
Contents
Insulin and Autophagy in Neurodegeneration
要旨
代謝性疾患と神経変性疾患の病態生理過程におけるクロストークは、インスリンシグナル伝達とオートファジー障害が両疾患の共通因子であることが示されており、広範な研究が行われてきた。
これらの経路を制御する薬理学的・遺伝学的戦略は、タンパク質の集合体のクリアランス、ひいては疾患の発症や進行を遅らせるための有望なアプローチであると考えられているが、このような経路の制御は、疾患の発症や進行を遅らせることにつながる。
しかし、オートファジーの調節による反応は時間依存的であることから、オートファジーの適切な調節を見つけることが神経変性疾患の治療薬開発のターゲットとなる可能性がある。
このように、本総説では、神経変性疾患におけるインスリンシグナル/抵抗性とオートファジーの役割に焦点を当て、これらの疾患における薬理学的介入と非薬理学的介入について考察した。
キーワード
肥満、2型糖尿病、インスリン、オートファジー、神経変性疾患
序論
肥満や2型糖尿病(2型糖尿病)などの代謝障害は、平均寿命の短縮や医療合併症の増加をもたらすだけでなく、神経変性疾患の発症の危険因子であると言われている21世紀の世界的なパンデミックの課題である(El Sayed et al 2012)。文献によると、肥満と2型糖尿病の両方が、認知症および認知機能低下の有病率の増加に寄与している(Arvanitakis et al 2004;Kopf and Frölich 2009;Nameni et al 2017;Zhang et al 2017)。2017)およびインスリン抵抗性の発症(Schneeberger et al 2015)と同様に、代謝障害と中枢神経系(中枢神経系)障害との関連において重要な役割を果たすことが提案されている(Schneeberger et al 2015; Nameni et al 2017)。現在では、インスリン受容体(IR)が中枢神経系に広く発現していることが知られており、脳インスリンと神経変性疾患との正確な関連性はまだ不明であるが、脳の健康維持には最適なインスリンシグナル伝達のホメオスタシスが重要であることが、多くの研究によって実証されている。
細胞の恒常性の維持は、オートファジーとアポトーシスの間の興味深いクロストークを含む複雑なプロセスであり、他のプロセスの間で、ホスファチジルイノシトール(3,4,5)-三リン酸キナーゼ(PI3K)-タンパク質キナーゼB(AKT)-哺乳類のラパマイシン標的(mTOR)およびAMP依存性タンパク質キナーゼ(AMPK)経路の制御が関与している。mTORは、ユビキタスに発現し、細胞増殖、タンパク質合成、および死または生存シグナルを調節するセリン/スレオニンキナーゼである。主に細胞質に局在するmTORは、mTOR複合体1(mTORC1)とmTOR複合体2(mTORC2)という2つの異なるタンパク質複合体から構成されており、タンパク質複合体の構成要素、ラパマイシンに対する応答性、活性化剤または下流のシグナル伝達経路など、多くの点で異なっている。しかしながら、両複合体間のクロストークは、AKTによって媒介される(Perluigi et al 2015)。AKTシグナル伝達経路は、インスリン様成長因子1(IGF-1)およびインスリンによって誘発され得、このインスリンは、膜貫通受容体に結合し、インスリン受容体基質1(IRS-1)を活性化し、PI3K活性化およびそれに続くホスファチジルイノシトール(3,4,5)三リン酸(PIP3)の生成を導く。AKTは、結節性硬化症複合体(TSC1/2)などのmTORの負の調節因子の阻害に関与し、一方、ピルビン酸脱水素酵素キナーゼ1(PDK1)は、mTOR活性化因子Rheb(脳に富むRasホモログ)を誘導し、それによってmTOR複合体を放出してその標的をリン酸化する(Perluigi et al 2015)。興味深いことに、高インスリン血症によるmTORの慢性的な過剰活性化は、インスリン抵抗性に寄与することが提案されている(上野 et al 2005)。
オートファジーは、タンパク質および小器官を含む細胞の残骸が、タンパク質のクラスターおよび凝集体の蓄積を防ぐために分解され、リサイクルされる重要な細胞プロセスである(Klionsky and Emr, 2000; Rubinsztein er al)。 このプロセスの間、細胞質や小器官の一部はオートファゴソームと呼ばれる二重膜構造に取り込まれ、オートファゴソームはリソソームや液胞と融合し、それらのカーゴ物質(小器官や高分子)の分解とその後の再利用のための細胞質への放出を可能にする(Kundu and Thompson, 2005)。この複雑なプロセスは、30以上のオートファジー関連タンパク質(ATG)PI3K、Unc-51様キナーゼ1(ULK1)微小管関連タンパク質1軽鎖3(LC3)経路によって制御されている。生理的条件下では、PI3KタイプIは、成長因子によって活性化され、mTORの活性化をもたらし、その結果、哺乳類オートファジーの開始因子であるULK1をリン酸化および不活性化することによってオートファジーのダウンレギュレーションを引き起こす(Axe et al 2008)。また、AMPKはmTOR以外にもULK1の異なるアミノ酸残基をリン酸化することでオートファジーを抑制している(Nishida et al 2009)。しかし、栄養飢餓などのストレス条件下では、プロテインホスファターゼ2A(PP2A)によってULK1が脱リン酸化され、複数の基質がリン酸化され、従来のオートファジーが開始される(Axe et al 2008; Nishida et al 2009; Mizushima and Komatsu, 2011)。その後、他の成分とともに、Beclin1を含むクラスIII PI3K複合体(Beclin1,VPS15とATG14,クラスIII VPS34)により、分離膜の初期膜成分であるファゴフォアの形成が誘導され、ATG5-ATG12とLC3の2つの共役経路により膨張、曲線、閉鎖し、完成したオートファゴソームへと変化する(水島・小松 2011;図1)。
図1 飢餓とインスリンシグナルによるオートファジーの調節
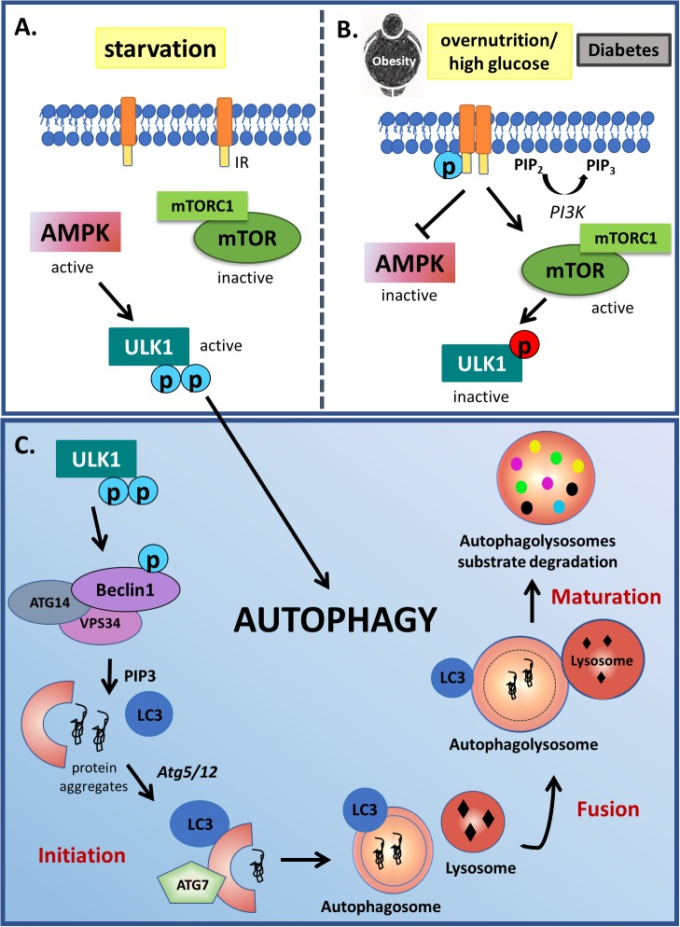
A)オートファジーは、飢餓状態ではULK1活性を介して古典的に活性化される。低栄養状態では、AMPKシグナル伝達が活性化され、2つの異なる活性化部位でULK1をリン酸化する。飢餓とは逆に、(B)栄養過多状態または糖尿病では、PI3KタイプIが成長因子受容体によって活性化され、mTORの活性化およびAMPKの不活性化をもたらす。(C) このプロセスでは、タンパク質を含む集合体や損傷を受けた小器官がオートファゴソームと呼ばれる二重膜構造に取り込まれ、リソソソームや液胞と融合し、オートファゴリソームでの貨物物質の分解を可能にする。この複雑なプロセスは、2つの異なる活性化残基(青丸で示した)でのULK1のリン酸化から始まる。活性化されたULK1はベクリン1をリン酸化し、他の成分とともに分離膜の初期膜成分であるファゴフォアの形成を誘導し、その後、ATG5-ATG12の作用に依存するLC3の脂質化への2つの共役経路によって拡張、曲線化、閉鎖し、完成したオートファゴソームへと変化する。
このように、本総説では、中枢神経系におけるインスリンの起源と役割をまとめ、アルツハイマー病(アルツハイマー病)パーキンソン病(PD)ハンチントン病(HD)筋萎縮性側索硬化症(ALS)前頭側頭型認知症(FTD)などのいくつかの神経変性疾患や、脊髄小脳失調症3型(SCA3)ラフォーラ病(LD)などにおけるインスリンとオートファジーの関係を論じている。オートファジーには、シャペロンを介したオートファジー、マイクロオートファジー、マクロオートファジーの3つの経路があることが知られているが、本論ではマクロオートファジーに焦点を当て、以下オートファジーと呼ぶことにする。
