
Contents
- Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines
Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines
www.ncbi.nlm.nih.gov/pmc/articles/PMC7177899/
要旨
アルツハイマー病とパーキンソン病(PD)は最も一般的な神経変性疾患(ND)であり、運動機能障害から心理行動症状まで幅広い症状を呈する。一般的な臨床経過は、プロテインオパシーに起因する神経機能障害が解剖学的に対応する神経障害をもたらすことである。しかし、現在の病理や症状に基づく診断基準は、病気の予防や薬剤開発のためにはほとんど価値がない。
本レビューでは、神経障害の病態メカニズムを概観するために、炎症性サイトカインとヒトサンプルのトリプトファン代謝産物であるキヌレニン(KYN)に関するシステマティックレビューを取り入れ、神経障害の背後にある潜在的な関連性を探るための推論手法を提示した。
その結果、炎症性疾患ではプロ炎症性サイトカインと神経毒性KYNが増加し、アルツハイマー病、PD、ハンチントン病(HD)クロイツフェルト・ヤコブ病、ヒト免疫不全ウイルス(HIV)関連の神経認知障害では抗炎症性サイトカインが増加し、アルツハイマー病、PD、HDでは神経調節性KYNが減少することが明らかになった。
これらの結果は、炎症と神経調節性KYNの間の強い関連性を補強し、適応免疫応答の活性化を確認し、神経調節性KYNの減少に関与している可能性を示唆した。多因子性神経変性疾患の共通点について議論し、現在の診断基準の限界、前臨床バイオマーカーの必要性、神経変性疾患の発症因子を探索するためのアプローチを提示した。
キーワード
神経変性疾患、アルツハイマー病、パーキンソン病、神経炎症、アミロイド、タウ、プリオン、サイトカイン、トリプトファン、キヌレニン、多因子因果関係
1. はじめに
神経変性疾患(ND)は、中枢神経系(CNS)に影響を及ぼす進行性で不治の病となることが多い疾患であり、神経変性、最終的には神経細胞死を引き起こし、運動機能障害から運動失調や認知症などの精神行動症状まで幅広い症状の原因となる [1]。アルツハイマー病とパーキンソン病(PD)は最も一般的なNDである。アルツハイマー病は世界で約5,000万人の認知症患者の約60~70%を占めており、高齢者の障害や依存症の主な原因となっている。世界では1,000万人以上がパーキンソン病を患っており、加齢とともに発症率が増加している[2]。その他のNDには、多発性硬化症(MS)ハンチントン病(HD)筋萎縮性側索硬化症(ALS)クロイツフェルト・ヤコブ病(CJD)ヒト免疫不全ウイルス(HIV)関連神経認知障害(HAND)脳卒中誘発性二次神経変性(SND)などがある。[3]. 神経疾患は、世界的に障害調整寿命の第一位の原因であり、心血管疾患に次ぐ第二位の死亡原因である[2]。著名な医師の名誉のために付けられた病名、病理学的・臨床的所見に応じたサブタイプ分け、重複する病理組織学的分類、多数の分子的発見など、すべてが疾患の根本的な観点を不明瞭にしており、神経疾患の病態解明のためのきめ細かな研究や新薬の開発を妨げる可能性がある[4,5,6,7,8]。
中枢神経系における神経細胞の構造的・機能的な喪失である神経変性は、運動神経系、自律神経系、認知神経系の機能障害や機能障害を反映した脳領域の一般的な病理学的所見である[9]。死後の脳標本の神経変性病変は、構造的および機能的イメージング研究とよく相関している。脳の収縮の局所パターンは、磁気共鳴画像法(MRI)や陽電子放出断層撮影法(PET)による疾患の診断や、患部の特定に役立つ可能性がある[10]。
神経細胞やその周辺における蛋白質物質の異常蓄積は、変性神経組織における病理組織学的特徴である。アミロイドβ(Aβ)タンパク質、タウタンパク質、α-シンヌクレイン、トランザクティブレスポンスDNA結合タンパク質(TDP-43),およびミエリン破片が一般的な沈着物である[7]。ほとんどの神経変性疾患は、自然免疫応答因子と適応免疫応答因子の活性化による炎症反応を呈している。神経炎症は、少なくとも神経変性につながる最終的な共通経路の一つである。退化した脳組織では、活性化されたグリア細胞やアストロサイトが異常なタンパク質の蓄積を引き起こし、さらに神経損傷を増強させる[11]。
本総説では、病理組織学的な障害によって引き起こされ、特定の徴候や症状を引き起こす神経変性と、神経変性によって増強され、蛋白質障害を悪化させる神経炎症を含む、変性疾患の分子的、病理学的、臨床的所見を概説する。文献検索は、PubMed/MEDLINE で、各セクションのテーマに応じて適切な検索用語とフィルターを用いて行った。システマティックレビューでは、炎症の状況を示すためにプロ炎症性サイトカインと抗炎症性サイトカインのヒトサンプル研究を、主要な神経変性疾患sにおけるKYN代謝産物の関与を調べるために生理活性キヌレニン(KYN)のヒトサンプル研究を合成し、付録Aに詳細を記載した。最後に、病理組織学的診断技術の現状の限界、疾患の進行を防ぐための前臨床および前駆期診断バイオマーカーの必要性、新たなバイオマーカーや薬剤の発見に向けたアプローチの可能性を提示するために、各疾患の共通性について議論した。
2. 共通テーマ タンパク質性沈着物、神経変性、神経障害
中枢神経系における神経変性は、健康な老化やNDの徴候や症状の原因となっている[9]。老化や病的なプロセスは、分子レベルから細胞レベル、全身レベルに至るまで、神経伝達の複数のレベルで観察される [12]。蛋白質物質の異常沈着、その細胞および解剖学的分布、およびそれに続く神経変性は、神経病理学的診断に不可欠な病理学的基準を構成する[7]。
アルツハイマー病は、進行性の認知機能障害、特に記憶障害の陰湿な発症を伴う最も一般的な慢性NDである。初期段階では、運動機能障害や感覚障害はみられない。運動機能障害および自律神経機能障害は併存疾患と関連しており、後期に典型的なものである [13]。無気力、抑うつ、攻撃性、睡眠障害に加えて不安が優勢である[14]。前頭、側頭、頭頂皮質の萎縮、側脳室の側頭角の拡大、および内耳皮質、扁桃体、海馬の萎縮は、アルツハイマー病患者の特徴的な所見である[15]。アルツハイマー病は、不溶性の繊維状タンパク質であるアミロイドが神経細胞やグリアに異常に沈着するアミロイドーシスの中で最も頻度の高いタイプである。アミロイド沈着の形態や神経解剖学的位置は、アルツハイマー病や病期に特徴的である[7]。アミロイドβの沈着は主にアルツハイマー病患者の大脳新皮質と海馬に存在する。アミロイドβ沈着の神経解剖学的分布は、認知症状や心理行動症状への進行とよく相関している[16]。
アミロイドβ沈着はミクログリアの活性化、サイトカイン放出、反応性アストロサイトーシス、および炎症の誘導につながる[17]。アミロイドβオリゴマーはまた、プロテアソーム依存性のカドヘリン1(Cdh1)の分解を引き起こし、グルタミナーゼをダウンレギュレートさせる[18]。グルタミン酸の増加は、N-メチル-d-アスパラギン酸(NMDA)受容体を含むグルタミン酸受容体において持続的な低レベルの活性化を引き起こす。慢性的な興奮毒性の障害は、神経細胞の死と認知障害をもたらす[19]。アミロイドβオリゴマーは、細胞膜を介したCa2+フラックスを直接引き金にして細胞内Ca2+濃度を上昇させ、ミトコンドリアのCa2+過負荷、スーパーオキサイドラジカル誘発性酸化ストレス(OS)およびプロアポトーシスミトコンドリアタンパクの産生を誘導する[20]。
タウ症は、神経細胞やグリアにおけるタウタンパク質の異常蓄積によって特徴づけられる、アルツハイマー病において非常に有病率の高い疾患である。タウの沈着は認知症状の重症度との相関性が高いことが報告されている[7,21]。しかし、アルツハイマー病のタウ症はアミロイドβの沈着が二次的なものと考えられている[22]。OSは、神経原線維のもつれを形成するために重合するリン酸化されたタウタンパク質の異常な増加をもたらす。タウタンパク質は、神経細胞における微小管の集合と微小管ネットワークの安定性に重要な役割を果たしている。タウタンパク質の機能不全は、細胞骨格の構造的・制御的機能に影響を与え、軸索輸送異常、シナプス機能不全、神経可塑性の障害、神経変性につながる[23]。
パーキンソン病は、筋硬直、振戦、発話や歩行の変化などの症状を伴う運動に影響を及ぼす進行性の神経系障害である。パーキンソン病の初期段階では、主な運動機能障害には徐脈、安静時振戦、硬直などがあり、これらの症状はドーパミン作動性黒質変性に現れ、大きく依存している[24]。パーキンソン病患者は、無気力、激越、性欲亢進、病的ギャンブルなどを含むいくつかの精神行動症状を経験することがある。精神病、幻覚、抑うつ、および不安はまれではなく、症状は疾患の初期段階で、時には古典的な運動症状が出現する前にも現れることがある[25]。全般性不安障害は一般的であるが、しばしば認識されないことが多い[26]。黒質質のパーコンパクトの神経細胞の喪失およびグリオーシス、およびルビー小体(LB)好酸球性から好塩基球性の同心円状構造で、末梢のハローを持つ構造物で、基底核などの色素核に存在することは、パーキンソン病の特徴である。LBsには、アルツハイマー病の老人斑の非アミロイド成分として発見されたα-synの異常凝集体が含まれている。その後、家族性パーキンソン病ではα-syn遺伝子の変異が発見された。α-synの病理学的凝集はシヌクレイン病と呼ばれている[7]。オリゴデンドロサイトにおけるα-synの凝集体の蓄積は、グリア細胞質内包物を形成し、多系統萎縮症に特徴的であるが、LB病理学を欠く。脳内のα-synフィブリル化とLB形成を支配するメカニズムは、まだ十分に理解されていない [27]。
