
Contents
Glycogen synthase kinase-3 signaling in Alzheimer’s disease
pubmed.ncbi.nlm.nih.gov/32006534/
概要
アルツハイマー病は、認知症を伴う神経変性疾患の中で最も一般的な疾患であり、全症例の約70%を占めている。現在、米国では580万人がアルツハイマー病患者として生活しており、2050年にはこの数が倍増し、社会経済的な負担が大きくなると予想されている。集中的な研究にもかかわらず、アルツハイマー病の引き金となる正確なメカニズムはまだ知られておらず、現在のところ治療法はない。
近年、グリコーゲン合成酵素キナーゼ-3β(GSK3-β)を含む、アルツハイマー病神経病理に関連する多くのシグナル伝達経路が、本疾患の治療のための可能性のあるターゲットとして探索されている。GSK3-βは、タウリン酸化、アミロイドβ産生、記憶、神経新生、シナプス機能など、この疾患の主要な特徴に影響を与えるため、アルツハイマー病の病態に重要な役割を果たしていると考えられている。
本レビューでは、GSK3-βの神経生物学に関する現在の理解を、アルツハイマー病の病態生理に関連する特定のシグナル伝達経路への影響に特に重点を置いてまとめている。さらに、GSK3-βを標的としたアルツハイマー病治療の実現可能性について議論し、前臨床試験および臨床試験におけるGSK3-β阻害薬の開発に向けた現在の研究努力の概要を示す。
1. 序論
アルツハイマー病は、認知症を伴う加齢性神経変性疾患の中で最も一般的な疾患である。現在、米国では、この疾患を持つ人の数は580万人と推定されており、団塊の世代の高齢化と治療法の欠如により、今後数年で急増すると予測されている[1]。実際、65歳以上のアメリカ人の数は 2018年の5,200万人から 2060年には9,500万人に倍増すると予測されており、将来の社会的・経済的負担が大きくなる可能性がある[1,2]。
臨床的には、アルツハイマー病は記憶と認知機能の進行性の喪失が特徴である。初期の症状としては、最近の会話、名前または出来事を思い出すことの困難さが挙げられる。病態の進行に伴い、患者はコミュニケーション能力の低下、混乱、行動の変化、判断力の低下を発症し、しばしば受動性や抑うつを経験する [1,3]。この障害の病理学的特徴は、アミロイドβペプチドで構成されるアミロイドβ(アミロイドβ)プラークの細胞外蓄積、高リン酸化タウタンパク質で構成される神経原線維のもつれ(NFT)脳の炎症および萎縮である。これらの変化は、臨床症状が顕著に現れる数年前から始まると考えられている[1,3]。アミロイドβ前駆体タンパク質(APP)プレセニリン1(PS1)PS2などの特定の遺伝子の突然変異の結果であり、全症例の約2-4%しか占めていない家族性の形態を除いて、アルツハイマー病は主に散発性の障害であり、その病因は現在のところ不明である[4]。実際、集中的な研究にもかかわらず、アミロイドβの蓄積やNFTsの形成・沈着のメカニズムは解明されておらず、有効な治療法はない。このため、過去数年の間に、いくつかの病態生理学的プロセスに関連するいくつかのシグナル伝達経路が研究され、アルツハイマー病治療のための新規治療アプローチの可能性のあるターゲットとして評価されていた。
