

Contents
概要
栄養過多によって引き起こされる主要なオルガネラやセンサーの変化は、細胞の生体エネルギー経路を書き換え、代謝状態の生合成への移行を促進し、その結果、代謝平衡の不均衡の下流の症状である種々の代謝異常の発症を本質的に引き起こす。これらのメカニズムに基づき、代謝異常の治療標的の可能性と新たな研究の方向性が提案されている。
イントロダクション
慢性的な栄養過多は、食事パターンやライフスタイルの変化によって引き起こされる現代的な現象であり、その主な症状は肥満である。1過去10年間だけでも、肥満やその他の関連疾患、例えば2型糖尿病、2 心血管系疾患および動脈硬化、3 そして癌、4 の有病率は、米国および世界の多くの地域で劇的に増加している。4これらの疾患は、個々人の反応性に応じて、単独、連動、または同時に現れることがあるが、5,6代謝とエネルギーのホメオスタシスに同様の変化が見られる。さらに、栄養バランスの維持に失敗すると、細胞のホメオスタシスに変化が生じる。7
細胞が栄養素や代謝物の増加に適応するために、複数の防御反応と経路が開始される。これには、ミトコンドリアによる活性酸素種(ROS)の産生とシグナルの増加、小胞体(ER)によるアンフォールドドタンパク応答(UPR)、ラパマイシン機構標的(mTOR)の活性化などが含まれる8。8しかし、慢性的かつ持続的な栄養過多は、ミトコンドリア9と ER に深刻かつ不可逆的なダメージを与え、10アデノシン一リン酸(AMP)活性化タンパク質キナーゼ(AMPK)のダウンレギュレーション、11およびサーチュインの不活性化による代謝感受性タンパク質の修飾、12が生じる。このレビューの目的は、動物実験および培養細胞を用いた試験管内試験の研究から得られた過栄養による代謝ストレスに関する最近の動向を研究し、統合することである。この総説では、脂肪酸、炭水化物、アミノ酸の過剰摂取によって促進される栄養過多に対する代謝反応について、細胞レベルでのエネルギー恒常性維持に重要な役割を果たすオルガネラと栄養センサーに重点を置いて論じている。
栄養過多によるオルガネラストレス
ミトコンドリアと小胞体は、それぞれエネルギー生成と生合成の機能を持つことから、栄養センサーに分類される。ミトコンドリアでは、トリカルボン酸サイクル(TCA)および電子輸送鎖(ETC)により有酸素呼吸が行われ、アデノシン三リン酸(ATP)の主な生成源となる13。13ERは、様々な転写因子、制御タンパク質、脂質生成や酸化を制御する酵素がその膜に存在するため、タンパク質合成と脂質代謝の両方に関与している。14ミトコンドリアと小胞体は、細胞の発電所および構築工場として、栄養と基質の利用可能性の変動により、ストレスや長期的な損傷を受ける可能性がある。
ミトコンドリアストレス
過剰な栄養摂取は、TCAサイクルとETCを飽和させることによってミトコンドリアストレスを引き起こし、その結果、ミトコンドリアが電子運搬補酵素NADHとFADH2をNAD+とFAD+に再利用する能力に影響を与える。例えば、Kimら15 は、高脂肪食を与えたモデルマウスの肝臓で、NAD+/NADH比が有意に(P< 0.029)減少していることを報告している。これらの結果は、高脂肪食を与えたマウスの尿中のNAD経路の最終生成物の一定の増加によって証明されるように、栄養過多に反応してNAD+/NADHのターンオーバーが加速されることを決定したBoulangéら(16)の研究によって裏付けられた。栄養過多はミトコンドリアの NAD+ 再生能力を低下させるので、Penke らによって示されたように、ストレスを受けた細胞は NAD+ サルベージをアップレギュレートすることを余儀なくされる。17Penke らによる研究では、高脂肪食を与えたマウスの肝臓で、NAD+ サルベージの律速酵素であるニコチンアミドホスホリボシルトランスフェラーゼが翻訳レベル(P< 0.01) と翻訳後レベル(P< 0.001) の両方で有意に増強された。17逆に、Cantóら18 は、ミトコンドリアは、より高レベルのNAD+の生成によって、慢性的な栄養摂取による異常から保護されている可能性があることを実証した。具体的には、高脂肪食を与えたマウスの骨格筋にNAD+前駆体であるニコチンアミドリボースを補充したところ、酸化的リン酸化が促進された。18
健康なミトコンドリアは、低いプロトン起電力とそれに伴う内膜の大きな電気化学的勾配を介してATPを生成している。栄養過多はミトコンドリアの電位とプロトン勾配を変化させることでこのメカニズムに影響を与え、高脂肪食を与えたラットの肝臓で見られたように、ATP生成速度を変化させる。19プロトンと電子の漏出はまた、NADHとFADH2からETC複合体IとIIIへの電子供給を伴う適応反応として細胞内で増加する(ミトコンドリアの生体エネルギー効率と定義される)。9
エネルギー需要の増加なしにミトコンドリア内の過剰燃料は、より高い酸化物質生産を刺激する。20 ETC複合体I、II、および/またはIIIに由来する可能性が最も高い。主に単離したミトコンドリアを用いた初期の研究では、ETC複合体IおよびIIIが生理的条件下での酸化種の主要な生産者であることが確認されている。21Nishikawaら22は、ETC複合体IIが高血糖細胞モデルにおいてその阻害により酸化物質レベルの上昇を抑制したことから、ETC複合体IIも主要なROS生成者であることを示した。Ruiz-Ramírez ら23は、さらに、高ショ糖慢性食を与えたマウスの肝臓から単離したミトコンドリアが、過酸化水素(H2O2)レベルの上昇とプロトン漏出を示すことを明らかにした。オレイン酸またはリノール酸を飼料に添加すると、H2O2の生成速度が増大した。23同様に、Vialら24は、高脂肪に曝された新鮮な単離肝細胞において、キノンプールの縮小やH2O2レベルの大幅な上昇などのミトコンドリア変化が起こることを報告している。さらに、ユビセミキノンなどのスーパーオキシド生成電子輸送中間体の寿命は、栄養過剰の条件下で延長され、スーパーオキシドレベルが高くなることが示された。22興味深いことに、多様な種類の脂肪は、ミトコンドリア機能に対する影響に違いがある。飽和脂肪の多い食事は、ミトコンドリア生合成マーカー(ペルオキシソーム増殖因子活性化受容体[PPAR] γ coactivator 1-α [PGC-1α] および mtDNA)、ミトコンドリア脂肪酸酸化酵素(アセチルCoAカルボキシラーゼ)、を減少させる。およびTCAサイクルとETCの酵素(44の酵素とタンパク質のパネル)により、マウスの副睾丸白色脂肪組織のミトコンドリア機能に深刻な損傷を与えるが、これはn-3脂肪酸を多く含む不飽和脂肪食(魚油)25により防がれる。この結果は、Lionettiらの研究、26、飽和脂肪を多く含む食餌は、不飽和脂肪酸を多く含む魚油食に比べ、肝ミトコンドリアにおけるプロトン漏出と活性酸素の産生が増加し、ミトコンドリア効率の低下が示唆されていることと一致する。
