
Contents
Inflammation, Nitro Oxidative Stress, Impaired Autophagy, and Insulin Resistance as a Mechanistic Convergence Between Arterial Stiffness and Alzheimer’s Disease
www.ncbi.nlm.nih.gov/labs/pmc/articles/PMC8039307/
オンラインで2021年3月29日に公開
概要
世界の高齢者人口の平均年齢は着実に上昇している。この未曾有の高齢者人口の増加は、心血管疾患(CVD)や神経変性などの加齢関連疾患の有病率を高める。近年、いくつかの血管危険因子がアルツハイマー病(AD)と関連していることから、CVDと神経変性症候群との相互作用の可能性について関心が高まっている。また、動脈硬化はCVDとADの独立した危険因子である。この総説では、急性組織特異的炎症、ニトロ酸化ストレス、オートファジー障害、インスリン抵抗性など、炎症に関連した疾患メカニズムについて考察し、動脈硬化とADの相乗効果を提案する。
キーワード:炎症性疾患、代謝、ニトロ酸化ストレス、オートファジー、神経変性、動脈硬化
はじめに
世界人口の平均年齢は着実に上昇しており、2019年には65歳以上の高齢者が約10億人に達している。2030年には、この数は14億人に増え、2050年には21億人に達すると推定されている(WHO, 2018)。このような世界の高齢者人口の未曾有の増加は、心血管疾患(CVD)や認知症などの加齢関連疾患の有病率を高める。世界的に見て、CVDは死因の第1位であり、2016年の死亡者数は1,790万人と推定されている(WHO、2017年)。同年、世界で4,380万人が認知症に罹患し、240万人がこの疾患で死亡し、世界の死因の第5位となった(GBD 2016 Neurology Collaborators, 2019)。歴史的には、CVDとアルツハイマー病(AD)は、臨床的な分類基準に基づいて別個のものと考えられてた。しかし、疫学研究の増加により、両疾患間の独立した収束が報告され、メカニズムの重複が示唆されている。本レビューでは、慢性低級炎症がCVDとADの間で大きな役割を果たしていることから、CVDとADの間のメカニズムの収束について議論する。
アルツハイマー型認知症
アルツハイマー型認知症は、昔から古文書に記述されている(例:”Be kind to your father, even if his mind fails him.”)。- 旧約聖書の Sirach 3:12)。認知症は、進行性の認知機能障害を伴う多数の神経症状から構成されており、患者の日常生活における自立した機能に影響を与える。これらの認知障害は、一般的に行動、気分、性格の変化を伴う(Tariot et al., 1995; Mega et al., 1996)。以前は、レビー小体型認知症、前頭側頭型認知症などの一次変性認知症とADを区別して考えることが多かった。しかし、1966年に開催されたLancet International Conference on Dementias(Eastwood et al.1996)以来、この単純な区別を、(エピ)遺伝的素因、生活習慣因子、心身症、神経病理学的変化の複雑な相乗作用として捉え直すことにより、認知症症候群の発生が解明された。現在では、認知症に対する病理の単数または複数の影響を考慮すると、年齢が包括的な共通因子であることは明らかである。現在の認知症の診断基準は、認知症および認知障害を伴う関連疾患の検出と理解における科学的知識と技術の進歩を取り入れたものとなっている。最新の「精神疾患の診断・統計マニュアル」(DSM-5)では、認知症の名称が大規模な神経認知障害(NCD)に変更されたが、認知症というラベルは、もちろん大規模なNCDと互換的に使用されている。また、DSM-5では、大規模NCDと軽度NCDを区別している。軽度NCDは、軽度認知障害(MCI)や前駆的認知症としてよく知られている(Vahia, 2013)。具体的には、皮質性認知症の原型であるADについて、米国国立老化研究所とアルツハイマー病協会は、神経画像診断と体液中の疾患関連化合物の測定に基づく生体内バイオマーカーも初めて考慮して、ADの前臨床期、MCI期、認知症期の別々の診断基準と関連勧告を記述した(Jack et al.)
ADの概念は、1907年にAlois Alzheimerが55歳のAuguste Deterの死後の脳分析の結果、神経病理学的なアミロイド斑と神経原線維絡み(NTF)の存在を報告したことに始まる(Maurer et al.) 過去数十年の間に、分子生物学と遺伝学の研究により、これら2つの重要な神経病理学的病変についての理解が深まった。その他の病変としては、脳アミロイド血管症(CAA)、ジストロフィー神経突起、神経糸、アストログリア症、ミクログリアの活性化などが挙げられる(Serrano-Pozo et al.) 認知症の最も顕著な形態として、ADは65歳以上の認知症患者の60~80%を占めると推定されている(Alzheimer’s Association, 2015)。ほとんどの患者は、中側頭葉の萎縮による社会的儀礼の保持以外に、前向性の記憶喪失症候群を呈する(McKhannら、2011年、Orgetaら、2015年)。そのため、世界保健機関はADを世界的な公衆衛生上の優先事項として認識している。遅発性ADの大部分は散発的に発症し、遺伝と環境要因が複雑に絡み合って引き起こされるが、そのうち70%は遺伝に起因すると考えられている。晩発性ADの最大の危険因子は依然として加齢であるが、遺伝的な危険因子も特定されている。最も一般的な遺伝的危険因子は、APOE遺伝子の3つの共通対立遺伝子(ε2、ε3、ε4)のうちの1つの多型であり、このうちε4対立遺伝子の変異は最もリスクを高める(Hardy, 1995; Verghese et al, 2011; AlzGene, 2020a)。このAPOE遺伝子型はコレステロール輸送動態と関連していることから、遅発性ADの血管危険因子であることが示唆されている(Kalariaら、2012)。APOE遺伝子の多型以外にも、コレステロール代謝(ABCA7)、炎症(TREM2)、アミロイドβ(Aβ)クリアランス(CLU)、免疫系(CR1)などに関連するいくつかの新規リスク遺伝子が示唆されている(AlzGene, 2020b)。あまり一般的ではない早期発症のADは、ほとんどがAPP、MAPT、PSEN1、PSEN2遺伝子の遺伝性変異に起因している(Haass and De Strooper, 1999; Bateman et al.) 最近では、前述の遅発性ADの遺伝的危険因子であるTREM2のmRNA発現レベルの年齢依存性の違いが、早発性ADの症例で発見されている。この知見は、TREM2遺伝子が早期発症ADの非遺伝性リスク因子である可能性を示している(Guven et al.) これまでの医療技術の進歩により、生存期間は長くなったものの、病気を改善する治療法はまだない。
アミロイドプラーク
