
Contents
Insulin Resistance in Alzheimer’s Disease
www.frontiersin.org/articles/10.3389/fnins.2018.00830/full
糖尿病、肥満、認知症の疫学的な関連性は、公衆衛生上の重要な課題であると同時に、これらの状態をさらに理解する機会でもある。この3つの疾患の間で重要な交点はインスリン抵抗性であり、これは糖尿病と肥満の末梢組織で起こることが古典的に記述されており、最近ではアルツハイマー病の脳で発症することが示されている。ここでは、障害されたインスリンシグナル伝達を抗糖尿病薬で標的にすることで認知症を治療できる可能性を示す、心強い前臨床および臨床データをレビューする。我々はさらに、末梢代謝異常が糖尿病、肥満、およびAD間の接続のための可能な説明を提供し、脳の機能不全につながる可能性があるを介して生物学的メカニズムを議論する。最後に、我々は簡単に生涯のアロスタティック負荷がどのように老化と相互作用して晩年の認知症のリスクを増加させる可能性があるかを議論する。
はじめに
アルツハイマー病(AD2型糖尿病(2型糖尿病肥満は、世界的に最も高価で障害のある疾患の一つである。長い間、認知機能障害と代謝性疾患との相関関係は検出されなかった。現在では、ますます疫学的証拠がこれらの疾患間の重要な関連を支持している(Razay et al 2006年;Baker et al 2011年;Crane et al 2013年;Ikram et al 2017)。したがって、実験的観察は、代謝異常のマーカーがADにも存在し、最も顕著なものはインスリン抵抗性であることを明らかにしている(Talbot et al 2012; De Felice、2013; Boles et al 2017)。しかし、このクロストークの根底にある分子メカニズムは、中枢および末梢インスリンシグナル伝達がADでどのように動作するかと同様に、まだとらえどころのないものである(BiesselsとDespa、2018)。
過去10年間の累積データは、脳がインスリン感受性の器官であることを補強してきた。インスリン受容体(IR)および関連するインスリン様成長因子受容体1および2(IGF1-R/IGF2-R)は、代謝制御のための重要な脳領域である齧歯類の視床下部だけでなく、大脳皮質、海馬、視床、嗅球、さらに低レベルでは小脳、線条体、中脳、脳幹でも発現している(Fernandez and Torres-Alemán, 2012; Kleinridders et al 2014)。
全身の代謝調節におけるインスリンの定型的な役割(Brüning et al 2000)を超えて、インスリンはまた、シナプス可塑性を促進するニューロン活動を修飾し(Wan et al 1997;Schmitz et al 2018哺乳類の脳における記憶機能を改善する(Park et al 2000;Benedict et al 2007)。グリアのインスリンシグナル伝達機能が注目されている。視床下部アストロサイトIRは、POMCニューロンのグルコース誘導活性化、グルコースの利用可能性に対する中枢および末梢応答、および血液脳関門を介したグルコース輸送を制御する(BBB、García-Cáceres et al 2016)。さらに、インスリンは、試験管内試験でミクログリアおよびアストロサイトにおける前炎症性サイトカイン分泌を調節する(Spielman et al 2015;Kurochkin et al 2018)。脳損傷下では、IGF1は活性化されたミクログリアによって産生され得、血管リモデリングに必要とされる(Walter et al 1997; Lopez-Lopez et al 2004)。異なる細胞型におけるインスリンシグナル伝達の複雑な調節を解読するためにはさらなる研究が必要であるが、これらのデータは、IR/IGF-Rが中枢神経系(中枢神経系)の生理学において役割を果たしていることを示唆している。
驚くべきことに、いくつかの研究は、インスリンシグナル伝達がアルツハイマー病患者およびAD実験モデルの脳で障害されていることを示している(Boyt et al 2000年;Craft et al 2003年;Steen et al 2005年;Bomfim et al 2012年;Hiltunen et al 2012年;Talbot et al 2012)。神経性インスリン抵抗性は、海馬ニューロンの初代培養物中のアミロイドβオリゴマー、およびマウスおよびサルにおけるアミロイドβOの脳内注射によって誘導され得る。