グリコーゲン合成酵素キナーゼ-3(GSK3)は、ユビキタスに発現し、構成的に活性なセリン-スレオニンキナーゼであり、多くの主要な細胞生物学的経路の制御に関与しており、そのうちのいくつかは神経変性にも関与している。GSK3には2つの異なる遺伝子によってコードされたGSK3-αとGSK3-βの2つのアイソフォームが存在する[5]。中枢神経系(CNS)ではGSK3-βが最も多く、その発現量は加齢とともに増加することが知られている[6]。GSK3-βはアルツハイマー病患者の脳内で高活性であることが明らかになっており,異なるメカニズムでアルツハイマー病の病態に寄与していることが証明されている[7].このキナーゼの調節障害は、試験管内試験および生体内試験のADモデルにおいて、アミロイドβおよびタウの代謝と毒性の両方に影響を与えることが観察されている。さらに、GSK3-β活性は、記憶の定着、神経新生、シナプス可塑性、長期増強、炎症と関連している(図1)[8]。
GSK3-βシグナル伝達とアルツハイマー病の病態との関連性を考えると、GSK3-βを阻害することによる治療の可能性は、前臨床試験や臨床試験で広く検討されてきた。本レビューでは、GSK3-βの生理機能、アルツハイマー病におけるGSK3-βの異常活性に関する現在のデータ、GSK3-βモジュレーションがアルツハイマー病治療薬として有効であることを裏付けるエビデンスに焦点を当てて解説する。
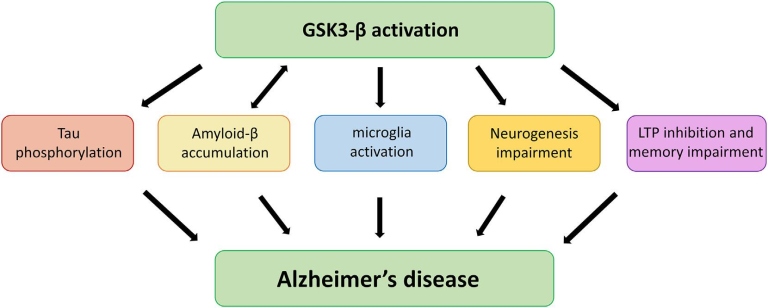
図1 アルツハイマー病病態におけるGSK3-βシグナル伝達の役割
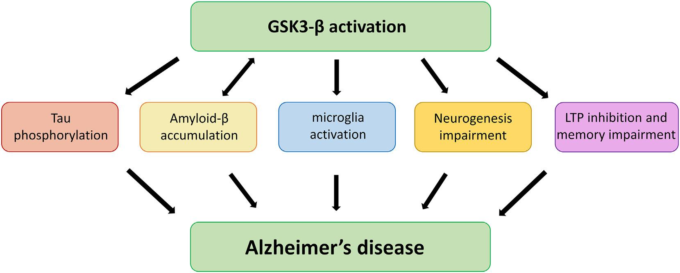
アルツハイマー病の発症・発症に関連する様々な側面や経路におけるGSK3-βの関与の可能性を模式的に示している。
- 1) GSK3-βの活性化は、タウの高リン酸化を直接的に促進し、神経変性に寄与する。高リン酸化したタウは微小管から解離し、軸索輸送障害、NFTsの形成、神経細胞の機能障害、シナプス機能障害などを引き起こす。
- 2) GSK3-βはアミロイドの産生と蓄積を促進し、アポトーシスや神経細胞障害を誘発する。
- 3) GSK3-βは、中枢神経系の一次免疫細胞であるミクログリアの生物学的反応を調節し、炎症性分子の産生を促進する。
- 4) さらに、GSK3-βは海馬の神経新生の調節にも重要な役割を果たしており、海馬の神経新生は学習や記憶を支える重要なプロセスであり、アルツハイマー病の病態に影響を与えることが知られている。
- 5) 最後に、GSK3-βはシナプス可塑性や記憶にも関与している。このキナーゼの過剰活性化は、記憶形成に必要なメカニズムである海馬長期増強(LTP)の阻害と関連している。
1.1. GSK3βの生物学的機能
GSK3 は、様々な基質のセリンおよびスレオニン部位をリン酸化するキナーゼである[9]。GSK3は1980年代に発見され、リン酸化によりグリコーゲン合成酵素の活性を調節し、その活性を阻害するキナーゼの一つとして同定された[10]。ウサギの骨格筋から抽出されたGSK3は、細胞のアポトーシス、分化、増殖、微小管の形態を含むいくつかの重要な細胞プロセスの制御に関与している[10]。GSK3は、中枢神経系では構成的に活性であり、豊富に発現している[9]。GSK3の発見後、研究が大きく進展し、19番染色体上と3番染色体上で異なる遺伝子がコードする2つのアイソザイム、GSK3-αとGSK3-βが同定された[12]。これらのアイソザイムは、構造が似ているにもかかわらず、異なる基質に作用することが知られている[13]。実際、生体内試験での研究では、GSK3-βの完全な除去は胚での死を引き起こすのに十分であることが示されているが[14]、GSK3-αの遺伝子欠失は生存に有意な影響を与えないことが分かっている[15]。
