
Contents
Insights into Potential Mechanisms of Injury and Treatment Targets in COVID-19, SARS-Cov-2 Infection
要旨
重症急性呼吸器症候群コロナウイルス2型(SARS-CoV-2)は、現代で最も深刻なパンデミックである。2020年1月に中国の湖北省武漢市で初めて報告されて以来、世界的に広がっている。ウイルスの急速な広がりと、それが医療システムを席巻した負担を考えると、SARS-CoV-2感染症における臓器不全の病態に関する現在の理解を簡潔に説明する必要がある一方で、まだ多くのことが明らかにされていないことを認識する必要がある。
このレビューは、ベッドサイドでの意思決定に役立つだけでなく、研究のための潜在的な治療目標を示すのにも役立つであろう。現在までに入手可能な文献を検索し、肺、心臓、腎臓におけるSARS-CoV-2感染症の罹患率と死亡率の増加の背景にある病態生理について、臨床家、研究者、学生にも理解しやすいように図解を多用して紹介した。
キーワードは以下の通り。COVID-19、病因、SARS-CoV-2、急性腎障害、心症状、多臓器不全
ウイルス学と宿主細胞への侵入
COVID-19パンデミックの起源を理解するためには、まず、2002年に出現した重症急性呼吸器症候群コロナウイルス(SARS-CoV)を思い出す必要がある。SARS-CoVとSARS-CoV-2という2つのウイルス株は類似しており、系統解析によりSARS-CoV-2はSARS-CoVと約76%のヌクレオチド同一性を有することが示されている[1]。
Hoffmanらの研究によると、SARS-CoV-2はSARS-CoVと同じスペクトルの細胞株に侵入し、その侵入メカニズムも類似していることが示されている。このことは、両ウイルスの侵入受容体の選択に類似性があることを示唆している。
両ウイルスは、宿主細胞への侵入のためにスパイク(S)タンパク質に依存している。Sタンパク質のS1ユニットは、宿主細胞の受容体に付着して結合する。細胞内への侵入は、その後、ウイルスと宿主細胞の膜の融合につながる宿主細胞の細胞プロテアーゼによるSタンパク質のプライミングを必要とする。
宿主細胞の受容体はアンジオテンシン変換酵素2(ACE2)であり、採用されているセリンプロテアーゼはTMPRSS2である[1]。この侵入は、ACE2の細胞内化とこれらの受容体のダウンレギュレーションを引き起こす:その重要性については後述する。
通常、ACE2は可溶性の形で循環中にほとんど存在しないが、ACE2は広く発現しており、その受容体は肺、心臓、腎臓、脳を含むほぼすべての臓器の動脈内皮細胞、静脈内皮細胞、動脈平滑筋細胞で発見されている。また、口腔粘膜、鼻粘膜、肺胞上皮細胞、小腸の腸球、心筋細胞、腎ポッドサイト、近位複雑管の細胞で豊富に見出されている[2-5]。
これは、部分的には、COVID-19で見られる全身症状の配列を説明するのに役立つかもしれない。
肺におけるSARS-CoV-2の提案された病因
我々は、ウイルスが最初に口腔粘膜または鼻粘膜から宿主に侵入し、その後、消化器系または呼吸器系に広がってから全身感染を起こす患者もいると考えている。SARS-CoV-2 に感染した患者の呼吸器症状の重症度は広範囲にわたっている。数日間は乾いた咳が続く患者もいれば、高呼気終末圧(PEEPs)を伴う機械換気を必要とする急性呼吸窮迫症候群(ARDS)に苦しむ患者もいる。