それは、TNF-α活性化およびIRS阻害によって媒介され、シナプス機能障害、シナプス可塑性の障害、およびシナプス損失に大きな影響を与える(Townsend et al 2007; De Felice et al 2009; Bomfim et al 2012; Batista et al 2012; De Felice et al 2009; Bomfim et al 2012; Batista et al 2007; De Felice et al 2009; De Felice et al 2009; Bomfim et al 2012; Batista et al 2012)。2018)驚くべきことに、我々のグループは、アミロイドβオリゴマーのICV注入もまた、末梢性インスリン抵抗性の古典的な特徴を有する末梢性グルコース不耐症を誘発することを発見した、このプロセスはまた、トランスジェニックADマウスモデルで観察され(Clarke et al 2015そしてそれは、ADにおける糖尿病のリスクの増加の根底にあるかもしれない(Janson et al 2004)。さらに、抗糖尿病薬は、認知、シナプス保護、インスリンシグナル伝達障害、および小胞体ストレスおよび慢性炎症などの他のAD関連の病理学的メカニズムに有益な効果を及ぼす(Lourenco et al 2013; Sebastião et al 2014; Batista et al 2018; Tai et al 2018)。
ここでは、2型糖尿病、肥満、ADに共通するインスリン抵抗性の類似のメカニズムに関するエビデンスをレビューする。末梢代謝緩和とアルツハイマー病との関連、これらの疾患間の因果関係、およびどのようにインスリン経路をブーストすることがADを治療するための治療的選択肢を提供するかを説明するのに役立つ最近の知見を議論する。
末梢代謝疾患と脳病理のクロストーク
糖尿病と肥満とアルツハイマー病との関連を理解するための重要な問題は、周辺部の代謝障害が脳の病理学的な変化を引き起こす可能性があることを知ることである。
一般的に脂肪酸と脂質は、肥満、糖尿病、およびアルツハイマー病の確立されたプレーヤーである。最近のメタ解析研究では、ADにおける脂質の異なるクラスの潜在的なメカニズム、診断、および治療的意味合いが明らかになった(Zarrouk et al 2017)。肥満と糖尿病は炎症性成分を呈する(Wang et al 2018)。実際、肥満患者はしばしば、脂肪組織における基底性低悪性度の全身性炎症を表示し、免疫介在性疾患への感受性を増加させる(Sun et al 2012年;MrazおよびHaluzik、2014)。摂食からの遊離脂肪酸(FFA)は、Toll様受容体4(TLR4)刺激によって開始される炎症性カスケードの引き金となり、TNF-αおよびインターロイキンIL-1βおよびIL-6などのプロ炎症性サイトカインを放出する(Weisberg et al 2003;Shu et al 2012)。高レベルのFFAは、インスリンの抗溶血作用をさらに阻害し、血流中へのFFA放出速度を増加させる(Guenther、2009)。BBB内皮細胞に結合したFFAは、脳へのFFAの浸潤を可能にする、透過性を変化させる(Rapoport、2001)。したがって、脳内FFAの増加は、セラミド産生、パターン認識受容体の活性化、炎症、およびERストレスなどの有害事象を誘発する(Spriegl、2005;Guenther、2009;Groop et al 2018)。脳がホメオスタシスの乱れを検出すると、ミクログリアが活性化され、神経炎症につながる(Jha et al 2016)。飽和脂肪酸および一価不飽和脂肪酸は、TLR4依存的な方法でミクログリアNF-kB経路を活性化し、プロ炎症性サイトカインおよび活性酸素種(ROS、Wang et al 2012; Arnold et al 2014; Button et al 2014; Carroll et al 2018)の産生の増加を導くことが示された。飽和脂肪酸はまた、サイトカイン産生につながる培養アストロサイトのTLR4依存性活性化を誘導することが示された(Gupta et al 2012;Wang et al 2012)。
高度な糖化最終生成物(AGEs)はまた、糖尿病とアルツハイマー病の共通の特徴を表し、この接続の下にある末梢と中枢神経系の間のクロストークに関与している可能性がある。AGEsは、糖とタンパク質や脂質の間の非特異的かつ制御されていない反応の産物である。AGEsは正常な老化の間に増加するが、高血糖症などのグルコースが豊富な環境下ではその形成が促進される。興味深いことに、AGEsレベルの上昇はAD脳でも観察されている(Shuvaev et al 2001; Choei et al 2004; Takeuchi et al 2007)。重要なことに、AGE受容体(RAGE)は、アミロイドβの可能性のある受容体として示唆され(Yan et al 1996ERストレスの誘導などのアベタ毒性メカニズムを媒介することが示唆された(Chen et al 2018)。