GSK3-βは構成的に活性ではあるが、リン酸化と脱リン酸化によって機能が制御されていることが知られている。例えば、チロシン279/216の自己リン酸化はGSK3-βの活性化を仲介し、AKT、プロテインキナーゼAおよびB(PKA-PKB)によるN末端のセリン部位21/9のリン酸化は抑制作用をもたらす[16]。さらに、リン酸化の際にGSK3-βに付加されたリン酸基は、プロテインホスファターゼPP2Aによって除去され、GSK3-βは再活性化される(図2)[17]。近年、死後の研究からin-vivoやin-vitroでの研究に至るまで、GSK3-βは神経変性に関連するいくつかのメカニズムにおいて、潜在的に活性化されていると考えられている[18-20]。
1.2. GSK3βとアミロイドβ
試験管内試験および生体内試験での証拠は、GSK3-βの活性化がAPPの切断を調節することにより、アルツハイマー病脳におけるアミロイドβの形成と蓄積に関与していることを示している。脳内では、非アミロイド原性とアミロイド原性の2つの異なる経路が、異なるプロテアーゼの作用を介してAPPの切断に関与している[21]。非アミロイド原性のAPPの切断は、ADAM-10と-17からなるα-セクレターゼとγ-セクレターゼの複合体によって行われる。α-セクレターゼによる開裂は、分解されやすいペプチドを産生する[22]。対照的に、アミロイド遺伝子経路では、APPは最初にβ-セクレターゼ(BACE-1)酵素によって切断され、続いてγ-セクレターゼ複合体処理によって、フィブリル化およびオリゴマーゼ化し、最終的にはアルツハイマー病脳内のアミロイドβ堆積物として蓄積する傾向があるアミロイドβペプチドの生成を引き起こす。これまでの研究では、APPとγ-セクレターゼ複合体の触媒成分の一つであるPS1がGSK3-β基質として同定されており[21]、APPの切断とPS1の機能がGSK3-β活性の上昇によって影響を受け、アルツハイマー病脳内でのアミロイドβ生成量の増加とそれに続く沈着を引き起こすことが示唆されている(図3)[23,24]。
PS1とGSK3-βの関係を調べたいくつかの研究では、GSK3-βの過剰活性化がPS1の機能や中枢神経系での局在化を変化させることが示唆されている[24]。PS1はゴルジ体膜、小胞体、形質膜に局在している。この遺伝子産物は、N-カドヘリン/β-カテニンと結合し、GSK3のリン酸化を受けるセリン残基からなるシナプス三量体を形成している。過剰に活性化されたGSK3-βは、PS1/N-カドヘリン/β-カテニン複合体の活性を低下させ、シナプスおよび神経細胞の生存性に障害を引き起こし、アルツハイマー病の病理学につながる[24]。プラーク形成におけるGSK3-βの関与は、BACE1の過剰活性化にも関連している。GSK3-βとBACE1の関連性を探る研究により、GSK3-βの過剰活性化または過剰発現がBACE1の酵素によるAPPの切断を促進することが明らかになった。さらに、BACE1の発現はGSK3-βによって媒介されることが示されているが、BACE1の転写にはNFK-βシグナル伝達が必要であることが示されている[16]。アルツハイマー病患者ではBACE1とNFK-βの両方の発現が上昇しており、NFK-βはシス作用を持つp65結合プロモーターを操作することで、GSK3-βによって誘導されたBACE1の転写を制御し、異常なアミロイドβ産生を引き起こすことが示されている(図3)[16,25]。これらの観察結果を裏付けるように、APP変異体細胞株におけるGSK3-βの阻害は、APPのBACE1切断を減少させる効果的な方法であることが証明されており、その結果、アミロイドβ40-42ペプチドの両方のレベルが有意に減少することになる[26,27]。
