現レベルの上昇を伴う、アルツハイマー病患者の脳内で大量の活性化アストロサイトおよびミクログリアを発見した(Cunningham et al 2005)。そこで、神経炎症仮説が提唱された。この仮説は、神経炎症が老人斑やNFTによって活性化されるアルツハイマー病における受動的なシステムではなく、プラークやタングルと同様に、疾患の発症に重要な役割を果たしていることを示唆している(Zhang et al 2013)。
興味深いことに、実質的な証拠は、生体内での金属イオン代謝の定常状態の調節障害がアルツハイマー病の病理に関与し得ることを示している(Kim et al 2018)。アルツハイマー病患者の脳内金属レベルの不均衡が同定されており、金属触媒による酸化的損傷を伴う(Nunomura et al 2001;Perry et al 2003)。
銅、鉄、亜鉛、マグネシウム、アルミニウムなどの金属イオンがアルツハイマー病の発生と発症に関与していることが多くの研究で示されている(Wang and Wang, 2017)。また、臨床研究では、アルツハイマー病患者の脳内で銅、鉄、亜鉛のレベルが上昇していることが示されている(Bush, 2013)。
金属イオンは、神経細胞の代謝に影響を与え、酸化ストレスを引き起こし、アミロイドβの沈着と老人斑の形成を促進する可能性がある(Lovell et al 1998)。同時に、脳内でのアミロイドβの沈着とその毒性は、大脳皮質および海馬での亜鉛、銅、鉄および他の金属イオンの代謝障害に直接関係していることも研究で示されている(Liu er al)。 また、金属のホメオスタシスのアンバランスは、神経細胞の機能不全を直接引き起こし(Myhre et al 2013年)神経細胞死につながることが研究で示されている(Wright and Baccarelli 2007)。
さらに、アルツハイマー病のいくつかの動物モデルおよび初期アルツハイマー病患者における金属(亜鉛、銅、鉄)キレート剤の適用の成功は、アルツハイマー病が遷移金属過負荷疾患であることの強い証拠を提供した(Guo et al 2013b 2015,2017;Dusek et al 2016;Giampietro et al 2018;川原 et al 2018;Zhang et al 2018)。
以上のような研究から、アルツハイマー病の病態における金属イオンの役割を強調する金属イオン仮説が提唱され、アルツハイマー病の病態をさらに補完することになった。
脳の鉄分異常とアルツハイマー病の病態生理
鉄は、アルミニウムに次いで地球上で2番目に豊富な金属であり、地球上のすべての生物の生存に不可欠な元素でもある(Crielaard et al 2017)。鉄の生物学的活性は、その効果的な電子移動特性に大きく依存しており、鉄の二価、第二鉄および四価の鉄の状態間の遷移の間に電子を受け入れるか、または提供することを可能にし、したがって、様々な生化学反応における触媒的補因子として機能する(Hohenberger et al 2012)。また、鉄は、鉄-硫黄クラスター(Fe-S)の形で、DNAの複製と修復の過程で様々な生物学的酵素の活性を促進する。同時に、鉄はヘモグロビンやミオグロビンの構成成分でもあり、生体内の酸素や二酸化炭素の輸送に関与している(武田 2004)。
脳の重要な生理活動の多くは鉄が関与している。脳の発達過程で鉄分が欠乏すると、不可逆的な発達遅延を引き起こすが、脳内の鉄分が過剰になると神経毒作用もあるため、脳の正常な生理活動が損なわれることになる。脳内の鉄分は加齢とともに徐々に増加していく。興味深いことに、磁気共鳴画像法(MRI)を用いて、アルツハイマー病患者の脳内の鉄分量が有意に増加していることが明らかになった(Du et al 2018)。この知見は、APP/PS1E9二重トランスジェニックADマウスと同年齢の野生型マウスの比較研究でも確認されている(Dwyer et al 2009)。また、鉄代謝障害がアルツハイマー病後期発症の重要な原因であると考える学者もいる(Corder et al 1995)。鉄に関する様々な発見をもとに、鉄がアルツハイマー病の発症に極めて重要な役割を果たしていることがわかっていた。そのため、鉄をターゲットにした研究は、徐々に研究者にとってアルツハイマー病の病態を探る新たな方向性を示すようになっていた。
鉄の吸収と脳内輸送
鉄は主に食品中の非ヘム鉄とヘム鉄に存在し、その90%を非ヘム鉄が占めている。非ヘム鉄は小腸上部でFe2+に還元され、小腸上皮の膜上にある二価金属イオントランスポーター(DMT1)を介して粘膜上皮細胞に入る。残りの10%のヘム鉄は、小腸近位部でヘムオキシゲナーゼ(HO)と反応してFe2+を放出し、DMT1を介して小腸粘膜上皮細胞に直接取り込まれる(Krishnamurthy et al 2007; Horl 2008)。
上皮細胞によって取り込まれたFe2+は、細胞によって直接利用され得るが、利用されなかったFe2+は、第一鉄酸化酵素(ヘファエスティン)またはセルロプラスミンによってFe3+に酸化される。
得られたFe3+は、腸粘膜上皮の基底側膜のフェルロポーチン1(FPN1)を介して細胞外に輸送される。移送されたFe3+は、主に血中のトランスフェリン(Tf)と結合して鉄-トランスフェリン複合体を形成し、また、ラクトフェリンと結合して非トランスフェリン結合鉄を末梢血循環中に形成することができる(Frazer and Anderson, 2005; Garrick and Garrick, 2009)。
末梢血中で脳に循環する鉄-トランスフェリン複合体は、脳毛細血管内皮細胞のエンドサイトーシスを介して細胞内に入る。このエンドサイトーシスは、主に内皮細胞表面のトランスフェリン受容体(TfR)によって媒介される。末梢血中の非トランスフェリン結合鉄は、ラクトフェリン/ラクトフェリン受容体経路を介して脳に入ることができる(Ke and Qian, 2007)。
鉄-トランスフェリン複合体はエンドサイトーシスを介して細胞内にエンドソームを形成する。エンドソーム膜のプロトンポンプの作用により、エンドソーム内のPHが低下し、鉄とTf/TfR複合体が解離し、同時にFe3+がFe2+に還元される。
Fe2+はエンドソーム膜上のDMT1を介して内皮細胞質に入る(図11)。分離した鉄Tf/TfR複合体は、小胞から内皮細胞の側腔に滲出する。pH7.4の環境下では、TfはTfRから解離し、血液中に再侵入する(Dringen et al 2007)。
図1 細胞内への鉄の輸送の模式図
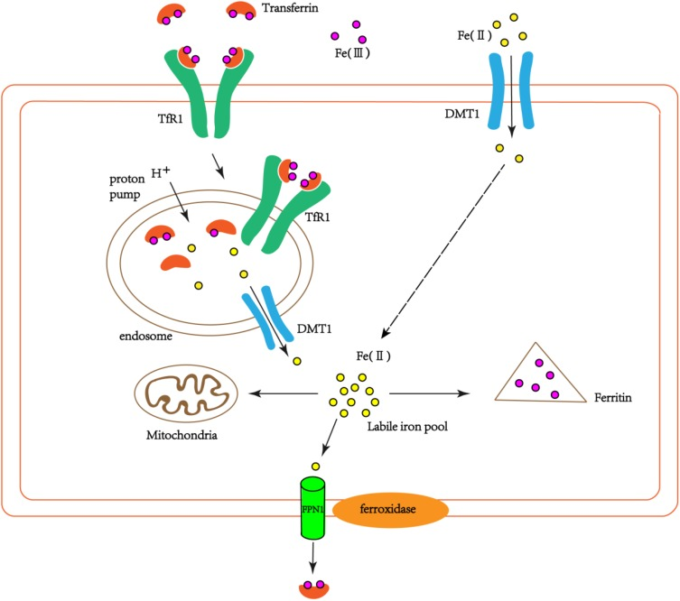
細胞外二価鉄の一部は、DMT1を介して直接細胞内に移行することができる。トランスフェリン結合鉄はエンドサイトーシスを介してTfR1と結合し、細胞内にエンドソームを形成する。エンドソーム上のプロトンポンプの作用により、3価の鉄はTf/TfR1複合体から解離して2価の鉄に還元され、DMT1を介して細胞質に入る。
細胞質に入った第一鉄の一部は細胞自体(ミトコンドリアなど)で利用され、その一部はフェリチンによって第一鉄に酸化されて貯蔵される。別の部分は細胞膜上のフェロキシダーゼによって第一鉄に酸化され、FPN1によって細胞外に輸出され、細胞外Tfと再結合する。
脳内の鉄の制御