さらに、RAGEは、アミロイドβ産生、タウ過リン酸化およびタングル形成、シナプス障害認知機能低下、および神経変性を促進することが示されている(Cai et al 2016)。ミクログリアにおいて特異的にRAGEシグナル伝達をダウンレギュレーションすることで、シナプス障害および認知機能障害を予防し、アルツハイマー病のマウスモデルにおいてストレス関連キナーゼの活性化を減少させた(Criscuolo et al 2017)。重要なことに、最近の証拠は、RAGEが、BBB完全性およびタイトジャンクション調節だけでなく、APOE4誘発BBB異常に対するAbetaの影響を媒介することを示している(Park et al 2014年;Wan et al 2014年、2015年;Alata et al 2015)。
集合的に、これらの観察は、代謝障害における脂質異常が、グリア細胞の活性化につながるイベントのカスケードを誘発し、FFAに対するBBBの透過性につながる可能性があり、神経炎症を引き起こすことを示している。並行して、高血糖症で増加したAGEレベルは、BBBの完全性のRAGE媒介の混乱に寄与し、さらにADにつながる脳病理を促進する可能性がある。侵害されたBBBを介して脳に到達する炎症性サイトカインは、ニューロンにとって有害な環境を形成し、ニューロンのインスリン抵抗性、およびシナプス機能不全を導く(Bomfim et al 2012年;Gupta et al 2012年;Lourenco et al 2013年;Kiernan et al 2016年;Vieira et al 2017年、図1)。この仮説と矛盾しないように、それはHFDがマウス海馬における炎症性反応を活性化することが示されている(Lu et al 2011; Almeida-Suhett et al 2017)とインスリンシグナル伝達を損なう(アーノルド et al 2014)一方、高AGE食は、アルツハイマー病のマウスモデルにおけるAD様表現型を悪化させる。
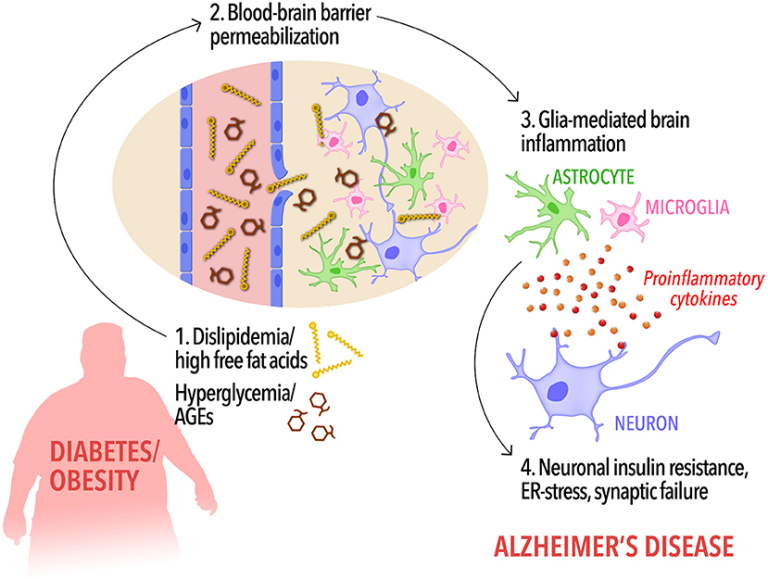
図1. 末梢代謝異常と認知症を結びつけるイベントのカスケードの可能性
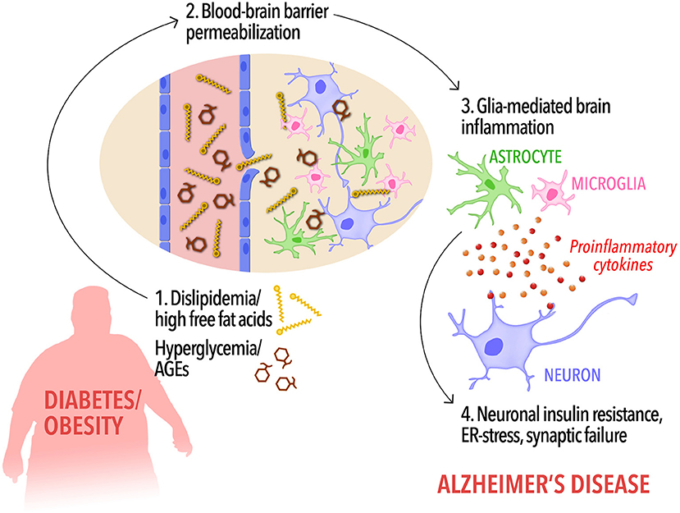
糖尿病や肥満の被験者では、脂質異常症や循環遊離脂肪酸の増加、高血糖や末梢性AGEsレベルの上昇(1)により、血液脳関門の透過性が上昇し、FFAの脳内への流入が可能になる(2)。脳FFAとAGEsの高レベルと一緒に破壊されたBBBは、順番に、ミクログリアとアストロサイトの活性化とプロ炎症性サイトカインの放出を引き起こすだろう(3)。低悪性度、慢性的な脳の炎症は、認知障害やアルツハイマー病に脳をプライミング、インスリン抵抗性(4)を含むニューロンの有害なイベントにつながる。
脳および末梢細胞におけるインスリンシグナル伝達障害の影響
興味深いことに、多くの研究は、アルツハイマー病の発生率が2型糖尿病患者および肥満者において高いことを示唆しており、これらの障害を推進する共通のメカニズムを示唆している(Kivipelto et al 2005年;Razay et al 2006年;Whitmer et al 2007年;Baker et al 2011年;Crane et al 2013年;Ikram et al 2017)。糖尿病、肥満、およびアルツハイマー病の間で共有される中核的な特徴は、インスリン抵抗性である(Kullmann et al 2016)。アルツハイマー病患者の脳では、インスリンシグナル伝達の障害は危険因子であるだけでなく、病理学を悪化させる(Matsuzaki et al 2010; Arnold et al 2018)。
過去数十年の間に、IR/IGF-Rシグナル伝達経路の変化とアルツハイマー病とを相関させるマウンティング研究が行われている(Boyt et al 2000年;Craft et al 2003年;Bomfim et al 2012年;Hiltunen et al 2012年;Talbot et al 2012年;Pitt et al 2017)。さらに、インスリンシグナル伝達に関与する他のタンパク質の変化も記載された。インスリン分解酵素(IDE)は、インスリンおよびアミロイドβクリアランスに必須である(Farris et al 2003)。アルツハイマー病や糖尿病のインスリン抵抗性は高インスリン血症を引き起こし、それにより、インスリンやアミロイドβ分解のためのIDEが飽和してしまう。さらに、IDEの機能は、散発性アルツハイマー病の主要な危険因子である加齢とともに低下する(Kurochkin et al 2018)。また、ホスファターゼおよびテンシンホモログ(PTENプロテインキナーゼB(Aktグリコーゲン合成酵素キナーゼ3β(GSK3β)がシナプスコンパートメントにリクルートされ、アミロイドβ曝露後のLTPを障害する(Knafo et al 2016)。