早期および後期発症のADの主要な病理学的特徴の1つは、異常に折りたたまれたAβペプチドが細胞外に蓄積したアミロイド斑である。Aβペプチドは、21番染色体に局在するアミロイド前駆体タンパク質(APP)の膜貫通ドメインと細胞外ドメインが結合した36〜42アミノ酸のタンパク質分解断片である。APPは、γ-セクレターゼとβ-アミロイド切断酵素(BACE)の両方の部位で切断されてAβペプチドを放出するアミロイド生成経路と、α-セクレターゼがAβ配列内を切断して神経保護作用のあるsAPPα断片を放出する非アミロイド生成経路のいずれかで処理される(Nunan and Small, 2000)。単量体のAβペプチドは、オリゴマー、プロトフィブリル、そして成熟したアミロイドフィブリルとして凝集する傾向がある。アミロイド斑は主にAβ1-40とAβ1-42のペプチドから構成されており、後者のタイプが最もアミロイド斑に凝集しやすい(Hardy, 1997)。APPの3つのセクレターゼ切断部位周辺の変異群は、家族性ADの原因となることが知られており、変異によってAβ全体の産生量が増加したり、Aβ1-42/Aβ1-40の比率が増加したりする(AlzForum, 2020)。
プラークにはいくつかの種類があり、非晶質のアミロイドが堆積したびまん性プラークから、典型的にはジストロフィーを起こした神経突起、反応性のアストロサイト、活性化したミクログリア細胞に囲まれた繊維状のアミロイドが堆積した濃厚コアプラーク、さらには燃え尽きたプラークまでが報告されている(Serrano-Pozo et al.2011)。アミロイド斑は主に大脳新皮質に蓄積し、大脳新皮質の6つの層すべてが関与することが多く、大脳皮質にも続き、最後には皮質下の領域にも進行する(Arnoldら、1991;Thalら、2002)。アミロイドプラークの負荷と広がりを段階的に評価するために、様々なスコアリングシステムが導入されている。当初は、3つのステージ(A〜C)が区別されていた(Braak and Braak, 1991)。Thalら(2002)は、アミロイド病理の時空間的な進行を表すために5つのステージ(1〜5)を提案した。
脳内アミロイド血管症
AD患者の約80〜90%は、実質的なAβの蓄積とは別に、CAAと呼ばれる脳血管へのAβの沈着を示す(Yamada et al., 1987; Yamada, 2002)。CAAは、主にレプトメンヘラや皮質の血管に観察される(Yamada et al.、1987)。アミロイド斑とは対照的に、脳血管のAβセグメントは主に40アミノ酸の長さである(Prelliら、1988年、Suzukiら、1994年)。神経細胞が放出された後、Aβ1-42は脳実質内でアミロイド斑に凝集するが、Aβ1-40は間質液の排泄を介して脳血管系に運ばれ、クリアランスされる。この際、Aβ1-40は脳血管の基底膜に凝集して沈着する(Wellerら、1998)。影響を受けた血管は、平滑筋細胞の減少、壁の肥厚、フィブリノイドの壊死、微小動脈瘤の形成、血管周囲の血液分解物の沈着などの二次的な血管病変を示す(Loveら、2015年)。
神経原線維のもつれ
神経原線維のもつれ(NFT)は、AD脳のもう一つの神経病理学的特徴である。細胞内NFTの主な構成要素は、リン酸化された微小管関連タンパク質タウ(MAPT)の対になったらせん状のフィラメントである(Serrano-Pozoら、2011年)。MAPTは、軸索の輸送に不可欠な微小管の細胞骨格の形成と安定化を介して、神経細胞の重要な構成要素を形成している(Paglini et al.) 細胞骨格の崩壊による軸索輸送の障害や、リン酸化されたタウの線維への凝集は、AD発症の初期の事象であり、神経細胞の機能に大きな影響を与える(Nagy et al., 1995)。タウ病の時空間的な分布は、ADのアミロイド負荷と逆のパターンをとる。ADのタウ病理は、おそらくタウ凝集体の超細胞的な伝播に基づいて、シナプスで接続された脳領域で徐々に進行する(Furmanら、2017年)。NFTは、側頭葉内側に位置する内嗅皮質と海馬で最も早く発生し、さらに連合体の大脳皮質に広がり、一次感覚、運動、視覚領域は相対的に除外される。NFTは特徴的な分布パターンを示し、Braakの6つのステージ(I~VI)に分類することができる(Braak and Braak, 1991)。
現在、ADの神経病理学的病期分類は、主にMontineのABCスコアリングシステムに基づいており、Thalら(2002)が記述したAβ沈着物の病期分類(Aスコア)、Braak NFTの病期分類(Bスコア)(Braak and Braak, 1991)、そして最後に神経斑の病期分類(Cスコア)を組み合わせている(Montineら、2012)。神経斑は、Aβ凝集体の中心核を、ジストロフィーを起こした神経突起を持つ変性した神経細胞と、反応性のアストログリアやミクログリアからなるコロナが取り囲んでいるのが特徴である。
アルツハイマー型認知症の危険因子としての動脈硬化
近年、高血圧、メタボリックシンドローム、高コレステロール血症、動脈硬化、高脂血症、ある種の冠動脈疾患、そして最近では動脈硬化など、いくつかの血管危険因子がこの認知症症候群と関連していることから、ADの病態における血管疾患の潜在的な寄与に関心が集まっている。これらのADの血管危険因子の中で、高血圧が最も強いと考えられている(Kennellyら、2009年)。ほとんどの臨床研究では、中年期の高血圧が晩年のAD認知症症候群の素因となることが実証されている(Skoogら、1996年、Kivipeltoら、2001年)。これは、長期にわたる血圧の上昇や血圧の変動が、白質の変化、動脈硬化による脳の低灌流、血管調節機能の破綻など、脳の解剖学的・機能的変化を誘発するためである(Kalaria、2010年、Lattanziら、2014年、2015年)。さらに、クラス特異的かつ用量依存的な降圧療法は、MCIおよび認知症の発生率を低下させ(Takeda et al.、2009)、ADの病態を抑制する(Hoffman et al.、2009)ことから、血圧は興味深い治療ターゲットとなる。しかし、18,017人の高血圧患者を対象とした国際的な臨床研究では、抗高血圧薬を投与した患者のうち、収縮期血圧(SBP)をコントロールできたのはわずか32%にすぎないと結論づけられている(Thoenes et al.、2010年)。その後、REASON試験(Protogerouら、2009年)では、SBPとCVDの強力な独立予測因子である動脈硬化との間に正の相関関係が認められ、この劣悪な臨床結果を説明することができた。
動脈硬化の概念は、すでに17世紀に医師のThomas Sydenham(1624-1688)が「a man is as old as his arteries(人はその動脈と同じくらい古い)」という有名な言葉で例証している。最近の老化理論では、血管構造の変化が生物の運命を決定する重要な要因であることが示唆されているので、この言葉は今でも有効である(Learoyd and Taylor, 1966; Bailey, 2001; Lakatta and Lévy, 2003; Greenwald, 2007)。心臓が動くたびに、栄養豊富な酸素を含んだ血液が心臓の心室から動脈樹に送り出される。血液の脈波の伝播と反射の周期的な性質は、血管系が経験する機械的な力を規定するだけでなく、各心室に課せられる負荷をも規定する(Minor, 1982)。この血行力学的な伝導挙動は哺乳類で維持されており、脳底部の平均脈圧はマウス、イヌ、ヒト、さらには最適な末梢灌流を確保するために心臓が多少働かなければならないキリンでさえも驚くほど似通っている(Noordergraafら、1979年、Kassら、1988年、Beyarら、1989年)。全体として、この心血管現象は、心血管循環が脈動性であるという、無視されがちだが重大な特性を強調している(Kass et al., 1988)。