剖検報告によると、ヒトの脳内の総鉄沈着量は年齢と正の関係があり、パタメン、掌蹠球、黒質の基底核に高濃度の鉄が含まれている(Connor et al 1995)のに対し、大脳皮質、脳幹、小脳には低濃度の鉄が含まれている(Zecca et al 2004; Ramos et al 2014)とされている。神経細胞における鉄の恒常性は、主に鉄代謝に関与するmRNAの転写レベルによって制御される。
脳の鉄代謝に関与するタンパク質には、主に鉄調節タンパク質(IRP)Tf、TfR1,フェリチン、FPN1,DMT1などがある(Crielaard et al 2017)TfR1,フェリチン、FPN1,DMT1をコードするmRNAはすべて、鉄調節エレメント(IRE)と呼ばれる特殊なアミノ酸配列を含んでいる。鉄は、IRPとIREの結合を制御することで鉄関連タンパク質の転写を制御し、それによって細胞内の鉄の恒常性を維持している(Zhou and Tan, 2017)。
継続的な綿密な研究の後、人々はさらに、抗菌ペプチドであるヘプシジンが鉄のホメオスタシス、特に脳の鉄のホメオスタシスにおいて重要な因子であることを理解した(Vela, 2018)。FPN1は、生体内でのヘプチジンの主要な受容体である。一連の研究は、ヘプチジンがヘプチジンとFPN1との直接的な相互作用を介して鉄のホメオスタシスを調節し、FPN1の内部化と分解を誘導して細胞が鉄を輸出する能力を低下させ(De Domenico et al 2008)、それによって細胞内鉄過負荷の可能性を高めることを示した(Daher et al 2017)。その後、細胞実験により、ヘプチジンはアストロサイトやニューロンにおけるFPN1の発現を低下させるだけでなく、TfRやDMT1の発現も低下させることが示された(Du et al 2011,2015年)。以上の結果から、ヘプシジンは、細胞の鉄出力を制御するだけでなく、細胞の鉄入力を制御することで、鉄の恒常性を調節していることが示唆された。
また、鉄を調節する鉄関連タンパク質に加えて、APPやタウも鉄を調節するように作用する。研究は、APPが鉄の恒常性のための特定の調節因子であり、FPN1と相互作用して鉄イオンの流出を調節することができることを示している(Kawahara et al 2017)。実際、APPノックアウトまたはハプロ不全は、マウスの脳鉄蓄積を優先的に媒介する(Duce et al 2010)。タウは、細胞内微小管関連タンパク質として作用し、生産されたAPPを細胞表面に輸送して鉄出力を促進することができる(Li et al 2015)。興味深いことに、タウノックアウトマウスは、年齢依存性の鉄蓄積と脳萎縮を発症し、初代培養ニューロンにおける鉄の滞留はAPPの表面トラフィッキングの減少によって引き起こされ、タウが媒介する鉄のホメオスタシスはAPP依存性である可能性があることを示唆している(Lei et al 2012; Tuo et al 2017)。
鉄はアルツハイマー病の発症に関与している
脳では、鉄はミエリンの合成や神経伝達物質の合成・代謝に関与するだけでなく、神経細胞の高い代謝能力の維持にも重要な役割を果たしている(Gerlach er al)。 正常な生理学的条件下では、鉄代謝は脳の恒常性を維持している。一旦、鉄代謝がバランスを崩すと、脳機能に異なる影響を及ぼすことになる。
早くもグッドマン(1953)は、アルツハイマー病患者の脳内老人斑で鉄が増加していることを発見した。その後、定量的感受性マップ(QSM)を用いて再び脳鉄沈着とアミロイドβプラークの共局在化が確認され、脳鉄沈着とアミロイドβプラークの共局在化が発症を促進することが示された(van Bergen et al 2016)。実際、脳の正常な加齢過程においても、特に黒質、淡蒼球、尾状核、大脳皮質には進行性の鉄沈着があり、これらの脳領域は神経変性疾患と密接に関連している(Rodrigue et al 2011;Callaghan et al 2014;Collingwood and Davidson et al 2014;Ward et al 2014)。同年齢の健常者と比較して、アルツハイマー病患者の鉄沈着は、これらの領域でより深刻である。さらに、アルツハイマー病患者の末梢血単核球のAPP mRNAは、蛍光定量PCRにより対照群に比べて有意に低かったことから、間接的にアルツハイマー病における鉄の不摂生を示唆していた(Guerreiro er al)。
鉄はアミロイドβプラークとタウタングルの沈着に関与している
鉄はAPP mRNAのIRE部位に作用し、それによって内因性APPの翻訳および発現を増強することができるため、鉄代謝障害はアミロイドβの産生および蓄積を誘導することができることが研究により実証されている(Cahill et al 2009)。また、APP/PS1E9二重トランスジェニックマウスにおける高濃度の鉄の長期投与は、脳内の老人斑の数の増加をもたらすことが明らかにされている(Smith et al 1997)。鉄の曝露を12ヶ月間延長すると、3,5,5-トリメチルヘキサノイルフェロセン食による脳内鉄の増加は、APPマウスにおける老人斑の形成とミクログリア鉄包接体の形成を加速させた(Peters et al 2018)。
鉄とアミロイドβプラークが共局在することは多くの証拠があるが、鉄がどのような形態でプラークに存在するかは知られていない。最近、Plascencia-Villa et al 2016)は、透過型電子顕微鏡(TEM)を用いて、鉄が酸化鉄(Fe3O4)マグネタイトナノ粒子の形で老人斑のコアに存在することを確認した。これは、鉄の蓄積とアミロイドβ凝集に関連した金属生物学の証拠を提供する。その後、in situ X線磁気円二色性を用いて、再びヒト老人斑中のマグネタイトの存在を明らかにした(Everett et al 2018)。マグネタイトは多結晶の酸化鉄として、ヒトの脳では正常な特徴ではなく、その含有量の上昇は、鉄の酸化還元化学の異常がアルツハイマー病に影響を与えることを示している(Ayton et al 2017b)。さらに、Telling et al 2017)は、X線顕微鏡の高度な亜ミクロ分解能を用いて、老人斑の形態と鉄との間の直接的な相関と、鉄アミロイド複合体の形成との間の直接的な相関の証拠を見出した。重要なことに、アミロイドβは、ペプチドの親水性N末端領域の3つのヒスチジン残基および1つのチロシン残基を介して鉄と結合し、これらの鉄イオンを安定化するのに役立つ(Lane et al 2018)。順番に、研究はまた、アミロイドβへの鉄イオンの結合がペプチドのらせん構造を減少させ、ペプチドのβシート含量を増加させることを発見し、鉄イオンがペプチド間の相互作用を増強することにより、アミロイドモノマーがオリゴマーおよびフィブリルを形成することを促進することを示している(Boopathi and Kolandaivel, 2016; Tahirbegi et al 2016)。アミロイドβ凝集を促進することを除いて、高い鉄レベルは、APPのアミロイド原性処理に影響を与え得る。初期の研究では、鉄がAPPのα-セクレターゼ切断活性に調節効果を有することが明らかになった(Bodovitz et al 1995)。その後の研究では、α-セクレターゼおよびβ-セクレターゼを不活性状態から活性状態に変換する過程がフーリンによって制御され、鉄が転写レベルでフーリンの発現を制御しうることが明らかになった(Silvestri and Camaschella, 2008)。過剰な鉄はフーリンの発現を阻害し、それによってβ-セクレターゼの活性化を促進し、それによってアミロイド経路からのアミロイドβの産生を促進する(Ward et al 2014)。別の研究では、γセクレターゼ成分であるプレセニリンエンハンサー2(PEN-2)がフェリチン軽鎖を介して鉄と結合し、γセクレターゼ活性を増強し、それによってアミロイドβの生成を増加させることを発見した(Li er al)。
