
Contents
- Drug Bioavailability Enhancing Agents of Natural Origin (Bioenhancers) that Modulate Drug Membrane Permeation and Pre-Systemic Metabolism
- 要旨
- 1. はじめに
- 2. 投与経路
- 3. 経鼻投与経路
- 4. 経口投与経路
- 4.1. アロエベラ
- 4.2. 胆汁酸塩
- 4.3. ブラッククミン
- 4.4. カプサイシン
- 4.5. キャラウェイ
- 4.6. Cylcosporine A
- 4.7. キトサンとその誘導体
- 4.8. クルクミン
- 4.9. ジオスミン
- 4.10. エモジン
- 4.11. ガリン酸エステル
- 4.12. ゲニステイン
- 4.13. ゴックル抽出物
- 4.14. グレープフルーツジュース
- 4.15. リコピン
- 4.16. リセルゴル
- 4.17. ナリンギンとベルガモットン
- 4.18. 塩化パルミトイルカルニチン
- 4.19. ピペリン
- 4.20. ケルセチン
- 4.21. キニジン
- 4.22. レスベラトロール
- 4.23. シノメニン
- 4.24. カプリン酸ナトリウム(脂肪酸)
- 4.25. Zonula Occludens Toxin (Zot)
- 5. 肺への投与経路
- 6. 結論
Drug Bioavailability Enhancing Agents of Natural Origin (Bioenhancers) that Modulate Drug Membrane Permeation and Pre-Systemic Metabolism
www.ncbi.nlm.nih.gov/pmc/articles/PMC6359194/
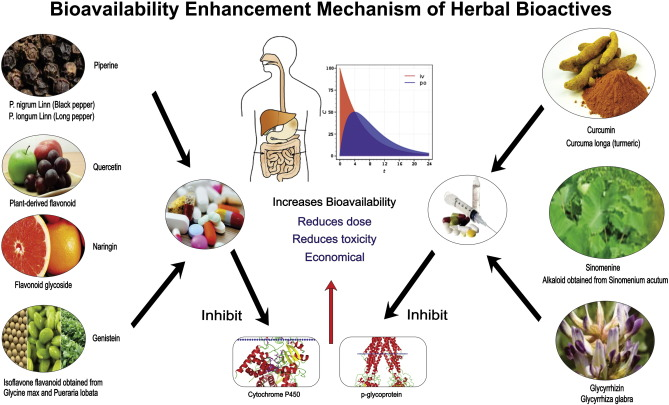
www.sciencedirect.com/science/article/abs/pii/S0367326X1400135X
要旨
多くの新しい化学物質が高い治療可能性で発見されているが、これらの化合物の多くは、溶解性が悪く、膜透過性が悪いために、薬物動態学的に好ましくない特性を示している。
後者は、主に上皮粘膜層によって課される脂質様のバリアに起因しており、治療効果を発揮するためには薬物分子が通過しなければならない。もう一つの障壁は、主に腸管腸球および肝肝細胞に存在するチトクロームP450酵素による薬物分子の全身性前代謝分解である。経鼻、頬、肺の投与経路はファーストパス効果を回避しているが、全身への薬物送達を達成するためには、粘膜表面を横切る薬物分子の吸収に依存していることに変わりはない。
膜透過および/または全身前代謝を調節することにより、全身血液循環に現れる未変化薬物の量を増加させることができるバイオエンハンサー(天然由来の薬物吸収エンハンサー)が同定されている。本論文の目的は、天然由来の吸収促進剤の概要と、経鼻、頬側、肺側、経口投与経路におけるそれらの主な作用機序を説明することである。
大型の親水性の治療薬のような生物学的利用性の悪い薬物は、しばしば注射によって投与される。バイオエンハンサーは、代替的な投与経路(すなわち、経口、鼻腔、頬または肺の投与経路)を介して、これらの生物学的利用性の悪い薬物の全身送達を可能にすることにより、患者に利益をもたらす可能性があり、また、低分子薬物の投与量を減少させ、それにより治療費を削減する可能性がある。
キーワード
バイオエンハンサー、チトクロームP450,薬物吸収促進剤、排出、代謝、P糖タンパク質、薬物動態相互作用、タイトジャンクション
1. はじめに
薬物吸収とは、薬物分子が投与部位から生体膜を越えて全身の血液循環に移行し、全身的な薬理作用を発揮する過程である。生体細胞膜は、そのリン脂質二重層構造により親油性を有している。したがって、分子は、生体膜を取り巻く水環境に溶解するのに十分な親水性を有するべきであるが、また、細胞間経路を介した受動的吸収を達成するためには、膜内に分割するのに十分な親油性を有するべきである[1]。隣接する上皮/内皮細胞は緊密な接合部によって接続されており、この接合部は水性チャネル/フェネストラエによって横断されており、小さな水溶性分子(<600 Da)のみが通過して傍細胞経路を介して吸収されることができる[2]。
多くの活性トランスポーター分子(取り込みトランスポーターと排出トランスポーターの両方を含む)が、様々な器官の様々な細胞タイプに存在している。腸管上皮細胞の漿膜に見られる薬物排出トランスポーターは、腸管上皮細胞内から構造的に多様な化合物を消化管内腔に送り出し、それによって薬物のバイオアベイラビリティを低下させることができる[3]。化合物の流出は、エネルギーを必要とする活性トランスポーターによって発生し、このプロセスは、アデノシン三リン酸(ATP)依存性である[4]。ATP結合カセット(ABC)トランスポータースーパーファミリーは、P-糖タンパク質(P-gp)多剤耐性タンパク質(MRP)乳がん耐性タンパク質(BCRP)で構成され、これまでに発見された最大かつ最も広く発現している流出トランスポーターの一つです[5,6,7]。上皮細胞を介した薬物透過以外に経口薬物のバイオアベイラビリティのもう一つの主要な決定因子は、オランザピン治療で観察されるように、薬物分子が全身循環に到達する前の取り込み中に行われる代謝である全身前代謝またはファーストパス代謝である[8]。全身性前代謝は、主に消化管上皮の腸球および肝臓の肝細胞で起こる。酵素のチトクロームP450(CYP)ファミリーは、全身性前代謝および全身性代謝の間の外来物質の酸化的代謝反応の大部分を占めている。30種類以上のヒトCYP酵素が同定されているが、その中でもCYP3A4はヒトにおける最も重要な薬物代謝酵素の一つであると考えられている[9]。
本論文では、バイオエンハンサーという用語は、共同投与された薬物分子が変化することなく全身循環に到達する速度および/または範囲を増加させる(すなわち、バイオアベイラビリティの向上)ことが可能な天然由来の分子に限定している。バイオエンハンサーが薬物分子のバイオアベイラビリティーを向上させる主なメカニズムとしては、受動的な細胞間薬物透過を増加させるための血漿膜の流動性の変化、傍細胞拡散を増加させるためのタイトジャンクションの調節、およびP-gp関連の排出阻害のような能動的な排出トランスポーターの調節などが挙げられる。腸管上皮および肝臓におけるCYP酵素の阻害は、全身の前代謝を減少させることにより、これらの酵素の基質となる薬物のバイオアベイラビリティーに大きな影響を与えうる[10,11,12]。
薬物投与の最も一般的な経路は、依然として経口経路である[2]。前述したように、薬物分子の経口バイオアベイラビリティは、その物理化学的特性(例えば、pKa、親油性、分子サイズ、電荷、溶解性および溶解性)[13]と、全身循環への移動中の酵素代謝の程度(全身前代謝またはファーストパス効果として知られている)とによって主に決定される消化管上皮膜への浸透能力によって決定される。薬物の経口バイオアベイラビリティに影響を与える他の要因としては、胃の空腹率、胃液のpH、他の化合物(例えば、他の薬物、食物またはハーブ)との相互作用、および活性トランスポーターへの親和性が挙げられる[2,14]。
経鼻経路を介した薬物投与は、局所的および全身的な薬物送達のために患者によって容易に達成することができ、これは非侵襲的であり、かつ無痛である。浸透性が高く、治療効果の迅速な発現を提供する比較的大きな上皮表面が利用可能である。中枢神経系を標的とする薬物については、鼻から脳への直接薬物送達が可能である[13,15,16]。さらに、経鼻的薬物投与は、肝臓の一次代謝をバイパスする[13,17,18]。しかし、保護粘膜層および毛様体クリアランスは、経鼻吸収に悪影響を及ぼす可能性がある[13,15]。
胃液中で不安定な薬物や、一次パス代謝の影響を強く受ける薬物には、頬腔投与ルートが良い代替手段となる。しかし、頬粘膜での吸収は、表面積が限られていること、頬上皮組織の透過性が低いこと、唾液による薬物の除去、および頬粘膜内のペプチダーゼの存在により、相対的に遅くなる[15]。したがって、この薬物投与経路は、ほとんどの場合、非常に強力で低用量の薬物に適している[15]。
薬物投与の肺経路(すなわち、肺を介した投与)は、大きな表面積と豊富な血液供給のため、迅速な薬物送達と関連している[2,15]。肺薬物送達は、局所的な治療(例えば、気管支拡張剤)または全身的な薬物送達の両方のために、エアゾールまたはネブライザーなどの異なる投与形態を介して起こり得る[2]。揮発性麻酔薬の場合、または揮発性薬剤の場合、吸入経路が好ましい投与方法である[2,19]。
本論文では、薬物吸収促進剤に関する議論は、天然由来のバイオエンハンサーに限定している(したがって、純粋に合成された化学的浸透促進剤は除外している)。選択された生物学的増強剤については、薬剤のバイオアベイラビリティに対する主な効果、および試験管内試験試験および生体内試験試験で明らかにされた作用機序の観点から論じている。さらに、口腔内投与、鼻腔内投与、肺内投与、経口投与の4つの選択された投与経路のそれぞれについて、バイオエンハンサーの包括的なリストを表1に示した。
表1 鼻腔、経口、頬、肺への薬物送達を強化するために選択された天然のバイオエンハンサーとその主な作用機序の概要
| 投与経路 | バイオエンハンサー(クラス) | 生物学的情報源 | アクションのメカニズム | 研究デザインモデル | 研究化合物 | 参照 |
|---|---|---|---|---|---|---|
| 頬側 | アロエベラ(ジェル、全葉) | 植物(アロエベラ) | 細胞間調節 | Invitro(フランツ拡散セル) | ジダノシン:抗ウイルス逆転写酵素阻害剤 | [ 20 ] |
| 頬側 | キトサン(生体高分子) | 甲殻類および真菌からの脱アセチル化キチン | 粘膜付着; 脂質組織の変化と細胞間フィラメントの緩み | インビトロ(T146細胞1) | FITC-デキストラン:親水性多糖類 | [ 21 ] |
| 頬側 | キトサン(生体高分子) | 甲殻類および真菌からの脱アセチル化キチン | 粘膜付着; 粘膜調節 | エクスビボ(ブタ頬粘膜) | ヒドロコルチゾン:コルチコステロイド TGF-ベータ:サイトカインポリペプチド |
[ 22 ] |
| 頬側 | キトサン–TBA(チオレートポリマー) | 甲殻類および真菌からの脱アセチル化キチン | 粘膜付着; 粘膜調節 | エクスビボ(ブタ頬粘膜); インビボ(豚) | PACAP:下垂体アデニル酸シクラーゼ活性化ペプチド | [ 23、24 ] |
| 頬側 | タラ肝油抽出物(脂肪酸) | 動物(タラ) | メカニズムが指定されていません | エクスビボ(ハムスターチークポーチ) | 酒石酸エルゴタミン:エルゴペプチンアルカロイド | [ 25 ] |
| 頬側 | メントール(アルコール) | 植物(コーンミント、ペパーミント、または他のミントオイル) | メカニズムが指定されていません | エクスビボ(ブタ頬粘膜) | ジデオキシシチジン:ヌクレオシドアナログ逆転写酵素阻害剤(NRTI) | [ 26 ] |
| 頬側 | オレイン酸、エイコサペンタエン酸、ドコサヘキサエン酸(脂肪酸) | 動物(タラ) | メカニズムが指定されていません | invitro(膜のない溶解試験)、invivo(ラット) | インスリン:ペプチドホルモン | [ 27 ] |
| 頬側 | グリコデオキシコール酸ナトリウム(胆汁酸塩) | 腸内細菌の副産物 | メカニズムが指定されていません | エクスビボ(ブタ頬粘膜) | ジデオキシシチジン:ヌクレオシドアナログ逆転写酵素阻害剤(NRTI) | [ 28 ] |
| 頬側 | TMC(カチオン性ポリマー) | 化学修飾キトサン(甲殻類、菌類) | 粘膜付着; 粘膜調節 | エクスビボ(ブタ頬粘膜) | FD4:親水性多糖類 | [ 29 ] |
| 鼻 | キトサン(生体高分子) | 化学修飾キトサン(甲殻類、菌類) | タイトジャンクション変調 | インビボ(羊) | sCT:内因性ポリペプチドホルモン | [ 30 ] |
| 鼻 | キトサン(生体高分子) | 甲殻類および真菌からの脱アセチル化キチン | 粘膜付着の増加; タイトジャンクション変調 | インビボ(羊、ヒト) | モルヒネ:アヘンアルカロイド | [ 31 ] |
| 鼻 | キトサン–TBA(チオレートポリマー) | 甲殻類および真菌からの脱アセチル化キチン | 粘膜付着の増加; タイトジャンクション変調 | インビボ(ラット) | インスリン:ペプチドホルモン | [ 32 ] |
| 鼻 | TMC(カチオン性ポリマー) | 化学修飾されたキトサン(甲殻類および真菌) | 粘膜付着の増加; タイトジャンクション変調 | In vivo (rat) | マンニトール:糖アルコール | [ 33 ] |
| オーラル | (-)-エピカテキン(フラボノイド) | 植物(木本) | 代謝(グルクロン酸抱合)阻害 | エクスビボ(ラット小腸) | アルファナフトール:有機蛍光化合物 | [ 34 ] |
| オーラル | アロエベラ(ジェルと葉全体) | 植物(アロエベラ) | タイトジャンクション変調 | エクスビボ(ラット腸組織) | アテノロール:ベータ受容体活性化合物 | [ 35 ] |
| オーラル | アロエベラ(ジェルと葉全体) | 植物(アロエベラ) | タイトジャンクション変調 | インビトロ(Caco-2細胞2) | インスリン:ペプチドホルモン | [ 36 ] |
| オーラル | アロエベラ(ジュース) | 植物(アロエベラ) | 局所粘膜組織調節 | インビボ(ヒト) | ビタミンCおよびE:アスコルビン酸、トコフェロール、トコトリエノール。 | [ 37 ] |
| オーラル | アロエベラ(ゲル多糖類) | 植物(アロエベラ) | 代謝阻害; タイトジャンクション変調 | In vitro(Caco-2、LS180細胞3)、In vivo(ラット) | インジナビル:抗ウイルスプロテアーゼ阻害剤 | [ 38 ] |
| オーラル | BHCl(フラボノイド) | ベタイン 植物の酸性化(ビートルート:Beta vulgaris) |
代謝増強(胃のpHの一過性の再酸性化) | インビボ(ヒト) | ダサチニブ:プロテインキナーゼ阻害剤 | [ 39 ] |
| オーラル | キャラウェイ(フラボノイド) | 植物(子午線フェンネル/ペルシャクミン:Carum carvi) | 局所粘膜組織調節 | インビボ(ヒト) | リファンピシン:半合成リファマイシン誘導体、イソニアジド:イソニコチン酸誘導体、ピラジナミド:ニコチンアミドピラジン類似体 | [ 40 ] |
| オーラル | キトサン(生体高分子) | 甲殻類および真菌からの脱アセチル化キチン | タイトジャンクション変調 | Invitro(HT-29クローンB6細胞4) | ヘパリン:抗凝固剤 | [ 41 ] |
| オーラル | キトサン(生体高分子) | 甲殻類および真菌からの脱アセチル化キチン | タイトジャンクション変調 | インビトロ(Caco-2細胞2) | キトサン–(リサミン–ローダミン標識) | [ 42 ] |