低レベルのミトコンドリア酸化物質は、抗酸化物質によって制御され、免疫経路に取り込まれる可能性がある。27本来、活性酸素は酸化的リン酸化(OXPHOS)の副産物と考えられているが、Collinsら28がレビューするように、活性酸素は細胞毒性を持ち、脂肪酸酸化およびOXPHOSの速度を遅くするなど、複数のミトコンドリア機能に影響を及ぼす。29活性酸素種はまた、ミトコンドリアの断片化を引き起こし、30 および/または脂質酸化を介してオルガネラ区画の膜を損傷することにより、その完全性を損傷することによって、細胞の構成要素に作用する。タンパク質やDNAなどの他の高分子も活性酸素の影響を受け、分子構造の変化、酵素機能の障害、あるいは早期の細胞死を引き起こす。このような傷害に対して、細胞は、炎症性シグナルの上昇などの生存メカニズムを開始することにより、さらなる酸化的傷害を防ぐ。例えば、Bekkeringら31 は、ヒト単球を酸化低密度リポ蛋白に曝した場合、炎症性サイトカイン産生の増加と泡沫細胞形成の増加を示した。酸化ストレスが長期の栄養過多によって引き起こされる場合、より多くの酸化性基質が循環および細胞内で利用可能となり、それによって細胞代謝により有害な影響を与えることが予想される。
PGC-1αは、ミトコンドリア生合成および酸化的代謝に直接関与し、外部刺激に応答してミトコンドリア機能を制御する多くの転写因子を調整するため、細胞エネルギー恒常性の中心的な制御因子として機能している32。肝臓のような酸化力の高い組織では、PGC-1αはTCAサイクルフラックス、脂肪酸酸化、肝グルコネシスの強力な正のレギュレータである。33そのノックアウト34または栄養過多条件下でのダウンレギュレーション35TCAサイクルおよびミトコンドリア脂肪酸β酸化によるフラックスの低下、TCAサイクルおよびOXPHOS遺伝子の発現低下、ミトコンドリアの酸素消費量の低下、これは実質的なミトコンドリア機能障害を反映し一致する、酸化ストレスとインスリン抵抗性に間接的に関連しうる36,37(Figure 1).25,38-40酸化還元バランスとのこの間接的な関連に加えて、PGC-1αはまた、ミトコンドリア抗酸化防御システムを調節することによって、細胞の酸化状態に直接影響を与える41; ROS代謝の強い調節因子として、PGC-1αはグルタチオンパーオキシダーゼ1およびスーパーオキシドディスムターゼ2などの多くのROS-解毒酵素の誘導に必要となる。42
図1 ミトコンドリア機能不全の図解

本総説では、ミトコンドリア機能障害とは、酸化効率の低下につながるミトコンドリアの変化を指す。これらの変化には、ミトコンドリア生合成および含有量の減少、25 トリカルボン酸(TCA)サイクルおよび電子輸送鎖(ETC)の障害、25 およびTCAサイクルとETCのデカップリングが含まれるが、これらに限定されない。38ETCの障害は、電子漏洩の増加や活性酸素の産生を引き起こし、酸化ストレスや細胞障害、さらには炎症反応の活性化の可能性もある。デノボ脂質生成は、未処理の基質を収容するために上昇し、インスリンシグナル伝達経路の複数の部位を阻害する。39,40,略語 Akt、プロテインキナーゼB;ETC、電子輸送鎖;IRS、インスリン受容体基質;PGC-1α、ペルオキシソーム増殖剤活性化受容体γコアクチベーター1α;ROS、活性酸素種;TCA、トリカルボン酸。
PGC-1αは、ミトコンドリアの制御とは別に、自身の翻訳後修飾を介してエネルギー恒常性の維持に関与している。Adenosine monophosphate-activated protein kinaseとSilent Mating type information regulator 2 homolog 1(SIRT1)は、それぞれリン酸化43と脱アセチル化 44 を通じてPGC-1αの正の制御因子である。エネルギーが枯渇した状態では、AMPKがNAD+レベルを増加させることによってSIRT1を活性化し、PGC-1αの脱アセチル化と活性化につながる。43カロリーの高い食事やエネルギーが豊富な場合、AMPKは細胞内の高いATPレベルによって抑制され、SIRT1とは逆の役割を果たすヒストンアセチルトランスフェラーゼGCN5がPGC-1αをアセチル化し、不活性化させる。mTOR は、mTOR-yin yang 1 (YY1) -PGC-1α 経路を通じて PGC-1α の上流活性化因子でもあり、46,47骨格筋のグルコースホメオスタシスとインスリンシグナルの調節に重要な役割を担っている。48PGC-1α活性を制御するこの代謝ネットワークにおける摂動は、細胞のエネルギー経路の再プログラミング、ひいては全身の代謝性合併症につながる可能性が十分にある。
小胞体ストレス
ERは、UPRを通じて、栄養センシングと細胞内シグナル伝達を結びつけている。49研究により、細胞の代謝的恒常性が変化すると、ERのフォールディング能力が低下し、その結果、フォールディングされていないタンパク質が蓄積し、ERストレスが引き起こされることが明らかにされている。Koumenisら50 は、ヒトおよびげっ歯類の細胞に存在するERは、適度な低酸素状態にさらされると急速にストレスを受け、真核 開始因子2α(eIF2α)およびプロテインキナーゼ様ERキナーゼ(PERK)が高リン酸化されて、タンパク質合成が減衰することを示していることを報告している。しかし、細胞が再酸素化されると、タンパク質合成は活性化されることから、酸素化はERの機能に大きく影響することがわかる50。50ERは、ホメオスタシスの乱れによるストレスに適応するために、UPRを開始し、過剰な栄養素や未変化タンパク質を管理し、環境の変化に適応している。UPRは、タンパク質の翻訳を抑制し、タンパク質のフォールディングを補助し、ミスフォールドしたタンパク質を分解することで、小胞体ストレスを緩和する49。10それでも、適応に失敗すると、複数の炎症経路の開始 (図2)、14、49、51Hotamisligilによるレビュー、52、さらにはアポトーシスカスケードの活性化など、病的状態に至る可能性がある。14
図2 Unfolded protein response (UPR) – 初期化された炎症反応

哺乳類細胞におけるUPRは、3つの枝からなり、3つの小胞体膜貫通型センサータンパク質、二本鎖RNA依存性タンパク質キナーゼ様ERキナーゼ(PERK)、イノシトール要求性酵素1(IRE1)、活性化転写因子6(ATF6)によって開始される。これらのセンサーの活性化は、ER常在シャペロンであるグルコース制御タンパク質78(GRP78、別名BiP)の内腔側ドメインからの解離に依存している。51ERに不適切に折りたたまれたタンパク質が蓄積すると、BiPがこれらのUPRセンサーから引き離され、同時に活性化される。活性化されたUPRセンサーは、複雑な下流シグナル伝達経路に関与し、転写因子を通じてERの恒常性回復に関連する遺伝子発現を制御する。49Mandl et al14より許可を得て複製。略語 ATF、活性化転写因子;BiP、グルコース制御タンパク質78;C12、カスパーゼ12;CHOP、C/EBP-homologous protein;eIF2α、eukaryotic initiation factor 2α;ER、小胞体。ERAD、ER関連タンパク質分解;IRE1、イノシトール要求性酵素1;JNK、c-Jun N-末端キナーゼ;Pc12、プロカスパーゼ12;PERK、タンパク質キナーゼ様ERキナーゼ;XBP1、Xボックス結合タンパク質1。