また、鉄がアミロイドβペプチドの凝集を促進し、細胞毒性を高めることも試験管内試験研究で明らかになっている(Tahmasebinia and Emadi, 2017; Galante er al)。 しかし、鉄とアミロイドβの役割については異なる意見がある。以前の研究では、Fe2+およびFe3+がAPPおよびアミロイドβと相互作用して、アミロイドβの繊維状形態への凝集を促進することが明らかにされている(Ha et al 2007)。Fe2+はまた、アミロイドβタンパク質のアミノ酸と相互作用することができ、これは、銅および亜鉛とは異なる方法でアミロイドの形態の変化を付与する可能性がある(Dahms et al 2012)。アミロイドβに結合したFe3+は、容易にFe2+に還元され、活性酸素種(ROS)産生を増加させ、これは、βセクレターゼが単量体アミロイドβ42をより毒性の高いアミロイドβオリゴマーに開裂させ、神経細胞死を加速させる(Cohen et al 2013; Balejcikova et al 2018)。重要なことに、アミロイドβは、ミトコンドリア機能を損傷し、酸化還元活性でFe3+をFe2+に変換し、酸化ストレスを誘発し、それによって鉄過負荷を悪化させ、アルツハイマー病状態を悪化させることができる(Everett et al 2014;Mena et al 2015)。さらに、鉄曝露は、培地中のβ-セクレターゼ活性およびアミロイドβ42の増加とともに、培養SHSY5Y細胞におけるAPPの蓄積を促進することが示されている(Banerjee et al 2014)。非矛盾的に、最近の研究では、神経細胞の鉄処理はAPP非アミロイド経路を促進し、sAPPαの分布を変化させ、細胞外に分泌されるのではなく細胞溶解物中に保持し、一方、鉄はβ分泌酵素の発現を変化させないが、その活性を有意に阻害することが示された(Chen et al 2018)。また、別の研究では、アミロイドβは鉄の酸化還元能を有意に低下させ、これはアルツハイマー病の発症期における神経保護とアミロイドβの金属キレーションを示す可能性があるが、特定の条件下では有毒になることがわかった(Lane et al 2018)。
神経原線維のもつれは、アルツハイマー病のもう一つの主要な病理学的特徴であり、リン酸化されたタウタンパク質はNFTの主成分である。研究では、NFTsを有するニューロンにおける鉄の沈着が発見されている(Smith et al 1997)。
アミロイドβペプチドに加えて、鉄はタウタンパク質に結合し、タウタンパク質のリン酸化を誘導し、リン酸化されたタウタンパク質を凝集させることができるが、この現象は鉄キレート剤によって逆転させることができる(Amit et al 2008)。また、Fe3+は高リン酸化タウタンパク質の凝集を誘導し、Fe3+をFe2+に還元すると、その誘導された凝集を逆転させることができる(Yamamoto et al 2002)。これらの結果から、鉄が高リン酸化タウタンパク質の蓄積に重要な役割を果たし、NFTを形成する可能性があることが明らかになった。
最近の研究では、タウタンパク質がアルツハイマー病の発症時に脳神経細胞における鉄イオンの伝達に間接的に関与していることが示されている(Lei et al 2012)。さらに、鉄がサイクリン依存性キナーゼ(CDK5)/P25複合体やグリコーゲン合成酵素キナーゼ-3β(GSK-3β)の活性化を介してタウタンパク質の過リン酸化に関与していることが試験管内試験および生体内試験実験で示されているが(Xie er al 2012; Guo er al 2013a)鉄がタンパク質ホスファターゼPP2Aの不活性化にも関与しているかどうかについては関連する実験は報告されていない(図22)。
さらに、Fe3+は、ミトコンドリアによって放出されるスーパーオキシドラジカルの還元を促進することができる(Kudin et al 2004;Aliaga et al 2011)。Fe3+の還元はスーパーオキシドの生成につながり、スーパーオキシドが一酸化窒素(NO)と反応して過硝酸塩を生成すると、正常に機能するチロシン残基が損傷することがある(Nakamura and Lipton, 2011)。アルツハイマー病では、タウの硝酸化は微小管格子の安定化を阻害し、タウタンパク質の硝酸化はタウの絡み合いや老人斑で観察されている(Reynolds et al 2006;Kummer et al 2011)。
興味深いことに、NFTsにおけるタウの蓄積はまた、HO1の増加した誘導と関連している(Wang et al 2015)。HO1は、損傷したミトコンドリアから放出されたヘムを代謝することができる強力な抗酸化物質であり、また、Fe2+の放出を促進し、これはフリーラジカルが追加の酸化ストレスを開始する原因となり得る(Ward et al 2014)。このように、鉄誘発性酸化ストレスはまた、タウの過リン酸化および凝集を促進する可能性がある(Lane et al 2018)。
図2 アミロイドβプラークとタウのもつれの沈着に鉄が関与していることを示す模式図
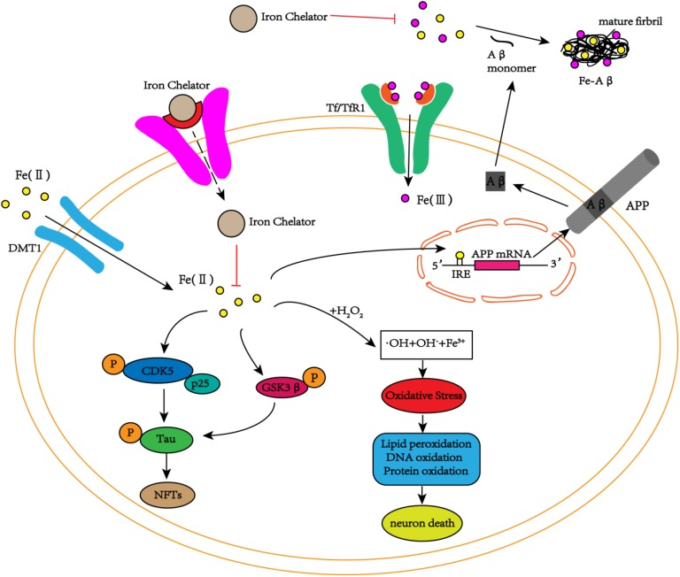
ニューロンでは、鉄はアミロイドβと相互作用し、アミロイドβの繊維状形態への凝集を促進する。鉄はまた、APP mRNAのIRE部位に作用し、内因性APPの発現を増加させる。アミロイドβとの相互作用に加えて、鉄はまた、CDK5/p25複合体およびGSK3βを活性化してNFTを形成することにより、タウのリン酸化を促進することができる。
同時に、鉄はフェントン反応を介して酸化ストレスを引き起こし、DNA、脂質、タンパク質を損傷し、最終的には細胞死に至る。鉄キレート剤は、鉄によるCDK5/p25複合体やGSK3βの活性化を阻害することで、タウのリン酸化を抑制し、NFTの生成を抑制する。同時に、鉄キレート剤は、鉄をキレートすることでアミロイドβモノマーの凝集を抑制し、細胞死を遅らせる。
これまでのところ、アミロイドβプラークおよび高リン酸化タウタンパク質の凝集における鉄の関与の具体的なメカニズムは明らかになっていないが、鉄が関連するシグナル伝達経路およびタンパク質の三次元コンフォメーションに影響を与えることにより、このプロセスを促進することが示されている。
それにもかかわらず、アルツハイマー病における鉄レベルの変化を強調する必要があり、ホメオスタシスの不均衡は、鉄の誘導を介して二重の効果を有する可能性がある。一方で、脳内の鉄過剰領域は、老人斑やNFT周辺の酸化ストレスや細胞死に寄与し、他方では、鉄欠乏による神経細胞機能の障害を受ける可能性がある(Belaidi and Bush, 2016)。
フェロプトーシスとアルツハイマー病
細胞死は、生物の成長・発達や組織の恒常性の維持に重要な役割を果たしている。アルツハイマー病では細胞死の制御が困難であることが研究で明らかになっている(Yang and Stockwell, 2016)。また、鉄過負荷による特異的な病理学的状態に基づいて、アポトーシス、壊死、オートファジーとは異なる第4の細胞死モード、すなわちフェロプトーシスが学者によって提唱されている(Dixon er al)。