| オーラル | キトサン–TBA(チオレートポリマー) | 甲殻類および真菌からの脱アセチル化キチン | 粘膜付着; タイトジャンクション変調 | エクスビボ(モルモット小腸粘膜) | セファドロキシル:セファロスポリン | [ 43 ] |
| オーラル | キトサン–TBA(チオレートポリマー) | 甲殻類および真菌からの脱アセチル化キチン | 粘膜付着; タイトジャンクション変調 | インビボ(ラット) | インスリン:ペプチドホルモン | [ 44 ] |
| オーラル | クルクミン(フラボノイド) | 植物(ウコン:クルクマロンガ) | 代謝(UDP-グルクロニルトランスフェラーゼ)阻害 | Invitro(ラットミクロソーム) | Mycohenolic acid: Immunosuppressant | [ 45 ] |
| オーラル | クルクミン(フラボノイド) | 植物(ウコン:クルクマロンガ) | 排出トランスポーター阻害; 代謝阻害 | インビボ(ウサギ) | ノルフロキサシン:フルオロキノロン | [ 46 ] |
| オーラル | クルクミン(フラボノイド) | 植物(ウコン:クルクマロンガ) | 代謝(CYP3A4)阻害 | Invitro(ヒト肝ミクロソーム) | ミダゾラム:ベンゾジアゼピン | [ 47 ] |
| オーラル | クルクミン(フラボノイド) | 植物(ウコン:クルクマロンガ) | 排出トランスポーター(P-gp)阻害; 代謝(CYP3A4)阻害 | インビボ(ラット) | ミダゾラム:ベンゾジアゼピン | [ 48 ] |
| オーラル | シクロスポリンA(免疫抑制剤) | 菌類(Tolypocladium inflatum Gams) | 排出トランスポーター(P-gp)阻害 | インビボ(ラット、犬) | クロピドグレル:血小板凝集阻害剤 | [ 49 ] |
| オーラル | ジオスミン(フラボノイド) | 植物(柑橘系の果物) | 排出トランスポーター(P-gp)阻害 | インビトロ(Caco-2細胞2) | ジゴキシン:ジギタリス配糖体 | [ 50 ] |
| オーラル | エモジン(アントラキノン誘導体) | 植物(センナ:Cassia angustifolia、アロエベラ(syn Aloe barbadensis)、ダイオウ:Rheum officinale) | 排出トランスポーター(P-gp)阻害 | インビトロ(MDR1-MDCKII細胞6、Caco-2細胞2) | ジゴキシン:ジギタリス配糖体 | [ 51 ] |
| オーラル | フルボ酸(有機酸) | 植物(分解物) | 代謝の向上(薬物の水溶性の向上) | インビボ(ラット) | グリベンクラミド:スルホニル尿素系糖尿病治療薬 インスリン:ペプチドホルモン ペンタゾシン:オピオイド鎮痛薬 |
[ 52 ] |
| オーラル | 没食子酸エステル(有機酸) | 植物(ガルナッツ、ウルシ、マンサク、茶葉、オーク樹皮) | 代謝(CYP3A)阻害 | Invitro(ヒト肝ミクロソーム) | ニフェジピン:カルシウムチャネル遮断薬 | [53] |
| オーラル | ゲニステイン(フラボノイド) | 植物(大豆:Glycine max、葛:Pueraria lobata) | 排出トランスポーター(MRP)阻害 | インビトロ(HT-29細胞4)、インビボ(ラット) | エピガロカテキン-3-ガレート(EGCG):フェノール系抗酸化剤 | [ 54 ] |
| オーラル | ゲニステイン(フラボノイド) | 植物(大豆:Glycine max、葛:Pueraria lobata) | 排出トランスポーター(P-gp、BCRP、MRP2)阻害; 代謝(CYP3A4)阻害 | インビボ(ラット) | パクリタキセル:四環系ジテルペノイド | [ 55 ] |
| オーラル | ゴクルエキス(ハーブ) | 植物(ハマビシ:ハマビシ) | 局所粘膜組織調節 | Invitro(ヤギ裏返し嚢) | メトホルミン:ビグアニド | [ 56 ] |
| オーラル | ゴクルエキス(ハーブ) | 植物(ハマビシ:ハマビシ) | 局所粘膜組織調節 | Invitro(鶏の裏返した腸) | メトホルミン:ビグアニド | [ 57 ] |
| オーラル | グレープフルーツジュース(柑橘系の果物) | 植物(グレープフルーツ:Citrus paradisi) | 排出トランスポーター(P-gp、MRP2); 代謝(CYP3A4)阻害; 腎取り込みトランスポーター(OATP)阻害 | いろいろ | いろいろ | [ 58 ] |
| オーラル | LSC(キトサン誘導体) | 修飾キトサン(甲殻類および真菌) | 粘膜付着の増加; タイトジャンクション変調 | In vitro(Caco-2細胞2)、In vivo(ラット)、Ex vivo(ラット腸) | インスリン:ペプチドホルモン | [ 59 ] |
| オーラル | リコピン(カロテノイド) | 植物(赤い果物と野菜) | 標的肝送達のためのデュアルカロテノイド/ LDL受容体メカニズム | インビボ(ヒト) | シンバスタチン:HMG–CoAレダクターゼ阻害剤 | [ 60 ] |
| オーラル | リゼルゴール(アルカロイド) | 植物(アサガオ植物:サツマイモ属) | 代謝阻害 | インビボ(ラット) | ベルベリン:ベンジルイソキノリンアルカロイド | [ 61 ] |
| オーラル | リゼルゴール(アルカロイド) | 植物(アサガオ植物:サツマイモ属) | 排出トランスポーター(BCRP)阻害; 代謝阻害 | Invitro(ラット肝臓ミクロソーム) | クルクミン:ショウガ 科スルファサラジン:アミノサリチル酸剤 |
[ 62 ] |
| オーラル | モリンガオレイフェラポッド(伝統的な漢方薬) | 植物(Moringa oleifera) | 代謝(CYP450)阻害 | インビボ(マウス) | リファンピシン:半合成リファマイシン誘導体 | [ 63 ] |
| オーラル | ナリンギン(フラボノイド配糖体) | 植物(グレープフルーツ、リンゴ、タマネギ、お茶) | 排出トランスポーター(P-gp)阻害; 代謝阻害 | インビボ(ラット) | ジルチアゼム:ベンゾチアゼピン誘導体 | [ 64 ] |
| オーラル | ナリンギン(フラボノイド配糖体) | 植物(グレープフルーツ、リンゴ、タマネギ、お茶) | 代謝(CYP3A4)阻害 | インビボ(ラット) | タモキシフェン:選択的エストロゲン受容体モジュレーター(SERM) | [ 65 ] |
| オーラル | ナリンギン(フラボノイド配糖体) | 植物(グレープフルーツ、リンゴ、タマネギ、お茶) | 排出トランスポーター(P-gp)阻害; 代謝(CYP3A4)阻害 | インビボ(ラット) | パクリタキセル:四環系ジテルペノイド | [ 66 ] |
| Oral | ナリンギン(フラボノイド配糖体) | 植物(グレープフルーツ、リンゴ、タマネギ、お茶) | 排出トランスポーター(P-gp)阻害; 代謝(CYP3A4)阻害 | エクスビボ(ラット裏返し腸嚢) | クロピドグレル:血小板凝集阻害剤 | [ 67 ] |
| オーラル | ナリンギン(フラボノイド配糖体) | 植物(グレープフルーツ、リンゴ、タマネギ、お茶) | 代謝(CYP3A4)阻害 | インビボ(ウサギ) | ベラパミル:カルシウムチャネル遮断薬 | [ 68 ] |
| オーラル | パルミトイルカルニチンクロリド(キレート剤) | カルニチンのエステル化植物/動物(各種) | タイトジャンクション変調 | インビトロ(Caco-2細胞2) | クロドロネート:ビスフォスフォネート | [ 69 ] |
| オーラル | ペパーミントオイル(ハーブ) | 植物(ペパーミント:ハッカピペルティタ) | 代謝(CYP3A)阻害 | エクスビボ(ラット腸組織) | シクロスポリン:免疫抑制剤 | [ 70 ] |
| オーラル | ピペリン(アルカロイド) | 植物(PiperlongumおよびPipernigrum) | 局所粘膜組織調節; 熱発生活性 | インビボ(ヒト) | B-カロテン:テルペノイド | [ 71 ] |
| オーラル | ピペリン(アルカロイド) | 植物(PiperlongumおよびPipernigrum) | 局所粘膜組織調節; 熱発生活性 | インビボ(ヒト) | コエンザイムQ10:ベンゾキノン | [ 72 ] |
| オーラル | ピペリン(アルカロイド) | 植物(PiperlongumおよびPipernigrum) | 排泄の減少(胃腸通過阻害;胃内容排出阻害) | インビボ(ラット、マウス) | フェノールレッド:スフェロイド | [ 73 ] |
| オーラル | ピペリン(アルカロイド) | 植物(PiperlongumおよびPipernigrum) | 代謝阻害 | インビボ(ヒト) | プロパノール:ベータ受容体活性化合物、テオフィリン:メチルキサンチン | [ 74 ] |
| オーラル | ピペリン(アルカロイド) | 植物(PiperlongumおよびPipernigrum) | 代謝(CYP450)阻害 | インビボ(ラット) | ニメスリド:非ステロイド性抗炎症薬 | [ 75 ] |
| オーラル | ピペリン(アルカロイド) | 植物(PiperlongumおよびPipernigrum) | 排出トランスポーター(P-gp)阻害 | インビボ(ラット) | フェキソフェナジン:テルフェナジン代謝物 | [ 76 ] |
| オーラル | ピペリン(アルカロイド) | 植物(PiperlongumおよびPipernigrum) | 代謝阻害 | インビボ(マウス) | レスベラトロール:ファイトアレキシン | [ 77 ] |
| オーラル | ピペリン(アルカロイド) | 植物(PiperlongumおよびPipernigrum) | 代謝阻害 | インビボ(ヒト) | ネビラピン:非ヌクレオシド逆転写酵素阻害剤 | [ 78 ] |
| オーラル | ピペリン(アルカロイド) | 植物(ヒハツとPiper nigrum) | 代謝阻害 | インビボ(マウス) | エピガロカテキン-3-ガレート(EGCG):フェノール系抗酸化剤 | [ 79 ] |
| オーラル | ピペリン(アルカロイド) | 植物(PiperlongumおよびPipernigrum) | 代謝阻害 | インビボ(ラット) | ペントバルビタール:バルビツール酸塩。 | [ 80 ] |
| オーラル | ピペリン(アルカロイド) | 植物(PiperlongumおよびPipernigrum) | 代謝(CYP3A4)阻害 | インビボ(ヒト) | カルバマゼピン:カルボキサミド誘導体 | [ 81 ] |
| オーラル | ピペリン(アルカロイド) | 植物(PiperlongumおよびPipernigrum) | 代謝(CYP450)阻害 | インビボ(ラット) | ナテグリニド:メグリチニド | [ 82 ] |
| オーラル | ピペリン(アルカロイド) | 植物(PiperlongumおよびPipernigrum) | 代謝(肝臓および腸のグルクロン酸抱合)阻害 | インビボ(ラット、ヒト) | クルクミン:ショウガ科の薬剤 | [ 83 ] |
| オーラル | ピペリン(アルカロイド) | 植物(PiperlongumおよびPipernigrum) | 代謝阻害 | インビボ(鶏) | オキシテトラサイクリン:細菌のタンパク質合成阻害剤 | [ 84 ] |
| オーラル | ケルセチン(フラボノイド) | 植物(柑橘系の果物、野菜、葉、穀物) | 排出トランスポーター(P-gp)阻害 | インビボ(ラット)、エクスビボ(ラットおよびニワトリの裏返した腸嚢) | ラノラジン:ピペラジン誘導体 | [ 85 ] |
| オーラル | ケルセチン(フラボノイド) | 植物(柑橘系の果物、野菜、葉、穀物) | 排出トランスポーター(P-gp)阻害 | インビボ(ラット)、インビトロ(Caco-2細胞2) | イリノテカン:細胞毒性アルカロイド | [86] |
| オーラル | ケルセチン(フラボノイド) | 植物(柑橘系の果物、野菜、葉、穀物) | 排出トランスポーター(P-gp)阻害 | インビボ(ラット)、エクスビボ(ラット腸裏返し嚢) | バルサルタン:アンジオテンシンII受容体拮抗薬 | [ 87 ] |
| オーラル | ケルセチン(フラボノイド) | 植物(柑橘系の果物、野菜、葉、穀物) | 代謝(CYP3A)阻害 | インビボ(ウサギ) | ベラパミル:カルシウムチャネル遮断薬 | [ 88 ] |
| オーラル | ケルセチン(フラボノイド) | 植物(柑橘系の果物、野菜、葉、穀物) | 排出トランスポーター(P-gp)阻害; 代謝(CYP3A)阻害 | インビボ(ウサギ) | ジルチアゼム:非ジヒドロピリジンカルシウムチャネル遮断薬 | [ 89 ] |
| オーラル | ケルセチン(フラボノイド) | 植物(柑橘系の果物、野菜、葉、穀物) | 排出トランスポーター(P-gp)阻害; 代謝(CYP3A)阻害 | インビボ(ラット) | ドキソルビシン:ダウノルビシン前駆体 | [ 90 ] |
| オーラル | ケルセチン(フラボノイド) | 植物(柑橘系の果物、野菜、葉、穀物) | 排出トランスポーター(P-gp)阻害 | インビボ(ヒト) | フェキソフェナジン:テルフェナジン代謝物 | [ 91 ] |
| オーラル | ケルセチン(フラボノイド) | 植物(柑橘系の果物、野菜、葉、穀物) | 排出トランスポーター(P-gp)阻害 | インビボ(ラット、犬) | クロピドグレル:血小板凝集阻害剤 | [ 49 ] |
| オーラル | ケルセチン(フラボノイド) | 植物(柑橘系の果物、野菜、葉、穀物) | 排出トランスポーター(P-gp)阻害; 代謝(CYP3A)阻害 | インビボ(ラット) | エトポシド:ポドフィロトキシン誘導体 | [ 92 ] |
| オーラル | ケルセチン(フラボノイド) | 植物(柑橘系の果物、野菜、葉、穀物) | 排出トランスポーター(P-gp)阻害; 代謝(CYP3A)阻害 | いろいろ | エピガロカテキン-3-ガレート(EGCG):フェノール系抗酸化剤 | [ 93 ] |
| オーラル | ケルセチン(フラボノイド) | 植物(柑橘系の果物、野菜、葉、穀物) | 排出トランスポーター(P-gp)阻害 | インビトロ(ヒトMCF-7 ADRr細胞7) | ドキソルビシン:ダウノルビシン前駆体 | [ 94 ] |
| オーラル | ケルセチン(フラボノイド) | 植物(柑橘系の果物、野菜、葉、穀物) | Efflux transporter (MRP) inhibition; metabolism (CYP3A) inhibition | インビボ(ラット) | タモキシフェン:選択的エストロゲン受容体モジュレーター(SERM) | [ 95 ] |
| オーラル | ケルセチン(フラボノイド) | 植物(柑橘系の果物、野菜、葉、穀物) | 代謝(CYP3A)阻害 | インビボ(ラット) | ピオグリタゾン:チアゾリジンジオン | [ 96 ] |
| オーラル | キニジン(クラスI抗不整脈薬) | 化学修飾:キニーネ 植物の立体異性体(キナの木:キナ属) |