中枢神経系のインスリン抵抗性と神経変性
膵臓の内分泌の糖低下特性は、バンティングとベスト(Banting and Best, 1922)によって記述され、その後まもなく1926年にインスリン自体が結晶化した(アミロイドβel, 1926)。インスリンとその機能を記述した科学的な出版物の数は1940年代に爆発的に増え、1960年代までは、ほとんどの科学界は、脳はインスリンに鈍感な器官であると考えてた。しかし、60年代の前半には、別のグループによる説得力のある研究が行われ、この議論の再開を余儀なくされた。Rafaelsenが1961年に発表した研究では、ラットの皮質脳スライスと小脳だけでなく、単離されたラット脊髄(Rafaelsen, 1961a)においても、インスリンによるグルコース取り込みの調節が示された(Rafaelsen, 1961b)。Butterfieldら(1966)は、5人の健康な男性ボランティアにインスリンを注入して脳のグルコース取り込みを測定した後、ヒトの脳は結局のところ、インスリンに敏感である可能性があることを示唆した(Butterfield et al 1966)。
最近の研究では、インスリンの効果は脳内で確認されているが、これらは周辺部での効果とはかなり異なることがあるが、インスリンが脳内で排他的に神経栄養作用を有するという考えは、いくつかの最近の出版物によって争われている。(2016,García-Cáceres et al 2016)は、視床下部アストロサイトにおけるIRを介したシグナル伝達が、グルコースを感知するプロオピオメラノコルチンニューロンの活性に影響を与える可能性があることを示し、脳内グルコース取り込みの調節におけるインスリンの重要性を示唆している(García-Cáceres et al 2016)。インスリン様成長因子I(IGF-I)は、IRのもう一つの重要な内因性リガンドであり、非カノニカル経路であるマイトジェン活性化プロテインキナーゼ(MAPK)/プロテインキナーゼD(PKD)経路を活性化するために、アストロサイトIRへのインスリン結合と並んで働き、最終的には、インスリン感受性であることが以前に知られていたグルコーストランスポーターであるグルコーストランスポーター(GLUT)1の膜トランスロケーションにつながる。したがって、インスリンおよびIGF-1の複合作用は、脳のグルコースを調節する(Fernandez et al 2017)。以下のセクションでは、インスリンの神経栄養作用に焦点を当てる。
脳インスリンの起源
インスリンは、1967年にMargolisとAltszuler(1967)によって、犬の脳脊髄液(脳脊髄液)中で初めて同定された。継続的なインスリンの静脈内注入と定期的な脳脊髄液と血液のサンプリングを通して、MargolisとAltszulerは、脳脊髄液中の絶食したインスリン濃度が血漿中のインスリン濃度の平均27%であることを初めて示した。さらに、これは、非常に限られた方法ではあるが、インスリンが脳のバリアを越えることができることを証明した最初の報告であった。インスリンが受動的な拡散ではなく、飽和可能な輸送システムを介して血液-脳および血液-脳脊髄液を横断することができるという概念は、重要なマイルストーンとなり、今でも脳内のインスリンを説明する最も受け入れられている仮説とみなされている。
膵臓がインスリンの主要な供給源であることは間違いないが、脳組織や脳脊髄液に見られるインスリンは専ら血液に由来するという考えは、1978年にHavrankovaら(1978)によって初めて挑戦された。この観察に続いて、同じグループは、肥満高インスリン血症および糖尿病性インスリン分泌不全ラットのモデルにおいて、彼らの所見を確認した。肝臓で観察された(そして予想された)結果とは対照的に、どちらの実験群でも脳全体のIRレベルは変化しておらず、末梢性の劇的なインスリン振動から中枢神経系を保護するために脳のバリアが関与していることが示唆された。さらに、中枢インスリンレベルもまた、試験したどのモデルにおいても変化しておらず、脳内でのホルモンの新規合成を示唆している(Havrankova et al 1979)。
視床下部以外での中枢性インスリン生合成については、まだ議論の余地がある。初代神経細胞培養物におけるインスリン産生の観察は、1986年にClarkeら(1986)によって最初に報告された。全脳初代ニューロン培養物から放出された培地を分析し、ラジオイムノアッセイとHPLC分析によって、分泌されたインスリンの存在だけでなく、K+とCa2+を介した脱分極による正の制御も示した。これらの観察が神経細胞の脱分極に特異的であることを証明するものとして、Clarkeは、培養グリア細胞ではそのような効果がないことを示した。プレプロインスリンのmRNAとタンパク質は、海馬と嗅球の錐体細胞で報告されている(Kuwabara er al)。 興味深いことに、最近の報告では、初代培養アストロサイトにおいてもインスリンの発現と産生が認められ、アミロイドβ(アミロイドβ)とリポ多糖(LPS)によって減少した(Takano er al)。 脳由来インスリンの別の仮説的な供給源は脈絡叢であるかもしれない(Lamotte et al 2004; Yong et al 2011)。
その起源にかかわらず、インスリンが脳機能に異なる影響を及ぼし、いくつかの病理学的状態において重要な役割を果たす可能性があることは明らかである。
インスリンの中枢作用
インスリンは、主にインスリン感受性細胞のグルコース取り込みの調節に役割を果たしており、筋肉、脂肪組織、肝臓などの末梢組織への影響は非常によく似ている。受容体が活性化されると、AKTやERK経路がリン酸化・活性化され、グルコーストランスポーター4(GLUT4)が細胞膜に動員され、これらの細胞によるより大きなグルコースの取り込みを可能にする。しかし、脳では、主にインスリン感受性のないグルコーストランスポーターGLUT1(アストロサイトと血液脳関門内皮細胞)とGLUT3(ニューロン)を発現しているため、全く異なる挙動を示す。したがって、脳内の細胞によるグルコース取り込みの古典的なモデル化では、これはインスリン非感受性のプロセスであると考えられているが、これは上記のようにいくつかの論争の対象となっている。対照的に、中枢性インスリンの作用は主に神経栄養作用とみなされ、シナプス生理学に影響を与え、したがって、記憶と学習に影響を与える。
インスリンおよびその受容体は、ショウジョウバエ(Song et al 2003;Gu et al 2014年)マウス(Grote et al 2011)およびヒト神経細胞(Liu et al 2013;Roloff et al 2015)において実証されたように、PI3K/AKT経路の活性化を介して、神経突起の伸長および軸索誘導に関与している。IRS p53は、樹状突起のアーボライゼーションに不可欠な役割を果たしているようである。IRSp53は、ニューロンのシナプス後膜に発現し、シナプス後密度と共局在し、細胞骨格を構成するタンパク質と相互作用する(アミロイドβbott et al 1999; Chiu and Cline 2010)。IRSp53の過剰発現は、神経細胞培養において、より高いレベルのアーボライゼーションと相関することが示されている(Govind et al 2001)が、その阻害は樹状突起の密度とサイズを減少させる(Choi, 2005)。
インスリンは、いくつかの異なるメカニズムによってシナプス活性および可塑性を調節することができ、海馬細胞培養物における長期的な抑うつの生成のためのAMPA受容体のエンドサイトーシスを誘導し(Beattie et al 2000)およびシナプス強化に関連するシナプス後膜のNMDA受容体の調節を誘導する(Skeberdis et al 2001)。これらのグルタミン酸受容体の調節は、インスリンが神経細胞の活性に依存したシナプス可塑性に参加することを可能にする(Van Der Heide et al 2005)。全体として、このようなデータは、中心的なインスリン効果が認知機能を支える神経細胞の可塑性プロセスと明確に結びついている。