さらに、心血管システムは、心臓の断続的な末梢へのポンプ作用によって生成される高圧血流が、抵抗の高い、または低い脆弱な血管床で安定した灌流を維持するために捕獲されるようにしている(Vlachopoulosら、2011)。このダンピング効果は、Windkesselモデルでは、大動脈が変化する噴出血液量に即座に適応し、収縮期には過剰なストロークボリュームを蓄え、拡張期には排出する能力として正確に評価される(Chau et al., 1982; Safar et al., 2003; Ben-Shlomo et al., 2014)。生理的条件下では、動脈の剛性は心臓から末梢に向かって徐々に高くなり、これは動脈径が近位側に狭くなることが一因となっている。この硬さの勾配がインピーダンスの不整合を引き起こし、部分的な波の反射を誘発することで、脈動エネルギーの伝達を抑え、微小循環を保護する(London and Pannier, 2010)。波の反射の概念は、上行大動脈の速度波と脈波を区別することで最もよく理解できる。速度波が心臓からの単一の噴出であり、拡張期サイクル全体の間に切片でゼロになるのに対し、脈波は2つの局所的なピークで反射され、1つ目は流れのピークに対応し、2つ目は体の特定の部分での反射の合計に対応する(London and Pannier, 2010; O’Rourke et al., 2014)。
Windkesselモデルが動脈硬化の非伝搬モデルであるのに対し、より適切なアプローチとして、Adriaan Isebree Moens(Dow, 1940)とDiederik Korteweg(Tijsseling and Anderson, 2012)により、伝搬と速度の両方を記述するMoens-Korteweg方程式が導入された。硬い動脈では脈波が速く伝わることから、脈波伝播速度(PWV)の検証は動脈硬化のより信頼性の高い測定法として提案された(Safar and London, 1987)。最近では、スカンジナビアの生理学者であるBjorn Folkowが加齢によって動脈機能の調節がどのように歪むかを説明しており、動脈硬化が進行すると動脈のインピーダンスと動脈樹の脈動圧が増加する(Folkow and Svanborg, 1993)。動脈組織は加齢とともに弾性を失うため、拍動性の歪みの増加は脳血管系などの最も脆弱な微小循環にダメージを与え(O’Rourke and Safar, 2005)、重度の病変や最終器官の不全、例えば、認知機能の低下や脳出血などを引き起こす(O’Rourke and Hashimoto, 2007)。
生理的条件下では、大動脈の近位部が遠位部よりも遠位にあるため、動脈硬化は心臓から末梢に向かって徐々に増加する。動脈径の近位-遠位方向の先細りとともに、硬さの勾配は脈波の反射を誘発するインピーダンスの不整合を決定する。部分的な波の反射は、末梢への脈動エネルギーの伝達を減少させ、その結果、微小循環を保護する。近位大動脈の硬化が進み、生理的勾配が減少または元に戻ると、脈波の伝播は十分に減衰できず、より細い動脈に伝わり、微小循環に影響を与える(Noordergraaf et al., 1979)。さらに、反射の少ない波は中心大動脈に戻り、入射脈波に重畳して収縮期BPおよび脈圧を増加させる(Tariot et al., 1995)。
動脈硬化とアルツハイマー病の関係
本質的に、動脈硬化は心臓から脳への脈動に関係する。広範な微小血管系を持つ脳は、低抵抗であるだけでなく、心臓の脈動圧力と機械的な力に継続的にさらされる高流量の器官でもある(O’Rourke and Safar, 2005)。研究デザイン(縦断的 vs 横断的)、認知スクリーニング、対象集団、およびその異質性の点で研究間に食い違いがあるにもかかわらず、PWVの上昇がより早い認知機能低下、精神運動速度の変化、および意味流暢性と言語学習の困難に関連するというコンセンサスがある(Rabkin, 2012; Hughes et al., 2015; van Sloten et al., 2015; Iulita et al., 2018)。これに関連して、ARIC-NS研究では、主に白人の参加者において、高い動脈硬化および脈動性がMCIおよび認知症と関連すると結論づけている。また、最近のASCEND研究では、ADのリスクがある健康な中年コホートにおける末梢血管の健康に対する民族性の影響が報告されている(Meyer et al.、2017年)。この研究では、アフリカ系アメリカ人は非ヒスパニック系白人に比べて、末梢血管の健康状態と認知機能が悪いと結論づけている(Kumar et al.) さらに、身体的運動は、高齢者の動脈硬化と認知の両方の強力な予測因子として報告されている。一般的に、運動量が多いと、体力が低い人でも認知力が高いことが予測され、動脈硬化の測定値は逆に個人の体力と認知力の状態を示していた(Asamoahら、2013年、Nascimentoら、2019年、Pereiraら、2019年、Kennedyら、2020年、Masonら、2020年)。さらに、APOE4遺伝子に関連する遺伝的素因は、非認知症高齢者の認知障害を予測するために動脈硬化と相乗的に作用することが最近提案された(Rodrigueら、2013年、Cambroneroら、2018年、Rivera-Riveraら、2020年、Smirnovら、2020年、図1)。
図1 動脈硬化とアルツハイマー病を関連付ける疫学的知見のまとめ
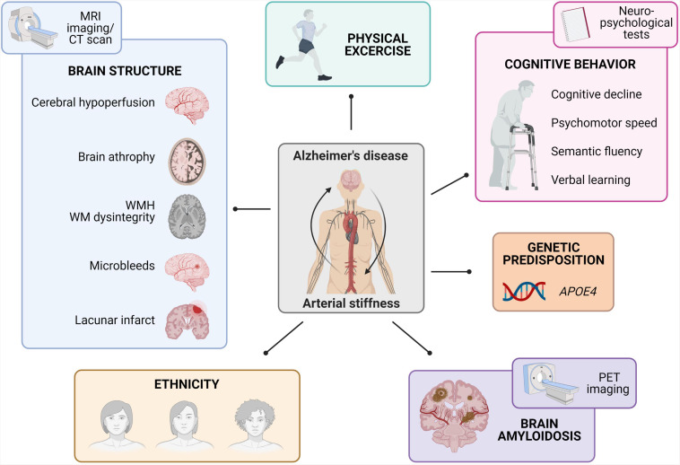
動脈硬化とアルツハイマー病との間に観察された関連性は、脳構造の変化(MRIを用いた画像診断技術で調査)、体力、認知行動(神経心理学的検査で調査)、APOE4遺伝的素因、脳アミロイドーシス(Aβ-PET画像で調査)、民族性の違いなどで構成されている(MRIは磁気共鳴画像、MWHは白質高濃度、WMは白質、PETはポジトロン・エミッション・トモグラフィー)。
動脈硬化による脳構造の変化の評価は、MRI画像および/またはCTスキャンを用いた横断的および縦断的な疫学研究によって広範囲に調査されている。最も特異的な繰り返し得られた知見の1つは、PWV測定値の増加と、より大きな白質高濃度および白質崩壊の高い発生率との間に相関関係があることであった(Henskensら、2008a,b;Coutinhoら、2011;Mitchellら、2011;Poelsら、2012;Tsaoら、2013;Paseら、2016;Araiら、2018;Suriら、2020)。また、他の脳スクリーニングプログラムでは、PWV値が脳の萎縮と関連することが報告されている(Tsaoら、2013;Paseら、2016)。脳血管レベルでは、PWV値の上昇は、脳梗塞(Matsumotoら、2007年;Henskensら、2008b;Tsaoら、2013年)、微小出血(Henskensら、2008a,b)、および脳低灌流の存在と相関していた(Mullerら、2007年;Watsonら、2011年;Jeffersonら、2018年;Suriら、2020年)。他の研究グループは、動脈硬化とADの間の収束を調べるために、大規模なヒトコホートにおいて、Aβ-PETイメージングをPWV測定と組み合わせて適用した。全体として、動脈硬化は、より大きな脳内Aβ負担およびCAAの存在と独立して関連していた(Hughesら、2013、2014、2018年、Moonら、2019年、Pashaら、2020年)。これらを総合すると、症状のある被験者と無症状の被験者の両方を対象とした画像ベースの横断的および縦断的研究では、PWV測定値の増加と、脳血管障害、神経変性、Aβ負担の増加およびCAAの観点からの脳障害との関連性が強調されている(図1)。