小胞体には、デノボ脂質合成に重要な役割を果たすステロール制御因子結合タンパク質(SREBP)ファミリー14など、多くの酵素や制御タンパク質が存在するため、脂質のホメオスタシスに深く関与している。53さらに、小胞体ストレスが肝脂質蓄積に関与していることを示す証拠が増えてきた。54-56小胞体ストレスは、トリグリセリド、コレステロール、ERクライアントタンパク質であるアポリポプロテインB100からのリポタンパク質の合成に悪影響を与えることによって、肝臓からの超低密度リポタンパクの分泌を減少させることが知られている。57一方で、ERストレスは肝臓での脂肪酸の酸化を抑制する。DeZwaan-McCabe ら58は、ATF6α-/-マウスにおいて、脂肪酸酸化とその活性化因子であるPPARαがともに有意に(P< 0.01) 抑制されることを示した。PPARαを活性化して脂肪酸酸化を持続させると、肝脂質蓄積は改善されるが、ERストレスを完全に抑制することはできないことが示された。Chanらによる研究()59では、高フルクトース食を与えたマウスにおいて、PPARα活性化剤は脂肪酸酸化を増加させることで肝脂質蓄積を完全に改善したが、UPRを部分的に活性化してもERストレスを軽減することができないことが示された。
興味深いことに、動物モデルや細胞モデルが摂取する脂質の脂肪酸組成は、ERの機能やストレスに重要であり、異なる脂肪酸プロファイルは、非常に多様な結果をもたらした。Zhao ら()は、ラード油または大豆油で強化した高脂肪食を60匹ラットに与え、体重増加を同程度にすることを目的とした。2つの食餌の間に体重増加の統計的差異は認められなかったが、ラードオイルを強化した食餌は、肝Toll様受容体(TLR)4、TLR2および腫瘍壊死因子α mRNAの発現上昇などの炎症反応を有意(P< 0.05)に高くする結果をもたらした。さらに、ラード食に反応した ER のストレスは、UPR マーカーである PERK、phospho-eIF2a、および CHOP の発現の上昇によって証明された60。60この研究では、大豆油はラード油ほどの劇薬効果を示さなかったが、ウサギ初代肝細胞においてかなりのERおよびミトコンドリア障害を引き起こし、最終的に脂質の蓄積とERストレスをもたらした。61 それは、多くの脂質滴が存在する急激なERの拡大とミトコンドリアの膨張によって証明された。n-3系多価不飽和脂肪酸を豊富に含む魚油は、対照的に、ERにそのような損傷を与えなかった。61実際、Yangら62 は、ドコサヘキサエン酸が分化した3T3-L1脂肪細胞のパルミチン酸トリガーERストレスを抑制することを示した。AMPK阻害剤の投与はこの有益な効果をほとんどブロックするので、おそらくAMPKのリン酸化を増加させることを介するのだろう。これは、Begumら63が、ドコサヘキサエン酸がイノシトール三リン酸受容体(InsP3R)を介したER Ca2+枯渇をブロックすることによりERストレスを改善するとまとめた後に発展したドコサヘキサエン酸のER保護作用の新しい説明である。さらに、Okadaら64は、非アルコール性脂肪肝炎の被験者において、n-3多価不飽和脂肪酸治療後にER機能が改善するプロテオーム・マーカーを示した。
ERは、膵臓でのインスリン産生に関与していることから、グルコースのホメオスタシスに関与している。Elouil ら65は、生理的な摂食状態に近い急性のグルコース刺激により、培養ラット膵島におけるインスリン合成の増強と UPR の発生を報告している。UPR は、容量と需要のバランスの維持と回復を助ける適応メカニズムとして開始された。49過剰なグルコース66および脂肪酸67は、PERK/eIF2α/ATF4/CHOP 経路を活性化し、膵臓β細胞に長時間の ER ストレスを誘発し、それによってインスリン発現が低下しβ細胞のアポトーシスを増加させることも報告されている。68Bacharら69は、グルコースは、イノシトール要求タンパク質(IRE)1αレベルを増加させ、c-Jun N-末端キナーゼ経路を活性化することによって培養膵臓β細胞におけるパルミチン酸誘発ERストレスを増幅させ、β細胞のアポトーシスを増加させることを明らかにした。最終的に、インスリン分泌と循環血中濃度が低下する。70
栄養センサー
栄養センサーは、細胞が栄養素や細胞内代謝物の濃度上昇を感知し、それに適応することを可能にする8。このような栄養センサーには、細胞エネルギーセンサーであるAMPK、NAD+依存性タンパク質脱アセチル化酵素であるサーチュイン、および栄養/成長因子感受性キナーゼであるmTORが含まれる8。これらのセンサーは、栄養に関連した細胞内外の刺激に応答するだけでなく、クロストークを調節して細胞のホメオスタシスを達成する複雑で高度なシステムを形成している( Figure 3)。71培養骨格筋管および筋芽細胞を用いて、Cantó ら72および Fulco ら73は、AMPK が NAD+ のサルベージおよび生合成に影響を与えることによって SIRT1 を制御することを明らかにした。さらに、Lanら74は、培養HEK293T細胞を用いて、SIRT1がその上流活性化因子である肝臓キナーゼB1(LKB1、セリン/スレオニンキナーゼ11[STK11]としても知られる)のアセチル化状態を調節することによりAMPK活性を調節することを明らかにした。mTOR の活性は、上流阻害因子である結節性硬化症複合体(TSC)75 または mTOR 複合体 1(mTORC1)結合サブユニットのラプターのリン酸化を介して AMPK によって負の影響を受ける可能性がある。76一方、活性化されたサーチュインは、TSC77を介して直接的に、あるいは TSC の上流阻害因子であるプロテインキナーゼ B/Akt を阻害することによって間接的に mTOR を制御することができる。78これらの酵素は細胞のエネルギー恒常性の維持に関与しているため、その機能障害は細胞のエネルギー欠乏を引き起こし、それによって細胞の好気性解糖への移行を促進する(Warburg効果)可能性がある。
図3 アデノシン一リン酸活性化プロテインキナーゼ(AMPK)、SIRT1(Silent Mating Type Information Regulator 2 homolog 1)、mTOR(Mechanistic Target of rapamycin)の相互作用。

栄養、酸素、エネルギー、成長、ストレスなどの細胞内外のシグナルの感知と統合には、mTOR、AMPK、SIRTの複雑なクロストークが必要である。72,Abbreviations:Akt, プロテインキナーゼ B; AMP, アデノシン一リン酸; AMPK, AMP 活性化プロテインキナーゼ; ATP, アデノシン三リン酸; LKB1, セリン/スレオニンキナーゼ 11; mTOR, ラパマイシン機構標的分子;NAD、ニコチンアミドアデニンジヌクレオチド;Nampt、ニコチンアミドホスホリボシルトランスフェラーゼ;SIRT1、サイレント交配型情報調節因子2homolog1;TSC1、結核性硬化複合体1;TSC2、結核性硬化複合体2。