フェロプトーシスとは、重度の脂質過酸化を伴う活性酸素産生と鉄の利用可能性に依存する鉄依存性脂質過酸化誘発細胞死を指す(Dixon, 2017)。フェロプトーシスの特徴の一つは、他の金属よりもむしろ細胞内の鉄に依存する死の形態である脂質ROSの鉄依存性蓄積である(アミロイドβdalkader et al 2018)。
フェロプトーシスの形態的特徴は、主に細胞内ミトコンドリアに反映される。正常細胞のミトコンドリアと比較して、フェロプトーシス細胞のミトコンドリア容積は小さく、ミトコンドリア膜の密度は低下し、ミトコンドリア痔は減少または消失し、ミトコンドリア外膜は破裂している(Xie et al 2016)。
さらに、フェロプトーシスの発生は、細胞内抗酸化物質であるグルタチオン(GSH)の不活性化をトリガーとした脂質活性酸素の蓄積からなることが研究により明らかにされている。したがって、フェロプトーシスは、細胞内酸化還元恒常性の不均衡によって引き起こされる(Gao er al)。
脂質の酸化損傷を修復する抗酸化防御酵素であるグルタチオンペルオキシダーゼ4(GPx4)は、フェロプトーシスの中心的な内因性抑制因子である(Chen er al)。 研究は、GPx4遺伝子ノックアウトマウスが、フェロプトーシスの3つの顕著な特徴(鉄の調節障害、脂質過酸化、炎症)およびアルツハイマー病の前駆指標(行動障害、海馬神経変性)の両方に関与し、これらの病理学的変化は、フェロプトーシス阻害剤によって改善または予防することができることを発見した(Seiler et al 2008; Hambright et al 2017)。
興味深いことに、フェロプトーシス誘引剤であるエラスチンは、フェロプトーシスに伴う神経細胞死を誘発することができる(Hirata et al 2018)。逆に、鉄キレート剤および抗酸化剤は、フェロプトーシスから細胞を保護することに特異的に関与している(Hambright et al 2017)。まとめると、フェロプトーシスの生理的機能はまだ明らかになっていないが、加齢に伴う神経変性疾患(アルツハイマー病を含む)における役割は確立されている。このことから、フェロプトーシスを中心に考えると、フェロプトーシス阻害薬の開発は、アルツハイマー病の症状を緩和するための新たな方向性を示しているのではないかと考えられる。
アルツハイマー病進行の臨床診断のためのバイオマーカー
アルツハイマー病の病態はまだ十分に解明されていない。特殊な方法を用いてアルツハイマー病を早期に発見することができれば、疾患の理解を深め、積極的な予防につなげることが非常に重要である。現在、アルツハイマー病の臨床診断は、主に家族歴と特定の認知機能障害に基づいて行われている(Alzheimer’s, 2016)。他の方法では、主に脳脊髄液中のアミロイドβ1-42,リン酸化タウおよび総タウのレベルを検出して、アルツハイマー病の発症および重症度を予測することが行われている(Bulk et al 2018)。アミロイドβプラークを検出する一般的な方法として、ポジトロン断層撮影(PET)がある。研究の深化に伴い、アルツハイマー型認知症の脳内変化を特定するための新しい技術が増えており、勾配エコー多重造影検査(GEPCI)技術もその一つである。今回の研究では、GEPCI脳組織指標とPETで定義されたアミロイドβ負荷との間に強い相関関係が確認され、アルツハイマー病の前臨床および初期症状段階におけるアルツハイマー病関連の病理組織を評価するための新たな方法を提供した(Zhao er al)。
アミロイドβおよびタウ変性はアルツハイマー病の重要な因子と考えられているが、鉄の異常安定性はアルツハイマー病の病態生理の潜在的な原因として報告されることが多くなっている。鉄は、アルツハイマー病の発症を検出し、アルツハイマー病の重症度を反映させるためのバイオマーカーとして、アルツハイマー病の進行に利用できるのであろうか?このため、アルツハイマー病のバイオマーカーとしての鉄は、科学者にとって研究のホットスポットとなっている。現在、脳の鉄分濃度を検出するのに有効な方法はQSMである。QSMはより優れた特異性を持ち、非侵襲的に組織の磁化率を評価するために使用することができ、これは脳の鉄レベルと良好な相関関係を持つことが確認されている(Du et al 2018)。QSMを通じて、Du et al 2018)は、脳の左側にある尾状核の磁化率値が、軽度・中等度アルツハイマー病疾患の重症度のバイオマーカーとして利用できる可能性を発見した。早くもMoon et al 2016)は、アルツハイマー病患者の中心核と尾状核の磁化率値が対照群と有意に異なることを発見している。その後、van Bergen et al 2018)はQSMとPETを共同で用いて、アミロイドβプラーク負荷と鉄沈着の局所的な相関が健康な高齢者の認知パフォーマンスに関する関連情報を提供できることを示した。これまでの研究では、アルツハイマー病患者では前頭前野の鉄蓄積が高く、その程度はアミロイドβプラークの数やNFTと関連しており、異なる鉄の分布があり、これらの変化はアルツハイマー病病理学的マーカーの発達後に起こるようであることが示されている(van Duijn et al 2017)。これらの知見は、鉄をベースとした磁気共鳴(MRI)造影の変化が、アルツハイマー病病理の程度を間接的に決定するために使用できることを示唆している。鉄濃度は脳内QSMの主な比較対象として同定されたが、O’Callaghan et al 2017)はQSMとタウ濃度の間に相関関係を見出しており、QSMがアルツハイマー病におけるタウ病理の早期発見に有用なバイオマーカーである可能性を示唆している。
体内の主要な鉄貯蔵タンパク質であるフェリチンは、アルツハイマー病と密接な関係がある。脳脊髄液中のフェリチンの上昇は、認知機能の低下と関連し、軽度認知障害からアルツハイマー病へのリスクを高めることが示されている(Ayton et al 2015)。さらに、前臨床アルツハイマー病の血液バイオマーカーパネルに寄与するフェリチンの可能性が研究で示されている(Goozee et al 2017)。脳脊髄液フェリチンレベルは認知パフォーマンスと負の相関があり、脳脊髄液中のアポリポタンパク質E(ApoE)と強い相関があり、脳の鉄負荷の指標としての脳脊髄液フェリチンがアルツハイマー病認知機能のバイオマーカーである可能性を示唆している(Ayton et al 2015)。しかし、QSMとPETを組み合わせて縦断的な認知機能悪化の値を予測する別の研究では、アミロイドβ病理の存在下では鉄が認知機能を悪化させる可能性があることがわかった。アミロイドβ病理は存在しない場合、鉄は認知機能と相関がない(Ayton et al 2017a)。さらに進んで、Ayton et al 2018)は、高濃度の脳脊髄液フェリチンが296人の参加者の脳脊髄液中のアミロイドβの減少を加速させることを発見し、これは鉄がアミロイドβの沈着を促進し、疾患の進行を加速させる可能性を支持している。これは、鉄がアミロイドプラーク形成と関連していることを示す初めての臨床的証拠である。