排出トランスポーター(P-gp)阻害 | エクスビボ(裏返したラット腸嚢) | シャクヤク:シャクヤク誘導体 | [ 97 ] |
| オーラル | レスベラトロール(ポリフェノールファイトアレキシン) | 植物(ベリー、ブドウの皮、赤ワイン) | 代謝(CYP2C9、CYP2E1)阻害 | インビボ(ヒト) | ジクロフェナク:NSAID | [ 98 ] |
| オーラル | レスベラトロール(ポリフェノールファイトアレキシン) | 植物(ベリー、ブドウの皮、赤ワイン) | 排出トランスポーター(P-gp、MRP-2)阻害; 除去の減少; 腎取り込みトランスポーター(OAT1、OAT3)阻害 | インビトロ(Caco-2細胞2、モック-MDCK 、MDR1-MDCK 6、MRP2-MDCK 6、モック-HEK293、hOAT1-HEK293 8、hOAT3-HEK293 8細胞)、エクスビボ(ラット裏返し腸、ラット腎臓スライス) 、in vivo(ラット) | メトトレキサート:免疫抑制剤 | [ 99 ] |
| オーラル | シノメニン(アルカロイド) | 植物(Sinomenium acutum) | 排出トランスポーター(P-gp)阻害 | エクスビボ(裏返したラット腸嚢) | シャクヤク:シャクヤク誘導体 | [ 97 ] |
| オーラル | シノメニン(アルカロイド) | 植物(Sinomenium acutum) | 排出トランスポーター(P-gp)阻害 | インビボ(ラット) | シャクヤク:シャクヤク誘導体 | [ 100 ] |
| オーラル | カプリン酸ナトリウム(脂肪酸) | 化学修飾:カプロン酸の塩漬け 動物(油脂) |
タイトジャンクション変調 | インサイチュ(再循環腸灌流)、エクスビボ(裏返したラット腸嚢)、インビボ(ラット) | ベルベリン:抗糖尿病植物アルカロイド | [ 101 ] |
| オーラル | コール酸ナトリウム/リン脂質混合ミセル(胆汁酸塩) | 腸内細菌の副産物 | 粘膜調節 | インビボ(犬) | シリマリンの主成分であるシリビン(オオアザミ、マリアアザミから分離された抗肝毒性ポリフェノール物質) | [ 102 ] |
| オーラル | 大豆ホスホチジルコリン/デオキシコール酸ナトリウム(SPC / SDC)(胆汁酸塩) | SPC:植物(大豆:Glycine max) SDC:化学修飾:デオキシコール酸(腸内細菌の代謝副産物)の塩漬け |
粘膜調節 | インビボ(犬) | フェノフィブラート | [ 103 ] |
| オーラル | タマリキセチン(ケルセチンの代謝物)(フラボノイド) | 植物(ホグウィード/カウパースニップ:Heracleum stenopterum) | 代謝(CYP2Cアイソザイム)阻害 | In vitro(ラット肝ミクロソーム)、In vivo(ラット) | フルバスタチン:HMGCoAレダクターゼ阻害剤 | [ 104 ] |
| オーラル | TMC(カチオン性ポリマー) | 修飾キトサン(甲殻類、菌類) | 粘膜付着; タイトジャンクション変調 | インビトロ(Caco-2細胞2) | マンニトール:糖アルコール PEG 4000:ポリエチレングリコール |
[ 105 ] |
| オーラル | TMC(カチオン性ポリマー) | 修飾キトサン(甲殻類、菌類) | タイトジャンクション変調 | インビトロ(Caco-2細胞2) | マンニトール:糖アルコール FITC–デキストラン:親水性多糖類 ブセレリン:ゴナドトロピン放出ホルモンアゴニスト |
[ 106 ] |
| オーラル | TMC(カチオン性ポリマー) | 修飾キトサン(甲殻類、菌類) | タイトジャンクション変調 | インビトロ(Caco-2細胞2) | クロドロネート:ビスフォスフォネート | [ 69 ] |
| オーラル | ZOT(毒素および毒抽出物) | バクテリア(コレラ菌) | タイトジャンクション変調 | インビトロ(Caco-2細胞2) | PEG 4000:ポリエチレングリコール FITC–デキストラン:親水性多糖類 イヌリン:天然多糖類 パクリタキセル:四環式ジテルペノイド アシクロビル:HSV指定DNAポリメラーゼ阻害剤 シクロスポリン:免疫抑制剤 ドキソルビシン:ダウノルビシン前駆体 |
[ 107 ] |
| 肺 | アプロチニン、ベスタチン(プロテアーゼ阻害剤) | 動物(ウシの肺組織)、細菌(Streptomyces olivoreticuli) | 代謝阻害 | インビボ(ラット) | rhG-CSF:顆粒球コロニー刺激因子 | [ 108 ] |
| 肺 | キトサン(生体高分子) | 化学修飾:キチン 動物(甲殻類)、真菌の脱アセチル化 |
タイトジャンクション変調 | インビトロ(Calu-3細胞5); インビボ(ラット) | オクトレオチド:ソマトスタチン類似体 | [ 109 ] |
| 肺 | クエン酸(キレート剤) | 植物(柑橘系の果物と野菜)、菌類(Aspergillus niger) | 局所粘膜組織調節; 代謝阻害 | インビボ(ラット) | インスリン:ペプチドホルモン | [ 110 ] |
| 肺 | HPBCD、クリスメブ(シクロデキストリン誘導体) | 植物(でんぷん) | タイトジャンクション変調 | インビトロ(Calu-3細胞5) | マンニトール:糖アルコール | [ 111 ] |
| 肺 | ランタン、セリウム、ガドリニウム(ランタニド) | 自然の要素 | 薬物標的化 | インビボ(ラット) | インスリン:ペプチドホルモン | [ 112 ] |
| 肺 | グリココール酸ナトリウム(胆汁酸塩) | 腸内細菌の副産物 | タイトジャンクション変調 | エクスビボ(ウサギ気管および空腸) | 甲状腺刺激ホルモン放出ホルモン(TRH):視床下部三 ペプチドホルモンインスリン:ペプチドホルモン |
[ 113 ] |
| 肺 | タウロコール酸ナトリウム(胆汁酸塩) | 腸内細菌の副産物 | 代謝増強(インスリン六量体の解離); タイトジャンクション変調; 代謝(酵素分解)阻害 | In vitro(Caco-2細胞2)、In vivo(犬) | インスリン:ペプチドホルモン | [ 114 ] |
| 肺 | ジデオキシシチジン:ヌクレオシドアナログ逆転写酵素阻害剤(NRTI) | 植物(でんぷん) | タイトジャンクション変調 | インビトロ(Calu-3細胞5)、インビボ(ラット) | エノキサパリン:抗凝固剤 | [ 115 ] |
| 肺 | TMC(カチオン性ポリマー) | 化学修飾:キチン 動物(甲殻類)、真菌の脱アセチル化 |
タイトジャンクション変調 | インビトロ(Calu-3細胞5); インビボ(ラット) | オクトレオチド:オクタペプチド | [ 109 ] |
1 T146細胞:頬側上皮細胞。2 Caco-2細胞:ヒト上皮性大腸腺癌細胞。3 LS180細胞:腸管ヒト大腸腺癌細胞。4 HT-29クローンB6:ヒト大腸癌細胞。5 Calu-3細胞:哺乳類気道上皮細胞。6 MDR1-MDCKII/MRP2-MDCK細胞。多剤耐性1(MDR1)または多剤耐性関連タンパク質2(MRP2)遺伝子を持つマディン・ダービー犬腎臓細胞。7 MCF-7 薬物有害反応r(再指定NCI-薬物有害反応-RES)細胞:卵巣腫瘍細胞。8 hOATP1/3-EK293細胞:ヒト有機陰イオン輸送ポリペプチド1または3をトランスフェクトしたヒト胚性腎細胞。
2. 投与経路
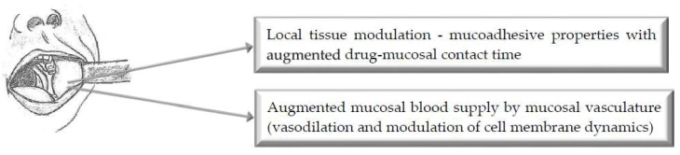
図1は、投与の頬側ルートを介して改善された薬物送達のための選択されたバイオエンハンサーの主な作用機序を示す図である。
図1 強化された口腔内薬物送達のためのバイオエンハンサーの主な作用機序の図示
2.1. アロエベラ
アロエベラゲルがブタの頬粘膜を横切るジダノシン(ddI)の透過性に及ぼす影響をフランツ拡散細胞を用いて調べた。対照液はpH7.4のリン酸緩衝生理食塩水(PBS)中のddIを単独で(5, 10, 15, 20 mg/mL)試験液はA. vera gel(0.25, 0.5, 1, 2, 4, 6% w/v)の存在下でddI(20 mg/mL)を含有していた[20]。
0.25~2% w/vの濃度では、A. vera gelは5.09 (0.25% w/v)から11.78 (2% w/v)までの範囲でdddIの口内透過性を有意に高めた。しかし、A. vera gelの高濃度(4及び6% w/v)では頬組織全体でのdddIの透過性が低下した。これは、A. vera gelの粘度が高く、薬剤の拡散に抵抗があったためと考えられる。A. vera gel は、HIV/AIDS 治療における ddI の口腔内浸透促進剤として使用できる可能性があると考えられる。
2.2. 胆汁酸塩
豚の頬粘膜を横切る2′,3′-ジデオキシシチジン(ddC)の試験管内試験浸透を、グリコラートナトリウムの非存在下および存在下で、インラインのフロースルー拡散細胞を用いて研究した[28]。10 mg/mL ddC,0.01% (w/v) ゲンタマイシン,0.6~50 mMの濃度のグリココール酸ナトリウムを含む、酵素を含まない歯肉液を模擬した新鮮な等張性マッキルバイン緩衝液(IMB、pH 7.4)をドナーチャンバーに添加した。0.8 mL/hの流量を維持し、サンプルを90分ごとに22.5時間採取した。その結果、ドナー濃度を1から 20mg/mLに増加させると、頬粘膜へのdddCの浸透が濃度依存的に増加することが示された。これは受動的な拡散を示している[28]。グリココール酸ナトリウム(4 mM)の存在下では、ddCの透過性は5.11±1.46×10-6 cm/sの見掛けの透過係数(Papp)値にまで有意に増加した(〜32倍)。一方、低濃度のグリココール酸ナトリウム(4mM未満)では、限定的な透過性向上効果が観察されたが、高濃度のグリココール酸ナトリウム(10mM及び50mM)では、Papp値は5.61±1.06×10-6 cm/sに増加しただけであった。これまでの研究では、4 mMのグリココール酸ナトリウムがグリココール酸ナトリウムの臨界ミセル濃度(CMC)に近いことが示されている[116,117]。グリココラートナトリウムは膜脂質をグリココラートナトリウムミセルに取り込むことで可溶化するため、4mM以下の濃度では膜脂質の増強効果が低いことが期待されていた。一方、CMC を超える濃度では、グリコ コリン酸ナトリウムミセルと脂質との界面飽和により増強効果が制限されている可能性がある [28]。
2.3. キトサンとその誘導体
キトサンは、大きな生理活性ペプチドであるトランスフォーミング成長因子-β(TGF-β)の頬粘膜組織への吸収を促進することが示された。2%キトサン-H(MW:1,400,000,脱アセチル化度:80%)を希釈乳酸溶液に溶解したゲルを調製した。I125標識TGF-b(MW:〜25Kda)を、PBSのコントロール溶液と同様にキトサンゲルに組み込んだ。連続フロー灌流チャンバーを用いて、約700μmの厚さのブタの頬粘膜皮膚を介したTGF-βの透過性を研究した。さらに、頬粘膜内でのTGF-βの局在を決定するために、水平切片化と計数を行った[22]。結果は、口腔粘膜はその大きさのためにTGF-βに対して比較的不透過性であるにもかかわらず、キトサンは、頬粘膜におけるTGF-β生理活性ペプチドの透過性を6~7倍に高めたことを実証した[22]。
さらに、対照のPBS溶液と比較して、TGF-βの量の増加が上皮の表層に認められた[22]。頬粘膜へのTGF-βの浸透性の向上は、キトサンの粘接着性による塗布部位での薬剤の保持力の向上の結果である可能性がある[22]。キトサンが頬粘膜上皮での薬物輸送を改善するもう一つの可能性のあるメカニズムは、上皮の細胞間領域での脂質組織への干渉である[22]。
別の試験管内試験研究では、キトサンをペプチドとタンパク質吸収のためのバイオエンハンサーとして使用した場合、頬部上皮TR146細胞培養モデルの経上皮電気抵抗(TEER)が減少したことが実証された。本研究では、キトサングルタミン酸濃度20μg/mL以上のキトサンは、pH6で大型親水性化合物である3H-マンニトールとフルオレセインイソチオシアネート標識デキストラン(FITC-dextrans)の輸送が増強されたことを示した。3H-マンニトールは最も高い細胞透過性を示し、FITC-デキストランでは分子量(MW)が4000 Da(FD4)FD10(MW10,000 Da)FD20(MW20,000 Da)と分子量の増加に伴って透過性の低下が観察された[21]。FD20を除くすべての被験物質のキトサンによる透過性の向上は、統計的に有意であった。未処理細胞と比較して、TR146細胞培養モデルのTEERは、20μg/mL以上の濃度のキトサン・グルタミン酸の存在下では、〜30%まで劇的に低下した[21]。鼻や腸の粘膜とは異なり、頬粘膜の細胞間バリアはタイトジャンクションに基づいていない[22]ため、タイトジャンクションの変調が透過性を高めるメカニズムであるとは考えられない。したがって、頬粘膜の脂質組織への干渉が、キトサングルタミン酸の存在下での薬物輸送の改善、または細胞間フィラメントの潜在的な緩みに関与していることが示唆された[21]。
チオール化キトサンは粘接着性と薬物透過性を大幅に改善する能力を示している[118]。チオール化キトサンを用いた粘接着性のある口唇ペプチド薬物送達システムは、下垂体アデニル酸シクラーゼ活性化ポリペプチド(PACAP)の口唇送達のためのアプローチとして、試験管内試験および生体内試験で設計され、評価された[23,24]。キトサン-4-チオブチルアミジン(キトサン-TBA)を合成し、酵素阻害剤および浸透メディエーターであるグルタチオン(GSH)でホモジナイズし、凍結乾燥し、平板状のディスクに圧縮した。すなわち、キトサン-TBA(69.5mg)GSH(3.75mg)ブリジ35(0.75mg)およびPACAP(1mg)からなる製剤A;一方、製剤Bは、キトサン-TBA(50mg)GSH(2.5mg)ブリジ35(2.5mg)PACAP(1mg)の最内層、およびキトサン-TBA(50mg)の最外層を含んでいた。さらに、製剤Bは、口腔内での損失を回避しつつ、頬粘膜への薬剤の一方向放出を確実にするために、片側にパームワックスコーティングを含んでいた [23]。対照製剤は、未修飾キトサンおよびPACAP(製剤C)または未修飾キトサン、Brij 35およびPACAP(製剤D)を含んでった。これらの試験用製剤は、ブタに6時間、口唇投与で投与された。製剤AとBでは1%の絶対的なバイオアベイラビリティが得られたが、対照製剤(製剤CとD)ではPACAPが全身循環に到達することさえできなかった[23]。
別の研究では、異なる四級化度(QDが4%、35%、90%)のトリメチルキトサン(TMC)は、粘接着特性の増加と、ブタの頬粘膜へのFD4(分子量(MW)4000 DaのFITC-デキストラン)の透過性の向上を示した[29]。豚頬粘膜の上皮を下層組織から剥離し、フランツ拡散細胞にマウントした。TMCポリマー溶液を、室温で穏やかに撹拌しながら4%(w/w)の濃度で調製し、次いで0.2%(w/w)の濃度でFD4を添加した[29]。引張応力試験機を用いて、頬頬粘膜および顎下ウシムチンに対するポリマー溶液の粘接着特性を評価した。試験管内試験浸透試験の結果から、切除した頬粘膜組織へのFD4の浸透は、浸透促進剤がない場合には不十分であり、困難であることが示された。粘接着性能は、使用される媒体または生物学的基質に関係なく、QDの増加とともに増加した。しかし、FD4の透過性の増加はpH6.4の緩衝液でのみ観察され、これはポリマー溶解度の増加に起因することができる。最高の浸透促進結果は、高QDの低分子量TMCで得られた[29]。