アデノシン一リン酸活性化プロテインキナーゼ
アデノシン一リン酸活性化プロテインキナーゼは、細胞のエネルギー状態を感知するセンサーであり、全身のエネルギー恒常性を調節している。アデノシン一リン酸活性化プロテインキナーゼは、ATP、アデノシン二リン酸(ADP)、AMPのレベルを感知し、それによってエネルギー不足の細胞にATPレベルを回復させるシグナルを送る。このプロテインキナーゼは、触媒αサブユニット、制御βおよびγサブユニットから成るヘテロ三量体複合体として存在する11。AMPKの活性化は、AMPがγサブユニットにアロステリックに結合するか、2つの上流活性化因子であるLKB1およびCa(2+)/カルモジュリン依存性プロテインキナーゼβによってThr172がリン酸化されることによって達成される。11
活性化すると、細胞の燃料計として機能するAMPKは、グルコーストランスポーター4型によるグルコース取り込みの増加、79、80 解糖、81-83 CD36による脂肪酸取り込み、84 アセチルCoAカルボキシラーゼ阻害による脂肪酸酸化、85 およびPGC-1α活性化によるミトコンドリア生合成などの細胞のATP供給を補充する異化経路を促進する。43逆に、AMPK は、タンパク質、86 脂肪酸、87 トリアシルグリセロール、88 およびコレステロールの合成を含む同化経路を阻害する役割を担っている。89
栄養過多条件下でのアデノシン一リン酸活性化プロテインキナーゼの阻害
摂食後の自然な反応は、AMPK活性の低下と異化作用から同化作用への切り替えである。実際、Assifiら(90)およびWilsonら(91)は、空腹時ラットモデルで肝および筋肉のAMPK活性が上昇し、再給餌後に有意に(P< 0.05)減少したことを報告している。AMPK活性は、栄養レベルが基礎レベルに戻ると再開されるが、複数の研究により、グルコース、アミノ酸、脂肪のいずれを主成分とする食事にかかわらず、動物(ラット、マウス)および細胞(HepG2細胞)モデルの両方で過剰な栄養暴露下でAMPKの低下および阻害に関するエビデンスが示されている。92-95
過剰なグルコース、アミノ酸、脂質は、さまざまなメカニズムでAMPKを阻害する。過剰な食事脂肪は、mTORC1を活性化することでAMPKの不活性化につながる。93 また、脂肪分解の調節障害および脂肪酸の酸化を阻害することでAMPK活性を阻害する可能性もある。Gaidhu ら96は、高脂肪食を与えたマウスモデルでは、脂肪のトリグリセリドリパーゼおよびホスホリパーゼ A2 の量が有意に増加し、クエン酸合成酵素活性および PGC-1α の発現、パルミチン酸酸化が抑制されたことを示した。さらに、Wuら97は、飽和脂肪酸のパルミテートが、デノボセラミド合成の増加およびセラミド依存性タンパク質ホスファターゼ2Aの活性化を通じて、組織98および細胞 99の両方においてAMPK Thr172リン酸化を低下させ、AMPK活性を低下させることを確認した。一方、不飽和脂肪酸は、そのような有害な効果を開始しなかった。97
過剰なグルコース供給は、主にインスリンを介してAMPK活性に影響を与える。グルコース過負荷に応答して、インスリン分泌が刺激され、その結果、Aktの活性化を通じてAMPK活性が抑制される。このことは、不死化マウス胚性線維芽細胞モデル100およびインキュベートラット骨格筋で示されている。101AktによるAMPK活性の阻害は、AMP/ATP比の減少、100 Ser485/491でのAMPKリン酸化の増加、101 またはmTORの活性化によって達成される。102
アミノ酸やタンパク質の過剰摂取は、mTORの活性化を通じてAMPKを阻害する。103研究では、過剰なロイシンと高タンパク食が、肝細胞とラット肝臓、104 C2C12筋芽細胞、105 膵臓β細胞、106 および視床下部におけるmTORリン酸化を促進し、AMPK活性を低下させることが実証されている。107Itani et al108and Kraegen et al109は、AMPK活性の低下はアセチル-CoAカルボキシラーゼリン酸化の低下を伴い、結果としてマロニル-CoAとジアシルグリセロール含量を増加させ、ラット筋肉と肝臓のインスリン抵抗性の原因となると報告している。さらに、Joseph ら110は、プロテインホスファターゼ 2A が Thr172 を脱リン酸化することで AMPK の活性制御に影響を与えることを提唱している。
Silent mating type information regulator 2 homolog 1/adenosine monophosphate-activated protein kinase cycle(サイレント交配型情報調節因子2ホモログ)
Suchankovaら111は、SIRT1がダウンレギュレートされるとAMPK活性が低下し、その逆もあるという結果を発表している。この同期した関係は、Rudermanら、112によってSIRT1/AMPKサイクルと呼ばれ、Coughlanらによって報告されたように、過剰なアミノ酸およびグルコース刺激で起こる。95Coughlanらは、ラット筋肉と培養したときにグルコースまたはロイシンがAMPKおよびSIRT1を同時に低下させることを実証している。驚くべきことに、Itaniら、108 Leeら、102およびSuchankovaら、111はすべて、AMPKの阻害がエネルギー状態(AMP/ATP比)の変化と直接相関していないことを報告している。むしろ、SIRT1活性の低下は、細胞の酸化還元(NAD+/NADH)の乱れが潜在的に関連する要因である可能性を示している。NAD+/NADHの酸化還元バランスがAMPKの活性化に影響を与えることを考慮し、Rafaeloff-Phailら113 は、ATPレベルの変化を伴わないメカニズムによってAMPKを活性化できることを提案した。具体的には、AMPK は NADH によって阻害されるが、NAD+ によって活性化される。この仮説は、Lan ら(74)によって部分的に支持された。彼らは、培養 HEK293T 細胞モデルを用いて、SIRT1 が脱アセチル化によって主要な AMPK 活性化因子である LKB1 の活性を高め、それが AMPK を活性化することを実証したのである。したがって、NAD+/NADHの比率が低下すると、SIRT1およびLKB1の活性が低下し、AMPK活性の低下を部分的に説明できる可能性がある。このようにAMPKと細胞の酸化還元状態(NAD+/NADHあるいは乳酸/ピルビン酸比)の間にSIRT1を介した相関関係が存在するとすれば、AMPKは好気性解糖よりも酸化的代謝を促進することによってワールブルグ効果を調節する存在である可能性がある。
Adenosine monophosphate-activated protein kinase as the anti-Warburg effect target.