MSは、脳や脊髄の神経細胞が損傷を受ける自己免疫性脱髄疾患である。MSのより一般的な症状は、運動機能や自律神経機能障害から、歩行困難、麻痺、痙縮、視力障害、めまいやめまい、失禁、便秘、性的問題、痛み、認知や感情の変化、およびうつ病などの心理行動障害に広く範囲がある。メタアナリシスでは、MS患者では不安や抑うつの有病率が高いことが示されている[28]。MSの神経病変は、白質や脊髄に発生するプラークと呼ばれる多数のグリア瘢痕によって表される[29]。活動性プラークは、リンパ球、ミクログリア、マクロファージなどの活性化した単核細胞がミエリンやオリゴデンドロサイトをミエリンの破片、タンパク質、脂質に破壊することを特徴としている。その結果、プラークはグリア瘢痕組織を通る脱髄軸索を発達させ、不活性プラークを形成する。オリゴデンドロサイトが部分的に再髄化したプラークは影プラークである。中心部では不活性だが、周辺部では拡大しているプラークはくすぶりプラークである[30]。
HDは常染色体優性疾患であり、進行性で不可逆的な運動機能障害を引き起こし、協調性や歩行障害、認知や行動の変化をもたらす致死的な疾患である。軽度のHDでは起立性低血圧、過度の発汗、頻脈などの自律神経症状がみられるが、進行期には植物性症状が最も顕著である[31]。不安はHDに共通しており、有病率は13%~71%であり、有病者と有病前HD遺伝子保有者の間に差はない[32]。線条体、特に大脳皮質、淡蒼球、視床、脳幹、小脳を標的とした尾状核の変性と神経消失は、HDにおける特異的な神経病理学的所見である[33]。病理的変化の程度は障害の程度と相関している。小脳、視床、脳幹ではバルーン化した神経細胞が多く観察されている[7]。ハンチンチンタンパク質の変異体は、細胞が風船のように膨らんで膜が破裂する風船細胞死(BCD)と関連している。BCDとその結果として生じる壊死は、転写抑制による非定型細胞死(TRIアルツハイマー病)のメカニズムで引き起こされ、転写共活性化因子YAP(Yes-associated protein)と転写エンハンサー因子TEF(transcriptional enhancer factor)のレベルが低下する[34]。
ALSは、随意筋を制御するニューロンの機能不全をもたらす変性疾患である。ALSはしばしば、筋痙攣、筋無力症、または構音障害から始まる。その後、体を動かす、話す、食べる、呼吸を行う筋肉が関与するようになる [35]。ALS患者は、尿意切迫感、便秘、排尿運動などの自律神経系、消化器系、循環器系、神経精神系に影響を及ぼす軽度の症状を呈する [36,37]。ALS患者では不安障害およびうつ病が報告されている[38]。TDP-43プロテインオパシーは、細胞質、核、および細胞過程における核タンパク質の異常な包接に起因する。TDP-43は、ALSにおけるタンパク質凝集体の主要な構成要素であり、脊髄および脳幹の下部運動ニューロンおよび運動野の上部運動ニューロンに見られる。また、TDP-43は、末期のALSや認知症のALS患者の海馬、扁桃体、大脳皮質にも見られる[7]。TDP-43は、かなり高い配列特異性を持つRNA/DNA結合タンパク質である。TDP-43プロテインオパシーは、ユビキチン化および高リン酸化TDP-43の体形成、切り捨てられた毒性C末端TDP-43フラグメント形成、およびタンパク質凝集を含む、核から細胞質への誤局在化によって特徴づけられる[39]。
CJDの初期症状は、健忘症、人格変化、ミオクローヌスを伴う幻覚を含む急速に進行する認知症である。心理行動症状には、抑うつ、不安、被害妄想、精神病が含まれ、運動症状には、言語障害、運動失調、硬直した姿勢が含まれる[40]。疾患の持続期間に関連して、神経衰弱やグリア症などの皮質萎縮が観察されることがある。神経細胞の微細で合体した空胞を伴う灰白質の海綿状変化と星状細胞症はCJDの特徴である[7]。可溶性細胞性プリオンタンパク質(PrPC)の断片は、タンパク質分解によって生成され、ミエリンの恒常性維持や神経突起の伸長を誘導する働きをしている[41]。プリオン病の病態メカニズムは、プリオン蛋白質(PrPSc)のスクレイピーアイソフォームが、PrPCを病原性のあるコンフォメーション変化したβシートに富んだ自身の蛋白質へとテンプレート指示されたミスフォールディングを起こすことである。PrPScの蓄積はユビキチン/プロテアソーム系を乱し、オートファジー/リソソソーム経路に影響を与え、疾患初期の状態ではアンフォールドされたタンパク質応答経路を疲弊させ、神経細胞を弱らせ、シナプス喪失を引き起こし、細胞死経路を誘導する[42]。
HANDは、免疫系の悪化を特徴とする後期HIV-1感染による中枢神経系の合併症を指し、日和見感染や悪性腫瘍につながる。運動障害や行動障害を伴う進行性の認知症が代表的な症状である。主な運動症状は運動失調と振戦である。潜在期には、HIVに対する自己免疫反応で起立性失神、低血圧、下痢などの自律神経機能障害が現れることがある。認知機能障害は、記憶障害、構音障害、集中困難、判断力低下として現れることが多く、不安や気分転換もよく見られる[43]。死後の剖検や画像検査では、アルツハイマー病のような顕著な皮質萎縮が認められた。白質では灰白質よりも多核マクロファージや単核マクロファージが顕著で、多核細胞からは芽球性のウイルスが認められた。脳梗塞や虚血、出血、大血管アテローム性動脈硬化の部位は、抗レトロウイルス併用療法(cART)を受けているHIV感染高齢者によくみられる所見である[44]。
SNDは脳の血流不足が原因で二次的に引き起こされ、麻痺、関節痛、運動失調、失明、意識不明などの本来の障害以外の症状を呈する。自律神経系(ANS)の症状としては、動悸、血圧不整脈、非対称発汗、膀胱・腸機能障害、インポテンツなどがある[45]。虚血性脳卒中の急性期では、うつ病よりも不安の方が頻度が高い[46]。うつ病、不安、脳卒中との間には強い関連性がある。SNDや血管疾患とアルツハイマー病の複合疾患では、明らかなアルツハイマー病様の病理学的変化が観察された。
アミロイドーシス、タウポパシー、シヌクレイン病、および/またはTDP-43蛋白質障害のように観察される異常な蛋白質沈着は、解剖学的に対応する運動機能、感覚機能、および/または自律神経機能障害を示す神経変性の病因と進行に表裏一体の関係がある。ミクログリアやアストロサイトの活性化に伴う分子変化は、プロ炎症性サイトカインの放出を誘導し、高活性酸素および窒素種(ROSおよびRNS)の産生を増加させ、周囲の組織をさらに損傷させ、周囲のグリア細胞を活性化させる。認知領域が大きく影響を受け、うつ病や不安などの心理行動症状が神経変性疾患sでは一般的な所見となる(図1a,b)。
図1 神経変性疾患の開始進行仮説
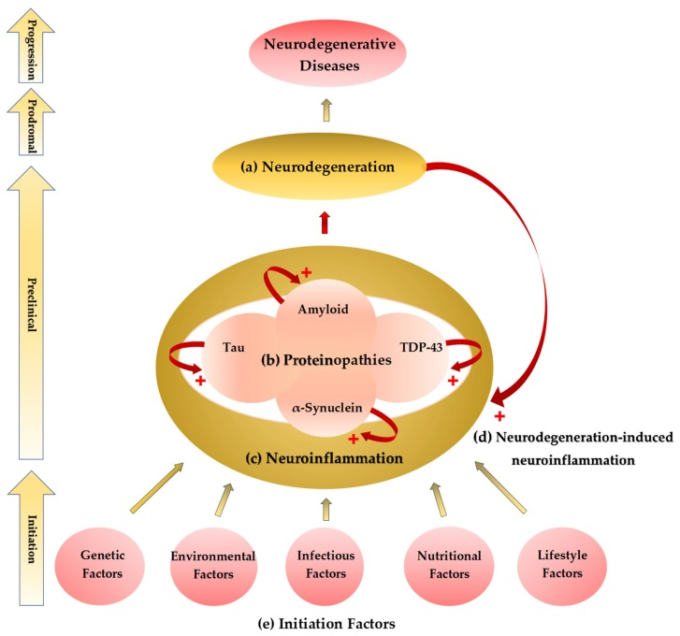
(a) 神経変性疾患の病理学的所見としては、神経衰弱や神経膠原病などの神経変性が一般的である。神経変性の場所や分布によって、神経学的、精神医学的な徴候や症状を呈する。
(b)病理組織学的検査では、変性神経組織に異常な蛋白質沈着が認められる。アミロイドーシス、タウオパチー、シヌクレアパチー、トランザクティブレスポンスDNA結合タンパク質(TDP)-43プロテインオパチーなどの蛋白質障害がある。
c)グリア細胞およびアストロサイトの活性化は、神経炎症を引き起こし、これはおそらく神経変性につながる最後の一般的な経路である。神経炎症は、脳組織内の異常な蛋白質性物質の蓄積を促進する。
(d) 神経変性もまた、神経炎症が周期的な増強を形成する。
(e)蛋白質障害-神経炎症-神経変性のサイクルは、まだ明確に定義されていない多因子性の開始因子によって引き起こされる可能性がある。遺伝的要素、環境的要素、感染症的要素、栄養的要素、生活習慣的要素などが神経変性疾患sの開始過程に寄与する主要な因子である。
3. 神経炎症。神経変性の共通の前奏曲
ミクログリア細胞とアストロサイトを媒介とする慢性的な中枢神経系の炎症は、NDDに共通する特徴である。プロ炎症性サイトカインで活性化されたミクログリア細胞、末梢由来の単球、血液脳関門(BBB)を介してリクルートされたマクロファージが炎症の主役である。ミクログリア細胞は、脳内のプロ炎症性サイトカインの主な供給源である[47]。ミクログリア細胞は、グルココルチコイド(GC)受容体とミネラルコルチコイド受容体の両方を発現することにより、コルチコステロンのピークに直接反応する。コルチコステロンはミクログリア細胞に影響を与えるが、海馬と前頭前野にはGC受容体が豊富に存在する[48]。
活性化したミクログリア細胞は、太く、短い突起、および膨潤した細胞体を特徴とし、より多くのサイトカインを産生し、末梢性単球をリクルートする。活性化したミクログリア細胞と末梢性単球は、インターロイキン(IL)-1β、腫瘍壊死因子(TNF)-α、IL-6などの炎症性サイトカインを分泌する [49]。