腰椎穿刺の侵襲性のため、この脳脊髄液バイオマーカーの使用は、広範な臨床応用を制限する一方で、血清または血漿バイオマーカーは比較的簡単に取得でき、侵襲性が低いため、アルツハイマー病での応用に大きな可能性を持っている。研究では、アルツハイマー病患者の血清および鉄関連タンパク質レベルが有意に上昇していることが示されている(Sternberg et al 2017)。アミロイドβペプチドとは異なり、鉄および鉄関連タンパク質は、認知評価試験、神経イメージングおよび臨床データと有意に関連している(Sternberg et al 2017)。これは、少なくとも部分的には、鉄がアルツハイマー病のバイオマーカーとして機能しうることを示している。
鉄を標的とした治療戦略
金属キレート剤は、キレート剤分子と金属イオンの強い結合作用により、キレート剤の内部に金属イオンを結合させ、分子量の大きい安定な化合物とすることで、金属イオンの作用を阻害することができる。鉄過多はアルツハイマー病の発生と発症に重要な役割を果たしているため、アルツハイマー病患者の脳の特定の領域における過剰な鉄を減少させ、アルツハイマー病を緩和する、あるいは治療するという戦略を達成するために、金属キレート剤を使用することが注目されている。金属キレート剤が効果的にキレーションを発揮するためには、以下のような特徴が必要である。
- (1)細胞膜や血液脳関門(BBB)に浸透しやすいこと、
- (2)血漿中のトランスフェリン結合鉄を枯渇させずに鉄富化部位を標的とすること、
- (3)キレート化した鉄を鉄蓄積部位から除去したり、循環トランスフェリンなどの他の生体タンパク質に移行させたりすること、
- (4)身体への副作用や軽度の副作用がないこと
(Boddaert et al 2007)などが挙げられる。
クリオキノール
クリオキノール(クロロキン)は、化学名が5-クロロ-7-ヨード-8-ヒドロキシキノリンであり、効果的な金属(鉄、銅、亜鉛)キレート剤である。アルツハイマー病の動物モデルをクロロキンで治療すると、脳内アミロイドの沈着を減少させ、記憶障害を改善できることが研究で明らかになっている(Cherny et al 2001;Grossi et al 2009)。この現象の推定される原因は、クロロキンの鉄、亜鉛および銅イオンへの高い結合親和性により、クロロキンがアミロイドβからこれらの金属を競合的に捕捉し、アミロイドβの凝集を防ぐことができることである(Opazo et al 2006)。別の研究では、クロロキンを投与された15ヶ月齢のAPP Tg2576ADマウスは、偽マウスと比較して脳内老人斑の数と大きさが有意に減少したことが示された(Cherny et al 2001)。同時に、生体内試験実験では、APP/PS1 ダブルトランスジェニックマウスの脳内β-セクレターゼ(BACE1)とγ-セクレターゼ(PS1)の発現を阻害することで、対照群と比較して、クロロキン が APP の発現を低下させることが明らかになった(Wang er al)。 また、小規模な第2相臨床試験であるアルツハイマー病の動物モデルでの結果と一致し、クロロキン経口投与後の中等度のアルツハイマー病患者では、対照群と比較して、認知機能の低下が遅く、脳脊髄液中のアミロイドβ42レベルが低下していることが明らかになった(Ritchie et al 2003)。しかし、クロロキンが生体内の鉄を標的にしてアルツハイマー病様表現型を救済するという直接的なエビデンスはまだない。タウ欠乏症による加齢に伴う鉄蓄積は、クロロキンの経口投与により予防できることが知られている(Lei et al 2012,2015)。さらに、クロロキン処置は、hA53Tトランスジェニックマウスにおける鉄-シヌクレイン相互作用を効果的に防止することができ(Billings et al 2016)また、Fe3+誘導フィブリン形成を試験管内試験で逆転させることができる(Pretorius et al 2013)。重要なことに、Fe3+およびCu2+の存在下でのアミロイドβ40およびアミロイドβ42凝集体の形成を調査したところ、Fe3+がアミロイドβ40およびアミロイドβ42の凝集を促進し、Cu2+ではなくFe3+がアミロイドβ40およびアミロイドβ42の凝集を促進し、クロロキンがFe3+誘発のアミロイドβ42凝集を有意に減少させることが実証された(Tahmasebinia and Emadi, 2017)。これらの研究は、クロロキンの抗アルツハイマー病能力は、少なくとも一部では、鉄を標的とすることを介しての可能性があり、確かに、基礎となるメカニズムをさらに解明する必要があるという証拠を提供する、それは今ではクロロキンが体に毒性があると考えられているが、少なくともそれは私たちのための有望な方向性を開く。
デスフェロキサミン、デフェラシロックス、デフェリプロン
デフェロキサミン
デスフェロキサミン(DFO)は、鉄やアルミニウムの毒性とそれが体内に誘導する活性酸素を抑制する、よく知られた鉄キレート剤である。当初、DFOはアルミニウムイオンのキレート剤であり、アルミニウムはアルツハイマー病のリスクを高める独立した因子であると考えられていた(Campbell and Bondy, 2000)。Crapper McLachlanら(1991)の結果は、DFOの筋肉内注射を与えられたアルツハイマー病患者の日常生活能力の低下の程度は、プラセボを与えられたアルツハイマー病患者と比較して緩和されたことを示した。また、試験管内試験実験では、DFOがアミロイドβ1-42のβシートの形成を抑制し、あらかじめ形成されたプラーク状のアミロイドプラークを溶解することが示されている(House et al 2004)。また、DFOがAPPのmRNAの翻訳およびAPP全タンパク質の発現を阻害し、アミロイドβペプチドの分泌を減少させることを示した研究もある(Rogers et al 2002;Morse et al 2004)。我々の研究では、DFOの経鼻給餌がADマウスの鉄誘発性記憶障害を逆転させ、APPの形成を抑制することが明らかになった(Guo et al 2013b)。APPへの効果に加えて、DFOはタウタンパク質のリン酸化にも影響を与える。Fine et al 2012)は、DFOがGSK-3βをリン酸化する能力を持ち、それによってリン酸化されたタウのレベルが低下することを発見したが、タウタンパク質のリン酸化を阻害するメカニズムはまだ明らかにされていない。DFOはADモデルマウスの脳内でタウタンパク質のリン酸化を抑制することができるが、鉄の存在下ではタウタンパク質のリン酸化に対するDFOの効果は決定されていない。我々の以前の実験では、APP/PS1トランスジェニックマウスに高濃度の鉄を給与した後、マウスにDFOを経鼻投与したところ、DFOの経鼻投与により、CDK5およびGSK-3β経路を介して鉄誘導タウリン酸化が阻害されることが示された(Guo et al 2013a)。また、DFOはまた、P38/HIF-1α経路を介してAPP/PS1トランスジェニックマウスの脳におけるシナプス損失を減衰させることができることを見出した(Guo et al 2015)。
DFOは、様々な実験マウスモデルにおいて一定の結果を得ており、鉄過負荷疾患の治療薬として連邦医薬品局(FDA)によって承認されているが、DFOの臨床応用には多くの問題点が残されている。第一に、DFOのバイオアベイラビリティが悪く、DFOの分子サイズと親水性が自由にBBBを横断することを妨げ、中枢神経系での利用可能性を低下させる;第二に、DFOは経口的に摂取することができず、注射によって与えられなければならない。1回の注射の時間は長く(10時間まで)注射の頻度は高く(週に5〜7回)結果として患者のコンプライアンスが低い(Crielaard et al 2017);繰り返しになるが、長期治療後の神経毒性および貧血を伴う全身金属イオン枯渇(Cuajungco et al 2000年)消化管の悪吸収および急速な分解(MayおよびBulman、1983)を含む多くの副作用がある。