神経衰弱性疾患におけるインスリンシグナル伝達とオートファジー:序論
文献データはまだ矛盾しているが、(Rotermund et al 2018)によって改訂されたように、最も有名な抗糖尿病薬の一つであるメトホルミンの使用は、例えば、パーキンソン病およびアルツハイマー病の動物モデルにおいて、いくつかの肯定的な効果を有することが実証された(Lennox et al 2014; Patil et al 2014; Lu et al 2016; Katila et al 2017)。文献によると、メトホルミンの急性および慢性投与は、インスリン分泌の誘導因子として知られているインクレチンであるグルカゴン様ペプチド-1(GLP-1)のレベルを増加させることが示されており(Maida et al 2011)これは、PI3K/AKTシグナル伝達の活性化および脳ATG7レベルの上昇につながる可能性があり、それによってオートファジーを促進する(Candeias et al 2017)。それに加えて、メトホルミンは、すでに2型糖尿病におけるインスリン抵抗性と関連しているAMPKシグナル伝達を調節することによって作用し(Xu et al 2012)オートファジープロセスにも関連している(図2)。
図2 PI3K-AKT-mTORシグナル伝達およびメトホルミンの標的
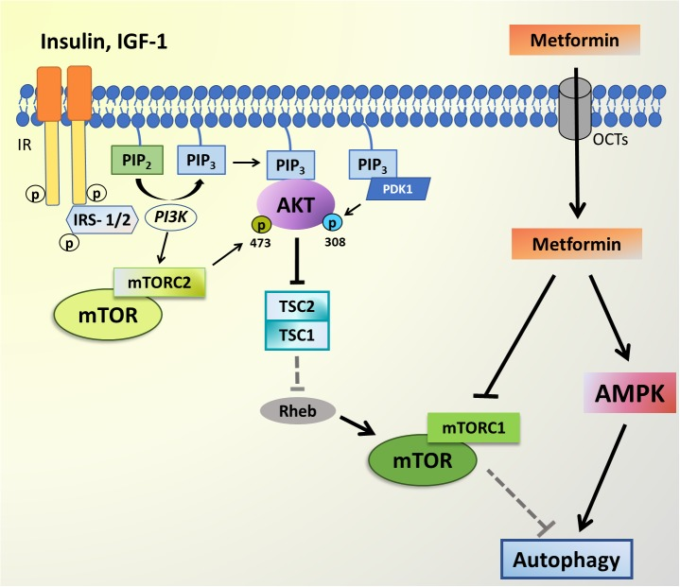
AKTシグナル伝達経路は、インスリン様成長因子1(IGF-1)およびインスリンによって誘発され得、このインスリンは、IRS-1/2リン酸化、PI3K活性化、およびそれに続くホスファチジルイノシトール-4,5-二リン酸(PIP2)からホスファチジルイノシトール-3,4,5-三リン酸(PIP3)への変換を導く膜貫通受容体チロシンキナーゼ(RTK)に結合する。AKTとPDK1は細胞膜でPIP3に結合し、PDK1はT308でAKTの活性化ループをリン酸化する。RTKシグナルはまた、未知のメカニズムによりmTOR複合体2(mTORC2)を活性化し、mTORC2はS473でAKTをリン酸化する。活性化されたAKTは、結節性硬化症複合体(TSC1/2)のようなmTORの負の調節因子の阻害に関与し、これによりmTOR活性化因子Rhebを誘導し、それによりmTOR複合体を放出して標的をリン酸化する。mTORC1活性はオートファジーを阻害する。メトホルミンは有機カチオン輸送体(OCT)を介して細胞内に入り、mTORC1複合体を阻害し、AMPKを活性化することでオートファジー活性化につながる。
(yamamoto et al 2018)が示したように、一過性のオートファジー阻害は、構成的に活性化されたオートファジー過程を示す高脂肪食を与えたBecn1F121Aノックマウスの耐糖能異常を救済することができ、オートファジーの適切な制御を見出すことが神経変性疾患の治療薬開発のターゲットとなる可能性を示唆している。また、神経変性疾患と古典的に関連している異常なタンパク質凝集体(Lasagna-Reeves et al 2011;Larson and Lesné 2012;Lashuel et al 2013)は、加齢とともに活性が低下するユビキチン-プロテアソーム系やマクロオートファジー系(Filimonenko et al 2010)の基質であることが知られている(Shibata et al 2006)。しかしながら、これらの凝集体と細胞毒性、および総プラーク負荷と認知状態との間の関係は、依然として議論のポイントとなっている(Koss et al 2016)。したがって、本節では、神経変性疾患のそれぞれにおけるインスリンシグナル伝達とオートファジーとの関係をまとめ、考察する。
アルツハイマー病(アルツハイマー病
脳インスリン抵抗性と神経変性を関連付けるいくつかの研究は、脳インスリンシグナル伝達の変化が、古典的なI型およびII型糖尿病の病態生理に由来するプロセスとは異なる、より複雑なプロセスである可能性があることを示している(Rivera et al 2005; Steen et al 2005)。上記のように、試験管内試験および生体内試験の両方の研究は、特にシナプス可塑性だけでなく、記憶および学習のその正の調節に、ニューロン細胞におけるインスリンの効果を示す。学習におけるインスリンのポジティブな効果は、1976年に初めて示され、海馬病変を受けたラットの記憶障害をインスリン投与で減衰させた(de Castro and Balagura, 1976)。これに続いて、海馬CA1領域の神経細胞の壊死組織を減少させながら、インスリンが空間学習と記憶を脳虚血後に積極的に調節することを示す同様の研究が行われた(Voll et al 1989)。CA1の錐体ニューロンにおけるその受容体を介したインスリンシグナル伝達は、水迷路テストにおける空間学習にも関与している(Zhao et al 1999, 2004)。インスリンの脳室内注射もまた、受動回避テストでテストされたように記憶力を向上させる(Park et al 2000)。
最近数十年の間に、ヒトの認知に対するインスリンの効果が記述されており、その中には、インスリン注入後数時間以内の健康な参加者における選択的注意および言語記憶に対する効果も含まれている(Kern et al 2001年)。インスリンの静脈内注入は、低血糖症や血液イオンバランスの不均衡など、様々な望ましくない効果をもたらすことを考えると、脳に特異的な作用を達成するためには、異なる投与経路を使用しなければならなかった。ペプチドおよび小タンパク質が嗅神経および嗅球の細胞間空間を介して鼻粘膜から脳脊髄液にアクセスできることは、10年以上前から知られていた(Balin et al 1986;Sakane et al 1991;Illum 2000)。このことから、Benedict et al 2004)は、8週間にわたって健康な参加者38人の記憶に対する経鼻インスリンの効果を調査した(Benedict et al 2004)。重要なことに、これらの効果は血糖値やインスリン値に障害を与えることなく達成された。したがって、経鼻的インスリンは、脳におけるインスリン効果を調査する研究において、インスリン投与の好ましい経路となっている。
近年のデータは、オートファジープロセスとII型糖尿病の発症との間の直接的な関係を示唆している。He et al 2013)は、ベクリン1と同様にベクリン2がオートファジーの重要なモジュレーターであることを示した。ベクリン2ヘテロ接合体欠損マウス(ベクリン2+/-)に、通常食または高脂肪食(60%脂肪)を8週間与え、いずれの条件でも体重、食物摂取量、インスリン抵抗性の増加を示す結果が得られた(He et al 2013)。糖尿病および非糖尿病C57BL/6マウスで行われた別の研究では、インスリン抵抗性の存在下でのオートファジー液胞形成の増加が観察された。膵β細胞においてATG7を選択的に不活性化することにより、オートファジープロセスが存在しない場合、ユビキチン化されたタンパク質や損傷を受けた小器官が時間の経過とともに蓄積し、膵β細胞の毒性を引き起こすことが示された(Ebato er al)。