圧倒的な証拠が(a)症状のあるヒト被験者における動脈硬化とADの関連性を強調しているが、これらの研究は相関的証拠によって制限されている。一方、大動脈硬化の動物モデルは、それぞれに長所と短所があるものの、両疾患の原因となるメカニズムをよりよく調べることができる。その例としては、eNOSを部分的または完全に欠失させたマウスモデル(Austinら、2013;Tanら、2015;Liら、2018;Austin and Katusic、2020)、ヘテロ接合のフィブリリン1(Leloupら、2018)およびエラスチンを表示したマウスモデル(Hughesら、2018。2014;Saidら、2018)、頸動脈の石灰化(Muhireら、2019;Neunerら、2019)、および横方向の大動脈の収縮(DemerおよびTintut、2008;Steppanら、2012;CooperおよびMitchell、2016)がある。これらのげっ歯類に共通する症状は、(神経)炎症状態の増加、脳血管構造および/または血流の変化、および動脈硬化と血圧の特徴の変化に伴う認知症の表現型からなる(図2)。概念実証が確立されているが、ADの動脈硬化の関連する原因メカニズムに関する知識はまだ不足している。したがって、これらの利用可能なモデル間の共通点を特定し、その継続的な特性を明らかにすることが重要だ。さらに、げっ歯類は自然にはAβを蓄積しないという事実も障害となっている。したがって、大動脈硬化モデルとトランスジェニックADモデルを組み合わせることが必要である。今後の展望としては、性差の影響や遺伝的背景の影響の可能性にもっと注意を払うべきである(Qosa and Kaddoumi, 2016; Neuner et al.) 動脈硬化とADの間の収束の根底にあるメカニズムが固まれば、ADの病態の発症および/または進行を遅らせるために、より正確な介入策を開発することができる。
図2 動脈硬化とそれに伴う大脳の所見のマウスモデルの概要
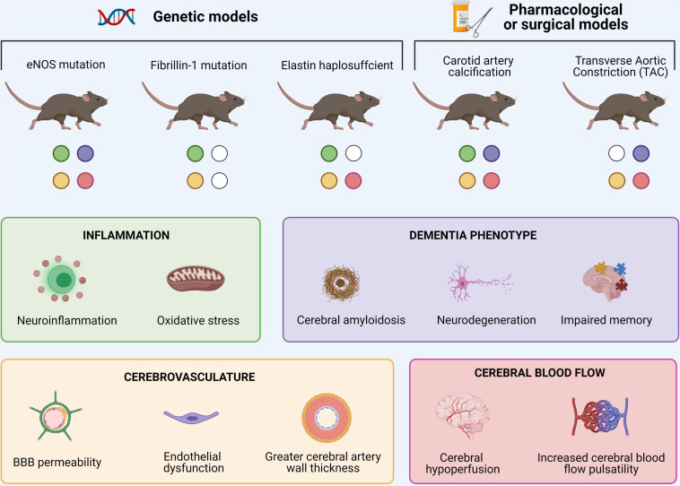
これらのネズミに共通する症状は、脳の神経炎症の増加、脳血管構造および/または脳血流の変化、および典型的な認知症関連の特徴である。
動脈硬化の原因
疫学研究では、年代的な加齢が動脈硬化の主な決定要因であることが明確に示されている(Ishidaら、2018年、Nambaら、2019年)。老化は、(エピ)遺伝的変化と個別のライフスタイル因子の微妙なブレンドによって引き起こされるユビキタスな複合現象である(LaRoccaら、2017年、Morrisら、2019年、Wahlら、2019年、Zhangら、2020b)。これらの刺激への長期的かつ累積的な曝露は、身体の主要なストレスシステムの1つである視床下部-下垂体-副腎(HPA)軸を調節不能にして過剰に活性化させ、コルチゾールなどのストレスホルモンの放出による全般的なストレス反応を引き起こす(Starr et al.、2019)。HPA軸の調節不全は、健康全般に悪影響を及ぼし、身体の免疫炎症系を活性化させる(Tappら、2019年、Ahmadら、2020年)。我々は、単純な「原因または結果」のつながりからなる個々の病態生理として考えるのではなく、ストレスに起因する加齢に伴う慢性的な低悪性度の炎症状態の摂動、すなわち炎症agingと呼ばれる状態(Franceschiら、2007年、Chadwickら、2012年)が動脈硬化の根底にあることを提案する。炎症が解消されていないと、寿命や老化のスピードが決まるため、ADを含む老化関連疾患(Giuntaら、2008年)だけでなく、動脈硬化(Fukamiら、2020年)、インスリン抵抗性(IR)(Itariu and Stulnig、2014年)、ニトロ酸化炎症(Miquel、2009年)、オートファジー機構の障害(Salminenら、2012年)とも高い関連性があるとされている。逆にグルココルチコイド濃度の上昇による悪影響は、AD(Khalsa, 2015; Justice, 2018)、動脈硬化(Vlachopoulosら, 2009)、急性炎症(Guerero, 2017)、オートファジー障害(Maら, 2019)、ニトロ酸化ストレス(Bernatovaら, 2018)、IR(Burkeら, 2017)などの炎症関連疾患で実証されている。動脈硬化とADとの間の圧倒的な関連性はさておき、最近の証拠は、動脈硬化それ自体がIRなどの前述の炎症性病態とも関連していることを示唆している(Cozma et al, 2018; Khoshdel and Eshtiaghi, 2019)、ニトロ酸化ストレス(Baileyら, 2013; Mozos and Luca, 2017)、炎症(Jainら, 2014; Peysterら, 2017)、オートファジーの低下(Chen and Sun, 2019; McCarthyら, 2019; 図3)などが挙げられる。以下のセクションでは、動脈硬化とADの間のメカニズムの収束として、ストレスによる炎症化について説明する。
図3 動脈硬化とアルツハイマー病の間のメカニズム的収束としての炎症化
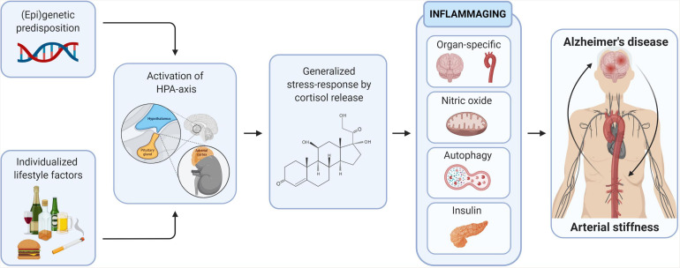
動脈硬化とアルツハイマー病のメカニズムを収束させるのは、(エピ)遺伝的素因と個別の生活習慣因子の長期的な因果関係と、その結果としての全般的なストレス反応であると考えられる。
組織特異的な炎症
人間の体は、常に多くの有害な生物学的、化学的、物理的刺激にさらされている。進化の過程で、人体は一般的な健康を維持するために、これらの刺激を認識し、それに対応するメカニズムを発達させてきた。炎症は、そのような脅威に対する身体の複雑な生物学的反応の一つである(Medzhitov, 2008)。炎症の概念は、一般的に急性炎症と慢性炎症に分けられるが、両概念は重複している。急性炎症は、可溶性の免疫媒介物質(ケモカイン、急性期タンパク質、サイトカインなど)によって促進される、損傷部位への免疫細胞の移動を具体化したものである。傷害の重症度によっては、急性の炎症では傷害を解決できない場合もある。その結果、刺激への長期的かつ持続的な曝露により、組織の損傷や線維化が生じる慢性炎症状態へと発展する(Germolecら、2018年)。
神経炎症 何十年もの間、脳は完全に免疫に恵まれた器官と考えられてたが、この概念は薄れ、洗練された免疫反応を伴う神経炎症の相対的モデルに取って代わられた(Louveauら、2015年)。臨床症状が多様であるにもかかわらず、神経変性疾患は共通の病原カスケードを共有している。多くの場合、疾患に特異的な時空間パターンで凝集するタンパク質のミスフォールディングが基盤となっており、疾患の発症と進行に寄与する重大な神経炎症と関連している。この発症経路は、ADにおいても極めて重要であると考えられる。まず、ADと自然免疫反応の間には強い関連性があり、脳内ではミクログリア細胞がその主役となっている。ミクログリアは主にアミロイド斑(神経斑)の近傍に存在し(Serrano-Pozoら、2011年)、Aβや細胞外のNFTと相互作用するため(Crasら、1991年、El Khouryら、1998年)、ADに伴う神経炎症は当初、これらのタンパク質沈着に対する受動的な反応と考えられていた。しかし、最近の知見では、自然免疫応答と適応免疫応答の両方が関与することで、ADの発症に炎症プロセスが積極的に寄与していることが認められ、神経炎症はADのもう一つの特徴となっている(Henekaら、2015年、Webersら、2020年)。