ARDSへの進行は、図1に示すように、以下のメカニズムによって促進される可能性がある。ACE2は肺胞上皮細胞、特に肺炎球Ⅱ型細胞に豊富に存在する[5]。正確なメカニズムは明らかになっていないが、現在の文献から、呼吸器系でのウイルスの細胞内化とその後の拡散は、以下のような過程を経ていると考えられる。
1) アンジオテンシン変換酵素1(ACE1)およびアンジオテンシンIIのレベルがACE2およびアンジオテンシン1-7のレベルと比較して増加する
2) 好中球、マクロファージおよびCD8+ T細胞によって媒介される有意な炎症反応が肺胞浮腫をもたらす
3) 血栓形成
4)肺炎球タイプII細胞の潜在的な破壊 [2,5-12]である。

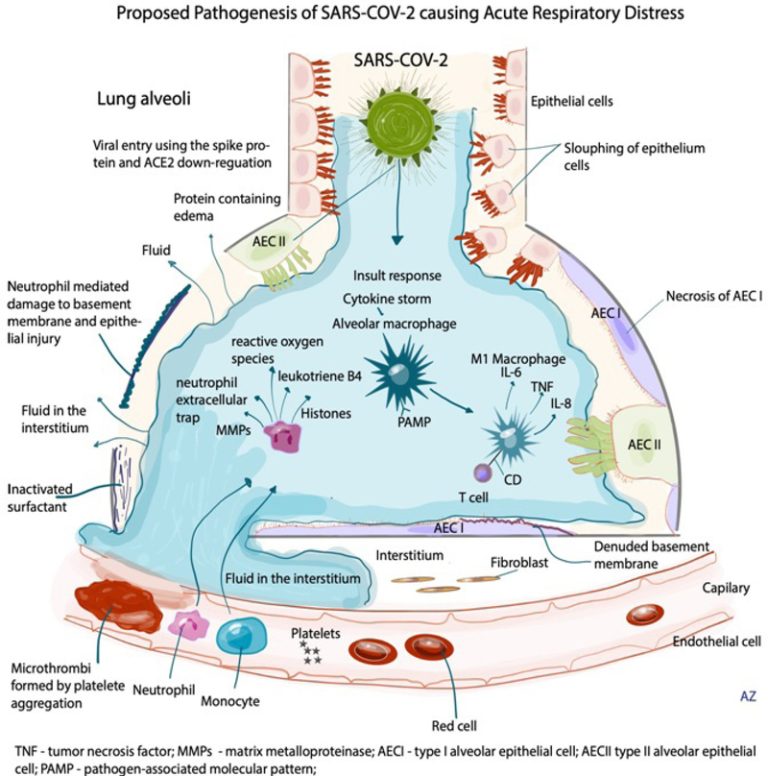
図1.
急性呼吸困難を引き起こすSARS-Cov-2の提案された病態。肺胞における急性炎症反応の画像である。SARS-CoV-2に対する炎症反応は、基底膜の変性と上皮バリアの破壊につながり、肺胞内の体液の蓄積につながる。
肺胞マクロファージは上記のように示され、好中球および単球の浸潤を増加させる炎症性サイトカインを放出する。肺胞マクロファージはIL-6、TNF、IL-8および炎症性カスケードおよびサイトカインストームに寄与する他のメディエーターを放出する。
好中球は、活性酸素種(ROS)、マトリックスメタロプロテアーゼ(MMPs)、ロイコトリエンB4(LTB4)、好中球細胞外トラップ(NET)およびその他のメディエーターを放出する。組織壊死因子(TNF)は、血小板凝集に寄与する組織因子の発現を増加させる。
血小板凝集は、微小血栓および血栓形成につながる。内皮傷害が増加し、凝固性が増加し、血小板凝集とともに高血栓状態につながる。AECI(I型肺胞上皮細胞)およびAECII(II型肺胞上皮細胞)。
ACE2レベルの低下はARDSへの進行において重要な役割を果たす。ACE2はアンジオテンシン1-7を生成しながらアンジオテンシンIIを不活性化するため、レニン-アンジオテンシン系(RAAS)をダウンレギュレートする[2,6]。その後に増強された血管収縮は、一部ではあるが、COVID-19で発症するARDSの非典型的な性質を説明しているかもしれない。
我々が我々の施設で目撃したことは、イタリアのGattinoniらによって観察されたことと類似している;すなわち、ARDSを発症する肺は比較的高いレベルのコンプライアンスを保持している。合理的な肺のコンプライアンスにもかかわらず、高PEEPと伏臥位[13]などの募集戦略を使用した場合でも、重度の低酸素血症が持続する。
肺血流の調節障害とACE2の消失による低酸素性血管収縮、およびその結果としての強力な血管収縮因子であるアンジオテンシンIIのアンジオテンシン1-7レベルへの増加が、低酸素血症の一部を説明するのに役立つ可能性がある。肺の炎症と凝固の増加は、アンジオテンシンIIの非作用として報告されている。12人の患者を対象とした研究では、SARS-CoV-2感染患者の血漿中のアンジオテンシンIIレベルが著しく上昇し、ウイルス負荷および肺損傷と線形に関連していることが明らかになった[14]。
アンジオテンシン1-7とアンジオテンシンIIの比率のこの変化をさらに悪化させるのは、アンジオテンシンIIがジスインテグリンとメタロプロテアーゼ17(ADAM17)をアップレギュレートすることであり、これは膜に固定されたACE2を切断する[5]。これにより、膜上のACE2の触媒活性が失われる。