定常的に活性なオートファジーは、Becn 1/Beclin 1の点変異(Becn1F121A/F121A)を介して達成することができる。高脂肪食チャレンジでは、そのようなオートファジー高活性マウスは、膵β細胞におけるインスリン顆粒の分解の増加にもかかわらず、改善されたインスリン感受性を示す(Yamamoto et al 2018)。Becn1F121Aマウスとアルツハイマー病のトランスジェニックマウスモデル(5×家族性アルツハイマー病)との交配により、オートファジーの亢進がアミロイドβ蓄積を有意に減少させ、認知機能の低下を予防することが示された(Rocchi et al 2017)。5 × 家族性アルツハイマー病 Becn1F121Aマウスモデルにおけるオートファジーの過剰活性化は、皮質および海馬の両方で可溶性および不溶性の両方のアミロイドβ42の低いレベルを導くが、アミロイド前駆体タンパク質(APP)レベルの変化は観察されなかった。Becn1F121AおよびAPPを発現するHEK293細胞を用いた細胞モデルにおいて、細胞は、ATG7をノックダウンするsiRNAでトランスフェクトされ、アミロイドβ42の増加をもたらし、アミロイドβ形成の調節におけるオートファジーの重要性を示した(Rocchi et al 2017)。このことは、ML246を用いた薬理学的治療または自発的運動などのオートファジープロセスを過剰に活性化させる戦略が、アルツハイマー病および認知機能低下のリスクを減少させる可能性があることを示唆している(Rocchi et al 2017)。
げっ歯類では、Caccamo et al 2010)は、対照食に対してラパマイシンを含む餌を10週間与えた3×Tg-ADマウスが、モリス水迷路課題でより良いパフォーマンスを持っていたことを観察し、初期の学習および記憶障害からマウスを救済するためのラパマイシンの役割を示唆した。ラパマイシンを8ヶ月間慢性的に投与すると、海馬のアミロイドβ蓄積とタウの過リン酸化の減少と関連してmTOR活性が低下する。興味深いことに、ラパマイシンを投与した3×Tg-ADマウスと対照の3×Tg-ADマウスでは、ATG7,ATG5/ATG12複合体、LC3-II(オートファジー誘導に必要な)のレベルが上昇したことで示されるように、オートファジー機能の向上がもたらされた。この効果は、オートファジー阻害剤である3-メチルアデニンによって阻止された(Caccamo et al 2010)。
軽度認知障害(MCI)アルツハイマー病、および健康なマッチドコントロールと診断された患者からの死後脳下頭頂葉組織を分析し、比較すると、Tramutola et al 2015)は、mTORの過剰活性化と相まって、AKTおよびPI3K(p85サブユニット)リン酸化の増加がMCIおよびアルツハイマー病患者で起こることを示した。PI3K-AKT-mTORシグナル伝達経路のこの増加は、オートファジーマーカーであるベクリン1およびLC3の減少と相まって、IRS-1の抑制部位のリン酸化の増加と同様に、初期のMCIで観察され、そのような変化がインスリン抵抗性と初期の認知症との関連を根底から支えていることを示唆している。重要なことは、オートファジーマーカーの減少は、アミロイドβレベルの増加と直接相関していたことである。前臨床アルツハイマー病患者のグループでは、剖検時にアルツハイマー病神経病理学と結合した正常な認知機能を持つ死後のグループでは、PI3K-AKT-mTOR経路の変化は見られなかったが、オートファジー障害の証拠があった、破壊されたオートファジーがアミロイドβ沈着の初期イベントである可能性があることを示唆している(Tramutola et al 2015)。
中前頭前野灰白質の死後分析では、重度アルツハイマー病(-30%)および軽度認知障害(MCI)(-70%)では、健常対照と比較してベクリン1レベルが有意に低下していた。しかし、ハンチントン病(HD)やLB変異型アルツハイマー病患者では有意な減少は認められなかった(Pickford et al 2008)。同じ研究では、広範なアミロイドβ沈着と高レベルの変異ヒトAPPを発現する2つの異なる系統の老化APPトランスジェニックマウスの皮質ベクリン1レベルを調査したところ、ベクリン1の発現は、非トランスジェニックで年齢をマッチさせた対照マウスと同じであることがわかった。このようなデータは、オートファジー障害がAPP病理学の前の上流事象で起こり得ることを示唆しているであろう(Pickford et al 2008)。しかしながら、オートファジープロセスによるAPPの分解の直接的な証拠は、いくつかの論争の対象であることに留意すべきである(Boland et al 2010;Jaeger et al 2010;Tian et al 2013;Rocchi et al 2017)。
いくつかのプロセスおよび行動は、カロリー制限および運動を含むオートファジーを調節することができ、これらはすべてインスリンシグナル伝達を改善し、神経変性プロセスを遅らせる。交互日絶食で数ヶ月間維持されたげっ歯類および霊長類において、神経細胞は、脳由来神経栄養因子(BDNF)CREB、およびオートファジーのより高いレベルと関連して、神経変性プロセスから保護され、また、mTORレベルの低下、ミトコンドリア機能の改善、および酸化ストレスの減少と関連している(Mattson et al 2017年にレビューされている)。前臨床データは、オートファジーを促進するサーチュインを増加させる断続的な断食と交互日断食が、アミロイドβ凝集体とタウ蓄積の存在下でも神経細胞を保護し、その結果、3×TgADマウスの認知障害に対する保護をもたらしたことを示している(Halagappa et al 2007)。このようなカロリー制限食のオートファジー促進効果の根底にあるメカニズムは、これらのメカニズムが、特にその初期段階では、神経変性過程に対して何らかの保護を与える可能性が高いため、明らかにする必要がある。注目すべきは、24時間の絶食は、オートファゴソームを増加させ、mTORシグナリングを減少させることが示されていることである(Alirezaei et al 2010)。変異型ヒトAPP770を発現し、生後3ヶ月で高レベルのアミロイドβ沈着を発症する5×家族性アルツハイマー病トランスジェニックマウスにおいて、48時間の絶食は、このモデルに存在する高レベルのアミロイドβをクリアするには不十分であるが、オートファジーを増加させることができた(Chen et al 2015)。以前に示されたように、運動は、オートファジー、アミロイドβクリアランス、および改善された認知の有意な正の調節因子であり(Rocchi et al 2017)有酸素運動は、食事制限と同様に、健康な参加者の認知パフォーマンスに正の影響を与え(CamandolaとMattson 2017でレビュー)およびアルツハイマー病の動物モデル(García-Mesa et al 2011; 図3)であった。
図3 アルツハイマー病におけるインスリン-オートファジーシグナルの変化のまとめ
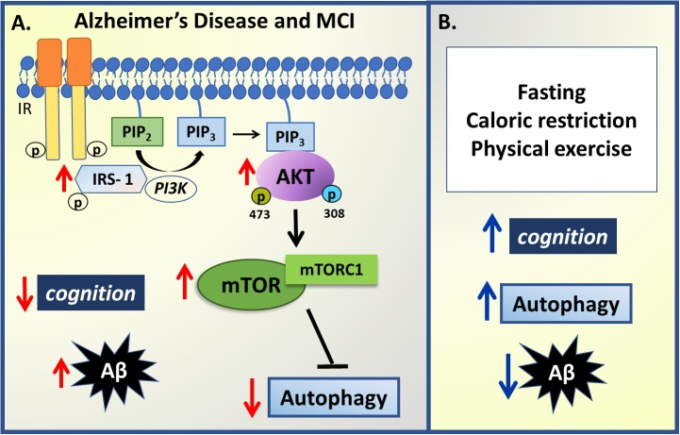
A)軽度認知障害(MCI)とアルツハイマー病では、認知レベル、インスリン感受性、オートファジー、アミロイドβ(アミロイドβ)凝集体の間に逆相関が見られる。IRS-1,PI3K、AKTのリン酸化の増加とmTORC1の過剰活性化は、オートファジー活性を低下させ、アミロイドβの分解を阻害し、その結果、アミロイドβの沈着が増加し、認知能力が低下することを示している。
B)しかし、いくつかの非薬理学的戦略は、認知パフォーマンスを改善し、オートファジーを増加させ、したがって、アミロイドβ凝集体を減少させることができる絶食プロトコル、カロリー制限や運動などのアルツハイマー病やMCIで観察されるいくつかの変化を調節するために提示されている。赤矢印は病態生理の変化を表し、青矢印は非薬理学的戦略によって促進される変化を表している。
パーキンソン病