ミクログリアは、脳の自然免疫系の主要なセンサーである。静止状態のミクログリアは、隆起した形態と弱い抗原提示活性を示す。組織の損傷や病原体によって活性化されると、ミクログリアの形態はアメーバ状に変化し、神経組織内を移動することができるようになる。神経細胞が損傷した後の局所再生は、抗炎症性サイトカインや神経栄養因子の産生が増加し、細胞破片の貪食が促進されることで、神経細胞の修復と生存が促進される(Kreutzberg, 1996; Nimmerjahn et al., 2005)。健康な加齢脳では、ミクログリアはより炎症的な表現型に進化し、炎症現象の一因となっている。このようなミクログリアのプライミングや感作は、炎症反応性が誇張され、AD脳の神経変性に大きく寄与する悪循環の基礎となっている(Norden and Godbout, 2013; Franceschi and Campisi, 2014)。
アストロサイトは、血液脳関門(BBB)の完全性、軸索の伸長と髄鞘形成の制御など、いくつかの重要な生理機能に関与するほか、自然免疫系の重要な細胞制御因子でもある。また、ADの脳では、特にアミロイド斑の近傍に反応性アストログリアが存在することが明らかになっている(Medeiros and LaFerla, 2013)。アストロサイトは、貪食とAβ分解プロテアーゼの分泌というプロセスによって、Aβプラークを除去する上で重要な役割を果たしている。ミクログリアと同様に、アストロサイトは、Aβにさらされるとサイトカイン、インターロイキン、一酸化窒素(NO)、および他の潜在的な細胞毒性分子を放出し、それによって神経炎症プロセスを悪化させる(Jensenら、2013年、Skaperら、2018年)。ミクログリアとアストロサイトの間の相互作用は、健康な脳と神経変性プロセスの両方で必須の役割を果たしている(Bouvier and Murai, 2015)。両方のグリア細胞タイプがADの神経炎症に寄与していると思われるが、その関与のタイミングとメカニズムは大きく異なる可能性がある。ミクログリアの細胞特性は、神経炎症カスケードの初期段階でより強い役割を果たすことを支持している。一方、初期にリクルートされたミクログリアからの腫瘍壊死因子α(TNF-α)の発現と放出は、例えば活性酸素種(ROS)の共同放出によって補完されるシグナルの波を起こし、近接するアストロサイトで反応経路を開始する可能性がある(Kreutzberg, 1996; Bouvier and Murai, 2015)。
また、ADなどの神経変性疾患には、適応免疫系の関与も推定される。末梢のTリンパ球の活性化と脳脊髄液や脳への浸潤は、ADの病理の重要な一因であると考えられる。浸潤したCD3+ T細胞は、主にCD8+サブタイプであり、ヒトの死後の脳実質に記載されている(Itagaki et al., 1988; Rogers et al., 1988; Togo et al., 2002)。グリオシスとは対照的に、CD3+ T細胞の存在は、アミロイド神経病理よりもタウと強く相関することが多い(Merliniら、2018年)。さらに、脳脊髄液や末梢血中のT細胞サブセットは、疾患関連の変化を示す。AD患者の血液中では、制御性T細胞の減少、Th17の増加、CD8+T細胞の増加が測定され(Obersteinら、2018年、Ciccocioppoら、2019年、Burgalettoら、2020年、Gateら、2020年)、一方、AD患者の脳脊髄液中には、クローン的に拡大した抗原特異的CD8+T細胞が存在し(Gateら、2020年)、中枢神経系へのT細胞のトラフィッキングを示している。自然免疫反応と同様に、適応免疫系は、ADにおいて「敵か味方か」の関係を築くことができる。様々な動物モデルに基づく研究により、制御された制御性T細胞の枯渇は、有害なT細胞サブセットの抑制、さらにはアミロイドβ沈着に対するミクログリア反応の変調を介して、神経保護的であり、ADの進行を緩和する可能性があることが示されている(Baruchら、2015年、Dansokhoら、2016年、Baekら、2018年、Mayneら、2020年)。
血管の炎症 動脈硬化の重要なメカニズムの1つは、”inflammaging “と呼ばれる慢性的な炎症促進状態によって特徴づけられる動脈壁の老化である。血管の炎症は、エンドセリン、レニン/アンジオテンシンII、ミネラルコルチコイド受容体シグナル伝達経路などの主要な分子シグナル伝達カスケードの障害による炎症性ストレス因子の産生を伴う(Lakatta、2013; Wang et al.、2014)。その結果、炎症性転写因子の発現が促進される(Wangら、2014)。血管炎症は、血管平滑筋細胞(VSMC)の老化と増殖、血管の線維化、エラスチンの破壊、細胞外マトリックス(ECM)の形成、血管の石灰化など、動脈壁の構造的リモデリングを引き起こす(図4)。
図4 動脈硬化とアルツハイマー病を収束させる炎症関連状態における組織特異的な細胞の形態と機能の概要
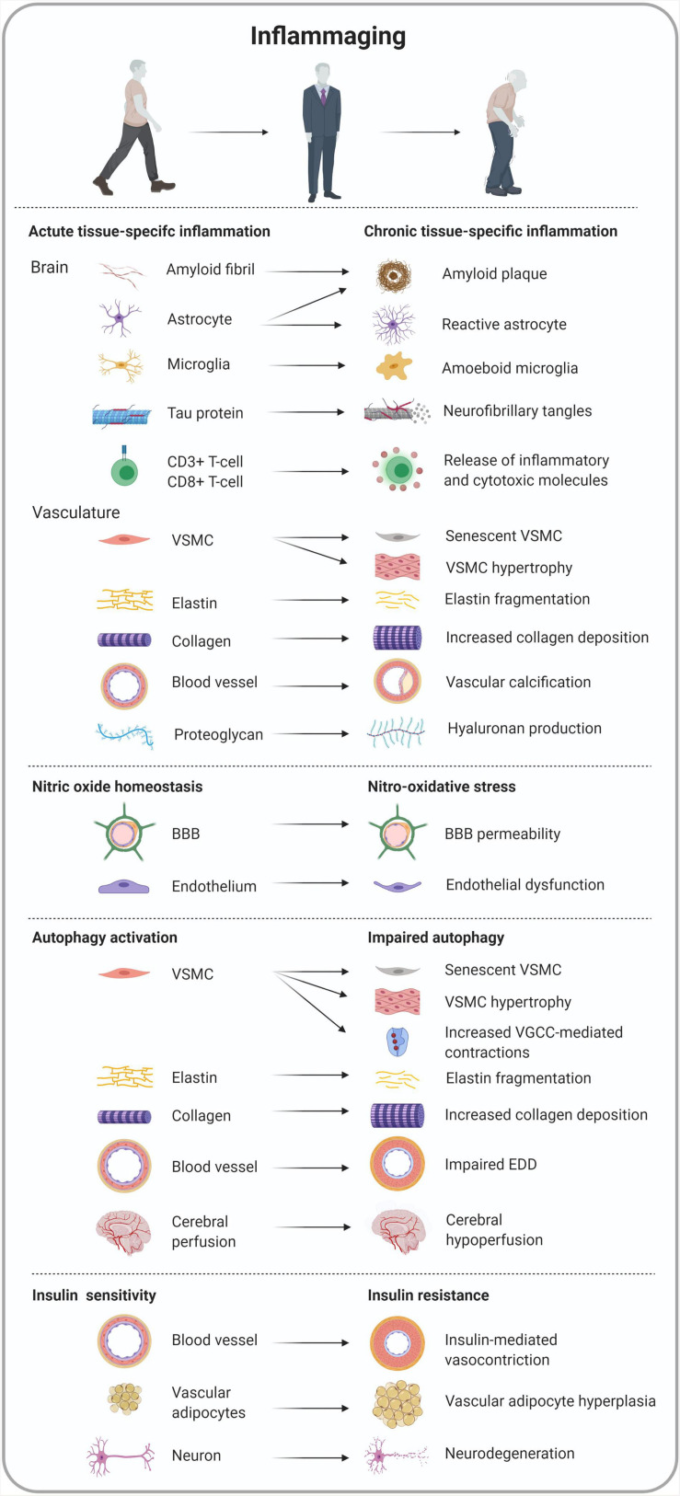
VSMCは、いくつかの表現型のスイッチを受けることができ、老化した動脈壁には、増殖性、老化性、硬化性のVSMCが共存している。老化したVSMCは、炎症性サイトカイン(IL-1、IL-6、IL-17)、単球化学吸引性タンパク質-1(MCP-1)、TNF-αを分泌することで、炎症促進状態に寄与している(Csiszarら、2012年、Accardiら、2016年)。この加齢に伴う動脈の分泌表現型は、隣接するVSMCを刺激して、表現型のスイッチを並立させているという仮説がある(Khanら、2010)。動脈壁に存在する老化したVSMCは、S期とG2/M期の細胞の割合が多く、G0/G1期の細胞が少ないことから、増殖能力が向上していることがわかる。in vivoおよびin vitroの研究では、乳脂肪球-EGF因子8タンパク質(MFG-E8)の発現が上昇し、下流のインテグリン/ERK1/2シグナルがVSMCの増殖と細胞周期を制御することが実証された(Wangら、2012年;Chiangら、2019年)。さらに、老齢のVSMCは、動脈中膜から内膜への誇張された移動/浸潤能力を示し、加齢に伴うびまん性内膜肥厚を引き起こす(Spinettiら、2004年、Fuら、2009年、Wangら、2012年)。MFG-E8は、VSMCの移動と浸潤に極めて重要な役割を果たしていることが、in vitroの結果から明らかになっている(Fuら、2009)。さらに、MFG-E8タンパク質が特異的に切断されると、アミロイドタンパク質であるメジンが形成される。このタンパク質は、50歳以上のヨーロッパ系アメリカ人の大多数の大動脈中膜に沈着している(Wangら、2013年、Migrinoら、2017年、Youngerら、2020年)。したがって、アミロイド原性のMFG-E8/medin複合体は、高齢化に伴う動脈硬化を伴う可能性がある。実際、高齢者ではPWVとMFG-E8の血清レベルの正の相関が確立されている(Cheng et al.)