2.4. 脂肪酸
不飽和脂肪酸(オレイン酸、エイコサペンタエン酸(EPA)ドコサヘキサエン酸(DHA)を含む)をプルロニックF-127(PF-127)ゲル製剤中にインスリンと同時に投与した場合の効果を調査した研究がある[27]。PF-127(MW:12,500 Da)を、不飽和脂肪酸の有無にかかわらず、インスリンゲル製剤を調製するために使用した。PF-127および不飽和脂肪酸の最終濃度は、それぞれ20および5%であった。対照製剤は、インスリンのみを含む20%のPF-127ゲルから構成されていた[27]。試験管内試験での放出試験は、膜なし溶解試験を用いて実施した。生体内試験試験は、麻酔をかけたラットに0.2 mLの製剤(インスリン投与量、25 IU/kg)を口内投与することにより実施した。血清グルコース値を測定するために、経口投与の前後に血液サンプルを採取した [27]。
不飽和脂肪酸を含むPF-127ゲル(インスリン投与量、25 IU/kg)では、インスリン遊離速度の低下と顕著で持続的な血糖降下作用が観察された。放出速度の低下は、親水性溶質が拡散する水性チャネルの数および寸法の減少に一部起因するか、または製剤の粘度に起因する可能性がある[27]。血清グルコースレベルは、不飽和脂肪酸を含むすべての製剤で有意に減少した。結果は、オレイン酸を含むPF-127ゲルが、皮下投与と比較してインスリンの最高のバイオアベイラビリティ(15.9±7.9%)をもたらしたことを実証した。一方、EPAおよびDHAはそれぞれ3.4±1.2%および4.1±3.4%のバイオアベイラビリティを示した。しかし、DHAの存在下で観察されたバイオアベイラビリティの増加は統計的に有意ではなかった[27]。
同様に、タラ肝油抽出物(CLOE)と水添ヒマシ油(HCO)のエルゴタミン酒石酸塩(ET)の口中浸透に対する効果を調査した研究もある[25]。ハムスター頬袋を、二室拡散セル(37℃)を用いた試験管内試験浸透研究のモデル膜として使用した。頬袋膜をリン酸緩衝液(PB、pH7.4)とプロピレングリコール(PG)(PB:PG、1:1)の溶液で1~3時間前処理し、それぞれ5%の浸透促進剤をドナー細胞に添加した。前処理後、両細胞の溶液を除去し、新鮮なPG/PB混合物を用いて細胞を複数回洗浄した。これに続いて、ドナー細胞をPG/PB中のETの懸濁液で満たし、レシーバー細胞をPG/PB混合液のみで満たした浸透実験を行った[25]。透過試験の結果から、ETの透過率は、透過促進剤のそれぞれの存在下で顕著に増加した(5%)ことが示された。HCOの存在下では、ETの溶解度が顕著に増加し、その結果、ETの粘膜への分配が減少し、ETのフラックスが相対的に低くなった[25]。一方、CLOEの存在下では、ETの溶解度は2倍に増加し、ETのフラックスは8倍に増加した。これらの結果は、CLOEがETに及ぼす可溶化効果に加えて粘膜に直接作用することを示唆している[25]。CLOEの濃度を高くしてもETの透過性は大きくは向上せず、3%の濃度で十分な透過性向上作用を示すと考えられた。さらに、前処理の有無にかかわらずETの透過量はほぼ一定であり、前処理の長期化はETの透過量に影響を与えなかった。これらの知見は、CLOEが一過性の増強作用を示すことを示唆している[25]。
CLOEに含まれる主要脂肪酸であるパルミチン酸、オレイン酸、エイコサペンタエン酸(EPA)ドコサヘキサエン酸(DHA)の4種類について浸透試験を繰り返した。ただし、ドナー溶液中の各脂肪酸の濃度は、CLOE中の組成比に応じて算出した。その結果、各脂肪酸は5%CLOEの場合に比べてETのフラックスに対する効果が有意に低く、その中でもオレイン酸が最大の増強作用を示したことが明らかになった。このことは、CLOEに含まれる個々の脂肪酸の相乗的な作用が、CLOEの優れた増強作用に寄与している可能性を示唆している[25]。
2.5. メントール
メントールをエンハンサーとして用いたジデオキシシチジン(ddC)の経粘膜透過性の研究では、ddCの透過性が有意に増加することが示された。透過実験には、37℃でサイドバイサイドのフロースルー拡散セルを用いて、豚の頬側組織を用いた。その結果、メントール濃度0.3 mg/mLでは、頬粘膜を横切るddCのPappは2.02倍に増加した。しかし、低濃度(0.1および0.2mg/mL)のl-メントールの存在下では、dddCの透過増強に有意な差は認められなかった。これは、研究された濃度の範囲にわたって細胞間脂質抽出に対するメントールの限定的な効果に起因すると考えられる[26]。観察されたddC透過の増強は、一部はl-メントールの分配係数増強効果によるものである可能性が示唆された[26]。
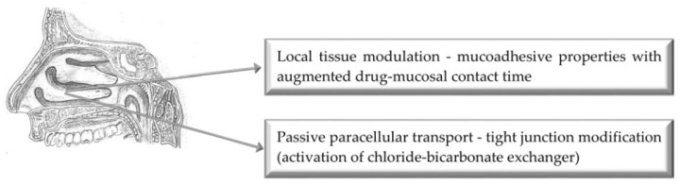
3. 経鼻投与経路
図2 強化された鼻腔薬物送達のためのバイオエンハンサーの主な作用機序の図示
3.1. 胆汁酸塩
5%のグリコフロール(GF)を含む経鼻製剤からのインスリンのバイオアベイラビリティーをウサギで研究した [119]。この研究では、亜鉛を含まないヒトインスリン、Novolin® Nasal(Novo Nordisc)およびglycofurol 75(Hoffman La-Roche)を使用した。経鼻投与用製剤の調製は、5%のグリコフロールを含む1mLのリン酸緩衝液(12.5mM、pH7.4)に溶解した6.6mgのインスリンで構成されていた。投与量は,0.66mgインスリン(15.8IU)に相当する各鼻腔内点鼻液50μLであった。血糖値は、予め決められた時間間隔で採取した血液中で決定した。Gizurarson, er al)。 [120]によって記述されたカエル口蓋モデルを使用して、リン酸緩衝液(12.5mM)をpH7.4および5%グリコフルールで使用して、粘膜クリアランスに対する局所毒性を試験した。その結果、グリコフロールを経鼻剤に含有させた場合、血漿中グルコースレベルの低下が大きいことが示され、これは以前に発表された結果[121]と同程度であった。グリコフロールの吸収促進機構は不明であるが、15 分後には初期グルコース血中濃度が 90%低下するなど、インスリンの吸収が速いことが示された。さらに、120分後の時間間隔でも血漿グルコースは初期値の85%で抑制されていた。この結果は、グリコフロールが血糖値を持続的に抑制しながらラットのインスリン吸収を促進する能力を持っていることを示している。
(Bagger, er al)。 [122]は、グリココラートナトリウムとグリコフロールを投与した場合のウサギにおけるペプチドTの絶対的な経鼻バイオアベイラビリティを調査した。ペプチドおよびペプチド様化合物の薬物吸収促進剤として胆汁酸ナトリウムを用いた以前の経鼻吸収試験では、明らかな生物学的増強作用が示されている[123,124,125,126,127]。本研究では,ペプチドTのTmax,Cmaxおよび時間依存濃度プロファイルを用いて,グリココラートナトリウムとグリコフロールの吸収促進作用を検討した。グリココール酸ナトリウムは、ペプチドTのバイオアベイラビリティーの最も高い増加(59%)を示し、一方、グリコフロールは、そのバイオアベイラビリティーを増加させた(22%)。一方、2つのバイオエンハンサーを併用した場合(すなわち、グリコフェロールとグリココール酸ナトリウム)ペプチドTのバイオアベイラビリティーは29%となり、さらに、ペプチドTをグリココール酸ナトリウムと共投与した場合、バイオエンハンサー効果は、吸収は速いが作用時間は比較的短いことが特徴であった。グリコフロールは吸収促進効果は低いが、作用時間が長いという特徴があった[122]。
3.2. キトサンおよびその誘導体
キトサンは、昆虫や甲殻類の外骨格に含まれる天然高分子であるキチンを脱アセチル化して得られる多糖類である。Illumと共同研究者らは、キトサンの鼻腔内薬物吸収促進効果を初めて実証した。キトサンは、例えば、羊のインスリンのCmaxを34mIU/Lから 191mIU/Lに増加させる能力を示し、AUCは7倍に上昇した[128]。ペプチド医薬品の新しい吸収促進剤としてのキトサンの使用については以前に述べられているが [129]、様々な治療薬の鼻腔内送達システムでの使用については最近包括的に検討されている [130]。天然ポリマーであるキトサンの鼻腔内薬物送達強化の可能性の説明として、選択された研究を以下に簡単に説明する。
Illum, Watts, Fisher, Hinchcliffe, Norbury, Jabbal-Gill, NankervisおよびDavis [31]は、溶液中のキトサンまたはマイクロスフィアに配合されたキトサンのいずれかの共同投与が、羊の経鼻投与後の塩酸モルヒネのバイオアベイラビリティーを向上させることを示した。キトサンはモルヒネのCmaxを対照群(モルヒネ単独)の151nMから657nMに上昇させ、モルヒネのバイオアベイラビリティーを10.0%(対照群)から26.6%に改善した。さらに、対照群では20分であったTmaxが14分であったことからもわかるように、吸収率が向上した。マイクロスフィアに配合されたキトサンは、さらに1,010 nMのCmaxと54.65%のバイオアベイラビリティーでモルヒネのバイオアベイラビリティーパラメータを改善した。吸収率も8分間のTmaxで改善され、コントロールとは統計的に有意に異なっていた[128]。
Hinchcliffe、Jabbal-Gill、Smith [30]は、ヒツジモデルにおけるサケのカルシトニンに対するキトサンベースの経鼻投与システムの薬物動態学的効果を調査した。サケカルシトニンを含む対照の鼻腔内溶液(2200 IU/mL)のみを、グルタミン酸キトサン(5 mg/mL)を含むサケカルシトニン溶液、および市販のサケカルシトニン含有鼻腔スプレーであるMiacalcin®と比較した。キトサン含有サケカルシトニン溶液では99pg/mL(範囲50~107pg/mL)のCmax値が得られたのに対し,サケカルシトニン対照溶液では33pg/mL(範囲13~49pg/mL),Miacalcin®点鼻スプレーでは42pg/mL(範囲15~79pg/mL)であった。さらに、キトサン含有溶液の平均AUC値3220 pg/mL/min(範囲1606-4972 pg/mL/min)は、対照のサケカルシトニン溶液(943 pg/mL/min、範囲198-2519 pg/mL/min)と比較して3.5倍、Miacalcin®点鼻スプレー(1636 pg/mL/min、範囲87-3792 pg/mL/min)と比較して2倍の増加を示した[30]。
キトサンはpKa値以下の酸性pH値でしか溶解しないため、N-トリメチルキトサンクロライド(TMC)のような化学的に修飾された誘導体が、より中性のpH値での溶解性を向上させるために合成されていた[131]。異なる四環化度(QD)を有するTMCポリマーを14C-マンニトールとともにラットに経鼻投与した研究では、TMCのQDが中性環境(pH7.4)での経鼻吸収促進効果において重要な役割を果たしていることが示された。14C-マンニトールの経鼻投与は QD の増加とともに 45%で閾値に達するまで増加した[33]。
3.3. 澱粉マイクロスフィア
ヒツジを用いた生体内試験研究では、生体接着性デンプン微小球送達システムの鼻腔内での薬物吸収が増強されていることが明らかになっており、他の吸収促進剤との併用により相乗的に改善される可能性があることが示された。インスリンおよびゲンタマイシンを含むデンプンマイクロスフィアと対照溶液を比較したところ、吸収効率がそれぞれ5倍および10倍増加した [132,133,134]。同様のデンプンマイクロスフィア製剤と薬物化合物は、ラットに経鼻投与した場合、薬物のバイオアベイラビリティが約30倍増加することを実証した[135]。
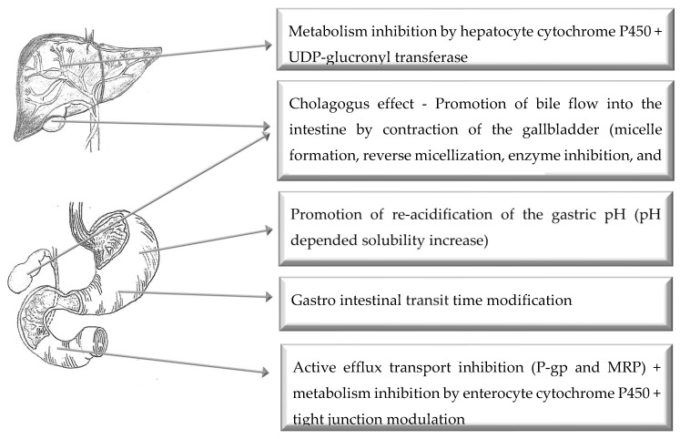
4. 経口投与経路
図3 強化された経口薬物送達のためのバイオエンハンサーの主な作用機序の図示
4.1. アロエベラ
アロエベラ葉材および抽出物は、試験管内試験での薬物輸送および生体内試験での薬物のバイオアベイラビリティーを修飾することがわかっている。アロエベラ液剤のビタミンCとEの吸収に対する効果を調べた二重盲検クロスオーバー臨床試験では、アロエベラゲル製品(AVG)とアロエベラ全葉製品(AVWL)の両方が調査された。その結果、対照(ビタミンCを水で投与した場合)と比較して、AVGはビタミンCのバイオアベイラビリティーが3.7倍、AVWLは2倍に増加した。ビタミンEのバイオアベイラビリティーへの影響については、アロエ製品はいずれも投与後6時間および8時間でビタミンEのベースラインレベルで統計学的に有意な増加を引き起こした。しかし、個人間のばらつきが大きいため、異なる治療法間の AUC 値は統計的に有意ではなかった。著者らは、A. vera製品によるビタミンCとEのバイオアベイラビリティの向上は、消化管での分解に対する保護作用に起因するとしているが、この研究では証明されていない[37]。
A. ベラゲルと全葉材の両方が、5.8 と 7.4 の 2 つの異なる pH 値で 0.1 から 5% w/v の濃度範囲で Caco-2 細胞の単分子膜を介して広範囲にインスリン輸送を増加させた。アロエ材料は,0.5% w/v 以上の濃度で Caco-2 細胞単分子膜の TEER を著しく低下させたが、これは可逆的であった [36]。A. ferox と A. vera のゲル材料、葉全体の材料、および沈殿した多糖類(これらの材料から得られた)を 2% w/v の濃度で、モデル薬物としてアテノロールと組み合わせて、ラットの腸組織を切除した拡散室で研究が行われた。その結果、すべてのアロエ材料は、対照(アテノロール単独)と比較して統計的に有意に(p < 0.05)また陽性対照(0.2% w/v ラウリル硫酸ナトリウム)よりも大きな範囲で、ラット腸管組織のTEERを低下させた。この研究では、いくつかの沈殿した多糖類は、対応するゲルや葉全体の材料よりも高いTEERの減少をもたらしたことも示された。このTEERの低下は、アロエ葉材がタイトジャンクションを開き、結果としてアテノロールのような親水性薬物分子の傍細胞輸送を促進する能力があることを示している。脱水したA. vera gel(Daltonmax 700®)材料からの沈殿した多糖類画分のみが、対照と比較して統計的に有意にラットの腸管組織におけるアテノロールの輸送を増強することができた。コントロールと比較して統計的に有意ではなかったが、A. vera全葉抽出物(Daltonmax 700®)から得られた多糖類画分は、アテノロールの輸送を大幅に増加させた。しかし、A. ferox材料は、切除されたラットの腸管組織を横断するアテノロールの輸送を増強することはできなかった[35]。