デフェラシロクス
デフェラシロックスは、日常的に使用できる最初のFDA承認の経口鉄剤である。その化学名は、4-[3,5-ビス(2-ヒドロキシフェニル)-1,2,4-トリスオキサゾール-1-イル]安息香酸であり、タラセミア鉄過剰症患者の治療薬として一般的に使用されている。デフェラシロックスの鉄との結合能力は限られている。この薬剤は、鉄の一部しか結合できず、細胞外および細胞内の鉄受容体に鉄を供給することができない。鉄欠乏症を誘発することは容易ではないが、鉄の蓄積を抑える効果も比較的低いとされている。現在のところ、ディフェラシロックスが脳内鉄蓄積を減少させる役割を果たしていることが研究で明らかになっている。しかし、ディフェラシロックスが脳内鉄蓄積を減少させたり、脳内鉄毒性を減少させたりすることはないという研究もある(Sripetchwandee et al 2016)。これは、ディフェラシロックスがBBBを通過しにくく、鉄と結合する能力も弱いためと考えられる。鉄の結合分子1個に対してデフェラシロックスの分子が3個必要となるため、より高い用量が必要となる。
デフェリプロン
デフェリプロンの化学名は、3-ヒドロキシ-L,2-ジメチル-4-(lH)-ピリドンである。デフェリプロンもデフェラシロックスと同様に、タラセミアの鉄過剰症患者の治療薬として承認されている。デフェリプロンは、体内のほぼ全ての鉄を結合させて活性酸素の産生をこれ以上誘導できないようにしたり、結合した鉄を細胞内外の鉄受容体に供給したりすることができる。デフェリプロンは鉄との結合能力が高いため、1分子の鉄が結合するのに2分子のデフェリプロンが必要となるため、鉄の凝集を効果的に抑えることができる。研究では、デフェリプロンとDFOは、鉄過負荷の存在下でBBB崩壊の速度を低下させ、脳の鉄蓄積を減少させ、脳のミトコンドリア機能障害を減少させることで保護効果を発揮することが明らかになっている(Sripetchwandee et al 2016)。デフェリプロンとDFOの両方が保護効果を発揮するが、デフェリプロンは経口投与によってより大きな利点を達成することができる。
現在、鉄過負荷の第一選択の鉄キレート剤としてDFO、デフェラシロックスおよびデフェリプロンが推奨されているが、いずれもアレルギー反応、肝機能障害および腎機能障害、および神経性難聴を含む一定の副作用を有する(Borgna-PignattiおよびMarsella 2015)。このため、鉄キレート療法のさらなる発展が求められている。
α-リポ酸
α-リポ酸(αリポ酸)は、哺乳類で自然に合成できる低分子化合物である。ミトコンドリアに存在する補酵素である。構造には水酸基とジスルフィド結合が含まれているため、脂溶性と水溶性の両方の性質を持ち、BBBを容易に越える。この研究では、マウスにαリポ酸を60分間静脈内注射した後、マウス大脳皮質にαリポ酸の痕跡が観察されることがわかった(Panigrahi et al 1996)。また、7〜14日にわたって連続注入した後、マウスの脳の複数の部位でαリポ酸の存在が検出された(Arivazhagan et al 2002)。しかし、驚くべきことに、マウスに毎日一定量のαリポ酸を経管投与した場合、一定期間経過してもマウスの脳内にαリポ酸の蓄積が検出されなかった(Chng et al 2009)。これは、αリポ酸が胃の中でジヒドロリポ酸(DHαリポ酸)に速やかに還元され、血液循環を介して全身の組織に運ばれて代謝に参加し、最終的に尿中に排泄されることが最も可能性が高いと考えられる。
ヨーロッパでは、αリポ酸は50年以上前から治療薬として使用されており、主に糖尿病性多発性神経障害の治療に使用されている。その後、臨床研究では、アルツハイマー病を患っている可能性のある129名の患者さんの認知能力の低下が、αリポ酸治療後(600mg /日)しばらくの間、効果的に緩和されたことが明らかになった(Fava et al 2013)。しかし、本研究のサンプルサイズは比較的小さく、無作為化サンプルは存在しなかった。また、研究対象者はアルツハイマー病患者である可能性が高く、神経病理学的にも確認されていなかった。そのため、結果にはあまり説得力がなかった。
いくつかの研究では、αリポ酸の補充はげっ歯類の脳内アセチルコリン転移酵素(ChAT)の活性を増加させ、認知障害を緩和することができることが示されている(デ・フレイタス 2010; Dwivedi et al 2014)。ChATの活性化は脳内アセチルコリンの含有量を正に調節しており、アセチルコリンの発現低下はアルツハイマー病の認知機能障害と密接に関連している。したがって、αリポ酸はChATを活性化することでアセチルコリンの発現を増加させ、アルツハイマー病患者の認知機能を改善する可能性がある。同時に、ChATの活性化はコリン作動性ニューロンの神経新生にも有益であることが研究で示されており(Park et al 2013年)αリポ酸が中枢神経系疾患の重症度を改善する可能性があることをさらに実証している。多くの研究により、αリポ酸はNF-κBおよびMAPKシグナル伝達経路の活性を阻害することにより、IL-1β、IL-6およびTNF-αの放出を減少させることが示されている(Bierhaus et al 1997年;Wong et al 2001;ZhangおよびFrei 2001)。同時に、αリポ酸はまた、中枢神経系への炎症性細胞の浸潤を阻害し、血管細胞接着分子-1の発現をダウンレギュレートする(Kunt et al 1999; Guo et al 2016)。金属イオンをキレートする能力に加えて、αリポ酸はまた、抗炎症、抗酸化、および活性化されたグルコースの取り込みおよび利用において役割を果たすことができる。これまでのところ、αリポ酸治療による重篤な副作用は観察されておらず、αリポ酸がアルツハイマー病治療の臨床薬開発のトレンドになる可能性も示唆されている。
試験管内試験での研究では、αリポ酸はCu2+、Zn2+、DHαリポ酸などの金属イオンと結合し、Cu2+、Zn2+、Pb2+、Hg2+、Fe3+と錯体を形成し、それによって金属キレートを発揮することが示されている(Ou et al 1995)。αリポ酸での水晶体上皮細胞の治療は、鉄の取り込み率と細胞内の動的な鉄プールの有意な減少を引き起こす(Goralska et al 2003)。これらの結果は、αリポ酸は細胞内に入る鉄のレベルを低下させるだけでなく、鉄の貯蔵量を増加させることにより、細胞内の動的な鉄プールを減少させることを示唆した。生体内研究では、高齢ラットにαリポ酸を2週間給餌すると、加齢に伴う皮質領域の鉄蓄積を減少させることが示されている(Suh et al 2005)。Fonte et al 2001)の結果は、αリポ酸がAPP過剰発現トランスジェニックマウスの前頭皮質におけるアミロイドβの溶解度を高めることを示し、αリポ酸は、他の金属イオンキレート剤のように、正常にアミロイドβを再溶解し、アルツハイマー病患者の脳におけるアミロイド沈着を減少させることができることを確認した。しかし、別の研究では、αリポ酸処理したADマウスの学習と記憶保持は有意に改善されたが、脳内の可溶性または不溶性のアミロイドβレベルには有意な変化は見られなかった(Quinn et al 2007)。αリポ酸の金属キレートは多くの実験結果で証明されているが、鉄をキレートすることでアミロイドβの沈着を抑制し、アミロイドβの溶解度を高めることができるかどうかについては議論がある。最も最近、我々のグループはまた、αリポ酸を慢性的に投与することで効果的にタウの高リン酸化を抑制し、P301Sタウトランスジェニックマウスの神経細胞の変性や異常行動を緩和することを発見した;その改善は、トランスジェニックマウスの脳内の酸化ストレス、炎症、フェロプトーシスの緩和を伴う(Zhang et al 2018)。