APPに対する直接的な効果の他に、GSK3-β活性はまた、インスリンシグナル伝達経路の破壊を介してアミロイドβ病理を誘導することができる。インスリン欠乏マウスを用いた生体内試験では、アミロイドβとGSK3-βレベルの間に正の相関があることが明らかになっている[28]。さらに、アミロイドβ分解の主要な担い手であるインスリン分解酵素タンパク質(IDE)は、糖尿病マウスのGSK3-βレベルと負の相関があることも明らかにされている[28]。さらに、他の知見では、インスリン欠乏性げっ歯類モデルの脳組織ではGSK3-βとアミロイドβのレベルが上昇しているが、IDEは有意にダウンレギュレーションされていることが示唆されている[29-31]。
図2 GSK3-β活性の制御
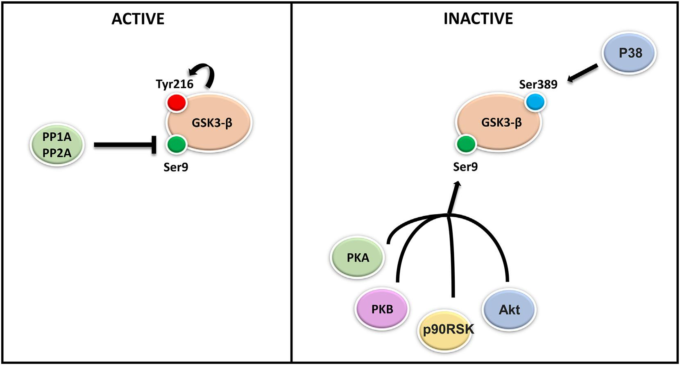
GSK3-βはそのリン酸化状態に応じて活性型と不活性型が存在する。GSK3-βの活性を調節する酵素には2つのファミリーがある。リン酸基をGSK3-βに付着させるセリン/スレオニン/チロシンキナーゼと、外部刺激に応答してGSK3-βタンパク質からリン酸基を除去するホスファターゼである。GSK3-βの活性化はチロシン216上での自動リン酸化を介して行われ、GSK3-βの活性化はセリン9上でのリン酸化を介して阻害されることが試験管内試験および生体内試験で明らかにされている。AKT、プロテインキナーゼA・B(PKA-PKB)P90RSKなどのキナーゼによるセリン9上でのリン酸化がGSK3-βの活性化を阻害している。代わりに、P38 MAPKキナーゼによるGSK3-βのセリン389上のリン酸化がGSK3-βを阻害することが示されている。さらに、リン酸化の際にGSK3-βに付加されたリン酸基は、プロテインホスファターゼPP2Aによって除去され、結果としてGSK3-βの再活性化をもたらすことが知られている。
興味深いことに、いくつかの研究では、アミロイドβとGSK3-βの活性化の間にフィードバックループが存在し、最終的にタウの過剰リン酸化につながる可能性があることが提案されている。さらに、アミロイドβは、WntがGSK3-βを阻害するのを防ぐ正常なWnt経路の活性を阻害し[32]、GSK3-βを直接活性化してタウのリン酸化を促進する可能性がある[33]。これらの知見は、GSK3-βが両方のアルツハイマー病神経障害(すなわち、アミロイドβとタウ)の生物学的リンクであると考えられるという仮説を支持するものである[21]。
図3 GSK3-βはアミロイドβとタウの代謝を阻害する
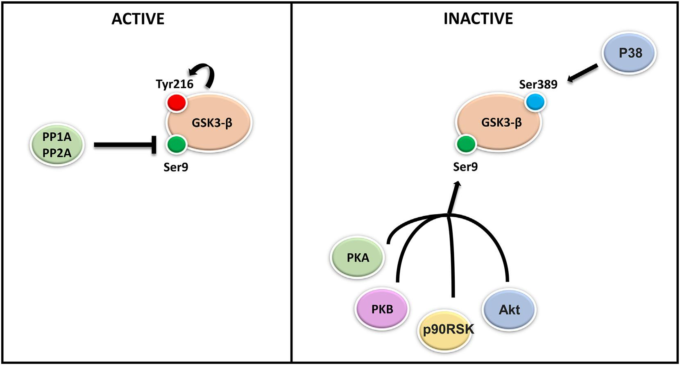
GSK3-βの活性の増加は、2つの異なるメカニズムを介してアミロイドβの形成を促進する。GSK3-βはNFK-βシグナルのアップレギュレーションを介してBACE1遺伝子の発現を誘導し、γセクレターゼ活性を調節する。1) ヒト BACE1 プロモーター領域には 2 つの機能的な NF-kB 結合部位が存在する。GSK3-βの活性化はNFK-β/p65核内転座とBACE1プロモーター部位への結合に関与しており、最終的にはBACE1タンパク質レベルの上昇とBACE1媒介のAPP処理とアミロイドβ産生につながることが、試験管内試験および生体内試験で証明されている。2) GSK3-βは、PS1活性と細胞内局在化を直接制御することにより、γセクレターゼ活性を調節する。GSK3-βがリン酸化されると、PS1はN-カドヘリンとの結合親和性が低下し、細胞表面での発現が低下し、PS1/γセクレターゼの基質特異性が変化することが明らかになった。
さらに、GSK3-βはタウのリン酸化に関与する主要なキナーゼの一つである。タウタンパク質の特定のThr231残基にリン酸基が付加されると、微小管の分解が起こり、タウオリゴマーやNFTの形成が促進され、神経細胞の機能障害や変性に寄与すると考えられている。