神経炎症の素因は、加齢、代謝性疾患、高血圧、脳卒中、うつ病、認知症などである。健康的な老化は慢性炎症と関連しており、不安やうつ病に対する脆弱性の増加に寄与している。肥満および高血圧における脂肪組織の機能不全は、インスリン抵抗性および心血管疾患の発症を促進する慢性低悪性度炎症に寄与する[50]。脳小血管疾患およびアテローム性動脈硬化症は、微小血管の低灌流、オリゴデンドロサイトの機能不全、脱髄を誘発し、永続的な低悪性度炎症を引き起こし、脳卒中のリスクを増幅させる。ウイルス感染は、BBB浸潤を介した血行性播種と神経細胞を介した逆行性播種によって神経炎症を引き起こす [51]。HIV感染は、サイトカインレベルの上昇、コレステロール、リポ多糖、アポリポタンパクE4,インスリン抵抗性、テストステロン欠乏症と関連しており、これらはすべて神経炎症に寄与する。HIV-1はマクロファージとR5 Tリンパ球で複製することができ、どちらも独立して認知症と関連している[52]。加齢は、代謝性疾患、高血圧、うつ病、認知症の有病率の高い危険因子である[53](図1c,e)。
3.1. 神経変性疾患における炎症性サイトカインに関する系統的レビュー
神経変性疾患sにおけるプロ炎症性サイトカインおよび抗炎症性サイトカインの状態を調べるために、システマティックレビューを実施した。選定基準とバイアス評価のリスクは付録Aに記載されている。合計23,206件の論文がデータベース検索に含まれた。次に、252のメタアナリシスとシステマティックレビュー、342の論文を適格性のために評価した。最終的に、システマティックレビューの対象となった論文は22本であった(図A1)。合成に含まれた論文、研究の種類、バイアス評価のリスクを表 A1に示す。エビデンスレベルは、アルツハイマー病、PD、ALS、CJDではバイアスのリスクが低いが、MS、HD、HAND、SNDではバイアスのリスクが高いと評価した(表A1)。
すべてのND(神経変性疾患)は、自然免疫応答因子とプロ炎症性サイトカイン、適応免疫応答因子と抗炎症性サイトカインの活性化によって証明される炎症反応の変化を呈している。サイトカインレベルの変化は、血漿と脳脊髄液(脳脊髄液)の両方でアルツハイマー病患者で観察された。プロ炎症性IL-1β、IL-6,およびTNF-α、ならびに抗炎症性サイトカイン、IL-1受容体アンタゴニスト、およびIL-10は、アルツハイマー病患者の脳脊髄液および血漿中の両方で上昇している[54]。C-Cモチーフケモカインリガンド5(CCL5)はアルツハイマー病で上昇している。C-X-Cモチーフケモカイン12はアルツハイマー病と負の関連がある。IL-2の血清レベルはアルツハイマー病の初期段階で上昇している[55]。
パーキンソン病では自然免疫と適応免疫の両方の活性化が観察される。パーキンソン病患者の血液サンプル中のサイトカインの状態をメタ解析したところ、パーキンソン病患者の血清中のプロ炎症性サイトカインIL-1β、IL-6,TNF-α、c-反応性タンパク質、CCL5のレベルが高く、抗炎症性サイトカインIL-2およびIL-10のレベルが高いことが示された。インターフェロン(IFN)-γ、IL-4,およびIL-8レベルには有意差は認められなかった[56]。パーキンソン病患者の脳脊髄液サンプルのメタ解析では、IL-1β、IL-6,およびトランスフォーミング成長因子(TGF)-β1のレベルが有意に高いことが報告されている[57]。プロ炎症性サイトカインであるIL-1,IL-12,IL-17,IL-22,TNF-α、およびインターフェロン(IFN)-γはMSで上昇しており、おそらく神経経路の脱髄に寄与していると考えられる。対照的に、IL-4やIL-10などの抗炎症性サイトカインはMSでは低い [58]。
HDの初期段階では、ミクログリアの活性化と神経炎症が特徴的である[59]。ミクログリア由来の炎症マーカーのうち、IL-6,マトリックスメタロペプチダーゼ9,血管内皮増殖因子(VEGF)TGF-β1のレベルが有意に上昇したのに対し、HDの血漿中のIL-18レベルは有意に低下した。血漿中のIL-6はHDの重症度と逆相関していた[60]。2年間の追跡調査では、TNF-αとIL-10レベルの上昇が認められた。運動症状のあるHD患者では、運動前症状のある患者よりもIL-6レベルが高く、IL-5レベルが低いことが観察された。IL-6とIL-8レベルはHDの進行と逆相関していた [61]。死後の研究では、HD患者の線条体ではC-Cモチーフのケモカインリガンド2やIL-10などの炎症性メディエーターの発現が増加し、大脳皮質と小脳ではIL-6,IL-8,マトリックスメタロペプチダーゼ9の発現が増加していることが明らかになった[62]。
TNF-α、TNF受容体1,IL-6,IL-1β、IL-8,およびVEGFレベルは有意に上昇したが、ALS患者の末梢血ではIL-2,IL-4,IL-5,IL-10,IL-17,およびIFN-γは変化しなかった [63]。顆粒球コロニー刺激因子、IL-2,IL-15,IL-17,単球化学戦術タンパク質-1,マクロファージ炎症性タンパク質(MIP)-1α、TNF-α、およびVEGFレベルは有意に上昇したが、顆粒球-マクロファージコロニー刺激因子は変化しなかった。IFN-γ、IL-4,IL-5,IL-6,IL-7,IL-8,IL-10,IL-12p70,IL-13,MIP-1β、および活性化時に調節される正常T細胞発現、および推定的に分泌される(RANTES)レベルは、ALS患者の脳脊髄液において有意な差はなかった[64]。これらの結果は、ALSにおける炎症反応の存在を確認するものである。IL-1β、IL-8,IL-17,および単球化学吸引性タンパク質のレベルは、CJD患者の脳脊髄液において有意に増加していた。また、CJD患者の脳脊髄液では、IL-4,IL-10およびIL-1受容体拮抗薬のレベルが有意に上昇し、TGF-β2のレベルが低下した[65,66,67,68]。
言語記憶能力とHIV陽性者との関連を調べるために、13種類のサイトカインの血漿中濃度をマルチプレックスビーズアレイ免疫測定法で測定した。IL-8およびIFN-γのレベルは正の相関があり、IL-10およびIL-18のレベルとC型肝炎感染は記憶力と負の相関があった[69]。HIV感染の神経認知障害患者の脳脊髄液では、IL-8,単球ケモタクチックタンパク、顆粒球コロニー刺激因子、誘導タンパク-10のレベルが上昇している[70]。TNF-αによって駆動される深部白質病変は認知機能の変化と関連しており、これはHANDの発症に寄与する脳内HIV感染の間接的な影響である[71]。
プロ炎症性サイトカインの産生量の増加および抗炎症性サイトカインの産生量の減少は、脳卒中の臨床転帰の悪化と相関している。TNF-α、IL-1β、IL-6,およびIL-10は虚血性脳卒中に関連する炎症性サイトカインである[72]。脳卒中患者では24時間以内に脳脊髄液 IL-6が上昇し、脳卒中後1週間は血清および血漿中で上昇する [73,74]。IL-10の低レベルは脳卒中後12時間以内に血漿中に示され、IL-10レベルは組織型プラスミノーゲンアクチベーター治療の24時間後に上昇する [74,75]。IL-10レベルは脳卒中24時間以内に低下し、脳卒中後72時間以上にわたって上昇する [76]。プロ炎症性サイトカインはすべての主要なND(神経変性疾患)で常に増加し、抗炎症性サイトカインはアルツハイマー病、PD、HD、CJD、およびHANDで増加し、アルツハイマー病、PD、およびCJDでは偏りのリスクが低かった。抗炎症性サイトカインはMSとSNDで減少し、ALSでは変化しなかったが、MS、HD、HAND、SNDではバイアスのリスクが高かった(表1)。
表1 神経変性疾患におけるプロ炎症性サイトカインレベルと抗炎症性サイトカインレベルの系統的な合成
神経変性疾患における炎症誘発性および抗炎症性サイトカインレベルの体系的な合成。↑:増加; ↓:減少; -: 変更なし。HIV-ヒト免疫不全ウイルス。
| 病気 | 炎症誘発性サイトカイン | 抗炎症性サイトカイン |
|---|---|---|
| アルツハイマー病 | ↑ | ↑ |
| パーキンソン病 | ↑ | ↑ |
| 多発性硬化症 | ↑ | ↓ |
| ハンチントン病 | ↑ | ↑ |
| 筋萎縮性側索硬化症 | ↑ | – |
| クロイツフェルト・ヤコブ病 | ↑ | ↑ |
| HIV関連神経認知障害 | ↑ | ↑ |
| 脳卒中誘発性の二次神経変性 | ↑ | ↓ |
4. 神経変性誘発性神経炎症、慢性炎症、アロスタティック負荷
神経変性、ミクログリアの活性化、そしてそれに続く慢性的な神経炎症は、神経変性疾患sの病態形成の中心的な役割を果たしている。活性化したミクログリア細胞は、アミロイドβプラークの形成と成長に積極的に関与していることが観察された。ミクログリア細胞はアミロイドβプラークを取り囲むようにアミロイドβを取り込んでいる。未解決のアミロイドβはクラスター状に発達する。死滅したミクログリア細胞は、蓄積したアミロイドβクラスターを細胞外に放出し、アミロイドβプラークの成長に寄与する[77]。活性化したミクログリア細胞は、神経原線維性のもつれを持つニューロンを取り囲み、樹状突起や軸索に損傷を与えることがわかっている。ファゴサイト化されたタウは、エクソソソームで放出され、隣接する細胞でのタウの伝播を促進することができる[78]。タウはプリオンのように凝集し、凝集したタウは、自然免疫応答の活性化に重要なセンサーの1つである結節様受容体P3-アポトーシス関連スペックク様タンパク質含有カスパーゼ活性化およびリクルートドメイン(CARD) (ASC) inflammasomeを活性化する[79]。LBを有する認知症患者では、初期のミクログリア活性化と中枢性および末梢性炎症が検出された[80]。さらに、動物を用いた研究では、あらかじめ形成されたα-シンフィブリルを注射することで、神経変性に先立って反応性ミクログリア症が誘発されたことが示されている[81,82]。細胞質TDP-43のレベルは加齢とともに増加し、TDP-43は炎症の調節因子であり、活性化B細胞経路の核因子κ-光鎖エンハンサーを活性化し、自然免疫応答の調節を緩和することが示唆された[83]。