Woods ら114と Wilson ら115は、Saccharomyces cerevisiae に存在する AMP-activated serine/threonine protein kinase の触媒サブユニットである AMPK ortholog SNF1 が、グルコース利用度が低いときに解糖(発酵)から OXPHOS へと呼吸を切り替えるために必要であると報告している。解糖系遺伝子は、二酸化シフトの間、SNF1によってダウンレギュレートされていた。116このオルソログはまた、酸化的代謝への移行を支援した。117酵母のオルソログと同様に、哺乳類のAMPKは、増殖細胞や炎症性免疫細胞で通常急速に起こる好気性解糖を犠牲にして、エネルギー効率の高いOXPHOSを促進することができる(Warburg効果)。118,119AMPKは抗ワルブルグ標的として考えられているため、AMPKに作用する薬剤、例えばメトホルミン、120サリチル酸、121およびメトトレキサート、122が、それぞれ2型糖尿病、炎症およびがんの治療のために開発されてきた。
サーチュイン
タンパク質のアセチル化レベルは、タンパク質アセチルトランスフェラーゼと脱アセチル化酵素という、相反する働きをする2つの酵素群によって制御されている。12318種類の脱アセチル化酵素が同定されており、4つのクラスに分類されている。クラスIIIのタンパク質脱アセチル化酵素は一般にサーチュインと呼ばれ、12、哺乳類のオルソログの代表でもあるセレビシエのSIRT (silent mating type information regulator 2) にちなんで命名された。124現在までに、SIRT1-7という7つの哺乳類サーチュインが同定されており、それぞれが同一のNAD+結合コアと異なるN-およびC-末端伸長を有している。 126 すべてのサーチュインは、タンパク質からアシル基を除去し、主にアセチル基、サクシニル基、マロニル基、パルミトイル基を除去する。127脱アセチル化反応では、NAD+が消費され、切断されてニコチンアミドとADP-リボースが生成し、これがアシル受容体となってアシル-ADP-リボース産物が形成される。
表1 哺乳類サーチュイン126
| サーチュイン | 活動内容 | 場所 | 基材 | 機能 |
|---|---|---|---|---|
| シルト1 | 脱アセチル化酵素 | 核、サイトゾル | p53、Foxo1、Foxo3、Bax、HIF-1α、HIF-2a、HSF1、Ku70、b-catenin、E2F1、Myc、STAT3、PGC-1α、NF-κB、TORC2、LXR、FXR、SREBP、PER2、CLOCK | エネルギー代謝、ストレス応答 |
| シルトツー | 脱アセチル化酵素 | サイトゾル | チューブリン、H4、Foxo3a | 細胞周期 |
| SIRT3 | 脱アセチル化酵素 | ミトコンドリア | OXPHOS複合体I、AceCS2、LCAD、HMG-CoAシンターゼ2、IDH2、MnSOD、SOD2 | ATP産生、抗酸化ストレス、サーモジェネシス |
| シルト4 | ADP-リボシルトランスフェラーゼ | ミトコンドリア | ジーディーエイチ | インスリン分泌、脂肪酸酸化 |
| サイアットファイブ | 脱アセチル化酵素 | ミトコンドリア | CPS1 | 尿素サイクル |
| シルト6 | 脱アセチル化酵素/ADP-リボシルトランスフェラーゼ | 核 | H3K9、H3K56、CtIP、SIRT6 | DNA修復、代謝、炎症 |
| シルト7 | デアセチラーゼa | 核 | P53a | rDNA転写 |
略号は以下の通り。AceCS2、アセチルCoA合成酵素2;AMP、アデノシン一リン酸;ATP、アデノシン三リン酸;Bax、Bcl2関連Xタンパク質;CPS1、カルバモイルリン酸合成酵素1;CtIP、C末結合タンパク質相互作用タンパク質;Foxo、フォークヘッドボックスO;FXR、ファルネソイドX受容体;GDH、グルタミン酸デヒドロゲナーゼ。H3K9、ヒストン3アセチル-リジン9;H3K56、ヒストン3アセチル-リジン56;H4、ヒストン4;HIF、低酸素誘導因子;HMG-CoA合成酵素2,3-ヒドロキシ-3-メチルグルタリルCoA合成酵素2;HSF1、熱ショック因子1;IDH2、アイソクエン酸デ水素酵素2;LCAD、長鎖アシルCoAデ水素分解酵素。LXR、肝臓X受容体;MnSOD、Mn-スーパーオキシドジスムターゼ;NF-κB、核因子カッパB;OXPHOS、酸化的リン酸化;p53、腫瘍抑制タンパク質53;PER2、周期概日タンパク質ホモログ2;PGC-1α、パーオキシソーム増殖剤活性化受容体ガンマコアクティブ化因子1α。SIRT1-7, silent mating type information regulator 2 homolog 1-7; SOD2, superoxide dismutase 2; SREBP, sterol regulatory element binding protein; STAT3, signal transducer and activator of transcription 3; TORC2/CRTC2, 転写因子CREBに対する転写共活性化因子.。
a SIRT7の活性や基質については、まだ明らかになっていない。
栄養過多時のサーチュイン活性の制御
前述したように、過剰な栄養素は2種類のストレス、NAD+/NADH比の低下15,16、酸化物質の過剰発生20をもたらし、いずれもサーチュインの活性に大きな影響を与える。様々な栄養素からのエネルギー(電子の形で)が解糖およびTCA経路を介してNAD+に移動し、それがNADHに酸化されるので、細胞質NADH生成の流れは非常に高くなり、持続的な栄養過多の場合には永久的に増加しうる。128NADHがその活性を阻害する一方で、サーチュインは脱アセチル化プロセスを制御するためにNAD+を消費するため、NAD+/NADH比の減少によりサーチュイン活性に悪影響が及ぶ。129