1.3. GSK3βとタウ
タウは微小管関連タンパク質(MAP)であり、チューブリンの集合体を確保することで微小管を安定化させることが知られている[34]。タウは17番染色体上に位置するMAPT遺伝子にコードされており、代替スプライシングによって生成された6つのアイソフォームから構成されている[18,19]。タウの機能や微小管への親和性は、主にそのリン酸化状態に依存する。アルツハイマー病では、タウは高リン酸化され、微小管の分解、神経細胞の完全性の損失、最終的にはNFTsの形成につながるサイト質に蓄積されている[35]。タウの高リン酸化に関する初期の研究の大部分は、脳内のキナーゼ-リン酸化酵素の不均衡を探求し、アルツハイマー病のタウ病理の発達に関与している可能性のある主要なタウキナーゼとしてGSK3-βを同定した[16,20]。タウタンパク質には、GSK3-βのリン酸化の対象となるアミノ酸残基がかなりの数存在する[25]。タウリン酸化部位の局在化もまた、アルツハイマー病研究の関心事となっている。GSK3-β媒介によるタウタンパク質のリン酸化は、微小管結合ドメインとそのアミノ酸残基に近接した領域で見られる[36]。これらの結合ドメインではタンパク質間の相互作用が起こることが知られているため、GSK3-βによって誘導されたタウリン酸化は毒性のある方法で自己凝集しやすい(図3)[33]。ショウジョウバエのトランスジェニックモデルを用いたある研究では、GSK3-βの異常なアップレギュレーションが毒性のあるタウ凝集体の蓄積と正の相関を示すことが実証されている[36]。生体内試験での知見と一致するように、試験管内試験研究では、ヒト神経芽腫細胞がGSK3-βを介したリン酸化に起因するタウフィラメント凝集体を発現していることが示されている。このような理由から、また、試験管内試験および生体内試験での研究でGSK3-βの阻害がタウの過剰リン酸化を回復させることが確認されているため、タウ病理に対する創薬および開発プログラムの大部分はGSK3-β活性を標的とする傾向がある[38-40]。
1.4. GSK3β活性とシグナル伝達経路の性差
このキナーゼの性差に関する現在の知見は不十分であるが、GSK3β活性とアルツハイマー病関連シグナル伝達経路に対する男性ホルモンと女性ホルモンの影響を検討した研究はほとんどない。C57Bl6/Jマウスの脳では、17β-エストラジオール経路の活性化は、セリン-9リン酸化によるGSK3βの不活性化と関連しており、その結果、アミロイドβ蓄積量の有意な減少と高リン酸化タウ蛋白質の減少を伴うことが明らかにされている[41]。さらに、17β-エストラジオールは、海馬スライス培養において、ホスホイノシトール-3キナーゼ(PI-3K)とMAPK3トランスダクションを誘導することで、GSK3βによる神経細胞のアポトーシスを抑制した[41]。注目すべきは、アルツハイマー病患者におけるBACEとGSK3βのmRNAレベルの変化を調査した別の研究では、女性の死後脳組織におけるBACE mRNAの7%のダウンレギュレーションが男性被験者と比較して報告されているが、残念ながらGSK3βを測定したところ、そのレベルは信頼できる検出限界以下であり、結論を導き出すことはできなかった[42]。したがって、現在利用可能なすべてのデータを考慮すると、GSK3βの神経生物学のこの側面に対処するための更なる研究が必要である。
1.5. GSK3β、シナプス可塑性と記憶