Wallis、Malan、Gouws、Steyn、Ellis、アミロイドβay、Wiesner、P OttoおよびHammanによる研究[38]では、A. veraゲルおよび多糖類(すなわち、粗多糖類および分子量に基づく分画多糖類)のインジナビルのバイオアベイラビリティに対する効果が、Sprague-Dawleyラットで調査された。この研究の一環として、選択されたアロエ材料がカコ2細胞単分子膜のTEERおよびLS 180細胞におけるインジナビルの代謝に及ぼす影響についても調査した。その結果、すべてのアロエ素材がCaco-2細胞モノレイヤーのTEERを低下させ、タイトジャンクションの開通を示唆していた。沈殿した多糖類は、ゲル材料よりも大きな範囲でTEERを低下させた。しかし、多糖類の分子量分率の違いとTEERの低下との間には明確な相関関係は認められなかった。アロエ材料は、対照(インジナビル単独)と比較して、統計的に有意ではなかったが、すべてのアロエ材料が酵素阻害作用を示した。AUCで示されるインジナビルのバイオアベイラビリティーへの影響については、すべてのアロエ材料は統計的に有意ではなかったが、バイオアベイラビリティーの増加を示した。粗沈殿多糖類および多糖類画分によるインジナビルのバイオアベイラビリティーの増加は、A. veraゲルのそれよりも高かった。本試験におけるインジナビルのバイオアベイラビリティーの上昇は、タイトジャンクションの開通(TEERの低下で示される)とインジナビルの代謝抑制(代謝物血漿中濃度で示される)を含むメカニズムの組み合わせに起因していることが示唆された。さらに、本試験の結果から、薬物動態や吸収の調節に関与する生理活性成分は、多糖類成分の分子サイズと直接相関することはできないが、A. veraゲル材料の多糖類成分に集中している可能性が高いことが明らかになった[38]。
4.2. 胆汁酸塩
化合物、特に水溶性の低い薬物の経口バイオアベイラビリティーを高める胆汁酸塩の能力は、何年も前に発見されている[136,137,138]。
Yu, Zhu, Wang, Peng, Tong, Cao, Qiu, Xuによる研究[102]では、胆汁酸塩ミセルと混合した場合のシリビンのバイオアベイラビリティを、シリビン-N-メチルグルカミン単独の場合と比較して、イヌへの経口投与後に調査した。調製した混合胆汁酸塩ミセルの平均粒子径は75.9±4.2 nmであった。また、リン酸緩衝液(pH7.4)中では17.5%(w/w)、塩酸塩(pH1.2)中では15.6%(w/w)と非常にゆっくりとした放出を示した。この遅い放出にもかかわらず、犬における混合ミセル中のシリビンとシリビン-N-メチルグルカミンの相対的なバイオアベイラビリティーは252%であった[102]。
糖尿病ラットを用いた生体内試験試験では、グリココール酸ナトリウムの共同投与によるインスリンの地域特異的な腸管送達を調査するために使用された[139]。ラットにインスリン(10 UI/kg)を外科的手技により腸管(十二指腸、空腸、回腸)に投与した。消化管から吸収されたインスリンについては、投与後45分および60分後のラットの血糖降下作用を測定することにより検討した。陽性対照の血糖降下作用(100%)は、45分後に77.9 mg/100 mLのブドウ糖、60分後に70.5 mg/100 mLのブドウ糖で規定された。十二指腸領域については、インスリン対照では45分後に71%、60分後に84%の血糖降下作用を示し、グリココール酸ナトリウムを添加したインスリンでは投与後45分後に90%、60分後に95%の血糖降下作用を示した。空腸領域については、インスリン対照では34%、54%の低血糖効果を示したのに対し、グリココール酸ナトリウムを投与したインスリンでは、投与45分後及び60分後にそれぞれ39%、60%の低血糖効果を示した。しかし、ラットの回腸内投与では、対照と比較して有意な血糖値プロファイルの低下は認められなかった。回腸内投与では、インスリン対照では9%と5%の血糖値低下を示したのに対し、グリココール酸ナトリウムを含むインスリンでは投与後45分及び60分でそれぞれ5%と8%の血糖値低下を示した[139]。
これらの効果は胆汁酸塩の存在下でのインスリン吸収の増加に寄与している可能性があり、粘液層の修飾やタイトジャンクションの調節などの機序によるものと考えられる[140,141]。最後に、グリココラートナトリウムのインスリンの腸管吸収促進効果は、十二指腸がインスリン経口送達に最適な部位であるという部位依存性の効果を示した[139]。
4.3. ブラッククミン
一般的にブラッククミン/キャラウェイとして知られるNigella sativaは、以前にアモキシシリンのバイオエンハンサーとして評価されている[142]。N. sativa 抽出物は、種子を洗浄、粉砕、ふるいにかけた後、メタノールとヘキサンで 6 時間抽出することで調製された [142]。N. sativa種子のメタノール(3 mg)およびヘキサン(6 mg)抽出物の有無にかかわらず、PBS(pH 7.4)中のアモキシシリン(6 mg/mL)の移行を研究するために、エバートラット腸嚢を使用した。腸内で輸送されたアモキシシリンの量を 273 nm で分光光度計で定量した [142]。生体内試験試験では、ラットにアモキシシリン(25 mg/kg)を N. sativa ヘキサン抽出物(25 mg/kg)と経口的に共投与した。投与後 0,0.25,0.5,0.75,1,1.5,2,4,6,8 時間後に血液サンプルを採取し、UPLC-MS/MS を用いてラット血漿中のアモキシシリン量を定量した [142]。
試験管内試験試験の結果、N. sativaのメタノール抽出物とヘキサン抽出物の両方がアモキシシリンの浸透を有意に増加させ、後者が最大の増加を示したことが示された[142]。そのため、ヘキサン抽出物を生体内試験評価のために選択した。また、生体内試験試験の結果から、ラットのアモキシシリン血漿中濃度が有意に上昇したことが示された。N. sativa抽出物はアモキシシリンの吸収速度と吸収範囲を増加させ、Cmaxは4138.251±156.93から5995.045±196.28 ng/mLに増加し、AUC0→tは8890.40±143.33から13483.46±152.45 ng/mL-hに増加した。このようなN. sativaの浸透促進効果は脂肪酸の存在に起因する可能性が示唆された[142]。これまでに、脂肪酸が先端膜と基底膜の流動性を高めることで、低透過性薬物の透過性を高めることが実証されている[143]。ウサギで行われた同様の生体内試験試験では、反対の結果が得られた。その研究では、ニゲラ(200mg/kg)で前処理した後、シクロスポリン(30mg/kg)のCmaxおよびAUC0-∞はそれぞれ35.5%および55.9%有意に減少した。しかし、シクロスポリンクリアランスの有意な増加(〜2倍)が観察されたことから、N. sativaの存在下で腸内P-gpおよび/またはCYP3A4が活性化されていることが示唆された[144]。
4.4. カプサイシン
フェキソフェナジンに対するカプサイシン前処理の効果の試験管内試験、in situおよび生体内試験での評価では、フェキソフェナジンの腸管吸収が有意に増強されたことが示された [145]。試験管内試験試験ではラットの非倒立性腸嚢を用い、in situシングルパス腸灌流試験ではラットの回腸セグメント(~8~12cm)を分離してカニューレした。生体内試験試験では、ラットで同じ前処理を7日間行った。非倒立嚢試験の結果、カプサイシンの存在下でフェキソフェナジンの腸管輸送とPappが有意に増加することが示された。カプサイシン前処理と標準的なP-gp阻害剤であるベラパミルで得られた結果が同程度であったことから、ラットの腸内におけるP-gp流出阻害が作用機序であることが示唆された[145]。In situ シングルパス腸管灌流の結果、カプサイシン及びベラパミルで前処理したラットでは、対照群と比較してフェキソフェナジンの吸収速度定数、吸収率及び有効透過率が有意に増加した[145]。生体内試験では、カプサイシン投与前のラットにフェキソフェナジンを経口投与した場合、AUC及びCmaxが有意に増加した。さらに、フェキソフェナジンの見かけの経口クリアランスは有意に減少したが、tmaxおよびt1/2は変化しなかった。このように、この研究から得られた知見は、カプサイシンがP-gp介在性薬物排出の阻害を介してフェキソフェナジンのバイオアベイラビリティーを増加させる可能性があるという生体内試験での証拠を提供している[145]。
4.5. キャラウェイ
キャラウェイは、植物Carum carviの乾燥熟した果実から得られる。本試験では、リファンピシン、イソニアジド、ピラジンアミドを合剤(FDC)として投与し、その薬物動態に対するC. carvi(キャラウェイ)抽出物(100 mg)の生物学的増強効果を検討した。キャラウェイ抽出物は、3剤ともバイオアベイラビリティの指標であるCmaxとAUCを統計学的に有意に増加させた。Cmaxの上昇率はリファンピシン、イソニアジド、ピラジンアミドでそれぞれ32.22%、36.01%、33.22%であり、AUC0-24時間の上昇率はリファンピシンで32.16%、イソニアジドで29.06%、ピラジンアミドで27.92%であった。著者らによると、キャラウェイ抽出物によるバイオアベイラビリティーの改善は、P-gpの排出に影響を与えるだけでなく、粘膜から血清への浸透を促進することに起因すると考えられる[40]。キャラウェイは P-gp の流出抑制剤として作用することが示され、主な有効成分はカルボンとリモネンであることが判明した[146]。
4.6. Cylcosporine A
シクロスポリンAは、11個のアミノ酸からなるポリペプチドであり、最初は真菌であるTolypocladium inflatum Gams[147]に由来している。シクロスポリンAは、他の適応症の中で、臓器移植中の免疫抑制剤として、自己免疫疾患を治療するために、またC型肝炎のような特定のウイルス性疾患の治療に使用されているが、いくつかの用途を挙げることができる。シクロスポリンAは、流出トランスポーターであるP-gpの阻害剤として作用することが示されており、その結果、この活性な流出トランスポーターの基質である薬物のバイオアベイラビリティを増加させる可能性がある[51]。シクロスポリンAは、ラットにおける抗血小板薬であるクロピドグレルのバイオアベイラビリティーを改善することが示されている。10mg/kgのシクロスポリンAと30mg/kgのクロピドグレルの共投与は、クロピドグレルのAUCとCmaxのそれぞれ3.48倍と2.83倍の増加をもたらした。著者らは、このバイオアベイラビリティーの増加は、P-gp介在性排出の阻害によるものであるとしている[49]。
4.7. キトサンとその誘導体
キトサンおよびその誘導体の経口薬物吸収促進効果は、以前に広範囲にレビューされている[148,149]。簡単に言えば、いくつかの研究で、キトサン(塩酸キトサンやグルタミン酸キトサンなどのキトサン塩を含む)が、試験管内試験細胞モデルおよび生体内試験動物モデルにおいて、効果的な経口薬物吸収促進剤であることが示されている[42,150,151,152]。薬物吸収促進の主なメカニズムは、親水性および高分子薬物化合物の傍細胞への取り込みを促進するためのタイトジャンクションの調節が関与していることが示されている。多くの研究では、F-アクチンとZO-1の再分配を介してタイトジャンクションを開くためのキトサンと上皮細胞との相互作用[153]が示されているが[148,154]、キトサンのタイトジャンクション開通効果は、塩化物-炭酸水素交換体の活性化によって引き起こされる細胞内pHの変化によるものであることが2パスインピーダンス分光法を用いてHT-29/B6細胞で示された[155]。
キトサンの特性の一部を改善するために、トリメチルキトサン、チオール化キトサン、カルボキシメチルキトサンおよびその誘導体、疎水性キトサン、コハク酸キトサンおよびフタル酸キトサン、PEG化キトサンおよびキトサン酵素阻害剤コンジュゲートを含む、いくつかの誘導体および化学的に修飾されたキトサンが薬物送達の可能性のために研究されており、それらは以前にレビューで発表されている[156]。
4.8. クルクミン
クルクミン(diferuloylmethane)は、Curcuma longa L. (ウコン)の根茎に含まれる主要なクルクミノイドであり、いくつかの生物学的・薬理学的活性を有することが示されている [157]。以前に発表された、従来の薬剤とクルクミノイドの薬物動態学的相互作用に関するレビューでは、CYP450および第二相酵素の調節、P-gpの排出阻害、および有機陰イオン輸送ポリペプチド(OATP)への潜在的な影響を介して潜在的な相互作用が明らかにされた。残念なことに、クルクミンによる薬物吸収の増強から薬物吸収の減少に至るまで、異なる研究によって対照的な結果が得られた[158]。
クルクミンが薬物のバイオアベイラビリティを有意に増加させることが判明した研究の例としては、薬物投与の4日前からクルクミン(60mg/kg)を投与されたSprague-Dawleyラットにおける生体内試験研究(前処置群)があり、一方、別の群は薬物との併用で1回のみクルクミン(60mg/kg)を投与され、対照群はクルクミンを投与されなかった。セリプロロールのバイオアベイラビリティーは、クルクミンを前処理したラットでは統計的に有意に増加したが、クルクミンと1回のみ併用投与したラットでは増加しなかった。ミダゾラムのバイオアベイラビリティーは、前処理群と共投与群の両方で増加したが、前処理群でのみ統計的に有意であった。ウェスタンブロット解析の結果、クルクミン投与4日後の腸内P-gp蛋白質発現は49%減少し、CYP3A蛋白質含量は42%減少した。一方、肝P-gpタンパク質発現は144%増加し、CYP3Aタンパク質含量は91%増加した。腎のP-gpタンパク質量は変わらなかったが、腎のCYP3Aは41%増強された。セリプロロールはP-gp基質であり、ミダゾラムはCYP3A酵素によって広範囲に代謝されることから、小腸でのP-gpとCYP3Aのダウンレギュレーションにより、これら2剤の薬物動態がクルクミンによって変化していることが本研究から推論された[48]。さらに、タモキシフェンと一緒にクルクミンを 10 mg/kg の用量でラットに投与したところ、タモキシフェンの AUC 値及び Cmax 値が有意に上昇した(すなわち、それぞれ 64%及び 71%)[159]。ブロイラーのニワトリを 100 mg/kg クルクミンで 10 日間前処理すると、これらの動物における marbofloxacin の絶対的および相対的なバイオアベイラビリティーが向上した [160]。クルクミンの P-gp 阻害効果は、ヒト P-gp 過剰発現細胞株(LLC-GA5-COL300)を用いた 試験管内試験 試験で確認され、カルセイン-AM の取り込みが増加した[161]。
4.9. ジオスミン
柑橘類に含まれる特定のフラボン(タンゲレチン、ノビレチン、ベルガモットンなど)は、P-gp阻害作用を示しており、この流出トランスポーターの基質となる薬物との薬物動態的相互作用を引き起こしていた。柑橘類に含まれるフラボノイドであるディオスミンをモデル化合物として、ロドミン123とジゴキシンを用いて、Caco-2細胞モデルでP-gp阻害作用を調べた。ジオスミンの非存在下および存在下で、Caco-2細胞におけるロドミン123の蓄積を調べた。50μMの濃度で、ジオスミンは対照と比較して494±8.4%のロドミン123の細胞蓄積の増加を引き起こした。ジオスミンによるP-gp媒介の流出阻害の最初の観察をさらに調査するために、Caco-2細胞単層を横切るジゴキシンの双方向輸送(すなわち、先端から基底側[A-B]および基底側から先端側[B-A]方向)を、ジオスミンの非存在下および存在下で実施した。ジオスミン濃度50μMでは、Papp(A-B)は2.8±0.07×10-6 cm/sから9.5±0.06×10-6 cm/sへと有意に増加し、一方、Papp(B-A)は41.1±1.20×10-6 cm/sから17.1±0.10×10-6 cm/sへと有意に減少した。これらのPapp値は、ジオスミン非存在下では15.2,ジオスミン存在下では2.3の輸送比値に対応していた。本研究の結果から、ジオスミンはP-gp流出輸送阻害剤として作用し、P-gpの基質となる薬物のバイオアベイラビリティーを改善する可能性があることが明らかになった[50]。