循環可溶性ACE2のレベルの上昇は、RAASシステムの活性の亢進の徴候であり、予後の悪化と関連している[5]。さらに、ACE2の喪失は好中球の蓄積を引き起こし、最終的にはARDSや血管透過性の亢進、ひいては肺水腫を引き起こす可能性がある[2]。このことは、将来の治療戦略に役立つ可能性があるため、臨床的に重要である。
例えば、鳥インフルエンザH5N1に感染したマウスを用いた研究では、組換えヒトACE2の投与がウイルス誘発性肺障害を減少させることが示されている[15]。さらに興味深いのは、アンジオテンシンIIはマクロファージやその他の免疫細胞を活性化させ、IL-6、TNF-α、およびその他の炎症性サイトカインの増加を引き起こすことで、適応免疫をさらに阻害することである[5]。
SARS-CoV-2感染は、末梢性CD4+ T細胞およびCD8+ T細胞のレベルを低下させるが、これらの細胞は依然として高活性化したままである [14,16]。CD8+ T細胞は非常に細胞毒性が高い。T細胞の調節障害は肺胞バリアの破壊をもたらす。
SARS-CoVの研究では、ウイルスはリンパ球に親和性があり、CD4+ T細胞とCD8+ T細胞の両方の数が減少することが示された[14]。191人の患者のコホートにおいて、Zhouらは、COVID-19の非生存者は生存者よりもベースラインのリンパ球数が有意に低いことを発見した[17]。
免疫応答のゲートキーパーであることが多いCD4+ T細胞の数の相対的な障害は、免疫系全体の調節障害をもたらす [7]。バイオマーカーであるインターロイキン-6(IL-6)、腫瘍壊死因子α(TNF-a)などのレベルの上昇は、重篤な疾患の徴候であり、死亡率の増加と関連している[7]。
CD8+ T細胞は、標的細胞の細胞質に入り込み、カスパーゼカスケードを誘発する顆粒を含む細胞傷害性顆粒を分泌することで肺胞上皮細胞にダメージを与え、最終的にはアポトーシスを引き起こす[18]。また、アポトーシスは、感染細胞上のFasリガンドがT細胞上に発現するFas分子に結合することで、TCと感染細胞との細胞表面相互作用によっても誘導される[18]。
さらに、肺の肺胞Ⅱ型細胞への損傷は、界面活性剤のプロデュクションの不活性化を含む複数の結果をもたらし、これはバリア破壊と肺胞浮腫を悪化させ、間質と肺胞内のタンパク質に富んだ液体の蓄積をもたらす[5,18]。さらに、界面活性剤の損失は、肺組織の弾力性に有害な影響を与える。
SARS-CoV-2に感染した一部の患者で起こる免疫異常は、「サイトカインストーム 」と呼ばれている。パンデミックが始まって以来、この用語は頻繁に使われていたが、理解されてはいなかった。
サイトカイン放出症候群(CRS)は、この障害の別称であり、キメラ抗原受容体(CAR)T細胞療法および血球貪食症候群で治療された患者の細菌性敗血症を含む様々な状態で発生する[19,20]。
CRSは、全身性炎症の増加、胸水および浮腫の形成に寄与する血管透過性の増加を引き起こす;CRSはまた、第三空間液減少および低血圧を介した血管内枯渇を引き起こす[20]。
IL-6はおそらくCRSにおいて最も重要なサイトカインである。SARS-CoV-2に感染した患者では、IL-6のレベルが上昇するとARDSの発症、臨床的に有害な転帰、死亡に相関することがわかっている[19-21]。
COVID-19に感染した一部の患者では、d-ダイマー濃度の上昇によって特徴づけられる凝固活性の上昇が、酸素化をさらに阻害している。高濃度のdダイマーは死亡率の増加との関連が報告されている[17,22]。炎症と内皮の損傷はさらにプロコアグラント因子を増加させ、さらなる血管内損傷を引き起こし、最終的には血栓症と虚血を引き起こす可能性がある [8-12]。