神経グリア細胞と末梢マクロファージは、HIVにおける神経炎症の主な寄与者である。ミクログリア細胞およびアストロサイトは、HIV-1の細胞リザーバーであるだけでなく、HIV感染した末梢単球およびT細胞によって産生されるプロ炎症性サイトカインによっても活性化される[84]。神経変性は、遺伝的、脂質、タンパク質成分を傷つけて神経炎症を誘発する慢性OSと関連している。ミクログリア細胞は、アルツハイマー病、MS、PD、およびALSの場合のように、凝集したタンパク質を介してToll様受容体からの刺激により、有害なROSおよびRNS、プロ炎症性TNF-α、および興奮性グルタミン酸を分泌する[85](図1d)。
慢性炎症は、炎症性サイトカインの放出を維持するミクログリア細胞の持続的な活性化、ROSおよびRNSの増加、および神経毒性によって特徴づけられるが、これらはすべて、炎症サイクルを永続化させるために協働し、炎症をさらに長期化させ、これは神経変性疾患sの有害な病態である[86]。同様に、神経刺激性ウイルスは、ウイルス性疾患の根底にあるメカニズムへの長期的な神経免疫活性化を引き起こす [87]。
過度のストレス刺激は交感神経系を活性化し、GCの感度低下につながり、その結果、粘性のあるサイクルで免疫応答が増幅される。有害な刺激に対する生物の適切な応答は、増殖性不活性化と細胞の排除による損傷の修復とその後の回復をうまく導くことができる[88]。しかし、慢性的な免疫応答は有害で不適応な状態になり、結果として健康な状態に戻ることができなくなることがある。慢性ストレス応答の不適応期は、セリエの一般適応症候群またはアロスタシスの疲憊期を指す場合がある[89,90]。
有害な結果は免疫機構の疲弊によるものではなく、ストレスメディエーター自体が宿主にダメージを与えるものであり、特に疲弊段階では、ストレスメディエーターが宿主にダメージを与える。SterlingとEyerは、変化によって安定性を維持するプロセスを指すアロスタシスの概念を提案した[91]。アロスタシス負荷と呼ばれる、身体への不適切な反応への慢性的な曝露による適応コストを特徴とするアロスタシス系は、最終的には正常に機能しなくなり、その結果、疾患を引き起こし、最終的には死に至る[92]。
5. 神経変性疾患の背後にある病因論的リンク。トリプトファンと生理活性キヌレニン
トリプトファン代謝の乱れが神経変性疾患sで報告されている[93]。トリプトファンはタンパク質合成に用いられる必須アミノ酸であり、セロトニン、メラトニン、ニコチンアミド・アデニン・ジヌクレオチド(NAD+)などの生合成化合物の前駆体である。研究の大部分はセロトニン経路の領域に焦点を当てていたが、炎症、免疫系、神経系、精神疾患に関連する様々な生理活性化合物を産生するKYN経路に注目が集まっている[94]。トリプトファンの95%以上は、タンパク質合成を除いてKYN経路で代謝される。トリプトファン(TRP)は、コルチゾールによって誘導される肝性律速トリプトファン2,3-ジオキシゲナーゼ(TDO)およびユビキタス律速インドールアミン2,3-オキシゲナーゼ(IDO)1によってKYNに変換され、それぞれIFN-α、IFN-γ、およびTNF-αによって誘導される。KYNは、キヌレニナーゼによりアントラニル酸(AA)に変換され、KYN-3-モノオキシゲナーゼ(KMO)により3-ヒドロキシ-L-キヌレニン(3-HK)に変換され、KYNアミノトランスフェラーゼ(KATs)によりキヌレニン酸(KYNA)に変換される。また、KATは3-HKをキサントウレン酸(XA)に変換する。KYNAはNMDA受容体でのアンタゴニストである。XAは自己酸化によりシナバリン酸(CA)に変換される。AAと3-HKは3-ヒドロキシアントラニル酸(3-HAA)に変換され、さらにピコリン酸(PA)とキノリン酸(QA)に変換される。3-HKとQAはNMDA受容体でアゴニストである。QAは、TDO[95]のフィードバック阻害剤であるNAD+に変換される(図2)。
図2 l-トリプトファン代謝とキヌレニン経路
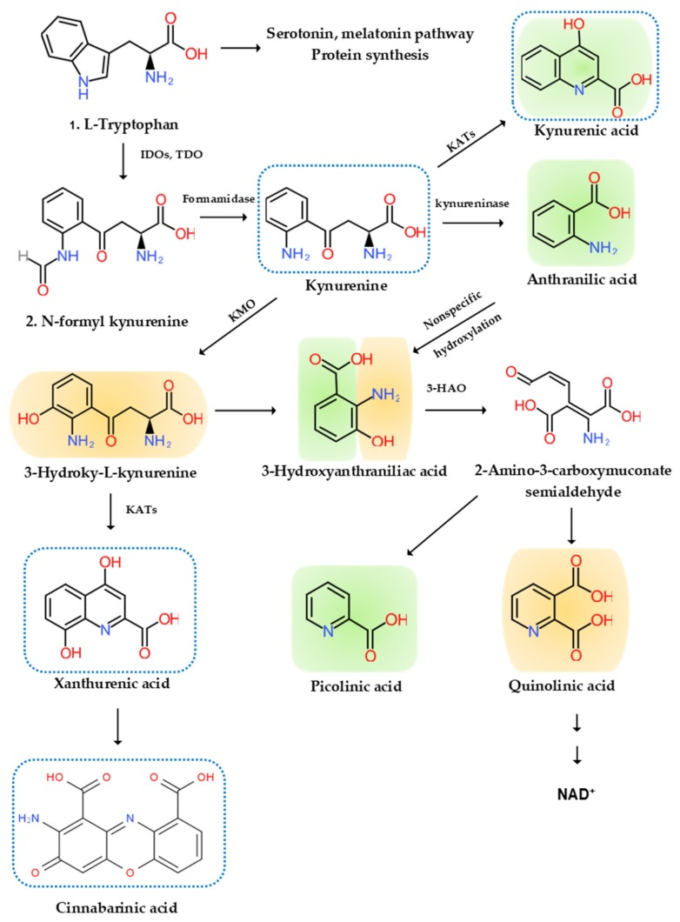
l-トリプトファン(TRP)のインドール環は、TRP ジオキシゲナーゼ(TDO)とインドラミン-2,3 ジオキシゲナーゼ(IDO)によって酸化され、N-ホルミル キヌレニン(KYN)を生成する。N-ホルミル KYN はフォルマアミダーゼによって KYN に変換され、KYN-3-モノオキシゲナーゼ(KMO)によってアントラニル酸(AA)KYN-3-モノオキシゲナーゼ(KMO)によって 3-ヒドロキシ-KYN(3-HK)KYN アミノトランスフェラーゼ(KATs)によってキヌレン酸(KYNA)の 3 つの下流代謝物の基質となる KYN に変換される。XAは自己酸化によりシナバリン酸(CA)に変換される。AAおよび3-HKは、3-ヒドロキシアントラニル酸オキシダーゼにより3-ヒドロキシAA(3-HAA)に変換される。3-HAAは、さらにピコリン酸(PA)およびキノリン酸(QA)に変換される2-アミノ-3-カルボキシムコン酸セミアリアルデヒドに3-ヒドロキシアントラニル酸ジオキシゲナーゼによって変換される。QAはさらにニコチン酸、ニコチン酸アデニンジヌクレオチド、ニコチンアミドジヌクレオチド(NAD+)に変換される。神経毒性、酸化性KYNはオレンジ色で、ニューロモデュラートリー抗炎症性、抗酸化性KYNは緑色で、アリール炭化水素受容体アゴニストは青色の点線で表される。
5.1. トリプトファン
TRPの異常なレベルとTRP代謝の障害は、ND(神経変性疾患)を患う患者で観察される。高齢者のアルツハイマー病、PD、HDなどの疾患患者では、循環TRPレベルの低さが関連している[96]。TRPサプリメントは認知機能を高めることが報告されている。短期的なTRP補給は、直列注意や反応時間、要約視覚記憶を改善し、慢性的な補給は顔認識記憶を増加させ、ベースラインの驚愕反応性を低下させた[97]。TRPで強化された食事は、MS患者の気分状態を改善することなく記憶プロセスを強化した[98]。
5.2. 生理活性キヌレニン
KYN 経路は、神経毒性、神経保護、抗炎症、酸化、抗酸化、免疫特性を含む幅広い活性を持つ、いくつかの小 型受容体アゴニストと生理活性分子を産生する。
5.2.1. 神経毒性のあるキヌレニン
QAは興奮毒性のあるNMDA受容体アゴニストであり、HDと同様の海馬病変を誘発する。ラットの線条体にQAを注射すると、線条体の萎縮と側脳室の拡大を伴う線条体内在性ニューロンの選択的な変性が誘導され、内在性グリア細胞と内被膜の脊髄軸索は免れる[94]。
5.2.2. 神経調節性キヌレニン
KYNAは、イオントロピー性α-アミノ-3-ヒドロキシ-5-メチル-4-イソオキサゾールプロピオン酸(AMPA)カイナイト、NMDA受容体の受容体アンタゴニストである[99]。しかし、用量に応じて、KYNAはAMPA受容体で異なる作用を発揮する。マイクロモル濃度では抑制的であるが、ナノモル濃度ではAMPA受容体のアロステリックな調節により促進的である[100,101]。α-7ニコチン性アセチルコリン受容体におけるKYNAの作用については議論の余地がある[102]。KYNAは、ミクログリア細胞、マクロファージ、単球のGタンパク質共役型受容体35(GPR35)に結合してグルタミン酸の放出を減少させる[94]。
PAは神経保護作用を有することが示された。PAは脳内のQAおよびカイニン酸誘発性神経毒性から保護する[103]。しかし、PAは神経毒作用はブロックするが、QAの興奮作用はブロックしない。PA の抗神経毒性作用の機序は不明であるが、亜鉛キレート化や一酸化窒素合成酵素の阻害に関与している可能性がある[104]。
5.2.3. 抗炎症性キヌレニン
KYNAはグリア、マクロファージ、および単球で発現するGPR35に結合し、細胞株でのプロ炎症性サイトカイン放出を減少させる[105]。AAおよびその5-水酸化代謝物は、潜在的な抗炎症特性を有している可能性がある。AAは、哺乳類の肝臓でミクロソームヒドロキシラーゼによって3-HAAに代謝される。AAとその関連代謝物の抗炎症反応は、AAがメフェナム酸やジクロフェナクなどのいくつかの非ステロイド性抗炎症薬の前駆体であるという事実と関連している[106]。AAおよび3-HAAは、プロ炎症性サイトカインIFN-γ、TおよびBリンパ球細胞増殖、Th1細胞活性、およびIL-1またはIFN-γによって誘導される神経毒性を抑制することが見出された。また、抗炎症性サイトカインIL-10を誘発する[107]。PAは免疫応答に影響を与え、抗真菌、抗腫瘍、抗菌活性を有する[108]。
5.2.4. 活性酸素種
3-HK、3-HAA、QAは神経毒性がある。3-HKと3-HAAは活性酸素を発生させ、OSの制御に関与している可能性がある[109]。3-HKレベルの上昇は興奮性障害と関連しており、神経障害患者で観察されている[110]。中間体である3-HKと3-HAAの神経毒性効果には、スーパーオキシドアニオンと過酸化水素の生成が関与しており、これらは髄膜炎の病態生理に関与する酸化的プロセスに寄与している[111]。3-HKはビタミンB6欠乏者の血漿中に増加する唯一のKYN代謝物であり、そのクリアランスにはピリドキサールリン酸(PLP)依存性酵素が関与していることを示唆している[112]。紫外線をろ過する3-HKや他のTRP代謝産物の産生は、加齢に伴う水晶体の黄色みを帯びた色素沈着の段階的な増加に寄与している[113]。