より最近の研究では、ラットにおけるフェキソフェナジンの生体内試験薬物動態に対するジオスミンの効果が調査された。この研究では、ラットをジオスミン(50 mg/kg)で 7 日間前処理し、8 日目にフェキソフェナジン(10 mg/kg)を投与した。その結果、ジオスミンの前処理により、Cmaxは2.5倍、AUC0-∞は2.2倍に増加した。本研究におけるフェキソフェナジンのバイオアベイラビリティーの向上は、ジオスミンが強力なP-gp阻害剤であるという事実に起因している[162]。
4.10. エモジン
エモジン(1,3,8-トリヒドロキシ-6-メチル-アントラキノン)のようなアントラキノン類は、リウマチ・パルマタム[163]、ポリゴナム・マルチフローラム[164]、ポリゴナム・クスピダタム[165]、カシア・オブツシフォリア[166]、アロエベラ[167]などの多種多様な植物やハーブに天然に存在している。エモジンの投与は、P-gp関連の流出および/または多剤耐性関連タンパク質1,2および3(MRP1,MRP2およびMRP3)の調節に対する相乗効果および/または抑制効果をもたらす可能性があり、これらの効果は、共投与された基質分子のバイオアベイラビリティーに重大な影響を及ぼす可能性がある[163,168,169,170,171]。
Minらによる広範な研究[172]では、エモジンとロダミン123の間の潜在的な相互作用が様々な試験管内試験モデルで調査された。この研究では、ヒト骨髄性白血病細胞(K562)P-gp過剰発現アドリアマイシン耐性K562細胞(K562/AD病M)およびCaco-2細胞を試験管内試験モデルとして使用し、様々な濃度でのエモジンの共投与がロダミン123(1μM)の細胞内蓄積に有意な影響を及ぼすかどうかを調査した。その結果、エモジンの添加は、Caco-2細胞モデルとK562/AD病M細胞モデルの両方において、濃度依存的にロダミン123の細胞内蓄積を増加させることが示された。エモジン(20μM)によるCaco-2細胞におけるロダミン123の蓄積の程度は、はるかに高いベラパミル濃度(200μM)と同等であった。エモジンとロダミン123のP-gp上での結合部位競合についても調べた。その結果、計算されたKi値は、ロダミン123濃度の増加(0~20μM)に伴って一貫して増加し、エモジンとロダミン123の間で競合的な相互作用が起こっていることが示された。エモジンとロダミン123はP-gp上の同じR結合部位を共有していると結論づけられた[172]。
MTT法もまた、エモジンの添加がP-gp介在性MDRを逆転させる可能性があるかどうかを調べるために、K562/AD病M細胞株と組み合わせて使用した。その結果、エモジン(20μM)の添加は、単独または10μMアドリアマイシンとの組み合わせで、対照群と比較してP-gp発現の調節を低下させることが示された。著者らは、エモジンはK562/AD病M細胞におけるP-gpの発現を阻害し、また、エモジンとP-gpのR結合部位であるロダミン123との競合的相互作用により、P-gpが媒介するMDRを逆転させる可能性があると結論づけた。その結果、エモジンはP-gpの効果的な阻害剤であり、その効果は、P-gpに直接結合してP-gpが介在する排出機能を弱めるか、あるいはP-gp発現の低下に関連した間接的なメカニズムによって媒介されることが確認された[172]。
別の研究では、Caco-2細胞モデルへのエモジンの添加がP-gp介在性の流出を減少させる能力を有することも示されているが、さらに、COX-2阻害によりP-gp発現がMAPK/AP-1経路を介して媒介されることも報告されている[173]。Ussing-chamber法を用いたラットの腸管組織を横断した試験管内試験輸送研究では、ジゴキシンの輸送はエモジンの存在下では減少し、テニポシドの輸送はエモジンの存在下では増加することが示された。著者らは、エモジンはP-gp関連排出の誘導剤であり、MRP3の阻害剤でもあると結論づけている[174]。
4.11. ガリン酸エステル
ガリン酸は水溶性フェノール酸であり、ブドウや様々な植物の葉に含まれており、ガロタニン[175]として知られる植物ポリフェノールのより大きなグループの一部を形成している。ガロタンニンは、消化管で加水分解されて遊離ガロン酸を形成する[176]。ガロン酸の摂取には様々な健康上の利点があり、文献では抗がん、抗炎症、心臓保護、抗糖尿病特性を発揮することが報告されている[175]。ガリン酸は、P-gpを介した薬物排出、およびCYP3A4などのCYP450関連アイソザイムに対して阻害効果を示すことが報告されている[177,178,179]。
ガロン酸がディルチアゼムの輸送に及ぼす影響を調べるために、試験管内試験およびin situ輸送試験が実施された。試験管内試験試験では、ウィスターラット腸管組織の非倒伏腸嚢を使用した。その結果、ガロン酸の存在下では、十二指腸、空腸、回腸の各部位において、ディルチアゼムの見かけの透過性が対照群と比較して4.4倍、5.1倍、4.9倍に増加していることが示された[180]。
in situシングルパス腸管灌流研究のために、ラットを4つのグループ(n = 5)に無作為に分割した(コントロール、標準インヒビター、およびガリン酸またはエラグ酸のいずれかで前処理)。カニューレ化された腸管セグメントの灌流は、ディルチアゼム(100μg/mL)およびフェノールレッド(50mg/mL)を含むリン酸緩衝生理食塩水(pH=7.2)を用いて行った。0.2 mL/minの一定流量を90分間維持し、10分間隔でサンプルを採取した。その場試験の結果、ガロン酸(50mg/kg)を7日間前処理すると、7日間のガロン酸による前処理後の対照群(ディルチアゼム単独)と比較して、ラットにおけるディルチアゼムの輸送が有意に(p<0.05)増加することが示された。ディルチアゼムのCmax、AUC0-t及びAUC0-∞は、それぞれ1.90-、2.06-及び2.08倍増加した(Athukuri and Neerati. 2018)。これらの結果から、ガロン酸はP-gpとCYP3A4の両方の二重阻害剤である可能性が高く、この二重作用がディルチアゼムのバイオアベイラビリティーの有意な向上につながっている可能性が示唆された[180]。
メトプロロールの輸送は、Wistarラットに経口投与したメトプロロールの薬物動態パラメータを評価するために、同様のin situシングルパス腸管灌流試験で評価された。ラットは試験開始前に7日間、ガリン酸(50mg/kg)で前処理した。その結果、メトプロロールのCmaxおよびAUC値の有意な上昇が認められた。ガテン酸は肝臓のCYP2D6活性を阻害することで経口バイオアベイラビリティを高め、メトプロロールの代謝を低下させたと結論づけられた[181]。
4.12. ゲニステイン
フラボノイドのゲニステイン(5,7-Dihydroxy-3-(4-hydroxyphenyl)chromen-4-one)は、植物由来(Glycine maxおよびPueraria lobata)のイソフラボンおよび植物性エストロゲンであり、ヒトが頻繁に摂取する [54,55,182]。ゲニステインはそれ自体のバイオアベイラビリティーが非常に悪く、ゲニステインが抗がん作用を持つ可能性を示唆する研究もあるが、バイオアベイラビリティーが低いため、臨床研究ではこれらの効果を得ることができなかった[183]。ゲニステインは、P-gp、BCRPおよびMRP2トランスポーターの流出を阻害することが示されているため、薬物のバイオアベイラビリティーを高めると考えられている。これらの特性は、Caco-2抗寄生虫剤の輸送試験で調査され、タキソール(P-gp基質)を33または100μMのゲニステインと組み合わせて適用した[184]。33μMゲニステインでは輸送阻害は測定できなかったが,100μMゲニステインでは20%の輸送阻害が得られた。その後、ゲニステインはP-gpを阻害するが、P-gpの基質にはならないと判断された。別の研究では、10-30μMのゲニステインは、用量依存的な方法(10-200μM)でローダミン123とビンブラスチンの流出を減少させながら、薬剤耐性ヒト子宮頸がん細胞株(KB-V1)のビンブラスチンとパクリタキセルに対する感受性を増加させることが示された[185]。しかし、ゲニステインへの暴露後にはP-gp発現の相関的な変化は観察されず、P-gp活性のみが調節されていることが示された。Li and Choi [55]は後に、3.3mg/kgまたは10mg/kgのゲニステインを摂取した30分後に30mg/kgのパクリタキセルを単回経口投与すると、雄性のSprague-DawleyラットにおいてAUCが54.7%有意に上昇することを示した。これは血漿クリアランス(35.2%)が減少した結果、全身曝露が増加したことによる。パクリタキセルを静脈内投与(3.3mg/kg)した場合も同様の効果が認められた。ゲニステインはパクリタキセルが基質となることが知られている排出トランスポーターおよび代謝酵素(おそらくP-gpおよびCYP3A)を阻害することが提案された[55,182]。
ゲニステインはまた、MRP1基質である2′,7′-ビス-(カルボキシプロピル)-5(6)-カルボキシフルオレセイン(BCPCF)のヒト赤血球からの流出を阻害した。ゲニステインはBCPCFの流出に対して50〜70μMのIC50濃度を示した。ゲニステインがMRP1流出トランスポーターを阻害することが報告されているが、この変調は基質依存性であることが報告されている[186]。
ゲニステインは、より最近の研究では、(-)-エピガロカテキン-3-ガレート(EGCG)の生体内変換および腸管流出を阻害することが示されている。EGCGは、様々な食品や飲料に含まれるフラボノイドであるが、特にCamellia sinensis(緑茶)に含まれており、抗がん作用、抗ウイルス作用、抗炎症作用などのいくつかの治療効果を有することが示されているが、経口的な全身吸収は非常に悪い[187]。Lambert、Kwon、Ju、Bose、Lee、Hong、HaoおよびYang [54]は、HT-29ヒト結腸癌細胞において、ゲニステイン(20μM)の存在下で2〜5倍の細胞質EGCGの増加を実証した。また、彼らは、CF-1マウスを200mg/kgのゲニステインと75mg/kgのEGCGで処理し、その結果、血漿半減期とEGCGの最大濃度が増加した。しかし、雄性腺腫性ポリポーシス大腸菌(APC)min/+マウスモデルでは、この組み合わせもまた腫瘍発生を増強した。
試験管内試験および生体内試験での研究では、マルディンダービーイヌ腎臓(MDCK-II)細胞およびアサフヒツジを抗菌性フルオロキノロンであるダノフロキサシンとゲニステインを併用して治療した[188]。ダノフロキサシンは動物専用であり、乳中に積極的に分泌される。ゲニステインはダノフロキサシンのBCRP輸送を効率的に阻害したが、試験管内試験輸送試験では、イソフラボン補充後のダノフロキサシン血漿中濃度に変化がないことが示された。しかし、大豆を長期間摂取すると、乳中の抗菌薬濃度が低下することが示された。
重要なのは、ゲニステインが第II相薬物代謝酵素であるスルホトランスフェラーゼを誘導できることを示したChenら[189]の知見である。形質転換したヒト肝細胞株(HepG2)と結腸癌細胞株(Caco-2)を用いて、酵素活性、タンパク質レベルおよびmRNA発現を評価した。その結果、遺伝子発現(SULT1A1及びSULT2A1)はゲニステインにより時間及び用量依存的(0-25μM)に誘導されることが結論づけられた。反対に、約0.1μMのゲニステインは、競合的にヒト肝臓のフェノールスルホトランスフェラーゼを50%阻害した[190]。
4.13. ゴックル抽出物
アーユルヴェーダ薬で使用されている人気のある植物エキスであるゴクルエキスは、Tribulus terrestris Linn (Zyygophyllaceae) [56,191]から得られたものである。T. terrestrisで以前に同定されたファイトケミカルの一部には、サポニン、ステロイド、フラボノイド、カルボリンアルカロイドが含まれている。ゴックル抽出物は、利尿剤、抗炎症剤、同化剤、鎮痙剤、筋弛緩剤、血圧降下剤、および血糖降下剤として伝統的に使用されてきたが、併用薬のバイオアベイラビリティーに影響を与えることが報告されている。
ある研究では、常在嚢モデルにおけるメトホルミン塩酸塩(HCl)の吸収に及ぼすゴクフル抽出物の影響を調査した[56]。メトホルミンは、溶解性は高いが膜透過性が悪い(BCSクラスIII)ことが知られている抗糖尿病薬である。本研究では、乾燥した植物素材(新鮮な果実、葉、茎)から高温エタノールを用いて抽出物を調製した。その結果、ゴックル抽出物の存在下でメトホルミンの吸収が増加したことが示され、著者らは抽出物に含まれる主要なサポニン成分がこの薬物輸送の増加に大きく寄与している可能性を示唆した。サポニンは、ステロイドまたはトリテルペノイドのアグリコンに接続された1本以上の糖鎖で構成されている。細胞膜の構造の大部分を維持しながら、コレステロールを可溶化し、それによって膜透過性を高めるために浸透させることができる[57]。
同様の研究では、メトホルミン塩酸塩錠(175~500mg)に濃度を変化させたゴクル抽出物(0~100mg)を配合し、鶏腸常在嚢モデルを用いて検討した。この試験では、ゴクフル抽出物の薬物吸収促進効果が確認され、ニワトリの腸管膜を通過するメトホルミンの透過率が29%から54%に増加した[57]。
乾燥したT. terrestrisの葉からメタノール抽出物を調製し、サリチル酸(アスピリン)とともに常法で使用したヤギ腸管組織の粘膜側に塗布した。ゴックル抽出物はアスピリンの輸送を増加させたことから、サポニンの膜への作用による透過性向上作用ではないかと提案された[191]。
4.14. グレープフルーツジュース
グレープフルーツ(Citrus paradisi)ジュースとカルシウムチャネル拮抗薬フェロジピンとの薬物動態学的相互作用は、エタノールとフェロジピンとの間の潜在的な相互作用を評価するために設計された1989年の生体内試験臨床試験中に偶然発見された。この研究では、エタノールの味を隠すためにグレープフルーツジュースが使用された。グレープフルーツジュースとフェロジピンを同時に摂取すると、フェロジピンの血漿中濃度が高くなるだけでなく、より高い降圧効果が得られた。この観察から、1人のボランティアを対象としたパイロット研究の追跡調査が行われ、フェロジピンの血漿中濃度が水よりもグレープフルーツジュースを摂取した場合の方が5倍以上高いことが判明した[192]。この最初の発見以来、85種類以上の薬物が薬物動態学的にグレープフルーツジュースと相互作用することが示されており、そのうちほとんどの薬物が血漿中濃度の上昇を経験している[193]。