同済病院でSARS-CoV-2に感染した3人の患者の臨床検査所見には、白血球症、プロトロンビン(PT)と部分プロトロンビン時間(PTT)の上昇、フィブリノーゲンとd-ダイマーレベルの上昇、抗カルジオリピンIgA抗体、抗β2糖タンパク質I IgA抗体およびIgG抗体の存在が含まれていた[12]。
これらの抗体のほとんどは抗リン脂質症候群の一部であり、リウマチ性疾患における血栓性イベントを引き起こす可能性がある。これらの抗体はまた、C型肝炎ウイルス、ヒト免疫不全ウイルス、サイトメガロウイルス、水痘帯状疱疹、エプスタインバーウイルス、アデノウイルス、およびパルボウイルスBなどの様々なウイルス感染症に反応して上昇することができる。これは、COVID-19に罹患している患者における血栓症の潜在的なメカニズムである。
IL-6は、他のインターロイキンの中でも、血小板の多動、凝集および凝集を引き起こすことが知られている[25]。さらに、腫瘍壊死因子(TNF)を媒介とした組織因子の発現は、血小板の凝集および多巣性血栓形成および肺胞間凝固を促進する [25] [25] これは、TNFが血管単球および血管内皮細胞を活性化し、それらの細胞表面に組織因子を発現させるために起こる。これにより、凝固系の外因性経路が活性化され、微小血栓形成が誘導される。
TNFが小血栓形成に寄与する別の経路は、凝固系を調節するトロンボモジュリンおよびグリコサミノグリカンの内皮発現を減少させることによるものである[23,25]。これらのすべての調節障害は、播種性血管内凝固(DIC)につながる可能性がある。TNFおよびその他のサイトカインは、好中球を活性化して、好中球細胞外トラップ、ヒストン、ロイコトリエンB4、MMPマトリックスメタロプロテアーゼ、MPOミエロペルオキシダーゼ、活性酸素種などの様々な炎症性メディエーターを放出し、内皮細胞をさらに損傷させる可能性がある[23]。
低酸素症のもう一つの潜在的なメカニズムは、Liuら[26]によって提案された。分子解析を行った結果、COVID-19はヘモグロビンのヘム成分と相互作用するORF8、ORF10、ORF1ab、ORF3a、表面糖タンパク質を保有していることが明らかになった。
各ヘム基にはポルフィリンに結合した鉄イオンが含まれている。これらの鉄イオンは、体内の酸素と二酸化炭素の交換に参加している。ORF8タンパク質は、ヘムのポルフィリンに結合する能力を持っている。ORF10、ORF1ab、ORF3aタンパク質は、β1ヘモグロビン鎖のヘム分子から鉄を解離させることができる(図2)。
このようにヘモグロビンは、効果的に酸素を送達する能力を失い[26]、したがって、循環ヘモグロビンの一部は、さらに低酸素に貢献し、非機能になる。これは一酸化炭素中毒に類似している。機能不全のヘモグロビンは、赤血球がレオロジーを失い変形する原因となる。
これは、潜在的に低酸素症および微小循環における血栓症への別の寄与因子である。ヒドロキシクロロキンはヘムからの鉄の脱鉄化を阻害し[20]、それによってヘモグロビンの酸素運搬能力を維持すると推測されている。最初の傷害は肺で起こるが、全身の炎症や低酸素は心臓や腎臓などの他の臓器にも深刻な影響を及ぼす。このメカニズムに関するエビデンスは現時点では限られており、実験と臨床試験の両方が必要とされている。

図2.