糖尿病および肥満において、プロ炎症性サイトカイン、特に腫瘍壊死因子α(TNF-α)のレベルの増加は、c-Jun末端キナーゼ(JNK)を活性化し、その結果、Ser312IRS1,Ser636IRS1の阻害性リン酸化を増加させ、Tyr465IRS1の活性化性リン酸化を減少させることにより、インスリン受容体基質1(IRS1)の抑制をもたらす(Hotamisligil et al 2016.1993; Rui et al 2001; Pedersen et al 2003)。) これらの変化は、アルツハイマー病の病態においても同定された。JNK活性化およびIRS1阻害は、アミロイドβオリゴマーの脳室内(ICV)注射後のADトランスジェニックマウスおよびシノモルグスサルの海馬に存在する(Ma et al 2009;Forny-Germano et al 2014)。注目すべきは、インスリンシグナル伝達経路におけるこれらの変化は、アルツハイマー病患者においても記載されていたことである。最初の証拠は、アルツハイマー病の脳は、IR mRNA、IRS関連PI3K、および活性化されたAktの発現の減少を伴う、インスリンとインスリン様成長因子(IGF)-Iと-IIの両方の顕著な減少したレベルを表示することを示す研究から来ている(Steen et al 2005)。この観察は、その後、非認知症からMCIからADへと患者が進化するにつれて、それらの活性化されたセリンキナーゼGSK-3,IKK、JNK、mTOR、およびPKCζ/λのレベルの増大を伴う、IRS-1の阻害性セリン残基におけるリン酸化の漸増を示す研究によって裏付けられた(Bomfim et al 2012年;Talbot et al 2012)。増加したIRS-1セリンリン酸化は、後に、タウパソロジーおよびタングルを呈するニューロンと共局在することが発見された(Yarchoan et al 2014)。さらに、AD、前頭型認知症、2型糖尿病患者の神経由来の血液エクソソームにおいて、Ser312IRS1のリン酸化が増加していることが観察された。本研究の最も興味深い点は、p-Ser312IRS1の増加が、10年後にこれらの変化を持続させたアルツハイマー病患者としての進行期アルツハイマー病患者に現れたことである(Kapogiannis er al)。 インスリン抵抗性のためのエクソソソームバイオマーカーは、AD脳の形態変化とさらに関連していた(Mullins et al 2017)。特筆すべきことに、アルツハイマー病患者の脳およびトランスジェニックマウスにおいて記載されたIRS-1およびIRS-2の抑制性リン酸化は、記憶障害と相関し、インスリン抵抗性状態を導く(Steeen et al 2005;Bomfim et al 2012;Talbot et al 2012)。
糖尿病および肥満モデルの脂肪組織などの末梢組織において、TNF-α放出はまた、ストレスキナーゼIκBキナーゼ(IKK)および二本鎖RNA依存性プロテインキナーゼ(PKR)を活性化し、炎症を促進し、インスリンシグナル伝達の調節障害に加えて小胞体ストレスを誘発する(Hotamisisligil et al 1993;WellenおよびHotamisligil et al 2005;Yang et al 2009)。逆に、TNF-α枯渇は、マウスを肥満誘発性インスリン抵抗性から保護する(Uysal et al 1997)。同様に、アミロイドβオリゴマーもまた、AD動物モデルにおいて、TNF-α分泌およびその結果としてのIKKおよびPKR活性化の誘導を誘導する(Bomfim et al 2012;Lourenco et al 2013インスリン抵抗性を促進する共通のメカニズムを示唆している。
興味深いことに、マウスにおけるアミロイドβオリゴマーのICV注射は、JNKおよびIKKおよびIRS-1阻害の活性化によって特徴付けられる末梢性グルコース不耐症、インスリン抵抗性、および炎症を誘発し、アルツハイマー病病理学の進行を糖尿病の発症に結びつける(Clarke et al 2015)。アルツハイマー病患者では、[18F]-fln アルツハイマー病患者、脳内のiポジトロン断層撮影(FDG-PET)スキャン、血中インスリン抵抗性のホメオスタシスモデル評価(HOMA-IR)を相関させることで、末梢性インスリン抵抗性と中枢性インスリン抵抗性の関連性が提案された。この研究では、ADに脆弱な関心領域(ROI)を評価し、すべてのROIでHOMA-IRが高く、FDGが低いことを予測し、中枢性低血糖と末梢性インスリン抵抗性が関連していることを示した。さらに、両者は内側側頭葉の即時記憶障害および遅延記憶障害と関連していた(Willette et al 2015)。