QAもまた、フリーラジカル代謝物である。プロ炎症性サイトカインIFN-γは、ヒトのミクログリア細胞およびマクロファージにおけるIDO、フォルマミダーゼおよびKMO活性を活性化し、結果としてQA合成の増加をもたらす。脳内のQA濃度は、免疫刺激後に抗炎症性ステロイド剤デキサメタゾンにより低下した[114]。
5.2.5. 抗酸化物質
KYNAは、様々な試験管内試験モデルで観察された活性酸素を消去し、組織損傷を防ぐオーバーシュート炎症を抑制する抗酸化代謝物である。KYNA は、主にスーパーオキサイドと過酸化水素の産生を含む FeSO4 をトリガーとした活性酸素の毒性を減少させることができる [109,115]。KYNA の産生が不十分な場合は、炎症反応における組織損傷や細胞増殖の一因となる可能性がある。
5.2.6. 免疫性キヌレニン
KYN、KYNA、キサンツレン酸、桂皮酸(CA)は、細胞質アリール炭化水素受容体(AhR)転写因子に結合し、適応免疫応答を抑制する[116]。AhRの活性化は、樹状細胞におけるIDOの発現と同様にTGF-β産生と関連しており、AhR-Src-IDO1経路を介して、ナイーブな分化クラスター(CD)4+ T細胞の免疫抑制性FoxP3+調節性T細胞(Tregs)への分化を促進するが、プロ炎症性Tヘルパー(Th)7細胞への分化は促進しない[117]。KYN経路の活性化はエフェクターT細胞を抑制し、Tregsを誘導し、免疫状態を寛容な状態に導く [118]。KYNの増加は、IL-2シグナル伝達の阻害を媒介してCD4+ T細胞の生存率を低下させる [119]。IDOを発現する細胞は、CD4+ T細胞の細胞傷害性Tリンパ球関連タンパク質4を発現するTregsへの分化を促進し、免疫応答をダウンレギュレートする [120]。さらに、KYNA産生の増加とKMO発現の低下は、エフェクターCD4+ T細胞応答の機能不全につながる[119]。NAD+は、CD4+ T細胞から分化したTh1,Th2,およびTh17を保護し、ナイーブなCD4+ T細胞のアポトーシスを誘導し、Tregsの数を減少させるが、誘導されたTregsをアポトーシスから保護する[121]。このように、KYN代謝経路の酵素および代謝物は、寛容性T細胞機能へのシフトを促進する。
PA はマクロファージを活性化する。PA はマクロファージの炎症性タンパク質-1αおよび-1βの産生を強力に誘導し、腫瘍抑制に寄与する [122]。IFN-γと相乗的に作用して、PAはマクロファージにおける誘導性一酸化窒素合成酵素メッセンジャーRNA(mRNA)の発現を増加させ、強力な細胞障害性および細胞静電作用をもたらす。PAで処理したマクロファージは、がん動物モデルにおいて腫瘍の増殖を抑制し、生存率を向上させる。PAは鉄の内因性金属キレート剤であり、試験管内試験および生体内試験では腫瘍細胞の増殖率を低下させるが、同量の投与では正常なヒト細胞には影響を与えなかった[123]。
5.3. キヌレニン経路酵素活性
炎症は、KYN経路のいくつかの主要な酵素を活性化する。肝臓のTDO、脳および末梢組織のIDO 1,肝臓、腎臓、および抗原提示細胞(APC)のIDO 2は、TRP代謝の第一律律速達酵素である[93]。GCストレスホルモンであるコルチゾールはTDOを活性化する。プロ炎症性サイトカインであるIFN-α、IL-1β、IFN-γ、およびTNF-αはIDO 1を活性化し、抗炎症性サイトカインであるIL-2,IL-4,IL-10,およびTGF-βは、IFN-γを介してIDO 1を阻害する[93]。IDO 2はプロ炎症性の役割を持ち、自己抗体産生に寄与している[95]。さらに、IDOで活性化された細胞は、APCのサイトカイン産生を、プロ炎症性サイトカインであるIL-12から抗炎症性サイトカインであるTGF-βおよびIL-10に変化させることができる[117]。
プロ炎症性サイトカインIFN-γは、ヒトミクログリア細胞およびマクロファージにおけるフォルマミダーゼを刺激し、KYN合成の増加につながる。KATの活性は、神経学的および認知症状や高齢者に関与している[124,125]。KATsの活性を高めるためには、親和性が低いため、より高い局所KYN濃度が必要である。KATsには、ビタミンB6の活性型であるPLPという補因子とα-ケト酸という副基質が必要である。[126]. PLPの主な供給源は食物であり、ヒトではサルベージ経路酵素によってPLP依存性酵素が分解される。サルベージ経路酵素の遺伝的機能不全およびPLPまたはピリドキサールキナーゼの薬物相互作用は、痙攣およびてんかん性脳症をもたらす。PLPの低レベルは、アルツハイマー病、PD、およびてんかんを含む神経学的障害と関連している[127,128]。高齢者の約20%は食事性ビタミンB6の摂取量が低いことが観察されており、ビタミンB6の補充は高齢者の認知パフォーマンスを改善する。葉酸、ビタミンB6,ビタミンB12が認知パフォーマンスと関連していることが提案されている[124,125]。
プロ炎症性サイトカインIFN-γは、ヒトのミクログリア細胞およびマクロファージにおけるKMO活性を刺激し、QA合成の増加につながる。マクロファージおよびグリア細胞の活性化は、QAの産生増加を誘導する[129]。より高いKYNA産生に加えて、より低いKMO発現は、エフェクターCD4+ T細胞応答の機能不全と関連しており、適応免疫応答の抑制につながる[130]。
KATを欠くが、KYNからQAへの逐次変換に関与するすべての酵素を含むミクログリア細胞は、3-HK、3-HAA、およびQAの局所的な合成を担っていると考えられている。さらに、免疫系が刺激されたときに観察されるこの主要なKP分岐の実質的なアップレギュレーションには、ミクログリア細胞が関与している。一方、KYNA合成は、KMO [131] を欠くアストロサイトでほぼ独占的に起こるようである(図2)。
5.4. 主要な神経変性疾患におけるキヌレニンに関する系統的レビュー
認知症(主要な神経認知障害)におけるKYNに関するシステマティックレビューが以前に報告されている [95]。この研究を補完するために、MS、ALS、HAND、およびCJDにおけるKYNのレベルについてシステマティックレビューを実施した。選定基準およびバイアスのリスク評価は付録Aに記載されている。我々のデータベース検索では、合計157の論文が見つかった。1つのシステマティックレビューと22の論文が適格性を評価された。最終的に 6 本の論文がこのシステマティックレビューに含まれた(図 A2)。方法論的品質とバイアス評価のリスクを表1
に示す。神経毒性および神経調節性KYNレベルのエビデンスレベルは、MS、ALS、HANDではバイアスのリスクが高いと評価されたが、CJDでは研究が見当たらなかった(表A2)。
KYN代謝物の不正確なレベルは、MS、ALS、CJD、およびHANDの患者で観察された。KYN/TRP比は、MS患者の血清中で有意に上昇していた。QAレベルは上昇し、NADHは低下していた。3-HKはMS群で有意に高かった。QA/KYNA比は、原発性進行性MS、二次進行性MS、再発寛解性MSで高かった。KYNA値は原発性進行性MSで最も高かったが、進行性MSでは低かった[132]。QA/KYNおよびQA/KYNA比の有意な上昇が、再発寛解型MSの再発サブグループの脳脊髄液で観察された。一次進行性MSではTRP、KYNA、QAのレベルが上昇したが、二次進行性MSではTRPとKYNAのレベルが低下した [133]。赤血球(RBC)では KAT I および KAT II 活性が有意に増加し、MS 患者の血漿では KYNA 濃度が有意に増加した [134]。
ALS患者では、血清および脳脊髄液中のTRP、KYN、QAが有意に増加し、血清中のPAが有意に減少していた。免疫組織化学では、ヒト白血球抗原DRアイソタイプ(HLA-DR)を発現する活性化ミクログリア細胞が有意に増加し、ALSの運動野と脊髄では神経細胞とミクログリアのIDOとQAの発現が増加していた[135]。KYNAは、健常対照群と四肢発症群に比べて、ALSのバルバー発症群の脳脊髄液で有意に高かった。また、重篤な臨床状態のALS患者の脳脊髄液では、対照群よりもKYNAが高かった。しかし、重度の臨床状態のALS患者の血清中のKYNAは、健康な対照群および軽度の臨床状態のALS患者よりも有意に低かった。ALS患者全体と健康な対照群との間には有意差は認められなかった[136]。ALSサブグループのKYNAレベルの違いは、ALSにおける疾患進行や異なる病態におけるKYNAの神経保護的役割を示唆している可能性がある。CJDにおけるKYNレベルについては、データベースに論文は見当たらなかった。神経精神症状と組織学的証拠が確認されたHIV-1に感染した患者の死後のヒト脳組織は、l-KYNとKYNAの上昇と関連しており、KAT Iは前頭皮質と小脳の両方でKAT IIと比較して顕著に増加していた[137]。
神経毒性のあるKYNはすべての主要な疾患で常に増加し、神経調節性KYNはMS、ALS、およびHANDではバイアスのリスクが高いまま減少した。神経調節性KYNは、バイアスのリスクが高いHANDで増加し、MSとALSではサブタイプに応じて混合していた(表2)。