ヘモグロビン障害とT細胞障害の提案された病態。この画像で示されているのは、SARS-Cov-2タンパク質の過程である。ORF8, ORF10, ORF1ab, ORF3aはヘモグロビンのヘム成分と相互作用して組織への酸素供給を阻害する。この画像では、細胞毒性、TNFおよびインターフェロン(INF)の放出および走化性に寄与するCD8 T細胞が表現されている。感染細胞上のFasリガンドがT細胞上で発現しているFas分子と結合することで、T細胞と感染細胞の間の細胞表面相互作用がアポトーシスを引き起こする。
SARS-CoV-2と心臓の発症機序の提案
COVID-19の心臓障害の正確なメカニズムは確立されていないが、以下に考えられる原因のいくつかを概説する。
心臓への影響は、ACE2の減少、炎症反応、低酸素、および凝固経路の混乱に部分的に起因している(図3)。ACE2は心筋細胞、心臓線維芽細胞、冠動脈内皮細胞に広く発現している[2]。
COVID-19によるアンジオテンシン1-7に対するアンジオテンシンIIの相対的な増加は、心臓に多くの悪影響を及ぼす。アンジオテンシン1-7は抗増殖、抗アポトーシス、軽度の血管拡張作用を示し、心不全、血栓症、心筋肥大、線維化、不整脈、アテローム形成などの保護作用を有し、またメタボリックシンドロームに関連した血管機能障害を減衰させる。
SARS-CoV-2に感染した患者に急性心筋梗塞を認めた剖検報告がいくつかある。Zhouらは、SARS-CoV-2に感染した患者191人のコホートにおいて、死亡した患者の半数以上で高感度心筋トロポニンIの増加が認められたと報告している[17]。

図 3.
SARS-CoV-2の心臓病変の提案された病因。レニン-アンジオテンシン-アルドステロン(RAAS)経路を説明する画像である。アンジオテンシノーゲンはレニンによりアンジオテンシンIに変換され、アンジオテンシンIはアンジオテンシン変換酵素1(ACE 1)によりアンジオテンシン2(Ang 2)に変換される。
ACE 2はアンジオテンシン変換酵素2(ACE 2)によってアンジオテンシン1-7(Ang 1-7)に変換される。ACE 2はウイルスによってダウンレギュレーションされているため、画像の「×」で示すように、ACE 2はAng 1-7に変換されない。Ang 1-7は抗線維化、抗増殖、抗アポトーシス、血管拡張作用があり、そのダウンレギュレーションは線維化の増加、血栓症の増加、肺損傷の増加、血圧の上昇につながる。
ウイルスによる直接的な心筋損傷が心筋炎を引き起こす可能性があると推測されている。プロ炎症性サイトカインの増加は、心筋細胞のアポトーシスと壊死を引き起こする。炎症の増加は、アテローム性動脈硬化性プラークの形成とその破裂の促進につながる。心膜の炎症性細胞の浸潤は、浮腫や心膜炎の増加につながる。
Shiらは、416人の患者を対象とした研究で、心損傷を受けた患者はそうでない患者に比べて死亡率が高く、51.2%対4.5%であったことを明らかにした[28]。ハザード比は4.26で、死亡率の独立した予測因子であることが判明した[28]。