完全に解明されていないが、それは今、アルツハイマー病と糖尿病のインスリンシグナル伝達の変化が関連付けられていることが認められている。そのため、細胞型と文脈における選択的インスリン応答を区別するために大きな努力がなされてきた。インスリン効果のほとんどはニューロンによって媒介されると考えられていたが、最近の知見は、グリア細胞の機能および全身の応答に影響を与えるグリア細胞に対するインスリン作用を示す(Bélanger et al 2011年;García-Cáceres et al 2016年;Fernandez et al 2017)。アストロサイトIRノックアウトマウスは、中等度のグルコース不耐症および抑うつ様行動を呈する。さらに、ニューロンへのアストロサイト媒介ATPトラフィックは障害されており、プリンリン作動性シグナル伝達に影響を与え、その結果、ドーパミン放出を減少させる。不思議なことに、これらの表現型は雄マウスでのみ観察され、アストロサイトにおけるIRの性特異的な役割の可能性を示唆している(Cai et al 2018)。臨床的証拠がうつ病とADをリンクさせているので、これはアルツハイマー病と他の精神疾患の間の新しい分子リンクを表す可能性がある(Ownby et al 2006; Modrego、2010)。これらの観察に沿って、マウスのアストロサイトIGF1-Rをノックアウトすると、ミトコンドリア機能が衰弱し、活性酸素産生が上昇し、ワーキングメモリが損なわれる。さらに、アストロサイトIGF1-Rのノックアウトは、アストロサイトによるグルコースおよびアミロイドβの取り込みを減少させ、脳内のアミロイドβ蓄積に寄与する(Logan et al 2018)。
活性化されたミクログリアによって誘導される神経毒性アストロサイトの表現型が最近記述された。このA1様アストロサイトは、老化およびいくつかの神経変性疾患の悪化と関連している(Liddelow et al 2017;Clarke et al 2018)。逆に、新規なグルカゴン様ペプチド1受容体(GLP1R)アゴニストであるNLYP01を用いてアストロサイトのA1型へのミクログリア媒介変換を標的とすると、パーキンソン病のマウスモデルにおいて、寿命を延長し、ドーパミン作動性ニューロンの喪失、および行動障害を逆転させることができる(Yun et al 2018)。将来的には、ミクログリアGLP1経路を標的とした有益な効果が、アルツハイマー病などの他の神経変性疾患に改善をもたらすかどうかを調べることは興味深いことであろう(Liddelow and Barres, 2017)。
遅発性アルツハイマーの最強の遺伝的危険因子は、コレステロール代謝に関与するアポリポタンパクE(ApoE4)タンパク質のアイソフォームApoE4である(Strittmatter et al 1993a,b)。ApoE4リスクと並んで、アルツハイマー病患者はまた、学習と記憶に重要なコレステロール酸化物誘導体である24-ヒドロキシコレステロール(24-OHC)の異常なレベルを血漿と脳脊髄液(脳脊髄液)中に提示する(Zarrouk et al 2017)。興味深いことに、ApoE4は、2型糖尿病を持つ人のAD神経病理を悪化させるように見える(Malek-Ahmadi et al 2013)。また、ApoE4キャリアにおけるインスリン抵抗性が、脳脊髄液中のリン酸化タウの高レベルと相関していることが観察された(Starks et al 2015)。ApoE4キャリア、アルツハイマー病の発症、および肥満および糖尿病に関連する代謝変化との関連を解明しようとする報告が増えている(Peila et al 2002年;Reiman et al 2005年;MoserおよびPike et al 2017年;Zhao et al 2017)。ApoE4遺伝子型は、高脂肪・高糖質食下のマウスにおいて、体重増加、グルコース代謝障害、アミロイドβプラーク負荷の増大、グリオシスを悪化させることが記載されている(Moser and Pike, 2017)。また、ヒトApoE4バリアントは、インスリン受容体と相互作用し、インスリン受容体を神経細胞内のエンドソームにトラップし、神経細胞表面でのIRsの利用可能性を低下させ、インスリン感受性を低下させる。驚くべきことに、これらの効果は年齢に依存しており、高脂肪食(HFD、Zhao et al 2017)の下で加速される。また、TREM2-ApoE経路は、アポトーシスニューロンのファゴサイトーシスによって誘導される神経変性ミクログリアへのスイッチを調節し、ADにおいて有害な役割を果たしていることが記載されている(Krasemann et al 2017)。しかし、インスリンシグナル伝達がこのプロセスに参加しているかどうか、またどのようにして参加しているかは、まだ決定されていない。重要なことに、TREM2多型もまた、遅発性アルツハイマー病の高リスクを決定するが、神経変性疾患におけるそれらの役割については議論の余地がある(Jay et al 2015; Wang et al 2015)。したがって、現在の課題は、ADリスクと発症を構成する複数の遺伝的・環境的背景において、インスリンシグナル伝達を選択的に修飾するために、インスリンシグナル伝達の明確な役割を同定することである。