動脈硬化の主な特徴は、動脈の内側層のラメラにおけるエラスチンの断片化である。時間の経過とともに、ECM内のコンプライアンスの低いコラーゲン繊維に大きなひずみが伝達される。長い間、受動的な現象と考えられてきたが、エラスチンの断片化は、瞬間的な炎症反応中のタンパク質分解エラスターゼ活性によって、急激に始まることがある。ある種のマトリックスメタロプロテアーゼ(MMP)は、炎症時に内皮細胞(EC)やVSMCによって構成的に発現されるが、これらは正常な状態ではTIMP-2(tissue inhibitor of metalloproteinases 2)によって相殺される(Park and Lakatta, 2012)。炎症性サイトカインの影響や細胞接着分子の活性化により、マクロファージや好中球はMMP(MMP-1、MMP-2、MMP-7、MMP-9を含む)を産生する(Galis and Khatri, 2002)。これらのMMPは、基底膜の分解とそのコラーゲン分解活性により、エラスチン-コラーゲンの架橋を悪化させ(Park and Lakatta, 2012)、それにより、巻かれていないコラーゲンが刺激され、動脈硬化が進行する(Zieman et al.) 臨床レベルでは、関連するMMPの発現がPWVレベルの上昇と相関していることが実証されている(Wykretowiczら、2005年、Yasminら、2005年、Vlachopoulosら、2007年)。ECM形成の顕著な特徴は強化されたコラーゲンの沈着である。ECMの複雑な網目構造(コラーゲンI、II、III)は、VSMCによって生産され、維持される。VSMCによるコラーゲン分子の産生は、MMP-2が活性化されたTGFβ1シグナルによって支配され、最終的には血管の線維化につながる(Wangら、2006; Jiangら、2012)。さらに、硬い動脈は圧縮に耐えるためにECMを構成している。ECMの炎症は、ヒアルロン酸のようなグリコサミノグリカンの合成を促進し、プロテオグリカンの構造を変化させる。ヒアルロン酸はECM内で水分を捕捉してゲルを形成し、動脈壁の圧縮能力を低下させる(Maki-Petaja et al., 2012; Lorentzen et al., 2016; Maki-Petaja et al., 2016)。
骨芽細胞と同様に、VSMCは、カルシウムおよび/または無機リン酸の細胞内濃度の上昇に曝されると、骨形成分化を行うことができる(Neutel et al.、2020)。カルパイン-1を過剰発現させると、オステオポンチンやオステオネクチンなどの石灰化阻害物質が減少する(Jiangら、2012年、Tangら、2015年)。また、組織トランスグルタミナーゼ(TG2)の増加は、石灰化阻害遺伝子(Opnなど)をダウンレギュレートし、石灰化促進遺伝子(Runx2など)をアップレギュレートすることから、その活性化が血管石灰化における重要な分子イベントであることが強調されている(Konoplyannikov and Nurminskaya, 2014; Chen W. R. et al, 2019)。
血管炎症と神経炎症の関連性 動脈硬化とADにおける局所的な炎症プロセスがよく研究されているのと同様に、近年、全身性の炎症が両病態に及ぼす影響が提唱されている。全身性の炎症が動脈硬化と関連しているか、先行していることを示唆する証拠が増えている(Mahmud and Feely, 2005; Durham et al.2018)。最近では、慢性炎症性疾患(関節リウマチ、全身性硬化症、全身性エリテマトーデス、炎症性腸疾患など)の患者は、対照被験者と比較して高いPWV値を示すことが報告されている(Dregan, 2018; Vargas-Hitos et al.) さらに、脳のレベルでは、循環性の炎症性サイトカインの上昇によって特徴づけられる全身性の炎症が、神経変性およびADを促進する脳の炎症性環境を形成する可能性がある(Paouri and Georgopoulos, 2019; Walker et al.) 実際、全身性の炎症は、認知機能の低下や行動の変化を誘発することが実証されている(Kahnら、2012年、Andersonら、2015年、Riaziら、2015年、Sankowskiら、2015年)。これらの知見から、動脈硬化とADの収束において、全身性の炎症が代償的、機構的、あるいは単に連想的な役割を果たしているのではないかという疑問が生じた。
Aβペプチド、特にAβ1-40は、末梢および脳血管系で炎症促進作用を発揮することを示唆する実験的および臨床的証拠が蓄積されており(Noguchi-Shinoharaら、2017年、Stamatelopoulosら、2015年、2018年、Visconteら、2018年)、これは興味深いバイオマーカーとなっている。スタチン、アンジオテンシン変換酵素阻害剤、アンジオテンシン受容体遮断剤、アンジオテンシン受容体・ネプリライシン阻害剤、β遮断剤、利尿剤、カルシウムチャネル遮断剤、血液透析などを用いたいくつかの介入により、APP/Aβのターンオーバー、Aβ1-40ペプチドの凝集、またはその炎症特性の遮断を操作できる可能性が示された(Stakosら、2020年)。さらに、加齢に伴うアミロイド生成タンパク質であるメディンは、内皮機能障害を誘発することから、近年、血管や脳血管の炎症との関連が指摘されている(Migrinoら、2017年、Degenhardtら、2020年、Migrinoら、2020年、Youngerら、2020年)。このように、メディンは、動脈硬化および/またはADの病態の初期段階における重要な炎症バイオマーカーとも考えられる。最近、炎症によってグルコースのジカルボニル誘導体であるメチルグリオキサールの産生が増加することが示唆されている。メチルグリオキサールの増加は、主に糖尿病患者において、動脈硬化と認知機能低下を悪化させる可能性がある(Dhananjayan et al.、2017)。
前述のバイオマーカー候補の根本的な作用機序を解明することで、全身および局所の血管と脳の炎症のクロストークを支配することができるかもしれない。長い目で見れば、この理解は、動脈硬化および/またはADをうまく標的とする新しい抗炎症治療アプローチの基盤となるかもしれない。
ニトロオキシダティブストレス
NOは最小のガス状のシグナル分子でありながら、無数の組織や分子経路でホメオスタシスを維持する役割を果たしており(Balaiya and Chalam, 2014)、諸刃の剣ではあるが、炎症の発症において重要な役割を体現している(Sharma et al, 2007)。正常な生理状態では、NOは抗炎症機能を発揮する(Sarkateら、2017;Sherikarら、2019;Zhangら、2020a)一方で、NOは動脈硬化などの病的状態では炎症促進メディエーターとも考えられている(Sindlerら。2011;Isabelleら、2012;Aroorら、2018;Chibaら、2019)やAD(Cifuentesら、2017;Tangら、2019;Austin and Katusic、2020;Dubeyら、2020;Stefanoら、2020)などの病態においては、主にニトロ酸化ストレスにおける寄与が原因と考えられている(Pérez-Torresら、2020;図4)。
血管内皮のNO産生は、適切な血管運動機能を維持するための最も重要な血管拡張メカニズムと考えられている。心臓から最小の毛細血管まで、血管系全体が内皮の単層で覆われており、循環血液と周囲の組織との間にバリアを形成している。血管内皮は、NOを放出することで、基礎的な血管径の変化と動的な血管径の変化を調節している(Bauer and Sotníková, 2010)。実際、動脈硬化の存在は、内皮機能障害のげっ歯類モデルにおいて、N(G)-Nitro-L-Arginine Methyl Ester(L-NAME)による内皮一酸化窒素合成酵素(eNOS)の阻害や、高血圧および頸動脈-大腿部PWVの増加を引き起こす遺伝的eNOSノックアウトによって以前から示されていた(Isabelleら、2012年、Leloupら、2014年)。一方、老齢のマウスに亜硝酸ナトリウムを用いた食事介入を行うと、大動脈の脱硬化が起こり、PWV値が正常化した(Sindlerら、2011)。
内皮NOの生成およびそのバイオアベイラビリティーは、上流の酸化ストレスの増大によって低下する。酸化ストレスは、スーパーオキシドが過剰に生成されることでNOが急速に酸化的に不活性化されるため、それ自体が内皮機能障害の原因となる。その結果、eNOSの活性が低下し、NOのバイオアベイラビリティーが低下して、ニトロソ-レドックスのバランスが崩れ、総合的なニトロ酸化ストレスが発生し、多くの血管病態を引き起こす(Taverneら、2012年、Senaら、2013年)。過剰な酸化ストレスとその結果としてのeNOSのアンカップリングは、マウスモデル(Xiaら、2010年、Herranzら、2012年、Varadharajら、2012年、Yinら、2017年)およびアテローム性動脈硬化症の患者で実証されている(Varadharajら、, 2012; Ismaeel et al., 2018)だけでなく、動物モデル(Adlam et al., 2007; Toral et al., 2018; Cheng et al., 2020; Dong et al., 2020)や高血圧症患者(Cengiz et al., 2015)でも使用されている。活性酸素種(ROS、例えばスーパーオキシド)は、特に、マルチサブユニットのNADPHオキシダーゼ(NOX)複合体によって、NADPHを電子供与体として分子状酸素から形成される。長年、NOXの発現は、浸潤している単球/マクロファージや食細胞でのみ起こると考えられていた。しかし、最近では血管壁成分でもNOXの発現が確認されている(Pandayら、2015)。このように、NOX2は、VSMC、逆流性線維芽細胞、EC、および血管周囲脂肪細胞にも発現していることから、最も顕著な血管NOXアイソフォームと考えられている(Brionesら、2011年、Koniorら、2014年、Chen J.ら、2017年、Lingら、2018年)。最近の研究では、Nox2+ミエロイド細胞の血管浸潤が血管炎症を引き起こし、心筋梗塞を引き起こす心不全マウスモデルにおいて内皮機能障害を引き起こすことが示された(Molitorら、2021年)。さらに、酸スフィンゴミエリナーゼのダウンレギュレーションを介してMRs/Nox2レドックスシグナル伝達経路を阻害することで、アンギオテンシンIIで処理したラット初代線維芽細胞の血管外膜リモデリングをポジティブに改善することが示された(Liら、2019年)。AD脳の脳血管系では、主に疾患関連ミクログリアのNOX2を介して活性酸素が生成される。これらのミクログリアにおけるNOX2の活性化の亢進は、神経炎症やアミロイドプラークの沈着の増加と関連している(Simpson and Oliver, 2020)。