ラクトフェリン
ラクトフェリン(ラクトフェリン)は分子量80 kDaの非ヘム鉄結合糖タンパク質であり、牛乳、唾液、尿などの様々な分泌物中に広く存在する。そのアミノ酸配列はトランスフェリンと60%同一であるため、トランスフェリンファミリーに分類されている(Metz-Boutigue er al)。 ラクトフェリンは、703アミノ酸のポリペプチド鎖から折り畳まれた2つの球状の葉から構成されている。各分子は、2つの鉄、亜鉛、銅または他の金属イオンと可逆的に結合することができる。結合部位は、タンパク質の2つの球状ドメインに位置している。ラクトフェリンの鉄に対する親和性はトランスフェリンの300倍であり、弱酸性環境下ではさらに親和性が高まることから、炎症が起こるとトランスフェリンからラクトフェリンに鉄が移行することが関係していると考えられる(Dhennin-Duthille et al 2000)。また、中枢神経系では、活性化したミクログリアもラクトフェリンを生成して放出することがわかっている(Xu er al)。 免疫組織化学的研究の結果、神経変性疾患患者の脳組織にラクトフェリンが蓄積していることが明らかになった。さらなる研究では、アルツハイマー病患者の脳内でラクトフェリンの含有量と分布に異常が見られ(Brown et al 1985)老人斑-およびNFT富化領域に大量のラクトフェリンが沈着していることが明らかになった(Valverde et al 1990)。Wang et al 2010)は、ADモデルマウスの脳にラクトフェリン沈着を検出したが、野生マウスではラクトフェリン沈着は認められず、老人斑の形成がラクトフェリンに先行していた。さらに研究を進めると、ラクトフェリン沈着は老人斑とアミロイド病変に局在していることが明らかになった。ラクトフェリン沈着はトランスジェニックマウスの年齢とともに増加したが、18ヶ月齢以降はほとんどの老人斑でラクトフェリン陽性の減少を示した(Wang et al 2010)。アルツハイマー病トランスジェニックマウスでレーザー共焦点技術を用いて脳内のラクトフェリンとアミロイドβをコロケーションさせたところ、ラクトフェリンはアミロイドβ上に発現し、アミロイドβ形成はラクトフェリンの沈着に先行し、アミロイドβプラークはラットの年齢が上がるにつれて、またラクトフェリンの蓄積の大きさと数の両方が増えるにつれて発達することがわかり、ラクトフェリンとアルツハイマー病との間には密接な関係があることが示された(van de Looij er al)。 金属イオン代謝への参加や細胞増殖・アポトーシスの調節など、ラクトフェリンの他の生物学的機能と組み合わせることで、ラクトフェリンは活動性アルツハイマー病の発生を遅らせる可能性があるという仮説を立てている。
ラクトフェリンはBBB血管内皮細胞の血膜上にラクトフェリン受容体(LFR)が存在することから、外因性のラクトフェリンはBBBを容易に越えることができ、近年、脳への薬物ターゲティングのキャリアとして広く利用されている。興味深いことに、Kamalinia et al 2013)は、ラクトフェリン-DFOコンジュゲートがアポトーシスを妨害することができることを実証するために、PC12細胞株を使用した。Atg7,Atg12-Atg5およびLC3-II/LC3-Iを含むオートファジーマーカーの発現レベルが増加し、ラクトフェリン共役腹腔は主にCapsase-3,PARP、Baxおよびbcl-2の発現レベルに影響を与えた。さらに、ラクトフェリン抱合体の腹腔内注射は、アルツハイマー病ラットの学習能力を有意に改善し、アミロイドβを減少させることができる(Kamalinia et al 2013)ことから、神経変性疾患の治療におけるラクトフェリンの使用のための理論的根拠を提供する。
そのため、外因性ラクトフェリンを治療薬として用いることで、アルツハイマー病におけるその役割を明らかにすることが検討されていた。私たちのグループは、外因性ラクトフェリンの投与がAPPとα-セクレターゼの触媒活性と発現の非アミロイド性処理を刺激し、結果的にアミロイドβ沈着を減少させ、ADモデルマウスの認知機能低下を改善するかどうかを初めて調査した(Guo et al 2017)。また、ラクトフェリンがAPPの処理を調節する分子機構にも取り組んだ。実際、ヒトラクトフェリンの機能は鉄キレート剤DFOと類似しており、低酸素条件下で低酸素誘導因子(HIF-1α)発現の神経保護効果を誘導できることが文献で報告されている(Kawamata et al 1993)。具体的には、我々の結果は、ラクトフェリンが試験管内試験および生体内試験でERK1/2-CREBおよびHIF-1α経路を介してα-セクレターゼ依存性APPプロセスを増強することができることを示唆した(Guo et al 2017)ラクトフェリンがアルツハイマー病の治療において積極的な役割を果たしていることを再び証明した。以前の観察と一致して、相澤 et al 2017)は最近、低密度リポ蛋白質受容体関連タンパク質-1(LRP1)へのラクトフェリンの結合がAMP活性化プロテインキナーゼシグナル伝達経路の活性化をもたらし、それによって細胞のオートファジーが促進されることを報告した。このことから、ラクトフェリンは抗炎症、免疫の調節、金属イオンのキレート、細胞のオートファジーの調節を介してアミロイドβやNFTの産生を調節し、最終的にはアルツハイマー病の発生や発症に影響を与える可能性があることが示された。
おわりに
アルツハイマー病の病態は数十年前から研究されており、アミロイドβ沈着によって形成される老人斑とタウタンパク質の高リン酸化によって形成されるNFTがアルツハイマー病の病態の2大特徴であることは明らかであるが、老人斑とNFTの誘導因子については未だ明確に解明されていない意見が多い。アルツハイマー病の複雑な原因に鑑み、多面的な視点からアルツハイマー病の病態を研究することが一般に受け入れられ、良好な結果を得ている。
鉄をターゲットにしたアルツハイマー病の病態研究は、近年研究のホットスポットとなっている。また、アルツハイマー病患者の脳内の過剰な鉄をキレートする鉄キレート剤の使用も、アルツハイマー病治療の新たな戦略となっている。
しかし、脳には他の組織や臓器とは異なり、薬物の進入に厳密な特異性を持つBBBが存在する。そのため、BBBを容易に越え、効果的に機能する鉄キレート剤を見つけるには、体内の自己合成物質から探さなければならない。鉄と結合するタンパク質や分子は体内に多く存在し、αリポ酸やラクトフェリンはこれらの候補薬の一つである。αリポ酸やラクトフェリンの直接投与がアルツハイマー病の症状を緩和することが試験管内試験や生体内試験実験で確認されているが(Guo et al 2017;Zhang et al 2018年)それらに関連する作用機序は明らかにされておらず、さらなる研究が必要とされている。同時に、ラクトフェリンベースのナノ薬物分子も研究されている。