アルツハイマー病の文脈におけるインスリンシグナル伝達を高めることの有益な効果
現在、AD治療に使用されている薬剤は、アセチルコリン作動性経路、NMDA型グルタミン酸作動性経路、およびグルタミン作動性経路を標的としている。これらの治療法は、記憶障害の症状緩和を提供するが、アルツハイマー病の進行の根底にある病理学的メカニズムには対処しない(Graham et al 2017)。アミロイド仮説に基づく治療法は、アミロイドβ産生を減少させ、脳内の有毒なアミロイドβ凝集体を減少させることを目的としている(BarageおよびSonawane、2015)が、重篤な副次的効果(Searfoss et al 2003;Wong et al 2015好ましくない薬理学的効果(Searfoss et al 2003;Wong et al 2015)のために成功していないことが証明されている。2004年好ましくない薬理学的特性(Vassar、2014年または臨床試験でアルツハイマー病患者の認知を改善することに失敗する(Salloway et al 2014年;Lasser et al 2015年;Kennedy et al 2016年;Egan et al 2018年;Honig et al 2018)。この文脈では、症状の緩和を提供し、軽微な副次的効果で病理を改善し得る代替ターゲットを探索することが重要である。
この問題に関して、抗糖尿病薬がADモデルや臨床研究において神経保護効果を発揮する可能性があることを示すデータが蓄積されている。私たちのグループは、臨床で使用されている抗糖尿病薬によるインスリンシグナル伝達の変調について、様々なADモデルを用いて研究していた。我々は、インスリンがアミロイドβオリゴマーによるシナプス欠損や表面IRの低下を抑制することを試験管内試験で確認し(De Felice et al 2009年また、PKRを介した小胞体ストレスを抑制することを明らかにした(Lourenco et al 2013)。また、アミロイドβオリゴマーに曝露した海馬培養物、トランスジェニックADマウス、およびアミロイドβオリゴマーをICVに注射したシノモルグス猿を用いて、エクセンジン-4やリラグルチドなどのGLP1-Rアゴニストの有益な効果を調べた。これらの薬剤は、IRとは無関係にGタンパク質依存性シグナル伝達を介してインスリン関連経路を活性化し、IGF-Rを活性化する(Andersen et al 2018)。Exendin-4は、Ser312IRS1,Ser636IRS1,JNKの阻害性リン酸化を減少させる一方で、活性化するTyr465IRS1のリン酸化を回復させ、APP/PS1トランスジェニックADマウスにおけるインスリンシグナル障害、記憶障害、アミロイドプラーク負荷の減少を打ち消すように作用した(Bomfim et al 2012)。リラグチドは、アミロイドβオリゴマーを含むICVを注射したシノモルグスサルにおいて、タウリン酸化を減少させ、c-AMP依存的な方法でIRの減少とシナプスの損失を防止した(Batista et al 2018)。他のグループはまた、リラグルチドがADトランスジェニックマウスにおいて炎症を減少させ、LTPを増強することができることを示し(McClean et al 2011;McCleanおよびHölscher、2014エクセンジン-4が神経変性ラットモデルにおいて神経細胞の興奮毒性を防止することを示し(Perry、2002icvインスリン注射が認知パフォーマンスを増強することを示した(Park et al 2000,表1)。
表 1.
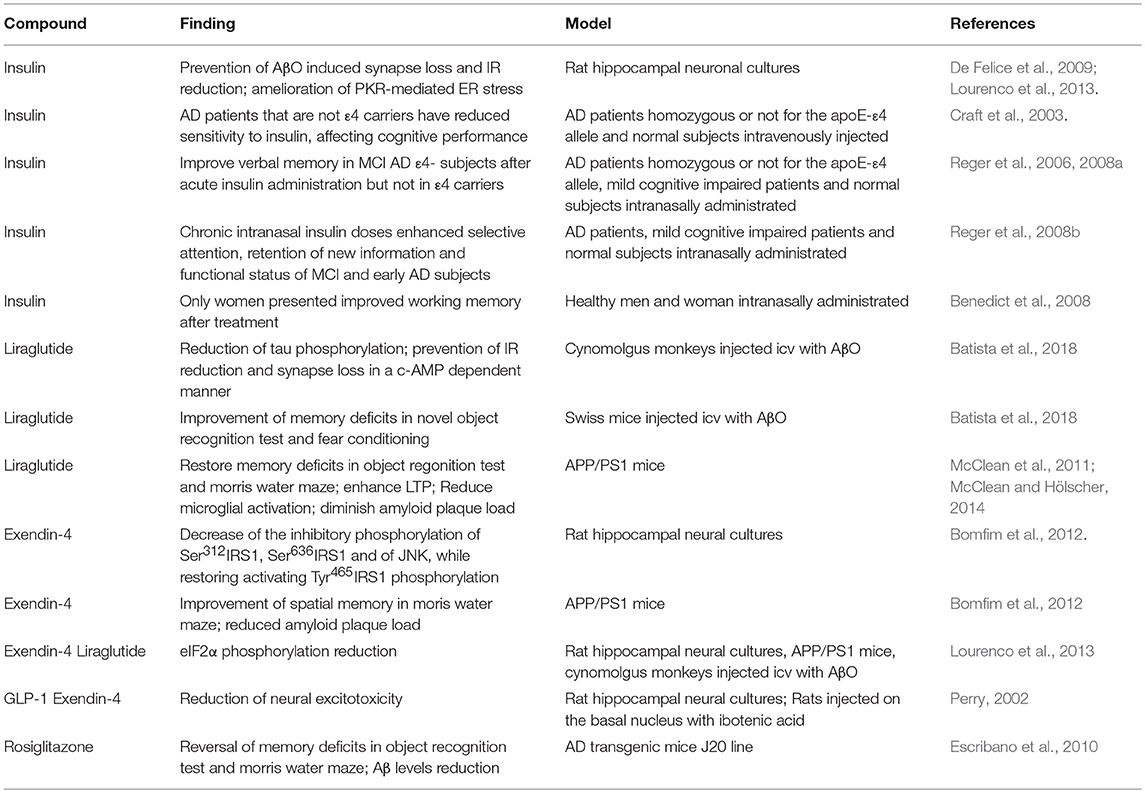
抗糖尿病薬、インスリン増感薬のAD病態の多面性に対する有効性に関する前臨床試験および臨床試験の概要をヒトおよび動物モデルで示した。