潜在的な原因は多岐にわたる。全身性の炎症反応は、冠動脈性動脈硬化性プラーク内の炎症性活動の亢進につながる可能性がある。これは内皮機能障害と相まって、プラーク破裂のリスクを増加させる[28]。逆に、SARS-CoV-2に感染した患者のトロポニン値が良好であった研究では、患者の転帰も良好であったことが明らかになっている[28,29]。
SARS-CoV-2に感染した一部の患者で発症する心筋炎では、炎症反応が大きな役割を果たしている可能性が高い。剖検報告では、マクロファージやCD4+T細胞を含む炎症性の浸潤が多数認められている[7]。この記事を書いている時点では、心筋組織内にSARS-CoV-2が存在するという報告はないが、ある研究では、SARS-CoVで死亡した患者の35%にウイルスゲノムが検出されている[30]。
COVID-19患者では定期的に心エコー検査を受けることが難しいため、駆出率温存(HFpEF)と駆出率低下(HFrEF)のどちらが心不全になりやすいかは自信を持って言えることではない。しかし、心不全症状はCOVID-19の患者では一般的である。Zhouらは、彼らのコホートの23%、および非生存者の52%に心不全が認められたと報告している。
COVID-19の潜在的な直接的な影響、炎症反応、疾患により生じる血栓症の状態に加えて、現在SARS-CoV-2に感染した患者に提供されている治療法を考慮に入れなければならないが、その多くはQTc間隔を増加させるものである。これには抗マラリア薬のクロロキンやヒドロキシクロロキン、抗ウイルス薬、アジスロマイシンなどの抗生物質が含まれるが、これらに限定されるものではない。
SARS-CoV-2と腎臓の発症機序の提案
SARS-CoV-2感染後の炎症、血管収縮、血栓症の傾向は全身性であり、腎臓に様々な影響を及ぼす。COVID-19に関する研究がより多く発表されれば、SARS-CoV-2感染者における急性腎障害(AKI)の発生率が明らかになるであろう。
ACE-2およびTMPRSS遺伝子の共発現は、腎臓では肺よりも少なくないことが明らかになっており、これは腎臓細胞がSARS-CoV-2の重要な標的である可能性を示唆している[3]。現在の文献では、SARS-CoV-2は腎臓へのトロピズムを示すことが示唆されている[31]。
この記事を書いている時点で、Yangらの研究では、52人の重篤な成人患者を対象とした研究で、23%がAKIを発症したことが示されている[32]。Zhouらは、彼らのコホート全体の15%、および非生存者の50%にAKIが認められたことを明らかにした。
ポッドサイトと近位直管では、ACE2とTMPRSSの共発現が比較的高い。ポドサイトおよび近位直管の破壊は、Chengらによる701人の患者を対象とした前向きコホート研究[33]において、COVID-19患者の多くで、発症時に蛋白尿および血尿がそれぞれ43.9%および26.7%認められた理由を説明することができる。
マウスモデルにおけるACE-2と腎細胞に関するこれまでの研究では、ACE-2の阻害は、アルブミン尿の増加と糸球体マトリックスの拡張、中膜マトリックスの沈着の増加、糸球体基底膜の肥厚と糸球体硬化症をもたらすことが示されている[34]。一方、ACE2の増幅は、マウスモデルにおいて腎症の影響を軽減することが示されている[4]。さらに、先に述べた炎症反応が腎臓に大きな影響を与えている可能性がある(図4)。

図4.