境界性高血圧患者を対象とした研究では、異なるフルーツジュースのフェロジピン薬物動態への影響が評価されたが、オレンジジュースではなくグレープフルーツジュースはフェロジピンのバイオアベイラビリティーパラメータを著しく増加させることが示された(CmaxとAUCの両方が増加した)。さらに、グレープフルーツジュースは、デヒドロフェロジピン(CYP3A4によって産生されるフェロジピンの一次代謝物)とフェロジピンの比率を減少させた。この効果は、グレープフルーツ果汁を用いたフェロジピンの静脈内投与では観察されず、これはCYP3A4が関与する全身前代謝の選択的阻害を示唆していた[194]。
いくつかの研究では、薬物の薬物動態パラメータに対するグレープフルーツジュースの効果に関して、「増加」、「減少」および「変化なし」の3つのグループに区別できることが示されている[195]。グレープフルーツジュースの取り込みおよびバイオアベイラビリティー向上効果は、試験管内試験および生体内試験の両方のモデルを使用した研究で示された。凍結乾燥した新鮮なグレープフルーツジュースは、ヒト子宮肉腫(MES-SA/DX5)細胞へのドキソルビシンの取り込みを有意に増加させ、ウサギにおけるマレイン酸チモールのバイオアベイラビリティーを有意に増加させた[196]。しかし、通常の強さのグレープフルーツジュース、倍強のグレープフルーツジュースのいずれも、Sprague-Dawleyラットに20mg/kgを28日間にわたって併用投与した場合、シンバスタチンの薬物動態パラメータに有意な影響を及ぼさないことが示された。一方、シンバスタチンを80mg/kgの用量で投与した場合、2倍強度のグレープフルーツジュースによって血漿中のシンバスタチン濃度が上昇した[197]ことから、用量依存的な薬物動態相互作用が示唆された。別の生体内試験研究では、グレープフルーツジュースはフェキソフェナジンの経口バイオアベイラビリティーを低下させた[58]。
さまざまな研究の結果から、いくつかの形態のグレープフルーツ(すなわち、全果、新鮮な果汁または冷凍濃縮物)の薬物動態効果は薬物特異的であり、場合によっては濃度依存性である可能性があることが、時間の経過とともに明らかになった。また、全身性前代謝による薬物の固有のバイオアベイラビリティが低いほど、グレープフルーツとの相互作用が危険である可能性が高いことも示されている[193]。
薬物(例えば、フェロジピン)のバイオアベイラビリティーが増加するグレープフルーツ果汁の主な作用機序の一つは、小腸の腸球および肝臓の肝細胞におけるCYP3A4のメカニズムベースの阻害によるものである[58,198]。また、グレープフルーツジュースがP-gp流出トランスポーターを阻害し、それによってこの流出トランスポーターの基質となる薬物(例えば、タリノロール)のバイオアベイラビリティーを増加させることも示された[199,200]。さらに、グレープフルーツジュースを1日3回6日間摂取した10名のボランティアを対象とした研究では、小腸のCYP3A4タンパク質発現は62%減少したが、肝臓のCYP3A4タンパク質発現は減少しなかったことが明らかになった。このCYP3A4の選択的なダウンレギュレーションは、グレープフルーツジュース摂取後のフェロジピンのCmax上昇と水との比較でよく相関していた[201]。一方、グレープフルーツジュースは、OATP、具体的にはOATP1A2およびOATP2B1などの取り込みトランスポーターを阻害する能力を有しており、それによって特定の薬物(例えば、フェキソフェナジン)の吸収を減少させることができることが判明した[202,203]。
4.15. リコピン
シンバスタチンを含むリコペン含有ナノ製剤の低密度リポタンパク質(LDL)に対する効果が、軽度高コレステロール血症患者を対象に調査された [60]。この研究の目的は、肝内輸送のベクターとしてのリコペンを評価し、シンバスタチンの肝内バイオアベイラビリティーを特異的に高めることであった。肝コレステロゲナーゼはスタチン作用の主要部位であるため、これは重要である。さらに、血漿脂質プロファイルは、HMG-CoA還元酵素の肝外阻害によって影響を受けず、肝外毒性はスタチン濃度の増加とともに観察される[60]。血漿中LDL値が中等度に上昇した患者10名(150~200mg/dL)に、非修飾シムバスタチンまたはリコソーム配合スタチン(ライコ-シムバスタチン)20mgを1日1回投与した。投与30日後に血漿サンプルを採取し、脂質の分析を行った。その結果、可溶化したリコソームナノ粒子は、未修飾シムバスタチンと比較して腸管吸収率を高め、肝細胞膜と結合できることが示された。リコペン含有シンバスタチン製剤は、高コレステロール血症患者のLDL値を未修飾シンバスタチンと比較して有意に(p = 0.0049)低下させた[60]。
リコピンの作用機序は、カロテノイド/LDL受容体の二重のメカニズムを介した肝内取り込みの亢進である [60]。リコピンの結晶やリコピン含有ナノ粒子(リコソーム)は、吸収されるとカイロミクロンに取り込まれることが示された。その後、カイロミクロンはリンパによって分配され、リコペンコアと関連した二重受容体媒介の取り込みを受ける可能性が高い。後者は、肝細胞によって発現されるカロテノイド受容体の強力なリガンドであり、したがってリコソームで形成されたスタチンの肝内送達を促進した[60]。リコスタチンの増強された肝内取り込みは、LDL受容体のメカニズムが関与する第二の肝内取り込み経路によっても可能である。チロミクロンとその酵素分解産物であるLDLと超低密度リポタンパク質(VLDL)には、LDL受容体を用いて肝細胞内での輸送を媒介するApoBが含まれている[60]。リコシンバスタチンの薬理に関連した更なる研究が必要であるが、リコピンはシンバスタチンの標的肝送達のための有望なバイオエンハンサーであると思われる[60]。
4.16. リセルゴル
リゼルゴル(9,10-ジデヒドロ-6-メチルエルゴリン-8-O-メタノール)は、いくつかの高等植物(例えば、Ipomoea violacea、I. muricataおよびRivea corymbosa)および低級真菌(例えば、Claviceps、PenicilliumおよびRhizopus)に存在する化合物である[182,204]。リゼルゴルは、伝統的に向精神薬、鎮痛剤、鎮痛剤、血圧降下剤、免疫刺激剤として使用されている。それは通常、その血管活性を介して正常な血流を維持し、消化管からの薬物吸収を促進することができる[205]。Shukla、Malik、Jaiswal、Sharma、Tanpula、GoyaniとLal [62]による生体内試験の研究では、リセルゴールは生物学的利用能を増加させ、クルクミンのクリアランスを有意に減少させることが示された。これに続いて、ラット肝ミクロソームとP-gpおよびBCRPトランスポータータンパク質のプローブ基質を用いた試験管内試験のメカニズム研究が行われた。その結果、P-gpではなくBCRPがリセルゴールによって阻害されることが示唆された。P-gp基質であるジゴキシンとBCRP基質であるスルファサラジンのin situシングルパス腸管灌流試験を、20mg/kgのリゼルゴルで前処理したSprague-Dawleyラットで行った。これらの研究は、P-gpはリゼルゴルの浸透促進効果には関与していないが、BCRPは阻害されているという結論を支持した[62]。
メチルアルコールと一緒にI. muricata種子から単離されたリゼルゴル(2-10μg/mL)は、抗生物質のバイオエンハンサーとして作用することが示されている。著者らは、リファンピシンおよびテトラサイクリンの腸管輸送がリゼルゴルによって試験管内試験で増強され得ることを示した(2.96~8.53倍)[204]。また、リセルゴールは、雄性スプラッグ・ドーリーラットにおける第四級プロテベリンアルカロイドであるベルベリンの経口バイオアベイラビリティーを増加させた。ベルベリンは、細菌感染、腸内寄生虫感染および下痢を含む多くの疾患に使用されているが、腸内での吸収が非常に悪く、広範囲に代謝される。リゼルゴル20mg/kgを投与することでベルベリンのバイオアベイラビリティーが向上することが示唆された[61]。
4.17. ナリンギンとベルガモットン
グレープフルーツに含まれるナリンジン(フラボノイド)やベルガモットン(フラノクマリン)のような特定の植物化学物質は、グレープフルーツジュースと特定の薬物との共投与で観察される薬物動態学的相互作用と関連している[195]。スプラague-Dawleyラットにパクリタキセルをナリンジンと経口的に共投与した場合、パクリタキセル血漿中濃度は統計的に有意に上昇した。ナリンジンはCYP3AおよびP-gpの阻害剤として知られるケルセチンと同様にパクリタキセルのバイオアベイラビリティーに影響を及ぼすことから、ナリンジンはCYP3A代謝およびP-gp排出トランスポーターの阻害によってパクリタキセルのバイオアベイラビリティーを高めたと推論された[66]。同様に、ニルチアゼムの薬物動態パラメータ(AUCおよびCmax)は、ナリンジン(ニルチアゼムの30分前に投与)をラットに前処理した場合、対照群と比較して2倍に増加した。また、ナリンジンの存在下では、AUC代謝物対親和比が対照群に比べて 30%減少した。このことから、ナリンジンがディルチアゼムの代謝を阻害することが確認された[64]。
ベルガモットンと6′,7′-ジヒドロキシベルガモットンのCYP3A4阻害活性をヒトの腸内マイクロソーム試験で調べたところ,テストステロンとミダゾラムという2つのモデル化合物を用いた。その結果,ベルガモットンは基質依存性の可逆的な阻害剤であるが,CYP3A4の基質非依存的なメカニズムに基づく阻害剤であることが確認された。一方、6′,7′-ジヒドロキシベルガモットンは、基質に依存しない可逆性のCYP3A4阻害剤であり、かつ機序に基づくCYP3A4阻害剤であることが確認された[206]。
4.18. 塩化パルミトイルカルニチン
カルニチンの脂肪酸エステルであるパルミトイルカルニチンの初期の薬物吸収促進研究では、タイトジャンクションの開通(傍細胞輸送)とブラシ境界膜脂質の破壊(細胞間輸送)によって腸内薬物吸収促進が可能であることが示された[207,208]。Caco-2細胞単分子膜の詳細な研究では、調査したパルミトイルカルニチンのすべての濃度において、親水性高分子の輸送促進に伴うリトミック効果と細胞生存率の低下が示された。その結果、TEER の低下と ZO-1 の共焦点レーザー走査顕微鏡による局在化によって示されたタイトジャンクショナルネットワークの変化は、パルミトイルカルニチンと膜との相互作用によって二次的に生じていると結論づけられた。TEERの完全な回復は得られなかったので、薬物吸収促進のためのパルミトイルカルニチンの使用は注意して検討する必要がある[209]。
4.19. ピペリン
ピペリンは黒胡椒(Piper nigrum)や長唐辛子(Piper longum)に含まれるアルカロイドである[11]。ピペリンの使用は紀元前7世紀にまでさかのぼるので、ピペリンはおそらく世界初のバイオエンハンサーの一つと考えられている[205,210]。ピペリンは伝統的に、抗炎症、抗解熱、抗真菌、抗下痢、抗がん作用のために使用されている[211]。ピペリンが生体強化効果を引き起こした可能性のあるメカニズムには、膜ダイナミクスの変化、P-gp流出の阻害、および消化管および肝代謝の阻害が含まれる[11,83,210]。
C57BLマウスに経口投与した場合のレスベラトロール(3,5,4′-トリヒドロキシスチルベン)の血清レベルに対するピペリンの効果を調査した研究では、AUCおよびCmaxがそれぞれ229%および1,544%増加したことが示された[77]。Jin and Han [76]は、ラットにフェキソフェナジンを経口投与(10mg/kg)または静脈内投与(5mg/kg)した場合のピペリンの経口投与によるバイオアベイラビリティ変化効果を調査した。その結果、フェキソフェナジンのCmax及び半減期に有意な変動は認められなかったが、フェキソフェナジンの経口曝露量(AUC)はほぼ2倍であった。この研究から、ピペリンの効果は静脈内投与後(肝代謝阻害による)よりも経口投与後(P-gp介在性の排出阻害と消化管上皮における代謝阻害によるものと思われる)の方が顕著であることが推論された[76]。
4.20. ケルセチン
CYP3A4およびP-gp阻害剤であるケルセチンは、ラットに経口投与されたタモキシフェンおよび4-ヒドロキシタモキシフェンのバイオアベイラビリティーを有意に向上させる能力を示している[95]。その研究では、タモキシフェン(10 mg/kg)を、ケルセチン(2.5,7.5および15 mg/kg)の有無にかかわらず、ラットに経口投与した。タモキシフェン(10 mg/kg)を対照液として 1.5 mL の蒸留水に添加し、必要量のケルセチンを 1 mL の蒸留水に溶解して経口投与用のケルセチン懸濁液を調製した。タモキシフェンの血漿中濃度は、タモキシフェン投与後 0,0.5,1,2,3,4,5,6,8,12,24,36 時間後に採取した血液サンプル(0.5 mL)を用いて、HPLC [95]を用いて測定した。
タモキシフェンの相対的なバイオアベイラビリティーは 1.35 倍と 1.61 倍で、絶対的なバイオアベイラビリティーは、それぞれ 2.5 mg/kg と 7.5 mg/kg のケルセチンで 20.2%と 24.1%であった。これらのバイオアベイラビリティの変化は有意であった(p<0.05)。興味深いことに、より高濃度のケルセチン(15 mg/kg)では、有意な変化は誘発されなかった。タモキシフェンの終末半減期(t1/2)とピーク濃度(Tmax)に達するまでの時間には、ケルセチンとの共投与では有意な変化は見られなかった。また、タモキシフェンの代謝物の一つである 4-ヒドロキシタモキシフェンとケルセチン(7.5 mg/kg)を併用投与すると、4-ヒドロキシタモキシフェンの AUC が有意に上昇した。これらの結果は、タモキシフェンの MDR の流出と一次代謝がケルセチンによって阻害されていることを示唆している [95]。
別の生体内試験研究では、フェキソフェナジンとケルセチンを併用した場合に、フェキソフェナジンのP-gp排 出を抑制する効果が示された[91]。健康な被験者12人に、ケルセチン(500mg)またはプラセボを1日3回、7日間経口投与した。7日目には、10時間以上の絶食状態でフェキソフェナジン60mgを240mLの水とともに単回経口投与した。投与後 4 時間後と 10 時間後には、フェキソフェナジンやケルセチンの吸収に影響を及ぼさないように、フラボノイドを少量しか含まない標準的な食事を摂取した。その後、フェキソフェナジン投与の直前,0.25,0.5,1,1.5,2,2.5,3,4,6,8,12,および24時間後に血液サンプルを採取した。尿サンプルは、投与後0-2,2-4,4-8,8-12,および12-24時間の時間間隔で採取した。血漿中および尿中のフェキソフェナジン濃度を HPLC [91] を用いて定量した。