一方、ミトコンドリアのOXPHOS複合体を通過するNADHのオーバーフローにより、周囲の環境は酸化状態になり、その結果、電子の漏洩が増加し、ROSが産生される。この酸化ストレスは、サーチュインの遺伝子発現、翻訳後修飾、サーチュイン-タンパク質相互作用に影響を与えるが、これについてはSantosらにより詳しくレビューされている。126 さらに、サーチュインの発現は栄養過多により直接変化することがある。Noriegaら() はマウスモデルを用いて、SIRT1の転写は低栄養状態のときにcyclic AMP response-element-binding protein (CREB) の活性化により正に制御され、carbohydrate response-element-binding protein (ChREBP) の活性化により負に制御されることを明らかにした。CREBとChREBPという2つの転写因子の結合部位は、SIRT1プロモーター部位上で重なり合い、同じ配列を共有していた。栄養欠乏時には、CREBを介してプロテインキナーゼA活性が上昇し、SIRT1の転写が促進された。栄養過多時には、ChREBPが核内に取り込まれ、SIRT1の転写が抑制された。130,131CREBとChREBPとは別に、RevolloとLiによってレビューされたように、PPARλと高メチル化癌1タンパク質は、栄養過多下でSIRT1の発現をダウンレギュレートする結果となりうる。132膵臓の島、133 β細胞、134 および脂肪組織において、栄養過負荷下でのサーチュイン発現の低下が記録されている。135これらのメカニズムの障害は、インスリン抵抗性および2型糖尿病、136 動脈硬化、136、137および非アルコール性脂肪肝疾患などの複数の代謝症候群と関連付けられている。138,139
NADHと活性酸素に加えて、アセチル-CoAも栄養過剰の条件下で広範囲に増加しうる代謝産物である。Wellen ら140は、グルコースで培養した HCT116 細胞では、アセチル-CoA とヒストンのアセチル化が共に上昇することを示した。サーチュイン不活性化を伴うと、アセチル-CoAレベルはさらに上昇し、Kendrickら(141)が高脂肪食を与えたマウスから分離した肝臓を用いて示したように、その大部分が糖新生、ミトコンドリア酸化代謝、メチオニン代謝、ERストレス応答に関わる酵素である多種類のタンパク質に結合しうる。
サーチュインとワールブルグ効果
栄養レベルが高いとき、インスリンなどの成長因子はグルコースの取り込みを促進し、NAD+をNADHに変換する解糖を行う。サーチュインはNAD+を基質として消費するため、解糖速度が高いとサーチュイン活性が低下すると予想される。サーチュイン活性が低下すると、解糖系酵素のアセチル化度が高くなり、より活発に解糖が行われるようになると考えられる。グリセルアルデヒド-3-リン酸デヒドロゲナーゼ(GAPDH)を例にとると、Liら142は、アセチル化型が解糖を促進することを示し、Guan and Xiong12とGuarente143は、脱アセチル化型GAPDHがより効果的にグルコゲン生成を促進することを報告している。
十分な栄養があれば、タンパク質のアセチル化を維持するために十分なアシル基(アセチルCoA)が供給される。解糖により生成されたピルビン酸は、乳酸に変換されるか、脂肪酸として貯蔵されるか、2つの運命をたどる。前者は、通常、嫌気性微生物や癌細胞で起こるプロセスである。後者の場合、ピルビン酸は正常な哺乳類細胞のミトコンドリア内でアセチルCoAに変換され、アセチルCoAはクエン酸の形でミトコンドリア外に運ばれて脂肪酸の合成に利用される144。Guarenteが論じているように、143、細胞質でのタンパク質アセチル化のためのアセチル基は、エネルギー貯蔵の一部を意味する。言い換えれば、エネルギー過剰下で細胞質での高いアセチル化は、細胞がエネルギーを貯蔵するための手段である。
一方、エネルギー不足は解糖を制限し145、サーチュインを活性化し146、サイトゾル内での脱アセチル化を促進する。147酸化的リン酸化が促進されるのは、グルコース投入量に対して高レベルのATPを効率的に生産し、グルコースを完全に二酸化炭素に異化することができるためである。また、脂肪酸の酸化は、エネルギー不足の下でTCAサイクルにアセチル-CoAを供給するために促進される。148さらに、サーチュイン脱アセチル化によって生成された酢酸は、TCAサイクルとOXPHOSを駆動するためのアセチル-CoA合成の基質として使用されることができる。明らかに、低エネルギーは、サーチュイン活性が高く、アセチルCoAが利用しにくいため、低アセチル化を維持するのに役立っている。
タンパク質のアセチル化/脱アセチル化とサーチュイン活性は、細胞の代謝およびエネルギー状態、生合成と異化の相対的バランスを表す尺度として機能する。エネルギーが過剰になると、解糖が促進され、サーチュイン活性が抑制され、生合成と貯蔵が促進される。がん細胞では、生合成の代わりに細胞増殖によって解糖が行われ、ワールブルグ機構によってエネルギーが獲得される。がん細胞ではサーチュイン活性が著しく低下しており、特にミトコンドリアのSIRT3やSIRT4、核のSIRT6 (図4) が、148-153 グルタミンの利用を増加させることが分かっている。148
図4 ワールブルグ効果におけるサーチュイン消光
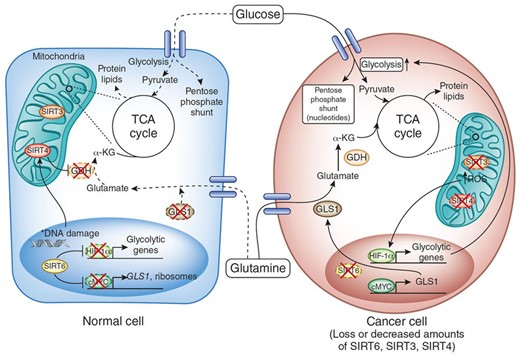
ワールブルグ効果では、トリカルボン酸(TCA)サイクルのダウンレギュレーションによって生じた細胞内のα-ケトグルタル酸の不足を補うために、グルタミンの利用がかなり増加することが分かっている。Silent mating type information regulator 2 homolog 3 (SIRT3)149,150および Silent mating type information regulator 2 homolog 6 (SIRT6)151は、解糖系遺伝子をアップレギュレートする低酸素誘導因子-αのコアプレッシャーとして機能する。また、SIRT6はcMYCのコアプレッシャーとしても働き、グルタミナーゼ1の発現を促進し、ひいてはグルタミンのグルタミン酸への変換を促進する。152Silent mating type information regulator 2 homolog 4 (SIRT4) は、グルタミン酸をα-ケトグルタレートに変換するグルタミン酸脱水素酵素を阻害する153。Guarente148より許可を得て複製。略語 α-KG、α-ケトグルタル酸;GDH、グルタミン酸デヒドロゲナーゼ;GLS1、グルタミナーゼ1;HIF-1α、低酸素誘導因子1α;TCA、トリカルボン酸;SIRT3/4/6、サイレント交配型情報調節因子2ホモログ3/4/6。
ラパマイシンのメカニスティックターゲット
ラパマイシンの機構的標的は、進化的に保存されたセリン・スレオニンキナーゼであり、成長因子、栄養素、エネルギー状態、酸素レベルなどの細胞外および細胞内の刺激を統合して、細胞の成長と増殖を促進する154,155(Figure 5).154,156-164このキナーゼは、mTORC1 と mTOR complex 2 (mTORC2) という 2 つの異なる機能・構造複合体として存在する。この 2 つの複合体のうち、mTORC1 は、栄養の利用可能性(特にアミノ酸)、インスリンと成長因子のレベル、アロステリック mTOR 阻害剤ラパマイシンに対する感受性という観点から、より広範囲に研究されている。155mTORC1 と異なり、mTORC2 は成長因子によって活性化されるが、栄養素によって活性化されることはない。165 そして、ラパマイシンによって急性に阻害されることはない。166栄養豊富な条件下では、mTORC1 は細胞代謝を制御し、特にタンパク質、脂質、ヌクレオチドの合成を促進し、オートファジーを抑制して成長と増殖を実現する。154