急性腎不全(AKI)を引き起こすSARS-CoV-2の発症機序の提案。SARS-CoV-2が腎臓に及ぼす急性炎症反応と全身的な影響を示す画像である。
ACE2受容体は主にポッドサイトと尿細管上皮細胞に存在している。細胞の内部化は糸球体および尿細管の損傷をもたらす。自然免疫応答と適応免疫応答は、全身の血管拡張と血管透過性の亢進をもたらし、糸球体と尿細管のさらなる損傷をもたらす。
腎尿細管細胞の損傷は、全身性の炎症性症状を悪化させる。免疫反応もまた血栓形成に寄与する。遺伝性感染症および重畳した細菌感染症はAKIの潜在的なメディエーターである。挿管した患者を高終息圧(PEEP)設定で挿管すると、高い腹腔内圧が発生し、腎コンパートメント症候群を引き起こす可能性がある。
さらに、腎尿細管上皮が損傷を受けると、サイトカインの過剰産生を悪化させる可能性がある[20]。このメカニズムはよくわかっていないが,ヒトおよび動物実験で尿細管上皮細胞の損傷がIL-6のアップレギュレーションを促進することが観察されている[20]。また、ポッドサイトはその表面に IL-6 レセプターを発現する数少ない細胞の一つであるため、IL-6 に直接反応することができ、このレセプターは炎症過程の間にアップレギュレートされ ることも興味深い。
我々は、この免疫応答のアップレギュレーションが肺損傷をさらに引き起こす可能性があり、その結果、より深刻な低酸素性腎髄質損傷につながる可能性があることを提案する。心筋症や心筋炎が最終的に腎静脈のうっ血や腎動脈の低灌流を引き起こす可能性があるため、心機能の悪化もAKIの一部を説明することができる。これらのメカニズムは、体液の不均衡、高い気道圧とともに、腎コンパートメント症候群につながる可能性がある[20]。
前述のような凝固経路の破綻および血栓形成もまた、COVID-19を有する多くの患者におけるAKIの発症に役割を果たしている可能性が高い。さらに、免疫反応が異常に制御されているすべての重症患者と同様に、特にフォーリーカテーテルを使用している患者では、重畳した細菌感染のリスクが高く、モニタリングと考慮が必要である。
治療目標および治療法の選択肢に関する現在の理解
図5は、SARS-CoV-2感染後に臓器損傷に至る傷害の4つの病態生理学的メカニズムと、現在治療法として検討されているものをまとめたものである。なぜ80%の人が発症時にウイルスを除去しても、他の人は強い炎症反応と血管収縮を引き起こし、血栓症や臓器損傷のリスクを高める下流の影響を受けるのかは明らかになっていない。それにもかかわらず、炎症反応と血管収縮反応は密接に関連しており、同時に起こるようである。
炎症性および血管収縮性の反応は、感染の結果として生成されたプロコアグラントによって誘導される高凝固状態とともに、微小循環血栓症のための完璧なセットアップであり、最終的には臓器損傷を終了する。
肺では、これが換気灌流のミスマッチを伴う急性呼吸窮迫症候群(ARDS)につながり、これらの患者のARDSが高圧の機械的換気支援システムに期待されるような反応を示さない理由を説明する。COVID-19に対処するために、抗ウイルス薬(レムデシビル)[36]、アジスロマイシンなどのマクロライド系抗生物質、抗マラリア薬のヒドロキシクロロキン37など、SARS-CoV-2複製を直接阻害する薬剤を慎重に使用して治療する機会があるが[38]、ベストプラクティスとしてこれらの薬剤の普及を促進するための無作為化臨床試験はまだ利用できていない。
回復期血漿の使用 [39] は有望であり、現在臨床試験が行われている。これらの患者に見られる発熱や下痢は、水分不足を引き起こすため、水分補給が必要であることに注意することが重要である;しかしながら、ARDSを悪化させる危険性があるため、これらの患者には過剰な水分補給を行わないように注意しなければならない。
SARS-CoV-2感染予防の究極の目標はワクチン開発であり、そのための研究が進行中である[40]。
炎症反応に対処するために、ステロイド [41] および選択的 IL-6 ブロッカー(トシリズマブ) [42] が使用されているが、反応は変動的である。インフルエンザH1N1の教訓からサイトカインストームがどのようにアプローチされるかについては、Liuら[43]によって詳しく述べられており、免疫抑制剤がJAKキナーゼ阻害剤の使用を含むCOVID-19の治療に対する反応にどのように影響するかについては、Russelら[44]によって徹底的に検討されている。