パーキンソン病は、嗅覚・自律神経機能障害、腸管機能障害、睡眠障害、疲労、認知機能障害、精神症状などの非運動症状を伴って、診断の何年も前から始まっているように思われるゆっくりとした進行の神経変性疾患である。疾患の進行に伴い、徐脈、安静時振戦、筋硬直、姿勢障害などの古典的な運動症状が現れる。危険因子には遺伝的および環境的因子が含まれており、古典的な運動症状は、黒質パーコンパクト(Snpc)内のドーパミン作動性ニューロンの喪失によって媒介されるが、他の神経解剖学的領域も影響を受ける可能性がある(Kalia and Lang, 2015)。
パーキンソン病では、他の多くの神経変性疾患と同様に、異常凝集体αシヌクレインタンパク(αSyn)からなる細胞内タンパク凝集体LBの増加が明らかになっている。LBの病態とパーキンソン病の病因との関係はまだ明らかにする必要があるが、インスリンシグナル伝達障害が関連するモジュレーターであることを示す疫学的証拠がある(Driver et al 2008; Schernhammer et al 2011)。
110人のパーキンソン病患者(パーキンソン病と認知症(PD/D)を持つ53人と認知症を持たない57人のパーキンソン病を対象とした研究では、2時間の経口ブドウ糖負荷試験後のブドウ糖値とインスリン値を評価した(Bosco et al 2012)。これらの著者らは、PD/D患者はグルコース代謝異常を伴うインスリン抵抗性の有病率が高いことを発見した(Bosco et al 2012)が、パーキンソン病と糖尿病の関連性は複雑で議論の余地があることに留意すべきである(Santiago and Potashkin, 2013でレビューされている)。しかし、2型糖尿病の最近の概念化は、長い間、PD(パーキンソン 2002)の初期の病態生理学的症状として評価されている腸内の変化と、脳へのα-Synの迷走神経輸送のための可能なサイトとして(アンダーソン et al 2016)と同様に、腸-肝臓-膵臓軸の重要性に重点を置いている。明らかにそのような全身的な変化は、さらなる調査が必要である。
4人のパーキンソン病患者の黒質からの脳ホモジネートと年齢をマッチさせたコントロールの死後研究において、SekarとTaghibiglou(2018)は、PI3K p85,AKT、PIP3,IRS-1,IRレベルの有意な低下を観察した。だけでなく、グリコーゲン合成酵素キナーゼ(GSK)3βの増加と腫瘍抑制ホスファターゼおよびテンシンホモログが10番染色体上に削除された(PTEN)の核内転座が増加し、パーキンソン病とインスリン抵抗性との関連を強く示唆している(Sekar and Taghibiglou, 2018)。セリン残基上でリン酸化されたIRS-1の高レベルは、インスリン/IGF-1のIRへの結合をブロックし、従ってAKTなどのダウンレギュレーションエフェクターをダウンレギュレーションし、これによりGSK3βのリン酸化がブロックされる。一度脱リン酸化されると、GSK3βは活性化し、タウタンパク質をリン酸化し、過リン酸化および神経原線維のもつれの沈着につながる(Athauda and Foltynie, 2016)。アルツハイマー病およびパーキンソン病の進行期では、タウ、アミロイドβおよびαSynのタンパク質凝集体が共存する(Yang et al 2018)。
パーキンソン病では、オートファジーの破壊は、PI3K/AKT/mTOR経路の変化とは少なくとも部分的に独立したプロセスであるように思われる(Heras-Sandoval et al 2014)。文献からのデータは、αSynの過剰発現が細胞培養および生体内試験でオートファジーを阻害し、凝集体を形成するタンパク質の蓄積につながることを示している(Winslow et al 2010)。αSyn過剰発現モデルのトランスジェニックマウスの脳で観察されるように、αSyn凝集体はBeclin 1の発現を増加させることで緩和され、オートファジーの増加と神経細胞の保護を引き起こす(Spencer er al)。 さらに、Dehay er al)。 (2010)のヒト神経芽腫細胞株および1-メチル-4-フェニル-1,2,3,6-テトラヒドロピリジン(MPTP)マウスモデルの研究により、パーキンソン病に見られるミトコンドリア活性酸素種(ROS)の増加は、リソソソーム膜の異常な透過性をもたらし、それによりリソソソームの枯渇とオートファゴソームの蓄積を引き起こし、非効率的なオートファジーを引き起こすことが示唆された(Dehay et al 2010)。2010). オートファジーにおける酸化ストレス誘発性障害の役割は、他の研究でも確認されている(Lin and Kuang, 2014; Navarro-Yepes et al 2014; Filomeni et al 2015; Teves et al 2018)。オートファジーフラックスの増加はまた、3時間6-ヒドロキシドパミン(6-OHDA)で処理したドーパミン神経芽腫細胞株(SH-SY5Y)およびマウス胚性線維芽細胞からの溶解物中で測定され、これはグルタチオン含有量の減少と相まって、6-OHDAでの処理は、mTORに依存しない経路において、活性酸素レベルの増加、およびオートファジーを活性化するAMPK/ULK1を導くことを示している(Urano et al 2018. 2018).
さらに、増加するエビデンスは、オートファジー障害の重要な因子として、パーキンソン病におけるリソソーム機能不全を暗示している。注目すべきは、パーキンソン病患者の脳において、コンフォメーションが異なる新しいタイプの非フィブリルリン酸化αSyn(pαSyn∗)が同定されたことであり、これはpαSynのオートファジー分解が不完全に行われた結果であると考えられる。pαSyn∗はミトコンドリアと結合し、高いマイトショック性を示し、構造障害を引き起こす。著者によると、pαSyn∗はLBとミトコンドリア障害の間のミッシングリンクである可能性がある(Grassi er al)。 さらに、Grassi et al 2018)は、事前に形成されたpαSynフィブリルはオートファジーを活性化するが、パーキンソン病で観察される不完全および/または異常なオートファゴリソーム分解は、フィブリルから離れて移動し、ミトコンドリアと関連付けるべきpαSyn∗の生成に関与していることを提案する。また、不完全なオートファゴリソーム分解は、パーキンソン病で観察されるATP13A2遺伝子とSYT11の変異によっても引き起こされる可能性がある(Bento et al 2016)。これは明らかに更なる研究が必要な領域である。
ヒトにおけるインスリンシグナル伝達に関しては、最近のPD試験では、II型糖尿病におけるインスリン経路の回復を指示されたグルカゴン様ペプチド1(GLP-1)の最初の合成アゴニストであるエクセナチドの効果が試験された(Sandoval and Sisley, 2015)。は、ドーパミン作動性ニューロンのミトコンドリア機能を温存し、アポトーシス死とリソソソーム枯渇を減少させ、タウの高リン酸化とプラーク負荷を減少させることが以前に知られていた(Athauda and Foltynie, 2018でレビューされている)。エクセナタイドPD試験は、エクセナタイドまたはプラセボを週1回48週間投与した60人の特発性パーキンソン病患者(男女とも)を分析し、その後12週間の休薬を経て分析を行った。血液サンプルから分離した細胞外小胞を用いて、脳内インスリンシグナル伝達経路を示した。その結果、エクセナタイド投与群では、プラセボ投与群と比較して、24週投与後のIRS-1リン酸化レベル、総AKTおよびpAKT S473の持続的な増加、48週投与時の総mTORおよびp-mTOR(S2448-mTORC1)の増加が認められた。総mTOR(mTORC1/2)レベルの増加は、60週目の運動スコアに関する持続的な臨床的改善と関連していた(Athauda et al 2019)。