前臨床試験および疫学的データから得られた知見は、インスリンをアルツハイマー病患者の治療薬として再利用する臨床試験を促進するものであった(表1)。抗糖尿病薬は、アミロイドβの毒性を緩和し、炎症を抑え、記憶障害を改善することで神経保護効果を発揮する(Bomfim et al 2012年;Lourenco et al 2013)。この文脈では、抗糖尿病治療は、アルツハイマー病の病理学的進行のいくつかの側面をカバーするマルチターゲットアプローチを提供している。しかし、インスリンシグナル伝達が様々な細胞プロセスをシステム的に制御していることから、オフターゲット化の影響についての懸念が提起されている。この問題を回避するために、一つの有望な解決策は経鼻投与であり、経口投与よりも効果的であることに加えて、中枢神経系への投与を制限し、低血糖などの主要な末梢作用を回避することである(Born et al 2002; Spetter and Hallschmid、2015; Schmid et al 2018)。軽度認知障害(MCI)および早期アルツハイマー病患者では、急性経鼻インスリン投与は言語記憶の想起を促進する。それにもかかわらず、ApoE4キャリアは、治療後により貧しい言語記憶想起を示し、インスリンの中枢作用におけるApoE4遺伝子型の役割を示唆した(Craft et al 2003; Reger et al 2006,2008a)。同じグループは、慢性的な経鼻インスリン投与により、MCIおよび早期AD対象者の選択的注意、新しい情報の保持、および機能的状態が増強されたことを報告した(Reger et al 2008b)。重要なことに、経鼻インスリン治療の選択的効果が観察された。女性は経鼻インスリン治療後、男性よりも認知改善のスコアが高いことが報告されている(Benedict et al 2008)。逆に肥満の人では、経鼻インスリンは体重には影響しないが、宣言的記憶と気分の改善が見られた(Schneider et al 2014)。現時点では、アルツハイマー病患者に対する経鼻インスリンの有益な効果にアクセスする10の進行中の未発表の臨床試験がある(NCT00581867,NCT02010476,NCT01436045,NCT01636596,NCT03038282,NCT01767909,NCT00018382,NCT01595646,NCT01547169,NCT02462161)。
インスリンに加えて、GLP1-Rアゴニストもまた、前臨床モデルでの先行する良好な結果を考慮すると、臨床試験に投入されるべき選択肢である(Duarte et al 2013年;Tramutola et al 2017)。少数の患者を対象とした1つのパイロット研究では、リラグルチドの皮下投与は脳内グルコース消費量の低下を防止したが、アミロイドβ負荷または認知には影響を及ぼさなかったことが指摘されている(Gejl et al 2016)。現在、ADにおけるリラグルチドの神経保護効果にアクセスする2つのより大きな臨床試験が進行中である(NCT01469351,NCT01843075)。代替経路を標的とする他の抗糖尿病薬もまた、ペルオキシソーム増殖因子活性化受容体γ(PPARγ)アゴニストであるロジグリタゾンおよびピオグリタゾンのようなADへの対応が検討されている(Miller et al 2011)。前臨床試験では、ロシグリタゾンが記憶力を改善し、リン酸化タウを減少させることが示された(Escribano et al 2010)。しかし、これらの研究も初期のものであり、今のところ有益な効果は報告されていない(Harrington et al 2011年;Miller et al 2011)。今後数年で、現在進行中の大規模臨床試験の結果は、アルツハイマー病患者の代謝状態が治療の成功にどのように干渉するか、また、認知症における抗糖尿病療法の可能性、特に、可能性のあるAD治療としてのインスリン経鼻投与についての更なる情報を提供することになるであろう(Femminella et al 2017)。
アルツハイマー病進行の累積仮説
現在、アルツハイマー病の進行に関するいくつかの謎は解明されているが、病気の発生を誘発する生物学的変化の順序は明らかにする必要がある。ADを説明するための最も受け入れられている仮説の一つは、脳内のアミロイドβオリゴマーとアミロイド斑の蓄積の結果として病状が進行するというものである(Hardy and Higgins, 1992; Haass and Selkoe, 2007)。家族性ADでは、APPやPSENなど、多くの変異がアミロイドβ処理に関連しており、この考えを支持している。しかし、アミロイドβオリゴマーの蓄積のメカニズムや、一見アミロイドβとは無関係に見える他の変異がどのようにして散発性ADリスクを増加させるのかについては、まだ解明されていない(Rao et al 2014)。アルツハイマー病が炎症とインスリン抵抗性によって引き起こされる可能性があるという理解は、新たな視点をもたらした。生活の中で蓄積された複数の環境影響であるアロスタティック負荷が、アルツハイマー病の発症を加速させる可能性がある。脳と全身の代謝恒常性の間のクロストークは、散発性アルツハイマー病のドライバーの1つであると考えられている(De Felice, 2013; Mattson and Arumugam, 2018)。