表2神経変性疾患における神経毒性と神経調節性のキヌレニンレベルの系統的な合成
神経変性疾患における神経毒および神経調節キヌレニンレベルの体系的な合成。↑:増加; ↓:減少; ↑↓:混合結果; ?: わからない。
| 病気 | 神経毒性キヌレニン | 神経調節キヌレニン |
|---|---|---|
| アルツハイマー病 | ↑ | ↓ |
| パーキンソン病 | ↑ | ↓ |
| 多発性硬化症 | ↑ | ↑↓ |
| ハンチントン病 | ↑ | ↓ |
| 筋萎縮性側索硬化症 | ↑ | ↑↓ |
| クロイツフェルト・ヤコブ病 | ? | ? |
| HIV関連神経認知障害 | ↑ | ↑ |
| 脳卒中誘発性の二次神経変性 | ↑ | ↑ |
5.炎症性サイトカインと生理活性キヌレニンの合成。徴候と症状
主要なND(神経変性疾患)のうち、運動機能障害はPD、MS、HD、ALS、HAND、SNDで有意であり、自律神経機能障害はMSで有意であり、PD、HD、ALS、CJD、HAND、SNDで比較的有意である。心理行動症状はアルツハイマー病、MS、CJD、HANDで有意であり、PD、HD、ALS、SNDでは比較的有意であった。すべてのNDは、プロ炎症性サイトカインの増加による自然炎症性活性化の証拠を示した。アルツハイマー病、PD、HD、日中韓、およびHANDは、抗炎症性サイトカインの増加による二次適応免疫応答の活性化を示した。しかし、MSおよびSNDは抗炎症性サイトカインの減少レベルを示した。二次適応免疫応答の活性化、不活性化にかかわらず、本レビューで紹介した主要なNDの炎症プロファイルは、健康状態から乖離していた。急性および慢性炎症の原因または結果として、生理活性KYNのバランスの変化が観察された。神経毒性のあるKYNはすべての主要な疾患で増加している。神経調節性のKYNは、HANDとSNDで増加し、アルツハイマー病、PD、HDで減少し、MSとALSでは混在したままであった(図3)。
図3 炎症性サイトカインとキヌレニンの状態

神経変性疾患の徴候と症状。すべての神経変性疾患でプロ炎症性サイトカインと神経毒性サイトカインが有意に増加した。神経変性疾患の代表的な3つの症状の関与について、運動機能障害、自律神経機能障害、精神行動症状のプロファイルの下軸に記載した。
6. テーマのバリエーション?症候学と組織病理学の共通点
ND(神経変性疾患)という用語は1965年に比較的最近導入され 2002年には加齢に関連した難治性で大部分が治療不可能な中枢神経系の慢性進行性疾患(遺伝性疾患および特発性疾患の不明確なグループを含む)として体系的に定義された。しかし、この大バケツ用語の使用には異議があり、その使用は難治性で進行性の神経疾患のみを対象に提案された。この用語は、加齢に関連した疾患(必ずしも遺伝するわけではない)との関連性を示唆するものではなく、急速に進行する疾患を含むものである;したがって、「神経疾患」が適切な分類用語である可能性があると提案された[138]。
神経疾患は、その基礎となる病理の解剖学的位置を反映していると考えられている症状によって大きく定義される。神経疾患は、主要な臨床的徴候および症状、神経変性の解剖学的分布、または病理組織学的および分子学的所見によって鼻腔内学的に分類される医学的疾患群である [6]。共通の特徴は、遅発性疾患、神経萎縮、シナプス機能不全、ミトコンドリア機能不全、OS、神経細胞のアポトーシス、およびタンパク質のホメオスタシスの障害である[139]。
神経変性疾患sの共通の病理学的経過は、トポロジー的に選択的な神経変性をもたらす脳内の異常なタンパク質蓄積であり、疾患の徴候や症状を特徴づけている[6]。それにもかかわらず、いくつかの疾患は、病理学的および臨床的症状において実質的に重複している。アルツハイマー病患者の30%、LBを有するアルツハイマー病患者の方がパーキンソニズムを発症するのに対し、パーキンソン病患者の30~40%は疾患経過中に認知症を呈している[140,図4a]。140,図4a)。α-synの異常沈着は、PD、認知症を伴うPD、LBを伴う認知症(DLB)を含むLB障害の病態的特徴である[142]。DLBと認知症を伴うパーキンソン病はアルツハイマー病型の病態を共有しており、認知症を伴うパーキンソン病の約50%はアルツハイマー病との併存疾患の進行と考えられている[143,144](図4b)。基底核におけるLBの存在はパーキンソン病の病理学的特徴であると宣言されていたが、ユビキチン免疫組織化学検査により特発性パーキンソン病と診断された患者の100%で検出された[145]。
図4 神経変性疾患の共通点
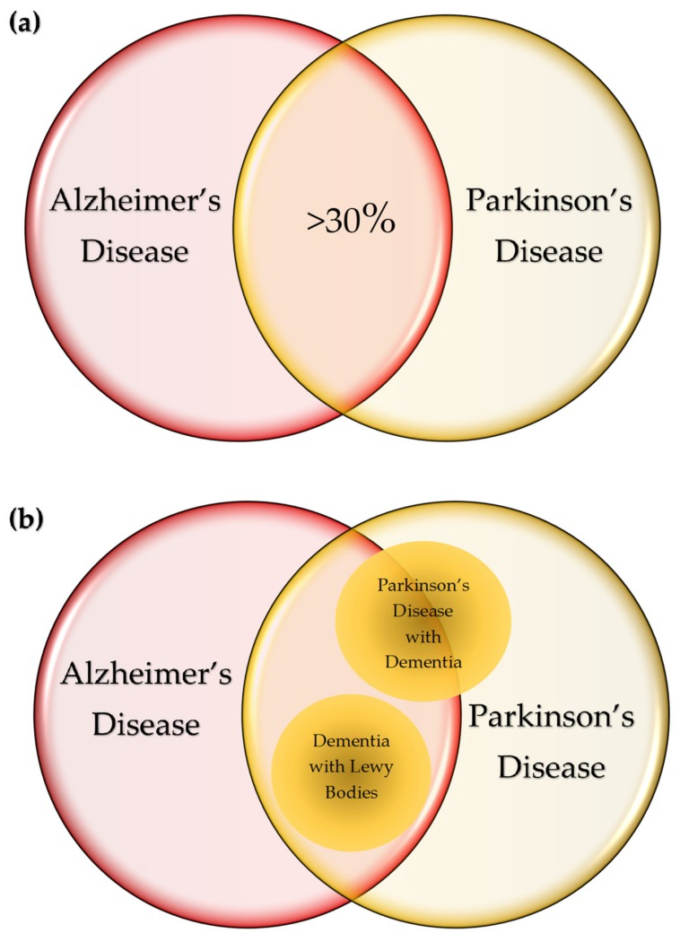
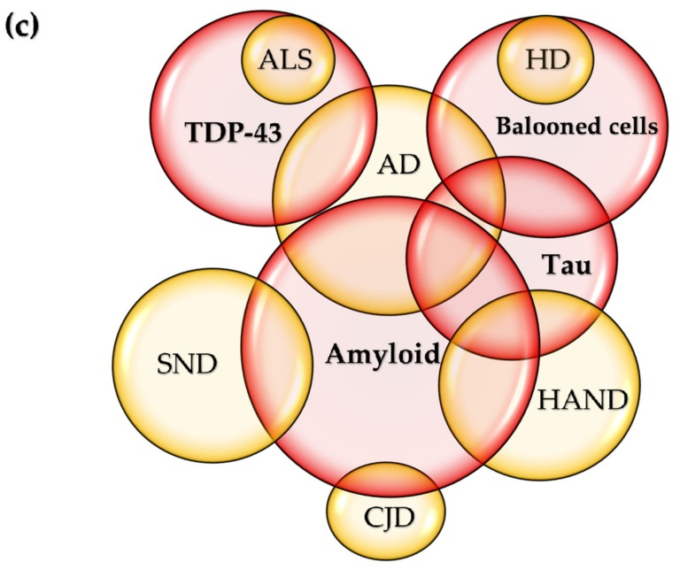
(a) アルツハイマー病(アルツハイマー病)では全体の30%以上の患者がパーキンソン病を発症するのに対し、パーキンソン病(PD)では30%以上の患者が疾患経過中に認知症を呈する。() 神経細胞やグリア細胞にアミロイドが沈着するのがアルツハイマー病の特徴であり、アルツハイマー病ではタウオパシーが最も多くみられる。アルツハイマー病のタウ症は、アミロイドβ(アミロイドβ)の沈着が二次的に起こると考えられている。筋萎縮性側索硬化症(ALS)に特徴的なTDP-43蛋白症は、アルツハイマー病患者の50%までに検出される。ハンチントン病(HD)に豊富に見られるバルーン化した神経細胞は、アルツハイマー病のようなタウロパチーに見られる。また、クロイツフェルト・ヤコブ病(CJD)の約10%にアミロイド沈着が認められ、アルツハイマー病様の認知症状から始まり、急速に認知症や死へと進行していく。びまん性細胞内アミロイド斑は、加齢やHIV関連神経認知障害(HAND)と関連している。高リン酸化タウの増加は、抗レトロウイルス併用療法(cART)を受けているHIV感染患者で報告されている。脳卒中誘発性二次神経変性(SND)は、アミロイド沈着異常などアルツハイマー病と多くの類似点を有している。
ALSに特徴的なTDP-43蛋白症は、アルツハイマー病患者の最大50%に検出される(図4c)[146]。プラークは異なるパターンを示し、臨床経過も変化するが、MSの亜種や臨床病期との相関は不明である[147]。脳および脊髄の灰白質および白質に複数のプラークが形成されることは、MSの特徴であるが、骨髄皮質MSと呼ばれるサブタイプでは、白質脱髄とは無関係に神経変性が起こりうることが報告されている[148]。
凸状輪郭の平坦化を伴う両側対称性の萎縮はHDに特有のものであるが、HANDにも萎縮変化が見られる。HDに豊富にみられるバルーン化した神経細胞はアルツハイマー病などのタオパチーにみられるが、その分布はHDとは異なる[149]。アミロイドβ沈着は、アルツハイマー病様の認知症状から始まるが、1年で認知症、昏睡、死亡へと急速に進行する稀な伝染性プリオン病であるCJDの約10%にも認められている[150](図4c)。アミロイドβ沈着物の進行性および非ランダムな広がりは、アミロイドβがアルツハイマー病の病態においてプリオンになるという仮説につながった[151]。タウもまたプリオンとして機能すると考えられている[152]。
側頭葉と前頭葉にびまん性の細胞内アミロイドβ斑が認められ、年齢やHIVの状態と関連していた。海馬における高リン酸化タウの増加がcARTを受けているHIV感染者で報告された(図4c)。このように、HAND患者におけるアルツハイマー病の鑑別診断は困難な問題である[153]。
SNDは、動物およびヒトの研究で観察された異常なアミロイドβ沈着、神経損失、およびグリア障害など、アルツハイマー病の病理学と多くの類似点を共有している。SNDは脳卒中の後遺症におけるアルツハイマー病やパーキンソン病の病態と関連しており、脳卒中が神経変性の引き金となることが提案されていた[154](図4c)。
共通点は分子レベルでも発見され、シリコで研究された[139]。疾患の家族歴はHDでは検出されるが、アルツハイマー病、PD、ALSの1~10%の症例でしか検出されない。共通疾患共通バリアント仮説は、単一核多型、コード化制御配列などの共通バリアントの遺伝子が、複雑な多遺伝子疾患の感受性につながることを提唱している[155]。多くのタンパク質が神経変性疾患sの制御に重要な役割を果たしている。危険因子や発症修飾因子については、遺伝子の重複が報告されている。19番染色体上に位置し、299-アミノ酸の糖タンパク質をコードするアポリポタンパクE遺伝子は、アルツハイマー病、PD、MS、SND、およびII型糖尿病の進行に影響を及ぼすと考えられており、共通の疾患における共通のメカニズムの仮説を支持している[157]。
7. 結論
科学者や医師は、疾患の原因を単一のユニークな出発点の結果に還元する傾向があり、たとえ因果関係との関連がないとしても、疾患の経過の中で生じた顕著な所見、徴候、症状には多くの注意が払われる。このような態度はしばしば誤りにつながる [158]。
神経変性疾患sは、病態生理学的交互性と臨床症状が時間の経過とともに一定の範囲で発現する動的な状態を持つ疾患である可能性がある。多くの慢性疾患は、遺伝的、環境的、社会経済的、文化的、および個人的なライフスタイルの素因と関連した多因子性の起源を持つ [159,160,161]。
遺伝的要因と環境要因との関連は、DNAメチル化、DNAヒドロメチル化、クロマチンリモデリング、ヒストン修飾、ノンコーディングRNA、マイクロRNAなどのエピジェネティクスのメカニズムによって明らかにされている。老化や中枢神経系疾患は、エピジェネティクスの乱れの影響を受けやすいと考えられている[162]。