一方、mTORの特異的阻害剤であるラパマイシンの効果は、PDモデルにおいてさらに明らかにする必要がある。6-OHDAラットのPDモデルにおいて、Jiang et al 2013)は、3つの異なる用量でラパマイシンを投与することで、酸化ストレスとミトコンドリア傷害を減少させ、結果的にアポトーシス的なドーパミン作動性神経細胞の喪失を回避することが観察された(Jiang et al 2013)。これらの効果は、mTORC2が急性ラパマイシン治療に無反応であるため、mTORC1複合体の調節に関与していた(Perluigi et al 2015)。6-OHDAモデルを用いた別の研究では、ラパマイシンによるmTORC1阻害、またはその下流標的であるS6K(PF-4708671による)の選択的遮断が、このモデルで明らかになった記憶欠損を改善することが示された。ラパマイシンの処置はまた、ラットPDモデルで観察された不安および抑うつ様行動を減少させることができた(Masini et al 2018)。
PD症状を改善するための非薬理学的戦略は、運動が有益な効果を有することを示している。6-OHDAモデルでは、週4日/週8週間のトレッドミルランニングを8週間行うと、6-OHDA誘発性の酸化ストレスから動物を保護した(Tuon et al 2012)。一方、MPTPマウスモデルでは、週5日/週8週間のトレッドミルランニングを行うと、線条体のαSynレベルが低下し、ドーパミン神経細胞の損失が減少した。重要なことは、運動期間が短いとあまり効果がないため、動物で観察された効果には運動期間が重要な要素であるように思われることである(Aguiar et al 2014)。
しかし 2014年にNational Parkinson Foundation Quality Improvement Initiative Databaseによって発表された大規模コホート研究では、運動の頻度が高いほど疾患の重症度が低くなり、認知機能の低下が遅くなることが示されているが、ヒトの研究では、パーキンソン病患者における運動の効果ははるかに決定的ではない(Ebersbach, 2015)。この研究では、すべての患者が米国のセンターオブエクセレンスで治療を受けていたにもかかわらず、異なる病期にある4,866人のパーキンソン病患者のデータを分析した(Oguh et al 2014)。運動の効果がパーキンソン病の後期段階で明らかになるかどうかは、まだ判断が必要である(Petzinger et al 2013)。
食事制限を含むオートファジーを調節することが知られている他の因子もまた、MPTPアカゲザルモデルで示されているように、黒質突起ドーパミンニューロンの数によって示されているように、パーキンソン病の進行を遅らせる可能性がある(Maswood et al 2004)。インスリン-AKT-mTORC1シグナル伝達経路を阻害するいかなる戦略も、オートファジー活性化を介した長寿の増加と関連しており(NtsapiおよびLoos 2016)したがって、パーキンソン病の発症率および病態生理をポジティブに変調することが期待されるであろう(Mattson 2010でレビューされている)。パーキンソン病患者における食事制限の有益性については、まだ明らかにされていない。
要約すると、インスリン抵抗性と異常なオートファジーとの相関は、アルツハイマー病とパーキンソン病の発症において重要なプロセスであるように思われる。しかし、これらの疾患間には、今後の研究で明らかにすべき重要な相違点がいくつかある。明らかに、オートファジーの亢進はアルツハイマー病では有益であるが、認知症を伴うパーキンソン病患者ではインスリン抵抗性を発症しやすいことから、認知症とパーキンソン病の相互作用を含めたパーキンソン病患者での更なる研究が必要である。また、パーキンソン病患者ではオートファジー異常の原因は明らかにされていないが、IRとその下流の蛋白質のレベルも低下している。パーキンソン病におけるオートファジーとmTORの役割についてはいまだに議論の多いところであり、これらのプロセスや経路を対象とした治療はかなり複雑になっている(図4)。
図4 パーキンソン病で観察されるインスリンオートファジー経路に関連する変化のまとめ
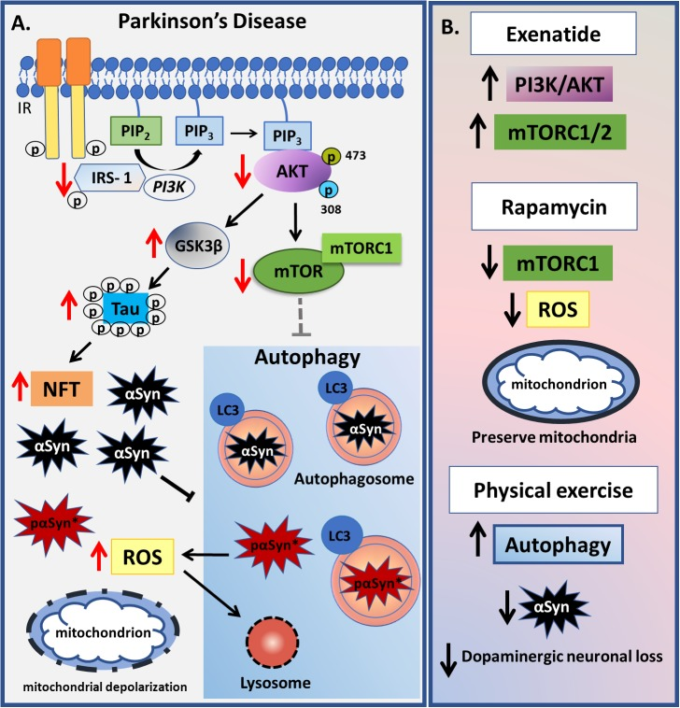
A)パーキンソン病では、PI3K-AKT-mTORシグナルの低下が観察され、GSK3βが活性化される。GSK3βはタウタンパク質を過リン酸化し、神経原線維のもつれの沈着を引き起こす。そうでなければ、高レベルのα-シヌクレイン(αSyn)がミトコンドリアの活性酸素種(ROS)の増加とともにオートファジーのプロセスを損なう。活性酸素は、リソソーム膜の異常な透過性をもたらし、それによってリソソームの枯渇とオートファゴソームの蓄積を引き起こし、非効率的なオートファジーをもたらす。αSynのオートファジー分解が不完全になると、オートファゴソーム内でpαSyn∗と呼ばれる新しい反応性物質が生成され、サイトゾルに放出されるとミトコンドリアと結合して高いマイトショック性を示する。損傷を受けたミトコンドリアを破線で示す。(B) PI3K-AKT-mTOR-オートファジーシグナル伝達に作用する薬理学的および非薬理学的戦略を示す。エクセナチドを投与された患者は、IRS-1リン酸化レベル、総AKTおよびpAKT、ならびにmTORのレベルの増加が持続的に増加し、パーキンソン病の病態生理で観察された障害されたシグナル伝達を回復させた。mTORC1活性を特異的に阻害するラパマイシンは、動物モデルにおいて酸化ストレスとミトコンドリア損傷を減少させることができ、一方、物理的な運動はオートファジーの増加とαSyn凝集体とドーパミン神経細胞の損失の両方の減少と関連している。
ハンチントン病
ハンチントン病は常染色体優性疾患であり、重度の運動機能障害や認知機能障害、精神障害、そして最終的には死を特徴としている。HDは、HDのN末端にポリQリピートをコードするシトシン-アデニン-グアニン(CAG)拡張を有するハンチントン(Htt)遺伝子の突然変異に起因すると長い間認識されてきたが、その結果、タンパク質の機能およびタンパク質の凝集体に障害が生じる(MacDonald er al)。 この状態に対する特異的な治療法はまだ存在しないが、HDマウスモデルにおける研究では、変異型Htt(mHtt)の発現を廃止することにより、症状の一部が救済され、脳内のタンパク質凝集が減少することが示されている(Yamamoto et al 2000;Harper et al 2005;Kordasiewicz et al 2012)。しかしながら、機能的なHttタンパク質は、ミトコンドリアダイナミクス、小胞トラフィッキング、軸索輸送、シナプス機能、および抗アポトーシス活性、ならびにオートファジーなどのいくつかの生理学的プロセスにおいて重要な役割を果たすので、この遺伝子の活性を抑制しようとすることには大きな問題がある(Choi et al 2014;Ismailoglu et al 2014;WongおよびHolzbaur 2014;WeissおよびLittleton 2016)。
Htt機能の喪失は、オートファジーの調節障害をもたらし、タンパク質の蓄積をもたらし、HDの病因に寄与している可能性が高い(Gelman et al 2015)。他のタンパク質凝集性疾患とは対照的に、ヒトおよびげっ歯類HDサンプルは、オートファゴソームのアップレギュレーションを示す(Kegel et al 2000;Petersen 2001)。しかしながら、これらのオートファゴソームは、カーゴ認識にいくつかの欠損を示し、その結果、凝集タンパク質の不十分な分解および損傷した小器官をもたらす(Boland et al 2008; Martinez-Vicente et al 2010)。それに加えて、Httまたはその相互作用因子であるHtt-associated protein 1 (HAP1)のいずれかをサイレンシングすると、軸索に沿ったオートファゴソームの輸送が損なわれることも知られている(Wong and Holzbaur, 2014)ことから、mHttもまた、内細胞性トラフィッキングを調節していることが示唆されている。