環境要因は、外傷性脳損傷、母体分離、心理的外傷が神経変性疾患のリスクを高める可能性がある最初の人生の最初の年であっても、認知パフォーマンスに干渉する(Barlow、2005)。この相関関係は、成人期に受けた脳損傷においても存在する(McKee and Robinson, 2014)。うつ病などの他の精神疾患は、アルツハイマー病の感受性を高める可能性がある(Ownby et al 2006)。この文脈では、脳は多くの種類の侮辱に従属しており、末梢組織の異常な機能を反映することもある。睡眠時の呼吸障害は、2型糖尿病につながる可能性のあるブドウ糖不耐症およびインスリン抵抗性と関連している(Punjabi et al 2004)。さらに、睡眠遮断は、アミロイドβ蓄積を調節するアルツハイマー病の病態にも役割を果たし(Bliwise, 2004; Kang et al 2009マウスのAD様の病態にも役割を果たす(Kincheski et al 2017)。
不健康なライフスタイルは、インスリン抵抗性、成長ホルモン、IGF-1,および性ステロイドの減少につながる内分泌学的制御の損失などの老化の有害な影響を加速する(Barzilai et al 2012)。最近の研究では、欧米の食生活(糖分と脂肪が大半を占める)が海馬の萎縮と関連していることが示され(Jacka et al 2015日本でのAD発症率の有意な上昇と関連していることが報告されている(Grant、2014)。喫煙、身体活動の欠如、アルコール乱用は、アルツハイマー病のリスクを高める他の不健康な習慣の一部である(Baumgart et al 2015)。
重要なことに、高齢の女性は男性よりも急速に認知機能の低下を示し、AD発症のリスクが高く、インスリンシグナル伝達異常を示す(Li and Singh, 2014; Duarte et al 2018)。マウスの遺伝子ネットワーク解析により、加齢に伴う転写変化は、男性の脳と比較して女性の脳では異なって早期に起こることが明らかになった(Zhao et al 2016)。さらに、腹部肥満は、高齢女性における死亡リスクの増加と相関し、インスリン抵抗性に寄与する(Folsom et al 1993)。その上、他の治療法はアルツハイマー病のリスクを高める可能性がある。Woman Health Initiativeでは、エストロゲンとプロゲスチンを組み合わせたホルモン補充療法が、いくつかの年齢関連マーカーをアップレギュレートし、認知機能の低下や心血管疾患のリスクを増大させる可能性があることが観察されている(Writing Group for the Women’s Health Initiative Investigators, 2002; Duarte et al 2018)。
このシナリオでは、アルツハイマー病の発生率を低下させ、および/または散発性アルツハイマー病の進行を遅らせる修正可能なリスクを特定することが不可欠である。代謝の変化が老化の引き金となるという考え方によれば、アルツハイマー病は代謝機能障害の結果として起こる可能性があり、それはライフスタイルや他の条件の生涯にわたる累積的な影響によって引き起こされる可能性がある。
おわりに
メタボリック障害と認知症を関連付ける臨床/疫学的証拠は、この接続の基礎となる生物学的メカニズムを探索するために科学者を奨励している。インスリンシグナル伝達がAD脳で障害されているという極めて重要な発見は、アルツハイマー病の生理学の現在の理解を大きく前進させたことを表している。さらに、説得力のある証拠は、アルツハイマー病の脳のインスリン抵抗性につながる分子メカニズムは、糖尿病や肥満の末梢インスリン抵抗性に関与するものに顕著な類似性を共有していることを示している。それらは、慢性的な低悪性度の炎症、TNF-αによるIRS-1の阻害、および小胞体ストレスを含む。重要なことに、これらの知見はまた、現在糖尿病の末梢インスリン抵抗性を克服するために使用される薬は、アルツハイマー病の脳のインスリン シグナルを救出するために再利用される可能性があるという命題につながった。インスリンシグナルは神経栄養、神経保護作用があり、シナプス可塑性や認知過程において重要な役割を果たしていることから、神経細胞のインスリンシグナルを増強することは、ADにおける疾患修飾的な治療アプローチとなる可能性がある。実際、インスリンを含む抗糖尿病薬のADに対する有効性を検証するための前臨床試験や臨床試験が行われており、有望な結果が得られている(表1)。また、神経細胞やグリア細胞におけるインスリンの作用を解剖することで、中枢神経系生理におけるインスリンや関連するシグナル伝達経路の役割についての知見が得られると考えられる。最後に、末梢エネルギー代謝と脳機能のクロストークをよりよく理解することは、治療標的やアルツハイマー病やその他の脳障害の修正可能な危険因子を特定し、公衆衛生政策や国民の意識を向上させることにも役立つかもしれない。