内因性因子と外因性因子の両方が相互に影響し合い、適切に制御された範囲内に留まり、一定の量と期間までは無害なままで恒常性を維持することができる[89]。しかし、適応コストを犠牲にした不適切な身体反応の長期化は、生理学的な機能不全を引き起こし、最終的には疾患に至る[91]。
多因子性疾患は、患者間では共通ではないが、類似の病態認知的徴候や症状を呈する因子群を介して、ある経路で脱落し、進行することがある[163]。このように、疾患のユニークな機序論的原因を発見しようとする試みは、根拠がなく誤解を招くと考えられている。
多因子性疾患の発症においては、因果関係が双方向性になることがある。絶え間なく変化する多数の要因が有効な因果関係複合体を形成し、それが複数の因果関係の相互作用の始まりの役割を果たし、一緒になって病気になりやすい状態に導くことがある[165]。
一連のメタボロームバイオマーカーを用いたレトロスペクティブなコホート研究は、前臨床段階の化学的フィンガープリントを特定するのに役立ち、疾患の進行を遅らせるための新薬の発見に大きな価値をもたらす可能性がある。
患者に必ずしも共通するとは限らない多数の因子からなる近位の原因が最終的な原因へと発展し、最終的には本格的なND(神経変性疾患)に至る [166]。このような複雑な病因と進行状況は、少なくとも部分的には多因子性NDの異質性に寄与しており、分析を困難にし、かなりの量の不確実性を残している。
図5 推定される発症段階の進行と多因子性神経変性疾患の因果関係探索のためのアプローチ
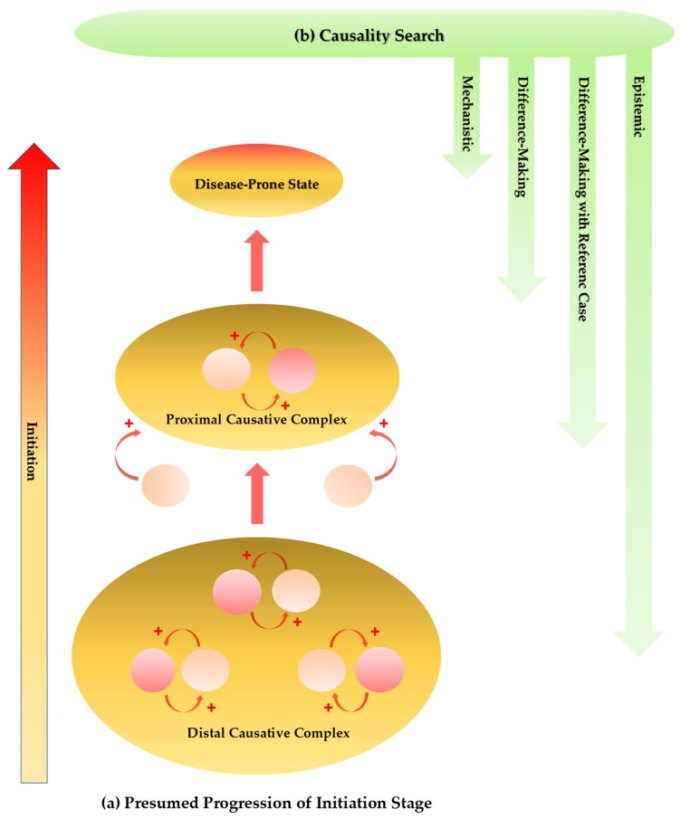
(a) 遺伝子、環境、感染症、栄養、生活習慣などの要素が発症段階の大きな要因となる。因果関係は双方向性になることがある。
原因が効果となり、効果が原因となり、複数の相互因果関係の中でイニシエーションの役割を果たす。絶え間なく変化する多数の要因は有効な因果複合体を形作る。
遠位因果複合体は、開始段階の進行で近位因果複合体に影響を与え、発展させ、最終的には病気になりやすい状態へと導く。
因果関係の評価は、多因性と確率論的決定論によって古典的な因果関係の基準を確立することによって行われる。
因果関係の評価には、機械論的解釈と差異化解釈が用いられる。参照ケースの組み込みは意味のある比較の解釈に達することを容易にするかもしれない。認識論的な因果関係の調査は機械論的な、相違作りの解釈によって減ずることができない証拠を査定する。
多因子性疾患の原因を探索するために、多原因性と確率論的決定論は、推定された原因と観察された効果との間の関係を証明する古典的な因果基準を精緻化している[167]。分析的近似は、因果関係における医学統計の役割の理解を深めることにも貢献している[168]。
参照ケースを組み込むことで、標準化を統合することで、意味のある比較の解釈に到達しやすくなるかもしれない [169]。さらに、因果関係は、単に因果関係の評価を知らせるだけの差異化や機械論的な解釈に還元されるものではない。
因果関係の要素で描かれた推論マップは、エビデンスを評価してエビデンスの階層を構築する認識論的因果性探索に従った説明・経営・予測の推論を可能にしている[170](図 5b)。
略語
| AA | アントラニル酸 |
| Aβ | アミロイド-β |
| 広告 | アルツハイマー病 |
| AhR | アリール炭化水素受容体 |
| AIDS | 後天性免疫不全症候群 |
| ALS | 筋萎縮性側索硬化症 |
| AMPA | α-アミノ-3-ヒドロキシ-5-メチル-4-イソキサゾールプロピオン酸 |
| APC | 抗原提示細胞 |
| ASC | カスパーゼ活性化および動員ドメインを含むアポトーシス関連スペック様タンパク質 |
| BBB | 血液脳関門 |
| CA | シンナバリン酸 |
| CD | 分化のクラスター |
| Cdh1 | カドヘリン1 |
| カード | カスパーゼの活性化とリクルートドメイン |
| カート | 併用抗レトロウイルス療法 |
| CCL5 | C–Cモチーフケモカインリガンド5 |
| CJD | クロイツフェルト・ヤコブ病 |
| CNS | 中枢神経系 |
| CSF | 脳脊髄液 |
| DLB | レビー小体型認知症 |
| GC | 糖質コルチコイド |
| GPR35 | Gタンパク質共役型受容体35 |
| 3-HAA | 3-ヒドロキシ-アントラリン酸 |
| 手 | HIV関連神経認知障害 |
| HD | ハンチントン病 |
| HIV | ヒト免疫不全ウイルス |
| 3-HK | 3-ヒドロキシキヌレニン |
| IFN | インターフェロン |
| ポンド | レビー小体 |
| IL | インターロイキン |
| KAT | キヌレニンアミノトランスフェラーゼ |
| KMO | キヌレニン-3-モノオキシゲナーゼ |
| KYN | キヌレニン |
| KYNA | キヌレン酸 |
| MIP | マクロファージ炎症性タンパク質 |
| MS | 多発性硬化症 |
| ND | 神経変性疾患 |
| NMDA | N-メチル-d-アスパラギン酸 |
| OA | 酸化ストレス |
| PA | ピコリン酸 |
| PD | パーキンソン病 |
| PLP | ピリドキサールリン酸 |
| PrP C | 細胞プリオンタンパク質 |
| PrP Sc | プリオンタンパク質のスクレイピーアイソフォーム |
| QA | キノリン酸 |
| ランテス | 活性化により調節され、正常なT細胞が発現し、おそらく分泌される |
| RBC | 赤血球 |
| RNS | 活性窒素種 |
| ROS | 活性酸素種 |
| SND | 脳卒中誘発性二次神経変性 |
| TDO | トリプトファンジオキシゲナーゼ |
| TDP | トランザクティブレスポンスDNA結合タンパク質 |
| TGF | トランスフォーミング成長因子 |
| Th | Tヘルパー |
| TNF | 腫瘍壊死因子 |
| 制御性T細胞 | 制御性T細胞 |
| TRP | トリプトファン |
| VEGF | 血管内皮増殖因子 |
| XA | キサンツレン酸 |
付録A
5.3 節および 5.5 節では、文献検索を行った。 5 では、データベース開始から 2020 年 2 月までの PubMed/MEDLINE および Google Scholar で文献検索を行い、検索語として「神経変性疾患」、「アルツハイマー病」、「パーキンソン病」、「多発性硬化症」、「ハンチントン病」などの同義語とその組み合わせを含む関連キーワードを用いた。”クロイツフェルト・ヤコブ病”、”筋萎縮性側索硬化症”、”(AIDS)腎症”、”脳卒中”、”サイトカイン”、”キヌレニン”、”キヌレニン酸”、”トリプトファン”、”インドールアミン2,3ジオキシゲナーゼ”、”トリプトファン2,3ジオキシゲナーゼ”、”キヌレニンアミノトランスフェラーゼ”、”キヌレニン3モノオキシゲナーゼ “などを検索した。検索フィルタには、「英語」、「レビュー」、「システマティックレビュー」、「メタアナリシス」などが含まれていた。第 3.1 節および第 5.4 節では、システマティックレビューの方法論として Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) [171]を採用した。文献検索は PubMed/MEDLINE で行った。M.T.が検索して適格性を評価し、さらにデータを抽出した(図A1,図A2)。
図A1 神経変性疾患における炎症性サイトカインに関する質的合成のフロー図
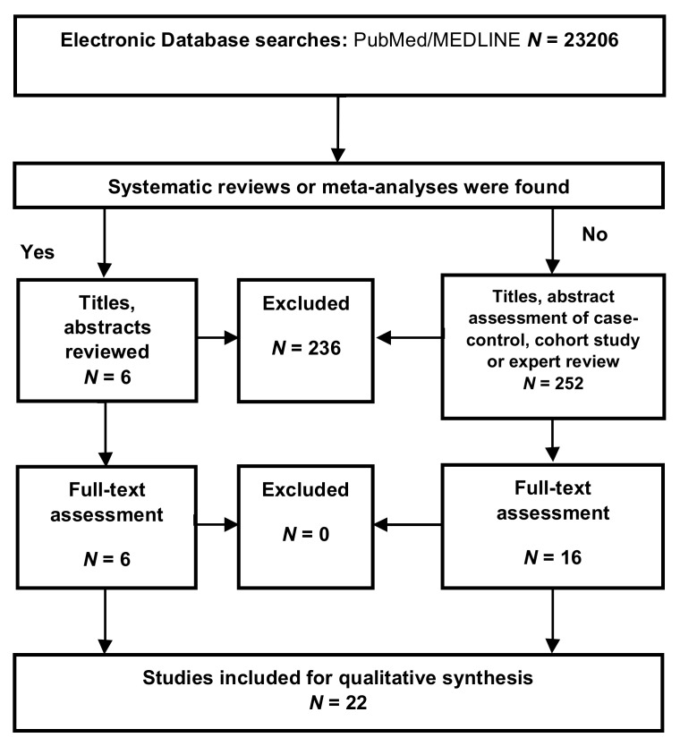
(Preferred Reporting Items for Systematic Reviews and Meta-Analyses(PRISMA)より採用
図A2 神経変性疾患におけるキヌレニンに関する質的合成のフロー図
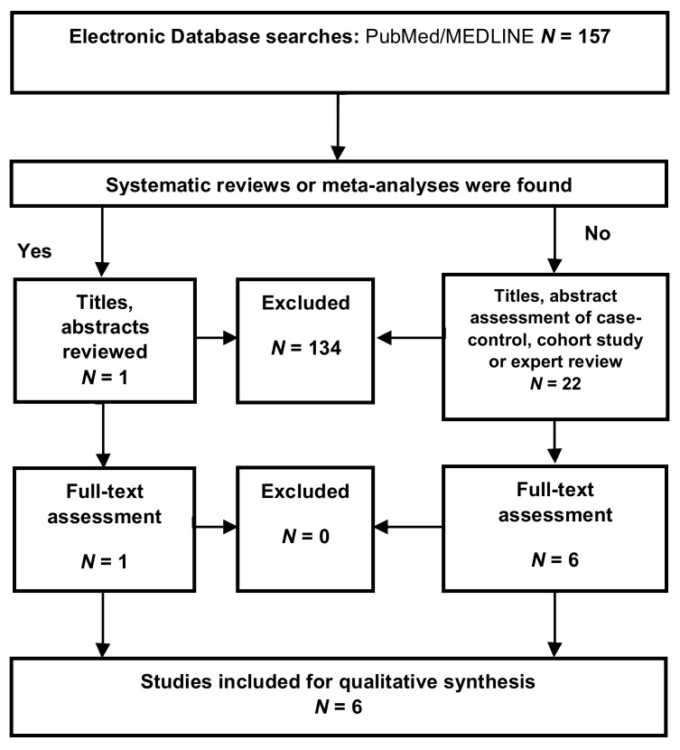
(Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA)より採用
付録 A.1. 包含基準
本レビューに含まれる論文は、以下の基準に基づいて選定された。(1)オリジナルの論文として発表された論文、(2)健康な対照群について十分な情報を提供している論文、(3)英語で書かれた論文、(4)オンラインで検索可能な論文。また、投薬中の患者を対象とした研究も含まれた。
付録 A.2. 除外基準
除外された論文は以下の通りである。(1)オリジナルデータがない論文、(2)動物実験の論文、(3)縦断的コホート研究を除いて対照がない論文。
付録 A.3. 選定プロセス
検索範囲は、データベース開始から 2020年2月までで、「神経変性疾患」「アルツハイマー病」「パーキンソン病」などの同義語や組み合わせを含む関連キーワードを検索語として使用している。”多発性硬化症”、”ハンチントン病”、”クロイツフェルト-ヤコブ病”、”筋萎縮性側索硬化症”、”エイズ関連腎症”、”脳卒中”、”サイトカイン”、”キヌレニン”、”キヌレニン酸 “など。重複を除外し、タイトルと要旨
をレビューした後、論文の全文を評価した。検索の優先順位は、メタアナリシス、システマティックレビュー、症例対照研究、コホート研究、レビューの順とした。メタアナリシスやシステマティックレビューが検索に見つからなかった場合は、症例対照研究、コホート研究、レビューのフルテキスト論文を検索対象とした。