文献によると、HD患者はIGF-1の基底血漿レベルが高く、これがこれらの患者に見られる認知機能の低下と関連しており、IGF-1抵抗性がこの病理学に関与する基礎的なメカニズムの1つである可能性があるという仮説につながっている(Saleh et al 2010; Salem et al 2016)。AKT-mTOR経路の上流の活性化因子であるIGF-1は、HttのAKTリン酸化を介して、mHtt誘発性神経細胞死を抑制し、mHtt核内包物の形成を減少させることができ、それによってHDにおける神経保護効果を提供することができる(Humbert et al 2002)。さらに、HD動物モデルのデータでは、組換えヒトIGF-1の経鼻投与によりAKTがアップレギュレートされ、変異型Httのリン酸化を増加させ、運動活性の改善、および末梢および中枢の代謝異常の改善につながることが示されている(Lopes er al)。 一方、山本 et al 2006)によれば、インスリンおよびIGF-1治療後のタンパク質集合体の用量依存的なクリアランスは、IRS-2活性化に関与し、IRS-1またはAKT活性には関与しない(山本 et al 2006)。興味深いことに、IGF-1受容体(IGF-1R)の枯渇は、オートファジーの減少およびmHttレベルの上昇をもたらし、これは、AKTに依存しない経路で、長期間(24時間)のIGF-1処置によってのみ救済された(Renna et al 2013)。別の研究では、IGF-1R欠損はHDマウスのmHttレベルには影響を与えず、サイズのみに影響を与えることが示されたが、この欠損は動物の性別によって異なる結果をもたらし、雄ではロータロッド性能の悪化や体重減少などのいくつかの有害な影響を引き起こすが、雌では振戦の発症が遅れることが観察された(Corrochano et al 2014)。それに加えて、mTORC1活性の基質である転写因子EB(TFEB)のウイルス過剰発現は、オートファジーの増加およびmHttのレベルの低下をもたらした(Vodicka et al 2016)PI3K-AKT-mTOR経路もまた、この疾患において重要な役割を果たしていることを示している(Wu et al 2009)。このように、IGF-1/インスリン経路の活性化は、関与するメカニズムを明らかにする必要があるが、二重的かつ症候性の有用性を有するように思われる。
栄養豊富な食事または飢餓状態の食事が、それぞれ、mTORのセリン/スレオニンキナーゼリン酸化を介してオートファジーを抑制または増加させることができることを考えると(Kim et al 2008;Wong et al 2015年)多くの研究がHDにおける食事介入の利用を検討してきた。突然変異タンパク質の蓄積は、蓄積されたタンパク質の分解が飢餓条件下での分解とは異なるmTOR非依存性のオートファジーを引き起こす可能性があるが(Yamamoto et al 2006)HDマウスモデルにおける食事制限は、オートファジーの増加、mHtt凝集体形成の減少、および血糖調節の正常化をもたらし、それにより疾患の進行が遅くなり、寿命が延びることに寄与した(Duan et al 2003; Ehrnhoefer et al 2018)。しかし、予定された給餌はオートファジーを増加させることを示しているが、8~24時間のような長期の血清欠乏は、オートファゴソーム形成の減少をもたらす(Renna et al 2013)オートファジーの調節は栄養飢餓時間に影響されることを示唆している(図5)。明らかに、食事制限がHDにおいてどのように利益をもたらすかを含めて、この分野の関連プロセスに関するさらなる研究が必要とされている。
図5 ハンチントン病(HD)におけるインスリンとオートファジーシグナルの変調
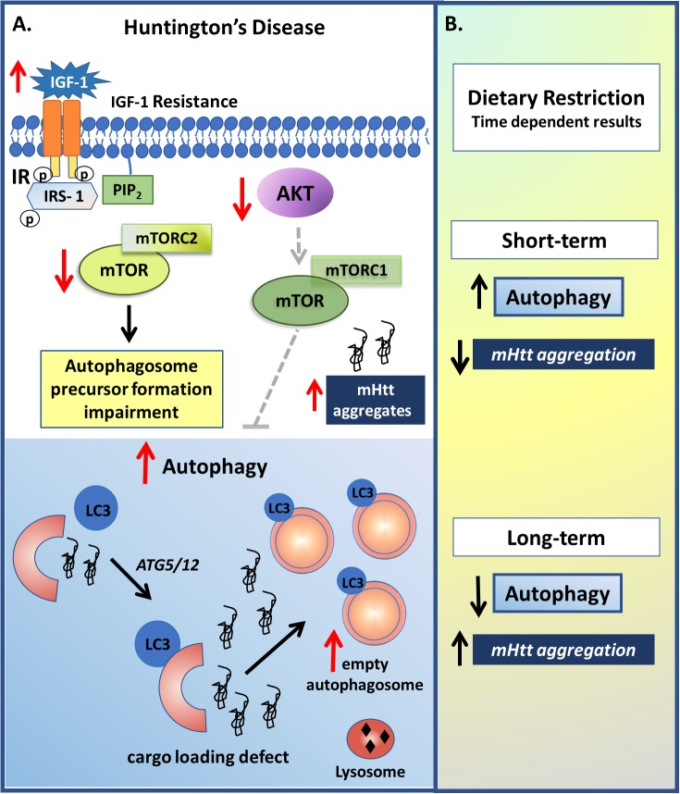
A)HDにおけるIGF-1の役割はまだ逆説的に思われるが、この病理学において重要な役割を果たしていることは明らかである。いくつかの研究で観察されたように、HD患者はIGF-1の高い基礎血漿レベルを示し、これはIGF-1抵抗性の状態とインスリン経路の低下に関連している可能性がある。IR活性の低下は、mTORC2およびAKT-mTORC1活性の低下をもたらし、それぞれオートファゴソーム前駆体形成の低下およびオートファジープロセスの増加をもたらするが、これは、カーゴ認識の障害を伴うオートファゴソームのアップレギュレーションと関連している可能性がある。しかしながら、例えば、受容体枯渇または長期的なIRの阻害による長期的なIRのダウンレギュレーションは、本質的なオートファゴソーム前駆体形成の欠如に起因する可能性のあるオートファジーの減少をもたらし、これは、mHtt凝集体の増加に関連している。B)食事制限などの非薬理学的アプローチも、時間に応じて二重の効果を示すことが示された。短期絶食ではオートファジーが増加し、mHtt凝集体が減少する一方、長期の血清欠乏ではオートファゴソーム形成が減少することが示された。
筋萎縮性側索硬化症と前頭側頭型認知症
筋萎縮性側索硬化症は、一次運動野と脊髄の上下運動ニューロンの変性を特徴とする神経変性疾患であり、筋力低下と最終的には麻痺を来し、死に至る(Al-Chalabi and Hardiman, 2013)。ALSの病因は不明であるが、前頭葉と前側頭葉の局所的な萎縮を特徴とするFTDとの併発も珍しくない(Van Langenhove et al 2012)。さらに、ALSとFTDは遺伝的危険因子を共有していることから、これらは神経変性疾患の連続体であることが提案されている(Van Langenhove er al)。
ALSは、高代謝性および脂質異常症の状態と関連しており(Dupuis et al 2008;Seelen et al 2014年)肥満および2型糖尿病と逆の危険因子の関連がある(Gallo et al 2013;Kioumourtzoglou et al 2015)。高脂血症を有するALS患者は平均寿命が増加しており、ALSの病態生理における脂質の神経保護的役割を示唆している(Dupuis et al 2008)。同様の結果は、SOD1変異マウスのALSモデルでも明らかであり、高エネルギー食が寿命を延ばしている(Dupuis et al 2004)。しかし、体格指数(BMI)強制生命力、年齢などの重症度の指標を調整した後、BMIは脂質異常症ではなく、ALS患者の生存率の増加の背景にあることが明らかになった(Paganoni et al 2011)。
ALSにおける脂質の役割は明らかにする必要があるが、ALS患者における高代謝異常の有病率が高いことは、より確実性が高く、それがより早い機能低下率と短い生存期間と関連している(Steyn et al 2018)。興味深いことに、高代謝はALSのいくつかの遺伝的危険因子と関連しているが、ALS-FTDと最も一般的に関連しているC9ORf72変異(Steyn et al 2018)には関連していない(DeJesus-Hernandez et al 2011)。驚くべきことに、C9ORf72遺伝子の変化は、オートファジー調節において二重の機能を有するようであり、C9ORf72はULK1複合体を制御することによりオートファジーの開始を正に調節し、その減少はオートファジー機能不全およびタンパク質凝集をもたらす(Webster et al 2016)。他の研究では、C9ORf72遺伝子がmTORC1調節を介してオートファジーを減少させることが示されている(Ugolino et al 2016)。しかし、Steyn et al 2018)は、C9ORf72と代謝亢進との関連を見つけることはできなかったが、Liu et al 2018)は、C9ORf72遺伝子の機能の損失が、細胞のグルコース飢餓条件下で、制御されたオートファジープロセスの異常を介して、脂質消化を障害し、de novo脂肪酸合成を増加させることを示した(Liu et al 2018)。このことは、飢餓または栄養豊富な条件が、オートファジーにおけるC9ORf72の機能を調節する上で重要な役割を果たしている可能性を示唆している可能性がある。