このような背景から、動脈硬化とADとの関連性は、主にBBBの完全性の喪失に起因するとされている(Taheriら、2011年、Wardlawら、2013年)。脳には燃料貯蔵庫がないため、脳はBBBを介して脳血管系から供給される血液からエネルギーを得ている(Iadecola, 2017)。BBBは、全身の血液循環に対する脳特有の微小環境の最初の防御線として機能している(Sweeney et al.、2019)。脳血管のeNOS発現のダウンレギュレーションは、特定の脳領域がBBBの完全性を失う素因となることが提唱されている(Soaresら、2015年、Al-Zaitiら、2018年、Munjiら、2019年、Zhangら、2019年、Bernsteinら、2020年)。脳血管系における無傷の内皮とその結果としての保存されたNO産生は、したがって、脳卒中、脳血管疾患および神経変性を予防するために重要である(Tabatabaei and Girouard, 2014; Khan et al., 2019; Haselden et al.) 代謝の観点からの研究では、1型糖尿病によるeNOSの欠損がBBBの透過性を高めると結論づけている(Mayhan et al.) ラットてんかんモデルを用いた別の研究では、脳内eNOS発現の増加が、てんかん重積状態の誘発を通じてBBBの破壊をもたらすことが示された。この観察結果は、eNOS阻害剤の投与によってさらに強化された(Koら、2015年)。逆の解釈は、チアミン欠乏マウスモデルのBBBにおけるeNOS遺伝子の欠損を調査した研究から得られた。ここでは、eNOSをノックアウトするとBBBの透過性が回復したことから、eNOS由来のNOがこの病態の脳血管系において重要な因子であることが示唆された(Beauchesne et al.) より具体的には、ADに見られるような脳血管機能障害の主な原因は、酸化ストレスと活性酸素の生成であると推測される証拠が蓄積されている。Aβペプチドの存在下での活性酸素の生成は、脳血管におけるNOのバイオアベイラビリティを低下させることが知られている(Millerら、2010年、Austin and Katusic、2020年)。活性酸素の上昇は、主にAβ主導のNOX2活性化によって引き起こされ、最終的に神経炎症を誘発すると思われる(Hanら、2015;Wyssenbachら、2016;Hwang and Kim、2018)。この神経炎症反応には、脳血管ECにおけるタイトジャンクションの発現変化が関与し、BBBの完全性の喪失とCAAにつながる(Carranoら、2011、2012)。
BBBでの役割に加えて、APPのアミロイド生成処理における内皮NOの重要性が、ヒトおよびネズミの脳血管系で報告されている(Austinら、2013年、AustinとKatusic、2016年、2020年)。加齢したeNOSヘテロ接合マウスを用いた研究では、脳内のアミロイド原性沈着の増加を伴わない脳血管組織でのAβ蓄積の増加が示されたが(Austin and Katusic, 2020)、これはeNOSの部分的な欠損のみが原因であると考えられる。しかし、より重症のeNOSノックアウトマウスを用いた同様の研究では、血管組織と脳組織の両方でAPPの発現とアミロイド生成処理が増加し、ミクログリアの活性化と記憶能力の低下が認められた(Austin et al.) その抗血小板作用と血管弛緩作用を考えると、NOX2による酸化ストレスのためにNOのバイオアベイラビリティが低下すると(Malkovら、2020年)、血小板の過活性化と凝固が促進され、AD患者の脳微小血管系で観察される血栓塞栓症の表現型を悪化させる可能性が高い(Veitingerら、2014年、Canobbioら、2015年、Boseら、2019年)。さらに、血小板がAPPの処理、ひいてはAβペプチドの生成の全身的な源であることが知られている(Chen M.ら、1995;Foidlら、2020)。全体として、脳血管におけるNOバイオアベイラビリティの低下は、脳血管血栓症を引き起こすだけでなく、脳卒中、脳出血および全体的な神経炎症の危険因子であるCAAにも寄与する。
現在のところ、動脈硬化とADの関連性におけるニトロ酸化ストレスの正確な役割についてはまだ議論中であり、より決定的な解釈を得るためにはさらなる調査が必要である。
オートファジー(自食作用)の低下
マクロオートファジー(以下、オートファジーと呼ぶ)は、50年前に、細胞の生存のために分子を再利用する細胞の恒常性維持プロセスとして記述された(De Duve and Wattiaux, 1966)。しかし、ここ数十年の間に、オートファジーの役割は、ほぼすべての種類の細胞、特に免疫細胞で機能することで、炎症などの生理的プロセスとも関連していることがわかってきた(図4)。これらの観察結果は、オートファジーの重要性を強調しただけでなく、炎症性疾患においてこの細胞プロセスを標的にしようという熱意を呼び起こした(Matsuzawa-Ishimoto et al.、2018)。オートファジーは、不要な細胞成分や機能不全の細胞成分をリソソームで分解することで、細胞内のホメオスタシスを維持する。このプロセスは、タンパク質凝集体、脂質小滴、完全なオルガネラなどの細胞質の小部分を取り込む二重膜の液胞またはオートファゴソームの形成から始まる。オートファゴソームはリソソームと融合することで、最終的にはオートリソソームとなる。この最終段階で、オートファゴソームの細胞質内容物は、リソソームのヒドロラーゼによって分解される。ECやVSMCにおける基礎的なオートファジーは、適切な血管機能を媒介する必須のプロセスであることを示すin vitroおよび前臨床の証拠が増えている(De Meyer et al.) 実際、血管生理学におけるオートファジーは、脂質代謝(Khawarら、2019年)、血管反応性(Michielsら、2015年、De Munckら、2020b)、ホメオスタシス(Ousephら、2015年)、および血糖値とアミノ酸レベルの維持(Ezakiら、2011年)において重要な役割を果たしている。興味深いことに、オートファジーは、栄養不足、酸化的傷害、小胞体ストレスなどのストレス関連シグナルによって活性化される。このようにして、オートファジーは、不利な条件での細胞の生存をサポートしている。また、オートファジーを誘導することで、さまざまな生物種で寿命が延びることから、オートファジーは修復的で生命維持的なプロセスでもある。しかし、加齢に伴い、動脈樹を含む様々な組織において、オートファジー関連タンパク質の発現低下によるオートファジーの低下が見られ(LaRocca et al.、2012)、その結果、動脈硬化や動脈形成の促進といった動脈疾患が引き起こされる。実際、VSMCのオートファジー欠損(必須オートファジー遺伝子Atg7の欠失)を持つマウスから分離した大動脈セグメントを用いたex vivo実験では、コンプライアンスが減衰し、動脈硬化が進行することが明らかになった(De Munckら、2020c)。コンプライアンスとスティフネスの違いは、外因性のNOを加えてVSMCを完全に弛緩させるとより顕著になることから、VSMCのトーンの違いではなく、エラスチンの減少とコラーゲン含有量の増加で示される受動的な大動脈壁のリモデリングが、これらの効果の原因であると考えられる(De Munck et al.、2020c)。大動脈壁の受動的リモデリングは、内側の壁の厚さの増加およびエラスチンの断片化の上昇を示す組織学的データによって支持される。VSMCsにおけるオートファジーの欠損はまた、Ca2+ホメオスタシスの重大な変化をもたらし、その結果、より高い基底Ca2+貯蔵量、およびより大きな電位依存性カルシウムチャネル媒介収縮をもたらす(Michielsら、2015;De Munckら、2020b)。さらに、VSMCにおけるオートファジーの欠損は、細胞肥大を誘発し、ストレス誘発性の早期細胞老化を増加させる(Grootaert et al.、2015)。老化した細胞は複製能力を失い、炎症促進性の分泌表現型(Senescence Associated Secretory Phenotype:SASP)を持つため、この状態は動脈硬化をはじめとする多くの加齢性疾患と関連している。VSMCにおけるオートファジーの障害と同様に、ECにおけるオートファジーの障害も血管機能に大きな影響を与える。ECにおけるオートファジーの低下は、eNOSのダウンレギュレーションや動脈内皮依存性拡張(EDD)の低下と関連しており、オートファジーがNOバイオアベイラビリティを高めることで内皮機能を維持していることを示している(LaRocca et al.) 重要なことは、オートファジーの誘導が動脈硬化や内皮機能障害の治療において画期的な変化をもたらす可能性があることを示す証拠が増えていることである(De Munckら、2020a)。例えば、天然のオートファジー増強剤であるスペルミジンは、NOを媒介とするEDDを回復させ、動脈のPWVを正常化し、血圧を低下させる(LaRoccaら、2013;Eisenbergら、2017)。この結果は、オートファゴソームマーカーのLC3-IIやコアオートファジー機械タンパク質のAtg3など、動脈壁におけるオートファジーマーカーの発現増強と関連している。オートファジー誘導物質であるトレハロースにも同様の血管保護作用と心筋保護作用がある(LaRocca et al.、2012)。