この薬物分子はラクトフェリン分子に付着し、BBBを容易に越えることができるため、病変部に治療効果を発揮し、より良い治療効果を得ることができる。今後の研究では、鉄を契機に、鉄の定常状態からアルツハイマー病の発生・発症メカニズムを研究し、アルツハイマー病の原因をより説明していく予定である。
新皮質アミロイドβ負荷が高い高齢者における血漿フェリチンの上昇
www.nature.com/articles/mp2017146?WT.feed_name=subjects_medical-research
考察
今回の研究から得られた知見は、血漿および血清フェリチン値の上昇が前臨床アルツハイマー病の特徴であることを示しているが、上昇した値はまだ主に正常範囲内である。さらに、血漿(および血清)フェリチン値とNALとの間には有意な正の相関が観察された。さらに、年齢、性別、APOE ε4対立遺伝子の状態からなるベースモデルに血漿フェリチンを加えると、特異度は62%から71%、感度は75%に上昇した。これは、フェリチンがベースモデル以上に統計的に有意なNALの追加予測因子であることを示しているが、図3からわかるように、フェリチン単独の寄与はベースモデルに比べて相対的に小さい。21-24 さらに、最近の研究では、アルツハイマー病におけるフェリチン(および鉄)のホメオスタシスの混乱が周辺部にも反映されていることが確認されている8,9,25,26。
現在の研究で観察されたフェリチンレベルの上昇が炎症と関連していたかどうかを調査するために、我々はCRP濃度を測定し、しかし、我々は血漿(および血清)フェリチンレベルとCRPの間に有意な相関関係を観察しなかった。CRP濃度の変化は以前にアルツハイマー病で報告されているが、今回の研究におけるCRP測定値は、前臨床アルツハイマー病、すなわちNALが高い場合と低い場合を比較して有意に変化していないように思われた。フェリチン濃度の上昇がCRP測定値を調整した後も有意な値を維持していたことを考えると、今回のフェリチン上昇の所見は、鉄の動員障害などの他のアルツハイマー病発症機序に起因する可能性がある。
本研究で得られた知見は、血漿(および血清)フェリチンの変化が、アルツハイマー病発症の非常に早い段階で、海馬の萎縮や認知機能障害の前に起こることを示している。アルツハイマー病のリスクが高い認知的に正常な人の血清フェリチンの変化は、最適でない鉄の動員がアルツハイマー病発症の初期のイベントであることを示唆しているかもしれない。興味深いことに、血清フェリチンレベルは2つのグループ間で変化したが、すなわち、高NALと低NALでは、これらの変化は血清鉄レベルの対応する変化を伴っていなかった;したがって、今回の研究で観察された血清フェリチンレベルの上昇は、組織の鉄負荷を反映している可能性があることを示唆している31,前臨床アルツハイマー病の脳組織における鉄の沈着を報告した先の研究によって裏付けられた4。前臨床アルツハイマー病における脳アミロイドβ沈着、循環フェリチン、組織鉄蓄積(MRIを介して)に関して、脳と周辺部での一連の事象に対処するためには、さらなる研究が必要である。
本研究で認められた限界には、両群間の性別の不均一およびAPOE ε4対立遺伝子のキャリア状態の分布が含まれる;しかしながら、本研究では、血漿(および血清)フェリチンレベルにおいて、男性と女性の間(補足図3)およびAPOE ε4キャリアと非キャリアの間(補足図4)で有意な差は観察されなかった。その他の制限事項としては、使用したサンプル数が控えめであること、及び本研究の横断的な性質が挙げられる。独立したコホートにおいて、現在の知見を縦断的に検証するためには、さらなる研究が必要である。さらに、フェリチンレベルの変化が他の神経変性疾患と関連していることが報告されているように、27,35 前臨床アルツハイマー病のバイオマーカーとしてのフェリチンの特異性は、さらなる研究が必要である。しかし、フェリチンは他の神経変性疾患と関連している可能性があるが、本研究で得られた知見は、アルツハイマー病のゴールドスタンダードバイオマーカーであるNALとの有意な正の相関を明らかに示していることに留意する必要がある。
本研究で得られた知見は、高NAL群では、低NAL群と比較して、明らかな認知機能障害や海馬萎縮の前に血漿(および血清)フェリチン濃度の上昇が観察されたことから、アルツハイマー病の前臨床血液バイオマーカーの探索に貢献する可能性がある。重要なことに、これらの所見は、APOE ε4対立遺伝子の存在自体が疾患の主要な危険因子であることを考えると、初期のアルツハイマー病バイオマーカーにとって有益な特徴であるAPOE ε4非キャリアにおいても有意なままであった。さらに、Aytonら6は、APOE ε4キャリアのベースライン脳脊髄液フェリチンレベルが非キャリアと比較して有意に高いこと、および脳脊髄液中のフェリチンの上昇が認知パフォーマンスの低下と関連しており、MCI患者がアルツハイマー病に移行するリスクを増加させるという彼らの知見に基づいて、「APOE ε4キャリアの状態はフェリチンレベルを増加させることでアルツハイマー病への感受性をもたらす」という概念を紹介している。
興味深いことに、今回の論文とAyton er al)。6の論文では、APOE ε4キャリアと非キャリアの間で血漿フェリチンレベルに有意な差は認められなかった。しかし、今回の研究から得られた知見は、高NALで観察されるフェリチン濃度の上昇がAPOE ε4キャリアに限ったものではないことを示している。しかしながら、本研究は、APOE ε4キャリアにおける高NALと低NALの間の血漿フェリチン濃度について声明を出すのに十分な力を欠いていることに留意すべきである。注意すべき重要なことは、本研究コホートのAPOE ε4非キャリアサブセット内で、低NALと比較して高NALのフェリチン濃度が有意に上昇していることである。
今回の発見は、世界中の病理検査室で日常的に検査されている血漿/血清フェリチンが、認知機能がまだ低下していない前臨床段階の個人の脳アミロイドβ負荷を表すマーカーのパネルに付加価値を与える可能性を示唆している。興味深いことに、O’Bryantら39は、血清フェリチンを含む30のタンパク質パネルを報告し,0.94というかなり高いAUCでHCからアルツハイマー病を予測した。
さらに、我々の知見はまた、アルツハイマー病発症の初期段階で金属のホメオスタシスの全身的な混乱を示すことで、本疾患の臨床発症に関与するメカニズムについての洞察を提供している。フェリチン値の上昇はパーキンソン病や筋萎縮性側索硬化症などの他の神経変性疾患と関連している27,35 が、本研究ではフェリチンとアミロイドβの相関性がアルツハイマー病発症との関連性を裏付けていることに注目すべきである。さらに、ベースラインでの我々の観察結果と一致しているのは、高NAL vs.前臨床アルツハイマー病患者ではフェリチンが高く、NALが低い患者ではフェリチンとNALの間の正の相関が12ヶ月間の時点で有意に保たれていた。アルツハイマー病の病態の複雑さを考えると、マーカーの「パネル」を使用することで、NALが高い人と低い人の間でより強い鑑別が可能になるだろう。