図5 Mechanistic Target of rapamycin (mTOR)シグナル伝達経路

上流シグナルは、主にRas homolog enriched in brain (Rheb)の制御を通じてmTOR複合体1(mTORC1)を制御する。Rhebは、グアノシン三リン酸(GTP)に結合すると、mTORC1と直接相互作用して活性化する小さなGTPaseである。156成長因子は、結節性硬化症複合体(TSC)/Rheb軸を介してmTORC1を活性化する157; グルコース、酸素、エネルギー状態は、adenosine monophosphate-activated protein kinase (AMPK) 依存経路を介してmTORC1に影響を与える154; アミノ酸は、リソソーム表面の常駐Rag A/B のGTP充電を促すことによってmTORC1を活性化する。158活性化されると、mTORC1 は、リボソーム生合成159および mRNA 翻訳160を、特に p70 S6 キナーゼおよび eIF 4E binding protein 1 (4E-BP1)161 をリン酸化することによりタンパク質合成を促進し、 SREBP-1 転写因子162 の活性化により脂質合成を行い、S6 キナーゼのリン酸化により核酸合成を行う。163活性化されたmTORC1はまた、オートファジー/ベクリン-1レギュレーター1(AMBRA1)をリン酸化することにより、オートファジーを活発に抑制する。164,Abbreviations:Akt、プロテインキナーゼB;AMP、アデノシン一リン酸;AMPK、アデノシン一リン酸活性化プロテインキナーゼ;ATP、アデノシン三リン酸;ERK、細胞外シグナル制御キナーゼ、マイトジェン活性化プロテインキナーゼとしても知られている;GDP、グアノシン二リン酸;GTP、グアノシン三リン酸。LKB1、セリン/スレオニンキナーゼ11;MEK、マイトジェン活性化ERKキナーゼ;mTORC 1/2、ラパマイシン機構的標的 1/2;PI3K、ホスホイノシチド3-キナーゼ。REDD1、発生とDNA損傷応答で制御される1; Rheb、脳に濃縮されたRasホモログ; TSC 1/2、結節性硬化症複合体1/2.
ラパマイシン活性化ターゲットとミトコンドリア酸化機能との相互作用
ミトコンドリア代謝とmTOR複合体活性の間の興味深い相互作用が、過去10年間に徐々に明らかにされつつある。より具体的には、ミトコンドリア代謝は、酸化還元環境を変化させることによって、mTOR-raptor複合体活性に影響を与える。167Sarbassov と Sabatini167は、HEK293T 細胞モデルにおいて、酸化剤が mTORC1 をうまく活性化し、還元剤が栄養による mTORC1 活性化を効果的に抑制することを明らかにし、それによって、酸化還元感受性メカニズムが mTOR 活性化に重要な役割を果たすことを示している。このレドックス依存的な mTOR 複合体の活性化には、raptor との相互作用が関与している。167,168TSC1-/-またはTSC2-/-マウス胚性線維芽細胞モデルでは、酸化剤または還元剤によってmTORC1活性が変化しないことから、TSCも必要であることがわかる。169一方、Schiekeら168は、ラパマイシンによるmTOR-raptor複合体の破壊が、HEK293T細胞モデルのミトコンドリア膜電位、酸素消費量、ATP合成能を低下させることを明らかにした。この研究により、mTOR複合体はミトコンドリアの酸化機能、膜電位、酸素消費量、ATP合成能を制御していることが明らかになった。このように、mTORの活性は、解糖系と好気性系のATP生成源の相対的なバランスを決定するのに有用であると考えられる。実際、Cunninghamら(46)は、骨格筋組織と細胞を用いて、mTORがYY1-PGC-1α転写複合体を介してミトコンドリア酸化機能を制御していることを明らかにした。Jiら47は、C2C12筋芽細胞において、mTOR-Y1-PGC-1α経路の活性化が、細胞内ATP合成(P< 0.05)、酸素消費率(P< 0.01)、クエン酸合成酵素活性(P< 0.01)およびミトコンドリアDNAコピー数(P< 0.01)を著しく増大することを示して、Cunninghamら46の結果に対する支持を提供した。
ラパマイシン活性化機構の小胞体ストレスへの寄与
mTOR経路の過活性化は、ERストレスを活性化し、UPRを引き起こす。これは、おそらくタンパク質合成の増加によって、ERのタンパク質折り畳み能力を圧倒するためである。170,171従来、mTORとUPRは別々の経路と考えられてきたが、Appenzeller-HerzogとHallによってレビューされたmTOR-UPR相互作用の特定により、比較的新しい研究領域が開かれた171。インスリンは、成長因子として、phosphoinositide 3 kinaseを介してAktにシグナルを送り、AktはmTORC1の上流阻害因子であるTSC2を複数のセリン/スレオニン残基でリン酸化して不活性化させる。172,173TSCの欠損は、mTORC1の構成的活性化をもたらし、170 c-Jun N-terminalキナーゼ経路を刺激する。活性化されたc-Jun N-terminalキナーゼは、インスリン受容体基質1のセリン残基をリン酸化して分解し、最終的にインスリン抵抗性につながる。174これは、mTORの下流のERストレス経路として最もよく知られており、mTORC1やERストレス促進性デノボ脂肪生成などの他のメカニズムと並行して起こる。175 これは、転写因子SREBP-1cの活性化を通じてインスリンが強く駆動している。53過栄養によるmTORの過活性化がインスリン抵抗性を引き起こすもう一つの方法は、S6キナーゼの活性化であり、これもインスリン受容体基質1のセリンリン酸化を引き起こす。176過剰なロイシンとグルコースは、mTOR/p70 S6キナーゼを活性化し、インスリン受容体基質1のセリン残基(S307、S635)をリン酸化させ、92,177それによってインスリンシグナルを障害することによってインスリン抵抗性を引き起こすことが示されている。mTOR-UPR相互作用によるインスリンシグナル伝達および脂肪生成への有害な影響は、mTOR活性化をアロステリックに阻害するラパマイシンの投与、またはAMPKを意図的に活性化することにより緩和することが可能である。92,93例えば、Li ら93は、ラパマイシンによる mTORC1 シグナリングの阻害が、HepG2 細胞において高グルコースおよびパルミチン酸によって誘発される ER ストレスおよび脂質蓄積を軽減することを明らかにした。