TANK結合キナーゼ(TBK)1表現型の機能喪失につながる変異もまた、オートファジー障害によるALSおよびFTDの発症リスクの増加につながる(Cirulli et al 2015; Freischmidt et al 2015; Cui et al 2018)。興味深いことに、肥満ザッカーラットで行われた研究は、インスリン抵抗性のメカニズムにおけるTBK1の関与の証拠を提供する(Muñoz et al 2009)。この研究は、肥満動物が肝内TBK1レベルを増加させ、TBK1によるSer994リン酸化によるIR活性の低下につながることを示している(Muñoz et al 2009)。さらに、高脂肪食は、肝臓および白色脂肪組織におけるTBK1遺伝子発現をポジティブに調節することができる(Chiang et al 2009)一方で、視床下部ニューロンの脂質ラフトへのTBK1の蓄積は、IR活性化およびAKTシグナル伝達を阻害し、脳のインスリン抵抗性におけるTBK1の潜在的な役割を示唆している(Delint-Ramirez et al 2015)。全体的に、このようなデータは、TBK1が肥満がALSおよびFTDのリスク低下と関連する基礎的なメカニズムの1つである可能性がもっともらしいことを示唆している。
カロリー制限下のALSの動物モデルは、脂質過酸化、炎症およびアポトーシスの増加、ならびにミトコンドリアのバイオエネルギー効率の低下のために寿命が減少している(Patel et al 2010)。同様に、ラパマイシン治療は有害であり、ALSのマウスモデルでは運動ニューロンの変性を増加させる(Zhang er al)。 オートファジーと代謝がALSの発症と進行を調節していることから、高脂肪食を治療に用いる可能性も含めて、食事療法がALSの管理に臨床的に有用であることが証明されるかもしれない(図6)。
図6 筋萎縮性側索硬化症と前頭側頭型認知症
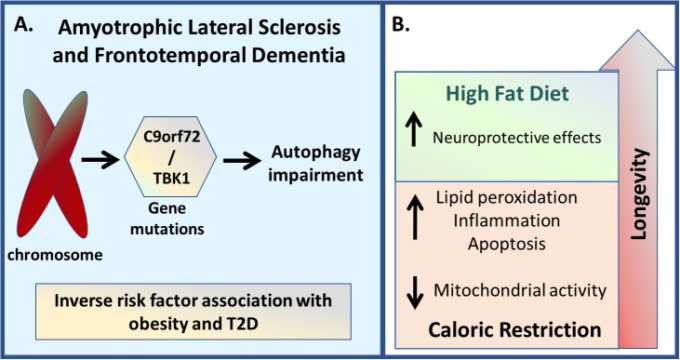
(A) C9orf72やTBK1などの遺伝子変異は、ALSとFTDの両方の疾患発症に関連しており、オートファジーの障害に関連している。また、高代謝状態や脂質異常症にも関連していることから、肥満や2型糖尿病とは逆の危険因子の関係があるように思われる。B)非薬理学的阻害については、カロリー制限は脂質過酸化、炎症、アポトーシスの増加、ミトコンドリア活性の低下による寿命の減少を示したが、高エネルギー食は寿命の増加を示した。
その他の神経変性疾患
3型脊髄小脳失調症
脊髄小脳性失調症3(SCA3)は、主にバランス、歩行および発話に影響を及ぼす進行性の失調を特徴とする神経変性疾患であり、プロテアソームタンパク質分解経路および転写の調節において重要な機能を有するユビキチン化酵素であるアタキシン-3タンパク質のC末端ポリグルタミン(PolyQ)領域の変異によって引き起こされる。文献によると、野生型アタキシン-3のpolyQストレッチは、プロテアソーム媒介分解からBeclin-1を保護することでオートファジーを誘導する(Ashkenazi et al 2017)。他の神経変性疾患と同様に、利用可能な治療法は、疾患症状の軽減を目的としたもののみである。しかしながら、基礎となるメカニズムは完全には理解されていないが、オートファジープロセスの調節は、疾患の進行を改善または停止するための潜在的なターゲットであることが実証されている(Menzies et al 2010; Nascimento-Ferreira et al 2011)。Menzies et al 2010)によって実証されたように、ラパマイシンの投与は、mTORの阻害とその結果としてのオートファジーのアップレギュレーションを介して、SCA3動物モデルにおける運動能力を改善した。さらに、Nascimento-Ferreira et al 2011)によると、Beclin-1の過剰発現は、内因性オートファジーマーカーの異常発現、オートファゴソームの蓄積、およびBeclin-1のレベルの低下を防ぎ、オートファジーフラックスの増加、変異型アタキシン-3のクリアランス、およびその他の神経保護効果をもたらした。同様の方法で、別の研究では、SCA3患者の線維芽細胞と健常者の線維芽細胞を比較したところ、オートファゴソーム産生の減少が観察された。しかし、Beclin-1の過剰発現はオートファゴソームのフラックスを増加させたが、オートファゴソーム産生の高レベル化には至らなかった(Onofre et al 2016)。まとめると、この結果は、オートファジーがSCA3病の調節にも重要な役割を果たしていることを支持するものである。
ラフォーラ病
ラフォラ病は、常染色体劣性遺伝性のまれな進行性致死性神経変性疾患であり、ミオクロニー性てんかんと持続的な神経衰弱を特徴としている。LD症例の約58%で報告されており,グルカンホスファターゼであるラフォリンをコードするEPM2A遺伝子の変異は,脳,脊髄,その他の末梢組織にラフォラ体(LB)として知られる細胞内包摂体の存在と関連している(Knecht er al)。 ラフォリンはオートファジーに絶対的に必要ではないことが示されたが、オートファジーを積極的に制御することが観察された(Knecht et al 2010)。本研究によると、LD は LC3-II のレベル低下と形成障害に関連しており、また AKT-mTOR 経路のシグナル伝達が変化していたが、ラフォリンを過剰発現させると、オートファジーに依存した方法でタンパク質凝集体の量が減少することが明らかになった(Knecht et al 2010)。さらに、オートファジー以外にも、LDはインスリン抵抗性や高血糖と関連しているようである(Nicolescu et al 2019)神経変性疾患におけるインスリングとオートファジーシグナル伝達の間のクロストークを強化している。
結論
中心的なインスリンシグナル伝達およびオートファジーの調節が多様な神経変性疾患のホストに関連していることは明らかであるが、それは薬理学的阻害、動物モデル、遺伝的戦略および患者を含むいくつかの研究によって支持されており、また代謝障害といくつかの神経変性疾患の発症との間のリンクによっても支持されているが、文献からのデータは議論の余地があり、基礎となるメカニズムおよびオートファジー調節の可能な利益はそれぞれの障害に固有のようであるため、声明を定義することはまだ困難である。
多くの重要な細胞プロセスがインスリンシグナル伝達およびオートファジープロセスと密接に関連しているため、薬理学的な阻害および/または刺激は、特に長期的な治療を考慮すると、いくつかの有害な効果をもたらす可能性があり、それが文献での相反する結果の一因となっている可能性がある。それに加えて、疾患の発症、年齢、性別が発症や治療成績に強い影響を与えることを考慮することが重要である。
このような観点から、代謝やオートファジーの制御を調節する遺伝子戦略は、これらの経路と神経変性疾患の根底にあるメカニズムの理解を深めることに貢献すると考えられる。また、神経変性疾患の発症・進行における運動や断続的な絶食などの非薬理学的介入の効果を研究することは、従来の治療法に貢献する可能性のある新たなアプローチだけでなく、文献上の新たな情報にも光を当てることができる。
このように、このようなプロセスが異なる中枢および全身の細胞でどのように統合されているかを理解することは、これらのまだ管理の行き届いていない状態に対して、より良い標的治療を提供することになるはずである。