付録A.4. データ抽出
対象となる論文からデータを抽出し、質的分析と批判的評価のために表にまとめた。各研究から収集されたデータには、発表年、研究デザイン、論文数または疾患集団と健常者集団、サンプルの種類が含まれる。
付録 A.5. 方法論の質の評価
方法論の質は、神経精神疾患ごとに、研究デザインの有無と数に応じて評価した。
表A1 システマティックレビューの合成、研究デザイン、リスクバイアス評価のためにサイトカインの状態に関する研究が含まれている
| 病気 | 研究タイプ | 参照番号 またはサンプル番号 (疾患/コントロール) |
サンプル | バイアスのリスク |
|---|---|---|---|---|
| アルツハイマー病[ 54、55 ] | ||||
| Brosseron et al。、2014 [ 54 ] | 系統的レビュー | 118 | 血漿、血清、CSF | リスクが低い |
| スチュアートとボーネ、2014年[ 55 ] | 系統的レビュー | 96 | 血液、血漿、血清、CSF | |
| パーキンソン病[ 56、57 ] | ||||
| Qin et al。、2016 [ 56 ] | 系統的レビュー | 25 | 血液 | リスクが低い |
| Chen et al。、2018 [ 57 ] | 系統的レビュー | 14 | CSF | |
| 多発性硬化症[ 58 ] | ||||
| Wang et al。、2018a [ 58 ] | 専門家によるレビュー | – | – | リスクが高い |
| ハンチントン病[ 60、61、62 ] | ||||
| Chang et al。、2015 [ 60 ] | ケースコントロール研究 | 15/16 | プラズマ | リスクが高い |
| Bouwens et al。、2015 [ 61 ] | コホート研究 | 124 | プラズマ | |
| Silvestroni et al。、2009 [ 62 ] | ケースコントロール研究 | 6/17 | 死後の脳組織 | |
| 筋萎縮性側索硬化症[ 57、63 ] | ||||
| Hu et al。、2017 [ 63 ] | メタアナリシス | 25 | 血清 | リスクが低い |
| Chen et al。、2018 [ 57 ] | メタアナリシス | 71 | CSF | |
| クロイツフェルト・ヤコブ病[ 64、65、66、67、68 ] | ||||
| Van Everbroeck et al、2002 [ 64 ] | ケースコントロール研究 | 19/19 | CSF | リスクが低い |
| Stoeck et al。、2005 [ 65 ] | ケースコントロール研究 | 20/20 | CSF | |
| Stoeck et al。、2006 [ 66 ] | ケースコントロール研究 | 23/111 | CSF | |
| Fujita et al。、2013 [ 67 ] | ケースコントロール研究 | 14/14 | CSF | |
| Stoeck et al。、2014 [ 68 ] | ケースコントロール研究 | 12/12 | CSF | |
| HIV関連神経認知障害[ 69、70、71 ] | ||||
| Correia et al。、2013 [ 69 ] | ケースコントロール研究 | 50/74 | プラズマ | リスクが高い |
| Yuan et al。、2013 [ 70 ] | ケースコントロール研究 | 64/43 | 血漿、CSF | |
| Seilhean et al。、1997 [ 71 ] | ケースコントロール研究 | 12/6 | 脳組織 | |
| 脳卒中誘発性二次神経変性[ 72、73、74、75 ] | リスクが高い | |||
| Lambertsen et al。、2012 [ 72 ] | 専門家によるレビュー | – | – | |
| Tarkowski et al。、1997 [ 73 ] | ケースコントロール研究 | 30/15 | CSF | |
| Perini et al。、2001 [ 74 ] | ケースコントロール研究 | 42/39 | 血清 | |
| Mazzotta et al。、2004 [ 75 ] | ケースコントロール研究 | 18/25 | プラズマ | |
表A2 システマティックレビューの合成、研究デザイン、リスクバイアス評価のために含まれたキヌレニンの状態に関する研究。
| 病気 | 研究タイプ | 参照番号 またはサンプル番号 (疾患/コントロール) |
サンプル | バイアスのリスク |
|---|---|---|---|---|
| アルツハイマー病[ 172、173、174、175 ] | ||||
| Guillemin et al。、2005 [ 172 ] | ケースコントロール研究 | 6/4 | 脳組織 | リスクが高い |
| ボンダ他、2010 [ 173 ] | ケースコントロール研究 | 12/7 | 脳組織 | |
| Gulaj et al。、2010 [ 174 ] | ケースコントロール研究 | 34/18 | 血清 | |
| Schwarcz et al。、2013 [ 175 ] | ケースコントロール研究 | 20/19 | 血清 | |
| パーキンソン病[ 176、177、178 ] | ||||
| Hartai et al。、2005 [ 176 ] | ケースコントロール研究 | 19/17 | プラズマ、RBC | リスクが高い |
| Lewitt et al。、2013 [ 177 ] | ケースコントロール研究 | 48/57 | CSF | |
| Chang et al。、2018 [ 178 ] | ケースコントロール研究 | 118/37 | プラズマ | |
| 多発性硬化症[ 132、133、134、135 ] | ||||
| Lim et al。、2017 [ 132 ] | ケースコントロール研究コホート研究 | 139/50 | 血漿、CSF | リスクが高い |
| Aeinehband et al。、2016 [ 134 ] | ケースコントロール研究コホート研究 | 30/6 | CSF | |
| Hartai et al。、2005 [ 135 ] | ケースコントロール研究 | 14/13 | CSF、RBC | |
| ハンチントン病[ 179、180、181、182 ] | ||||
| レイノルズとピアソン、1989年[ 179 ] | ケースコントロール研究 | 12/11 | 死後の脳組織 | リスクが高い |
| Beal et al。、1992 [ 180 ] | ケースコントロール研究 | 14–30 / 25–40 | 死後の脳組織 | |
| Jauch et al。、1995 [ 181 ] | ケースコントロール研究 | 17/17 | 死後の脳組織 | |
| Stoy et al。、2005 [ 182 ] | ケースコントロール研究 | 15/11 | 死後の脳組織 | |
| 筋萎縮性側索硬化症[ 135、136 ] | ||||
| Chen et al。、2010 [ 135 ] | ケースコントロール研究 | 35/140 | 血清、CSF、脳組織 | リスクが高い |
| Iłzeckaetal。、2003 [ 136 ] | ケースコントロール研究 | 32/30 | 血清、CSF | |
| クロイツフェルト・ヤコブ病 | ||||
| – | – | – | – | – |
| HIV関連神経認知障害[ 137 ] | ||||
| Baran et al。、2012 [ 137 ] | ケースコントロール研究 | 23/16 | 脳組織 | リスクが高い |
| 脳卒中により誘導される二次神経変性[ 183、184 ] | 不明 | |||
| ダーリントン他、2007 [ 183 ] | ケースコントロール研究 | 50/35 | 血清 | |
| Yan et al。、2015 [ 184 ] | ケースコントロール研究 | 28 / 20,11 | 血清、CSF | |
付録A.6. バイアス評価のリスク
バイアスのリスク評価は、Cochrane Handbook for Systematic Reviews of Interventions [185]を採用し、実施した。神経毒性KYNと調節性KYNの診断基準とレベルを、研究の有無、研究の種類、研究結果に応じて評価し、エビデンスレベルを高リスク、低リスク、不明瞭と判定した(表A3)。
表 A3
高リスク、低リスク、または不明確のエビデンスレベルを判断するために、メタアナリシスまたは系統的レビューの利用可能性の基準、研究タイプ、および矛盾する結果の有無にかかわらず、神経毒および調節KYNレベルを評価した。
| バイアスのリスク | 基準 |
|---|---|
| リスクが高い | メタアナリシスまたは系統的レビューなし、5つ未満のケースコントロールおよび/またはコホート研究、または専門家によるレビューのみの存在 |
| リスクが低い | 結果に矛盾することなく、少なくとも1つのメタアナリシスまたは系統的レビューが存在する |
| 不明 | ケースコントロール研究またはコホート研究、矛盾する結果を伴うメタアナリシス、または矛盾する結果を伴うケースコントロール研究のみの存在 |