ラパマイシンの中枢性炭素代謝への関与
いくつかの研究により、mTORの活性化とエネルギー恒常性との間に重要な関連性があることが示されている。Wagleら178は、インスリンによるグルコース-6-リン酸デヒドロゲナーゼ遺伝子発現の調節がラパマイシン感受性であることを示し、解糖の調節にmTORが関与していることを示唆した。グルコース-6-リン酸デヒドロゲナーゼは、ペントースリン酸経路を通る炭素の流れを制御し、mTORによって制御される生合成に必要な還元剤NADPHを供給している。178これらの結果は、Düvel ら(162)によって支持されている。彼らは、mTORC1 が転写因子 hypoxia-inducible factor 1α (HIF-1α) を介してグルコースの取り込みと解糖に関わる酵素(グルコース 6- リン酸脱水素酵素を含む)およびペントースリン酸経路に関わる酵素(6-ホスホグルコン酸脱水 素酵素を含む)の発現を誘導することを実証している。また、ステロール調節因子結合タンパク質を介したデノボ脂質/ステロール合成も重要である。162Hudsonらによって完成された研究179は、mTORの活性化が、PC-3ヒト前立腺癌細胞株における解糖系遺伝子発現のHIF-1α依存性調節と同様に、HIF-1αの蓄積をもたらすことを示した。これらの結果は、mTORがHIF-1αの上流活性化因子として作用していることを示している。Chenらによる最近の研究(180)では、さらに、mTORC1がインポータータンパク質であるKaryopherin subunit alpha-2を介して作用し、マウス胚性線維芽細胞モデルにおいて解糖をアップレギュレートすることが明らかにされている。
解糖とmTORの制御の間にはクロストークが存在することに注意しなければならない。Lee ら181は、以下のデータに基づいて、GAPDH がグルコースの利用可能性に応じて mTORC1 活性を調節していると仮定している。低グルコース条件下では、GAPDH は Ras homolog enriched in brain (Rheb) タンパク質と結合し、その結果、mTORC1 の活性化を抑制した。181 Lee らは、解糖系中間体であるグリセルアルデヒド-3-リン酸が、Rheb-GAPDH の結合を不安定にすることでmTORC1 の活性化を補助すると報告している。181ブラーら(Buller etal) 182によって行われた研究は、過剰なグルコースにさらされたメサンギウム細胞がグルコーストランスポーター1型の発現を増強して提示し、mTOR-Rheb結合を増加しGAPDH-Rheb結合を減少させることによってmTORを活性化したことから、リーら181の研究結果を支持した。
ラパマイシンのメカニスティックターゲットによるワールブルグ効果のポジティブな指標として
mTOR複合体の異常な活性化は、がん、肥満、自己免疫疾患を含む多くの代謝性疾患におけるバイオマーカーと考えられている。栄養によるmTORの活性化は、主にHIF-1αを介した転写調節を介して解糖系フラックスをアップレギュレートする。酸素の存在下でも解糖速度が上昇するのは、mTORシグナルの亢進により、細胞の生存を維持する手段として起こりうることである。Sunら183は、マウス胚性線維芽細胞モデルにおいて、mTORが発癌性Warburg効果の律速解糖酵素であるピルビン酸キナーゼM2の発現を、HIF-1αおよびc-Myc-異種核リボ核蛋白カスケードを介してシグナル伝達することを明らかにした。興味深いことに、ピルビン酸キナーゼ M2 は、デノボセリン合成を促進することでmTORC1活性を維持した184あるいはmTORC1阻害剤であるAkt1基質1をリン酸化した185明らかに、mTORの状態はWarburg効果の主要な陽性指標とみなすことができる。
結論
慢性的な栄養過多は、代謝の恒常性を乱し、生体エネルギー経路を書き換えて、細胞のエネルギー状態を変化させる。細胞内の過剰な代謝産物は、細胞内の小器官に負担をかけ、エネルギーの貯蔵と生合成を促進する経路を刺激し、エネルギーを消費する経路を阻害する。栄養の酸化を促進するために解糖が促進されるが、栄養を完全に分解する能力がないため、細胞内に半酸化状態の中間体が多く残り、細胞は生合成をしやすい状態にある。タンパク質のアセチル化度が上昇し、サーチュイン活性が低下する一方で、生合成のシグナルに答えるためにmTOR複合体が活性化する。ミトコンドリアは飽和状態になり、TCAサイクルと電子輸送鎖の両方が働き続け、高いエネルギーレベルが持続するとAMPKの活性が抑制され、最終的には酸化的リン酸化複合体が損傷して生体エネルギー効率が低下している。プロトン漏出と酸化ストレスはミトコンドリアを抜け出して細胞の他の部分に広がり、サーチュイン活性の抑制を倍加させ、炎症性シグナルを活性化させる。mTOR活性化の影響で、タンパク質のパッケージングやde novo脂肪生成の作業負荷が増大し、ERに負荷がかかる。ERストレスが持続すると、脂肪生成によって間接的に、あるいは炎症性経路の活性化を通じてインスリンシグナル伝達を阻害することによって直接的にインスリン抵抗性を引き起こす可能性がある。そして、ERストレスを改善するためにUPRが活性化され、それがうまくいかないと細胞のアポトーシスに至る。長期の栄養過多のもとでは、重要なオルガネラや栄養センサーにこうした変化が残り、代謝異常の発症の基礎となる可能性が高い。
メトホルミン、サリチル酸塩、メトトレキサートなど、主要な酵素や栄養センサーに作用する小分子は、さまざまな代謝異常の治療薬として研究されてきた。しかし、現在までのところ、臨床で有効性を示すには至っていない。レスベラトロールは、実験室で代謝のホメオスタシスに影響を与えることを実証した別の化合物である。レスベラトロールは、マウスモデルでSIRT1の活性化を通じてPGC-1αのアセチル化を減少させてPGC-1α活性を増加させた。186 これは、酸化的リン酸化およびミトコンドリア生合成の遺伝子の誘導とレスベラトロール摂取の関連を説明するものである。186しかし、これらの知見と矛盾する研究結果もあり、ネズミにレスベラトロールを与えてもミトコンドリア生合成には影響がないと結論づけている。187しかし、ハイスループットスクリーニングプロセスの利用が進むにつれ、新規低分子化合物 ZLN005188や SR-18292 などの薬剤の効果を効率的に測定できるようになり、新しい発見の可能性が広がっている。189。