動脈硬化に対するオートファジーの保護効果や、オートファジーとADとの相互作用以外にも、オートファジーは、脳内に蓄積されたミスフォールドタンパク質や機能障害を起こした小器官の毒性を除去する重要なメカニズムである。オートファジーの必須遺伝子であるAtg5の欠損は新生児の致死を引き起こすが、神経細胞でオートファジーが回復すればAtg5-nullの新生児は生存できること(Yoshii et al. この知見と一致するように、オートファジーの障害は、ADを含む神経変性疾患としばしば関連している(Fujikake et al.、2018)。興味深いことに、脳血管アミロイドの優勢成分であるAβ1-40は、オートファジーの誘導を通じてヒト脳血管ECの増殖を阻害し(Hayashi et al. この発見は、ADにおけるオートファジーの複雑な役割(病気の原因、保護、または単なる結果)を示しており(Liu and Li, 2019)、ADのさらなる発症を防ぐためには、病気の段階や標的細胞の種類に応じて、オートファジーを誘導または阻害する必要があることを示している。
インスリン抵抗性
インスリン抵抗性は、細胞がインスリンに反応しないために、グルコースの細胞への取り込みが起こらない病態である。IRは2型糖尿病(T2M)と密接に関連しているが(Sampath Kumarら、2019年)、IRは非糖尿病患者においても確立されている(Catenaら、2015年、Fuら、2017年)。さらに、IRは心血管疾患の罹患率および死亡率と関連している(Ormazabalら、2018年;Adeva-Andanyら、2019年;Markusら、2019年)。この関係を説明する基本的なメカニズムはまだ解明されていないが、動脈硬化が関与しているようである。実際、いくつかの観察研究および横断研究では、さまざまな年齢層の高血圧を有するまたは有さない(非)糖尿病患者において、IRと動脈硬化の間に相関関係があることが示されており、時には耐糖能異常を発症する前であっても同様である(Tounianら、2001年、Seoら、2005年、Agnolettiら、2013年、Fangら、2014年、Catenaら、2015年、Fuら、2017年、Wonら、2018年、Markusら、2019年)。さらに、肥満に起因するIRは、糖尿病性血管障害および動脈硬化の独立した危険因子であることが示されている(Jia and Sowers, 2014)。
IRによる血管機能の障害と動脈硬化の増加は、中膜、ECM、血管周囲組織の厚さの増加と内皮機能障害に関連している。動脈壁の内側の層では、インスリン代謝シグナルは通常、VSMCの血管拡張をもたらす。つまり、IRは血管弛緩の障害をもたらし(Olverら、2019年)、その結果、in vitro(Leeら、2012年)およびin vivo実験(Doronzoら、2004年、Leeら、2009年)の両方で強化された内皮機能をもたらす。より具体的には、後者の観察は、より大きな活性酸素の濃度とNOシグナル伝達経路の活性化の障害を伴っていた(Padillaら、2015)。さらに、アンジオテンシンおよびアルドステロンによるECおよびVSMCの刺激は、インスリン受容体基質1のリン酸化を介して、インスリン媒介性血管拡張の障害をもたらす(Coteら、2013)。また、IRに伴う食事誘発性肥満状態では、沈着量の増加やECMリモデリングが報告されている(Kangら、2014年、Williamsら、2015年)。動脈壁の外側では、ほとんどの動脈が血管周囲の脂肪組織を構造的構成要素として構成しており、分子のパラクリン機能の豊富な供給源となっている(Villacorta and Chang, 2015)。IRの文脈では、血管周囲脂肪組織における抗炎症性因子の発現低下と炎症性免疫細胞の浸潤により、脂肪細胞の過形成が起こる(Aroor et al.、2013)。このように、Framingham OffspringおよびThird Generationコホートでは、血管周囲の脂肪組織の体積、大動脈の寸法、および動脈硬化の間に相関関係があることが示されている(Thanassoulisら、2012;図4)。
インスリンの血管拡張作用はよく知られているが、インスリンは自律神経系を介して動脈を収縮させることもできる。インスリン濃度が適度に上昇した状態ですでに、インスリンの血管拡張作用と交感神経興奮作用のバランスが崩れることがある(Gordinら、2019)。T2Dに罹患している患者は、心血管自律神経障害が進行しているために、血管収縮作用が低下していると考えることもできる(Braffettら、2020年)。したがって、T2DとIRを持つ個人で観察された動脈硬化の測定値の増加は、インスリンの血管拡張作用がより顕著であるため、過小評価されている可能性があることを言及することが重要だ。
最もエネルギーを必要とする器官である脳は、体内のグルコース由来のエネルギーの最大20%を消費する(Mergenthalerら、2013年)。ADのような多くの神経疾患は、神経細胞のエネルギー代謝異常を特徴とする(図4)。最適なエネルギー管理を行うために、脳は主に全身の循環インスリンからインスリンを得ているが、脳の特定の領域ではデノボインスリン合成が行われており、脳を「インスリン感受性器官」にしているという証拠がある(Banksら、2012年)。末梢のインスリンが主に代謝調節因子として働くのに対し、中枢神経系におけるインスリンの機能は、分裂を促す成長因子であり、代謝調節ホルモンでもある祖先のインスリンタンパク質の作用に似ているようだ。Banksらは、インスリンの機能性の進化は、中枢神経系に比べて末梢では異なる経路をたどったのではないかと考えた(Banksら、2012年)。脳におけるIRの発生は、最近、III型糖尿病(T3DM)としてアノテーションされた(Leszekら、2017年、Candasamyら、2020年)。ADの全身IRの結果として、いくつかの神経変性の特徴が確立されており、代謝およびミトコンドリアの機能障害(酸化ストレスの増加)(Wilkinsら、2014年)、全身のインスリンシグナルとmiRNAの脱制御の障害(NFTの形成)(Liu et al, 2014)、神経炎症(ミクログリアと炎症性サイトカインの活性化)(Gasparら、2016;Chowdhuryら、2018)、レプチンシグナルの障害(シナプス可塑性の喪失)(Arnoldら、2018)、Aβプラーク形成の促進(Petrovら、2015;Sallamら、2015)。AD、末梢IRおよびT3DMの間の相互に関連するリンクは確立されているが、まだ十分に理解されていない(Willetteら、2015年;Pugazhenthiら、2017年;Denverら、2018年)。可変のホルモン抵抗性症候群が存在し、これらの症候群が異なる組織で互いに独立して発生する可能性があることに留意することが重要だ。
これまで、IR、動脈硬化、ADの関係についてはあまり研究されなかった。しかし、我々は最近、スウェーデン型二重変異を持つヒトAPPを過剰発現させた若いAD関連アミロイドーシスマウス(B6.Cg-Tg(Thy1-APP)3Somm/J、APP23と呼ぶ)において、疾患の初期の無症状期に末梢のIRを発見した。この代謝表現型は、最終的に高齢になると高インスリン血症の状態に発展した。この縦断的研究の間、動物は糖尿病の特徴を示さなかった(Hendrickx et al.、2021年)。以前には、同じ単一遺伝子導入ADマウスや、トリプル遺伝子導入ADマウスにおいて、体重の減少に加えてカロリー摂取量の増加が見られ、代謝亢進状態が原因であると考えられていた(Vloeberghsら、2008年、Knightら、2012年)。最近では、健常者の肥満に関連した代謝亢進と脳の萎縮との間にオーバーラップが見られた(Pegueroles et al.) 動脈硬化の文脈では、最近、比較的若い動物で心血管の表現型を整えた後、上記のAPP23マウスモデルでPWV値の上昇を測定した(Hendrickxら、2020a)。両方の研究(Hendrickxら、2020a;Hendrickxら、2021)において、動物は、対照同腹仔と比較して、全ての年齢で血清コルチコステロンレベルの上昇を示した。これまでに想定されてきたこと(Vloeberghsら、2008年;Knightら、2012年)に加えて、我々は、循環するストレスホルモンレベルが代謝亢進症の発症の原動力になっているという仮説を立てた。ADの病態へのストレスの関与は広く実証されており、現在では、この疾患の重要なリスク因子と考えられている(Bishtら、2018年、Carusoら、2018年、Dong and Csernansky、2019年)。代謝亢進は、IR(Willetteら、2015年;Gauglitzら、2008年)およびCVD(Cliftonら、1981年;Tullaら、1991年)と関連することが知られている。このようにして、動脈硬化とADの間のIRのメカニズム的収束は、ストレスによる代謝亢進の原因として説明することができる。
結論と今後の展望
老化はどこにでもある多面的な生物学的プロセスであり、CVD(例:動脈硬化)および神経変性疾患(例:AD)を含む多くの主要な疾患の高リスク因子となる。老化の過程では、コルチゾールレベルが上昇し、慢性的な低級炎症を引き起こす。このような加齢に伴う性質を考慮すると、動脈硬化とADの間のメカニズムの収束は、この炎症現象に基づいている可能性が高いと考えられる。生活習慣の改善を除けば、動脈硬化とADの疾患を改善する治療法はまだない。今後の展望としては、まず第一に、炎症の特徴を一貫してモニターすることが重要であり、動脈硬化、ひいてはADの進行を診断して止めるために、臨床の早い段階で動脈硬化を測定することが必要である。動脈硬化によるAD発症の背景には、複雑な一連の生物学的事象があると考えられるため、効果的な精密治療法を生み出すことは非常に困難である。精密医療はまだ初期段階にあるが、最近の科学的努力により、「オミックス」アプローチと高次元のデータ解析を組み合わせることで、病気の診断と進行の解明、バイオマーカーの発見がすでに促進されている(Hendrickxら、2020b)。最後に、異なる形態の炎症が動脈硬化とADを橋渡ししており、動脈硬化とADに見られる壊滅的な病理学的老化にタイムリーに介入するためには、それらの早期発見が重要であることを主張する。