非特異的な抗炎症剤や細胞枯渇剤の使用は、宿主の反応を減衰させ、ウイルス感染を悪化させる可能性があり、推奨されない。
最近では、間葉系幹細胞 [45] をCOVID-19感染者の免疫調節や再生効果のために使用するという概念が臨床試験のコンセンサスを得つつある。
血管収縮に対処するためには、ウイルスが細胞内に侵入するのをブロックするための融合阻害剤の研究や、アンジオテンシンIIの効果を改善するための外因性ACE2の投与が賢明であると思われる [15,45]。入院患者におけるアンジオテンシンII変換酵素阻害剤(ACEI)アンジオテンシン1型受容体拮抗剤(ARB)の予備的な観察では、死亡率の減少が示されており[46]、したがって、臨床試験を実施するための別の側面が開かれている。
血栓症に対処するためには、機械的支持に移行し、d-ダイマーが上昇した患者はすべて抗凝固療法を行うべきである[47]。最近になって抗凝固療法を支持するエビデンスが出てきて、これらの患者の管理が変わってくるまで、私たちがこれらの患者に遭遇した初期の段階では、これは考慮されていなかった。
COVID-19感染症におけるヘムの脱鉄化に起因する機能不全ヘモグロビンを交換輸血で置換する役割はまだ明らかにされておらず、ヒドロキシクロロキンがヘモグロビン中の鉄の安定化を助けるかどうか[48]、それによって酸素化の改善に寄与するかどうかは不明である。
最後に、末梢臓器障害を回避するためには、早期の介入とともに病態の理解を深めることが重要である。末端臓器損傷にはサポートが必要である。ARDSでは、肺保護換気(高呼気終末陽圧を避ける)と大流量酸素供給、臥位での理学療法 [49] が治療の主な滞在方法であるが、AKIでは透析が必要となる場合がある。
SARS-CoV-2感染患者の管理においては、心臓支持装置の使用は限られているが、各チェックポイントの管理が容易になり、陰性検査への転換が確認できるようになれば、肺移植や心臓と肺の複合移植への道を開く可能性のある心臓支持装置の研究の機会が増えるであろう。間葉系幹細胞[45]を、SARS-CoV-2感染患者の免疫調節効果や再生効果を利用して、ARDS後の肺の修復に利用するという概念は、臨床試験のコンセンサスを得つつある。

図5.
末端臓器損傷に至る4つの病原性経路の図解。各チェックポイントでの治療目標は、ボックスの外に示されている。この図は、COVID-19感染症患者の治療において、複数のチェックポイントに対応する必要性を強調している。また、早期介入の必要性と、まだ利用可能ではないが、各チェックポイントに対応するように設計された治療法の研究の必要性も強調している。
ここに記載されているSARS-CoV-2感染症に対する治療法は、いずれもランダム化臨床試験で試験されたものではなく、せいぜい治験的なものであるか、あるいは思いやりをもって使用されているものであることを強調しておくことが重要である。また、我々の試みは、SARS-CoV-2の臓器傷害の潜在的な機序と治療の対象となりうる場所を概説したものであり、COVID-19に対する利用可能な治療法の詳細なレビューではないことに注意することも重要である。
結論
COVID-19パンデミックの経験は短く、その病態の多くがまだ解明されていないことを認識しているにもかかわらず、傷害の病態に関する現在の理解には大きな進歩が見られ、それが治療標的の可能性を示す道を開いている。それぞれの治療標的には既存の治療法があるが、どれも広く使用することが承認されていない。
ウイルスの複製を遅らせる治療法や、予防のためのワクチン開発は確かに重要であるが、それにもかかわらず、提案されている治療標的のいくつかまたはすべてに対処する多面的なアプローチが、末梢臓器損傷を予防または遅らせるために必要であるように思われる。
現時点では、将来の研究により病態に関するより多くの知見が明らかにされ、無作為化臨床試験によりベストプラクティスのためのより構造化された方向性が示されるまでは、病態に関する現在の理解に基づいてCOVID-19患者の治療を開始するのが賢明である。