シナプス機能障害は、アルツハイマー病の病態における初期のイベントである[43]。記憶保存の基礎となる分子機構には、シナプス強度の長期的な増加を否定するLong Term Potentiation (LTP)と、その反対のプロセスであるLong Term Depression (LTD)を含むシナプス強度の変化が関与している[44]。また、GSK3-βがシナプス可塑性や記憶形成にも重要な役割を果たしていることが明らかになってきている(図1)[43]。GSK3-βは海馬で高発現しており、主に細胞質に存在するが、シナプトソーム、樹状突起、棘突起でも一部のキナーゼが検出されている[44,45]。興味深いことに、GSK3-βはAMPA受容体と結合し、LTPとLTDのバランスを制御する重要な役割を果たしている[43]。実際、抑制性のSer9部位におけるGSK3-βのリン酸化は、歯状回(DG)や海馬のCA1領域でLTPの誘導時に増加することが知られている[45]。代わりにGSK3-βの過剰活性化はLTPを阻害し、NMDA受容体依存性LTDを促進することが知られている[43]。この観察と一致するように、GSK-3βを条件付きで過剰発現させたトランスジェニックマウスでは、LTPが障害され、海馬依存性の空間記憶と恐怖記憶の形成に有意な障害が見られた[43]。
さらに、GSK-3-βを阻害すると、GSK-3-βリン酸化後にプロテアソームによって分解されてしまうβ-カテニンを安定化させることができる[46]。シナプス前・後シナプスの両方に存在するβ-カテニンは、カドヘリンの細胞質ドメインと結合し、細胞接着を調節し、シナプスの大きさや強さに影響を与える[47,48,49]。GSK3-β活性の増加によるβ-カテニンシグナル伝達経路のダウンレギュレーションは、アルツハイマー病脳で観察されており、学習と記憶障害の病態生理に示唆されている[50,51]。GSK3-βがこれらのプロセスの調節に重要な役割を果たしていることを考えると、遺伝学的および薬理学的なアプローチによりGSK3-βの活性を低下させることが広く検討され、いくつかのアルツハイマー病の動物モデルにおいて認知・記憶障害を救済することが報告されている[8]。例えば、GSK3-β阻害剤インディルビン-3′モノキシムは、モリス水迷路課題の障害を救済し、APP/プレセニリン-1ダブルトランスジェニックマウスにおけるアミロイドβ産生とタウの高リン酸化を減衰させた[52]。GSK3-β阻害剤である2-メチル-5-(3-{4-[(S)-メチルスルフィニル]フェニル}-1-ベンゾフラン-5-イル)-1,3,4-オキサジアゾール(MMBO)は、3xTgマウスのY迷路と新規物体認識テストの障害を改善した[53]。さらに、選択的GSK3-β阻害剤L803-mtsを投与した5X家族性ADマウスでは、アミロイドβペプチドレベルの低下と関連して、文脈的恐怖記憶が改善された [54]。
1.6. GSK3βと神経新生
また、GSK3-βは成体海馬の神経新生を抑制し、歯状回(DG)の神経原性ニッチにおいて神経細胞死を誘導することが報告されている(図1)[55]。神経新生は記憶の統合とパターン分離に必要とされる動的なプロセスであり、アルツハイマー病患者の中枢神経系や動物モデルにおいても同様である[56,57]。このキナーゼを過剰発現させたマウスでは、GSK-3-βが成人の神経新生の重要な制御因子であるとの仮説に基づき、新たに機能的なDGニューロンの増殖・成熟度が低下し、記憶障害が顕著に現れることが明らかになった。このGSK-3betaによる神経新生の低下は、ミクログリアの活性化と関連しており、変性をさらに促進させている[56,58]。したがって、この酵素の不活性化は、アルツハイマー病のような神経新生が障害された神経病理学において、新しい機能的ニューロンの誕生を促進し、記憶力を向上させるために非常に有益であると考えられる。試験管内試験および生体内試験のデータは、この仮説を支持しているようである。GSK3-βを阻害することで、試験管内試験では神経幹細胞の増殖、遊走、分化が促進されることが示された。さらに、NP03112化合物(ti-deglusib)との生体内試験でのその阻害は、成体ラットの海馬のDGにおける神経幹細胞の増殖と分化を促進した[59]。
1.7. GSK3-βと炎症
アミロイドβとタウの凝集の他に、神経炎症もアルツハイマー病脳病理のもう一つの主要な特徴である(図1)[60,61]。ミクログリアおよびアストロサイトの広範な活性化は、アミロイドβプラークおよび萎縮性神経突起の周囲で観察される[62-64]。これらの細胞が活性化されると、インターロイキン1(IL-1)インターロイキン-6(IL-6)インターロイキン-8(IL-8)腫瘍壊死因子α(TNF-α)などの様々な炎症性サイトカインが放出され、これらは神経毒性を促進し、神経細胞の死に寄与する可能性がある[65]。
驚くべきことに、GSK3-βは最近、複数の経路を介してミクログリアにおける炎症反応の重要な調節因子として同定された。GSK-3βの活性化は、IL-1,IL-6,TNF-αの産生を促進するだけでなく、JNK-、STAT3/5,NF-κBシグナルを活性化することが実証されている[65]。実際、GSK-3βは、p65サブユニットのffリン酸化によりNF-κBを活性化し、その結果、炎症性サイトカインおよびケモカインのさらなる発現をもたらすことが知られている[65]。さらに、GSK3-βのSTAT3への結合は、IFNγ活性化に必要なIFNγ受容体関連細胞内シグナル伝達複合体とSTAT3の結合を促進する[66]。いくつかの研究では、GSK3-βがミクログリア遊走の調節に関与していることも示唆されている。例えば、BV-2ミクログリア細胞をGSK3阻害剤で処理すると、トランスウェル遊走アッセイにおいて、試験管内試験でミクログリアの遊走が有意に減少することが示されている[67]。GSK-3βの抗炎症作用の実質的な証拠を考えると、その阻害は、アミロイドβ産生とタウリン酸化を減少させるだけでなく、アルツハイマー病脳で抗炎症作用を発揮する可能性がある。
1.8. 治療アプローチとしてのGSK3β
GSK-3βシグナル伝達は、タウリン酸化、アミロイドβ産生、神経新生、記憶、シナプス機能障害を含むいくつかのアルツハイマー病神経病理学的特徴と強く関連しているため、その阻害はアルツハイマー病治療のための潜在的に重要な治療アプローチとして浮上している。機能喪失の研究は、いくつかのADマウスモデルで実施されており、アルツハイマー病様病理学的表現型の多くの側面を打ち消すのに有効であることが証明されている[68,69] 気分安定剤のリチウムは、アルツハイマー病とタオパチーのいくつかのマウスモデルでリン酸化タウとアミロイドβレベルを減少させることが示されている[68-72]。非ATP競合性GSK-3β阻害剤であるチデグルシブは、ヒト変異APPを過剰発現させたAPPsw-tauvlwマウスにおいて、タウリン酸化レベルを低下させ、アミロイドβ沈着、アストロサイトの増殖を減少させ、記憶障害を改善した[73,74]。最後に、特異的なGSK3阻害剤SB216763を脳に注入したところ、ADモデルにおいてGSK3-β活性の低下による有益な効果が確認された。
GSK-3は高度に保存されたキナーゼであるが、前臨床段階で試験されたGSK-3阻害剤(合成および非合成)のうち、臨床試験に至ったものはわずかである。しかし、動物モデルではいくつかの好ましい結果が得られているにもかかわらず、GSK3-βは普遍的に発現しており、いくつかの重要な細胞生物学的プロセスに関与しているという事実から、前臨床試験および臨床試験の両方で長期的な治療試験には多くの懸念と重大な毒性学的課題が提起されている[75,76]。GSK3-β阻害剤としての第I相臨床試験の最初の候補はAZD2558であった[77]。この高選択性GSK3-β阻害薬は、試験管内試験および生体内試験でタウリン酸化とグリオシスの再抑制に成功した。しかし、生体内試験での投与に関連した標的外効果の重篤さが、この薬剤の慢性的なアルツハイマー病治療のための試験を妨げていた。2つ目の試みとして、AZD1080を用いた試験が行われたが、以前に試験管内試験および臨床試験前の研究でタウリン酸化を抑制する能力が確認されている。残念ながら、この薬の慢性投与には重大な副作用があり、第I相試験は中止された[77]。これまでのところ、GSK-3阻害薬のうち、アルツハイマー病および進行性上核麻痺の治療を目的とした第II相臨床試験に進出したのは、ティデグルシブの1つだけである[78-80]。Tideglusibは軽度から中等度のアルツハイマー病患者を対象とした2つの小規模な第II相臨床試験で試験され、良好な忍容性を示したが、有意な臨床的改善は認められなかった[76]。
軽度認知障害とアルツハイマー病の臨床診断を受けた患者を対象に、塩化リチウムを用いたパイロット臨床試験が実施されている。塩化リチウムは、双極性障害の治療薬としてFDAに承認されている[81]が、弱く非特異的なGSK3-β阻害薬である。治療用量では、GSK3-β活性を約25%低下させることができ、副作用はない[81]。この薬剤を用いた小規模な臨床試験では、認知機能の改善やタウリン酸化の減少など、いくつかの肯定的な結果が示されており、リチウムはアルツハイマー病の疾患修飾療法としての可能性を示唆している[76]。
2. 結論
近年、GSK3-βはアルツハイマー病の病態形成に重要な役割を果たすキナーゼとして認識されていた。また、GSK3-βが中枢神経系で過剰発現・過剰活性化していることが発見されて以来、GSK3-βがタウやアミロイドβの病態にどのような役割を果たしているのかが広く研究されていた。GSK3-βは主要なタウキナーゼの一つであり、タウの高リン酸化とNFTs形成を促進する。また、GSK3-βの活性が高まると、BACE1遺伝子発現の転写制御やPS1上での調節機能を介してアミロイドβのフォーメーションが誘導されることが強く指摘されている。アルツハイマー病発症には性差が存在することから、GSK3βの調節が性差的であるかどうかについては、いくつかの研究が試みられてきたが、これまでに得られたデータはすべて、GSK3-βの活性がアルツハイマー病発症に関連していると結論づけるには十分ではなかった。また、最近ではGSK3-βがシナプス可塑性、記憶、神経新生、炎症などの調節に関与していることが明らかになっており、GSK3-βはアルツハイマー病の疾患修飾薬として優れた候補となっている。しかし、これまでの臨床研究では、GSK3-βの有効かつ安全な阻害剤は発見されていない。また、GSK3-βの阻害には、そのユビキタスな発現と複数の細胞制御機能に関連した多くの課題があり、今後、GSK3-βの阻害を目的とした戦略が考えられる。将来的には、重篤なオフターゲット効果を回避するために、中枢神経系や関心のある細胞においてGSK3-βの阻害を特異的に誘導することが考えられる。したがって、前臨床試験での有望な結果を考えると、中枢神経系におけるGSK3-β活性を効率的かつ安全に制御する選択的かつ臓器特異的な阻害剤の開発に、より多くの研究が費やされる必要がある