その結果、ケルセチンはフェキソフェナジンの平均血漿中濃度を有意に上昇させることが示された。血漿中のフェキソフェナジンのAUCは、ケルセチンの存在下で2,005.3から3,098.6 ng.h/nL(p < 0.001)まで55%増加した。Cmaxも同様に、ケルセチンとの併用投与で295.3~480.3 ng/mL(p = 0.006)から68%増加した。ケルセチン投与後、フェキソフェナジンの経口クリアランスは61.4から38.7 L/hへと37%有意に減少した(p<0.001)。しかし、プラセボ投与群とケルセチン投与群の間では、腎クリアランスおよび半減期に差は認められなかった[91]。
要約すると、ケルセチンは、タモキシフェン[95]とフェキソフェナジン[91]の 腸管吸収を促進し、その結果、バイオアベイラビリティーを向上させるためのバイオエンハンサーとして使用できる。ケルセチンが採用する作用機序は、MDR トランスポーターの排出阻害と第一パス代謝阻害である [95]。ラットモデルのタモキシフェンで得られた結果が臨床試験で確認された場合、ケルセチンやケルセチンを含む栄養補助食品を本剤と同時に摂取する場合には、薬物相互作用の可能性を考慮して、タモキシフェンの用量を調整すべきである[95]。フェキソフェナジンのP-gp介在性排出に対するケルセチンの抑制効果は短期(7日間)で実証されたが、ケルセチンの長期使用でも同じ結果が得ら れるかどうかは不明である[91]。
4.21. キニジン
常時腸嚢を用いた研究では、キニジンがパエオニフローリン(20 µM)の吸収を有意に高めることが実証されている[97]。パエオニフローリンの吸収は培養時間の増加に伴って増加しており、この化合物の吸収は時間依存性であることが示された。また、パエオニフローリンの濃度を変えた吸収プロファイルから、濃度の増加とともに吸収が増加することが示されたが、~80 µMの濃度では試験管内試験の腸嚢系での飽和が観察された。この飽和は、パエオニフローリンの輸送が活性なトランスポーターまたはキャリアによって促進されている可能性を示唆している[97]。キニジン(1.3 mM)とパエオニフローリン(20 µM)の共投与では、45 分間のインキュベーション後にパエオニフローリンの吸収が 1.5 倍に増加した。腸内でのパエオニフロリンの吸収は、このようにキニジンの共同投与によって有意に強化される可能性がある[97]。
4.22. レスベラトロール
最近の研究では、レスベラトロールがラットのメトトレキサートの腸管吸収を生体内試験および試験管内試験で有意に増加させることが実証された[99]。さらに、レスベラトロールは、メトトレキサートの排出輸送を阻害し、腎クリアランスを減少させることもできた。模擬MDCK、MDR1-MDCK、およびMRP2-MDCK細胞単層を横切るメトトレキサートの双方向輸送を、メトトレキサート(10μM)および/またはレスベラトロール(10μM)を含む実験用緩衝液を用いて評価した。この実験用緩衝液をドナーチャンバー(それぞれ400μLのアピカルチャンバーまたは600μLのバソラテラルチャンバー)に添加した。0.5,1,2,および3時間の時間間隔で、50μLのサンプルを反対側(レシーバー)のチャンバーから取り出し、50μLの新鮮なハンクスのバランス塩溶液(HBSS、pH7.4)で補充した[99]。
さらに、腎臓切片およびCaco-2細胞におけるレスベラトロールの非存在下および存在下でのメトトレキサートの試験管内試験での取り込みを液体クロマトグラフィー-タンデム質量分析法(LC-MS/MS)によって決定した。レスベラトロールの作用機序を明らかにするために、トランスポーター取り込みアッセイは、模擬ヒト胚性腎(HEK)293細胞、ヒト有機アニオン輸送1(hOAT1)-HEK293細胞およびhOAT3-HEK293細胞を用いて同様に行った。輸送緩衝液には、10μMのメトトレキサート、p-アミノヒプル酸(PAH)ペニシリンG(PCG)またはレスベラトロールが含まれていた。PAHおよびPCGは、それぞれOAT1およびOAT3の特異的基質である[99]。
ラットの生体内吸収実験では、2%炭酸水素ナトリウムに溶解したメトトレキサート5 mg/kgをレスベラトロール(100 mg/kg,0.5%カルボキシメチルセルロースナトリウムに溶解したもの)を経口投与し、通常の生理食塩水で容量を増加させた。血液サンプルを5,10,20,30,60,120,240,360,480,および600分後に採取し、メトトレキサートの血漿中濃度を測定した。メトトレキサートの腎排泄を確立するために、メトトレキサート(5mg/kg)および/またはレスベラトロール(10mg/kg)をヒドロキシプロピル-β-シクロデキストリンの通常の生理食塩水に溶解した溶液をラットに静脈内投与した。ここでも、メトトレキサートの血漿中濃度を測定するために、所定の時刻に採血を行った。さらに、投与後2,4,6,8,10,12,16,24時間後に尿を採取し、尿中に排泄されたメトトレキサートの濃度を測定した[99]。
生体内試験での結果から、メトトレキサートを経口または静脈内で併用投与すると、メトトレキサートの血漿中曝露量が有意に増加することが示された。ラットの常在腸嚢モデルを用いた試験管内試験の結果では、メトトレキサートの血清中濃度は、レスベラトロール、P-gpおよびMRP2の阻害剤であるベラパミルおよびCDFの存在下で有意に上昇することが示された。これらの結果から、レスベラトロールがP-gpまたはMRP2を阻害することにより、メトトレキサートの腸管吸収を促進することが示唆された。さらに、メトトレキサートとレスベラトロールを併用投与した場合、メトトレキサートの累積尿中排泄量が37.3%減少したことから、レスベラトロールがメトトレキサートの腎排泄を抑制することが示唆された。後者の作用機序を明らかにするために、ラットの腎臓切片を用いてメトトレキサートの取り込み実験を行った。その結果、レスベラトロールとOATの阻害剤として知られるプロベネシドの存在下では、メトトレキサートの取り込みが有意に減少することがわかった。さらに、PAHおよびPCG(それぞれOAT1およびOAT3の基質)ならびにメトトレキサートの取り込みは、hOAT1-HEK293またはhOAT3-HEK293細胞において、レスベラトロールによって有意に阻害された。これらの結果から、レスベラトロールは腎臓におけるメトトレキサートとレスベラトロールの薬物-薬物相互作用に関与する標的輸送体であるOAT1/3を阻害できることが示唆された。また、MDR1-MDCK細胞およびMRP2-MDCK細胞では、レスベラトロールの存在下で、メトトレキサートの基底から上皮への輸送率がそれぞれ32.5倍および20.8倍に増加した。これらの結果から、P-gpとMRP2もまた、レスベラトロールによって阻害されるメトトレキサートの標的トランスポーターであることが示唆された[99]。
生体内試験および試験管内試験での結果から、レスベラトロールは腸内でのメトトレキサートの吸収を有意に増加させ、メトトレキサートの腎クリアランスを減少させることが示唆された。吸収、輸送および腎クリアランスの研究では、レスベラトロールの作用機序がP-gp、MRP2,OAT1およびOAT3の阻害を伴うことが確認された。これは、メトトレキサートが主にOAT1とOAT3によって未変化のまま尿中に速やかに排泄されることを示した先行研究と一致する[212,213]。
最近の研究では、MDR1を発現するMadin-Darby canine kidney(MDCKII-MDR1)細胞において、レスベラトロールがサンキナビルのP-gp排出を刺激したという矛盾した結果が示されている[214]。この研究では、MDCKII-MDR1細胞を50μMのsaquinavirおよび/またはレスベラトロール(1,10,33および100μM)またはベラパミル(50μM)と37℃で4時間インキュベートした。さらに、マイクロソームを用いた試験管内試験代謝実験により、レスベラトロールが腸内CYP3Aを介したサキナビルの代謝に及ぼす影響を評価した。サキナビルの薬物動態プロファイルに対するレスベラトロールの効果を評価するために、ラットに30 mg/kgのサキナビルをレスベラトロール(20 mg/kg)とともに、またはレスベラトロール(20 mg/kg)とともに経口投与した。サキナビルは溶媒(20%エタノール、30%プロピレングリコール、50%生理食塩水)に6 mg/mLの濃度で懸濁し、レスベラトロールは30%ポリエチレングリコール400の生理食塩水に20 mg/mLの濃度で懸濁した。その後,投与後0,0.25,0.5,1,2,4,8,12,24時間後に採血し,LC-MS/MSで分析した[214]。
その結果、レスベラトロールの存在下では、濃度依存的に有意に細胞内濃度が低下することが示され、レスベラトロールがP-gpを介したサンキナビルの排出を刺激していることが示唆された。代謝実験の結果、レスベラトロールの存在下では、用量依存的に残留サカナビルが有意に増加することが示された。このことから,レスベラトロールは腸内CYP3Aを介したサカナビルの代謝を濃度依存的に抑制する効果を有していることが示唆された。レスベラトロール(20 mg/kg)の経口投与により,平均AUC0-∞は31%減少し,平均見かけの全身クリアランス(CL/F)は51%増加した。しかし、これらの変化はいずれも統計学的に有意ではなかった(p > 0.05)[214]。
4.23. シノメニン
常時腸嚢を使用した以前の研究では、シノメニンを 16 µM および 136 µM の濃度で使用すると、パエオニフロリン (20 µM) の吸収がそれぞれ 1.5 倍および 2.5 倍に有意に増加することが実証された [97]。同様に、ジゴキシン(13 µM)の吸収は、シノメニン(136 µM)の存在下で2.5倍に有意に増強された。パエオニフローリンの吸収は培養時間の増加とともに増加し,この化合物の吸収は時間依存的であることが示唆された。一方、異なる濃度のパイオニフローリンの吸収プロファイルからは、濃度の増加に伴って吸収が増加することが示されたが、~80 µMの濃度では試験管内試験の腸嚢系の飽和が観察された[97]。この飽和は、パエオニフローリンの輸送がエネルギー依存性キャリアによって促進されている可能性を示唆している。シノメニン(90 mg/kg)の存在下でのパイオニフロリン(150 mg/kg)のバイオアベイラビリティーの向上は、以前にもラットの生体内で実証されており、12倍に増加している[100]。
観察されたパエオニフロリンの吸収増強の根底にある潜在的なメカニズムを調べるために、2種類のPg阻害剤(ベラパミルまたはキニジン)とP-gp基質(ジゴキシン)を用いた。ベラパミル(20μM)とパエニフロリン(20μM)の共投与により、45 分間のインキュベーション後にパエニフロリンの吸収が 2.1 倍に増加した。同様に、キニジン(1.3mM)を併用した場合、パエニフロリンの吸収は1.5倍に増加した。このように、腸内でのパイオニフロリンの輸送および吸収は、ベラパミルとキニジンによるP-gpの阻害によって有意に増強される可能性がある[97]。また、シノメニン(136μM)は、P-gp基質類似物質としてよく知られている13μMのジゴキシンの吸収を、45分間のインキュベーション後に2.5倍に有意に増強することができた。
4.24. カプリン酸ナトリウム(脂肪酸)
ベルベリンの腸管吸収に対するカプリン酸ナトリウムの効果は、in situ, 試験管内試験, 生体内試験 実験によって研究されてきた [101]。再循環灌流モデルの結果から、ベルベリン(50μmol/L)の4時間後の吸収率は、カプリン酸ナトリウムの存在下で9.3%から18.5%に増加した。一方,100μmol/L及び200μmol/Lのベルベリンにカプリン酸ナトリウム(0.2% w/v)を添加した場合は,それぞれ13.1%及び20.1%の吸収率の増加が認められた。また、試験管内試験でのエバーテッドラット腸嚢実験の結果、少量のベルベリンは腸内で吸収され、空腸と回腸でそれぞれ吸収率が最も高く、最も低いことが明らかになった。カプラーゼナトリウムの存在下では、ベルベリンの吸収は90分培養後に急速かつ有意に増加した。また、カプリン酸ナトリウムは、Pappの増加をもたらした。カプラーゼナトリウムの存在下でベルベリンの最大の吸収は回腸(ER = 3.49)であったが、カプラーゼナトリウムの不在下では最も弱い吸収を示した。十二指腸及び空腸ではそれぞれ2.08及び1.49の吸収促進比(ER)を示した。また,ベルベリンのバイオアベイラビリティーは,カプリン酸ナトリウムとの併用により改善されることを生体内試験試験で明らかにした。ラットにおけるベルベリンの血漿中ピーク濃度の有意な上昇(721.39±53.46から988.84±135.56ng/mL)が認められ,ピーク時間の遅延(30分から60分),AUC0-6時間の増加(28%)が認められた。また、ベルベリンとカプリン酸ナトリウムを併用した場合、ベルベリン投与群と比較して血糖値とグルコースカーブ下面積が著しく減少し、インスリンの抗糖尿病活性が増強された[101]。ベルベリンのバイオアベイラビリティーが低いのは、P-gpの流出によるものであることが示唆されている。カプ レートナトリウムがP-gpの流出を阻害することが以前に実証されている[215]。
4.25. Zonula Occludens Toxin (Zot)
研究では、高分子量マーカーや生物学的利用性の低い化合物の透過性は、吸収促進剤であるゾヌラ・オクルーデンス毒素(Zonula occludens toxin, Zot, MW: 45 kDa)(ビブリオ・コレラエ菌が産生する毒素)を併用することで、Caco-2細胞モノレイヤーを介して増加させることができることが実証された[107]。この研究では、親水性高分子量マーカー(すなわち、PEG4000,FITC-デキストラン10,000,およびイヌリン)および疎水性治療薬(すなわち、アシクロビル、シクロプソリン、パクリタキセル、およびドキソルビシン)の輸送を、Caco-2細胞モノレイヤーを用いてZotを用いて評価した。Zotは腸管内腔表面の特定の受容体に結合することで、腸管上皮細胞間のタイトジャンクションを可逆的に開くことが以前に確立されている[216]。細胞単層を30分間Zot(0,1,2,および4μg/mL)で事前にインキュベートした後、マーカーまたは治療薬を時間0で挿入物に添加し、サンプルを基底側チャンバから120分間にわたって収集した。輸送分析には、ラジオグラフ法またはHPLC法を使用し、TEER値を3時間にわたってモニターした。その結果、4 µg/mLのZotは、MW < 5 kDaのマーカーの輸送を6.2倍に増加させることが示された。同様に、Zotは4μg/mLの濃度で治療薬の輸送を有意に増加させ、アシクロビルおよびパクリタキセルではそれぞれ1.8および3.13の輸送増強比を示した。0.5~2時間の間に有意に低いTEER値が観察されたが、Caco-2細胞単層でのTEERに対するZotのこの効果は可逆的であった。
トランスセルラーマーカーであるプロプラノロールを用いた Caco-2 輸送研究では、Zot はトランスセルラーパスウェイを有意に修飾しないことが示された [107]。さらに、乳酸脱水素酵素(LDH)アッセイでは、Zot の存在下で LDH 活性に有意な差は認められず、3 時間の培養後の有効濃度レベルでは Zot は非細胞毒性であることが示唆された [107]。FITCの透過性は有意に向上しなかったが、イヌリンの透過性は7×10-7 cm/s(コントロール)から4.37×10-6 cm/s(4 µg/mL)へと有意な増加(6.3倍)が観察された。イヌリンが高分子量であるにもかかわらず、イヌリンのコンパクトな円筒形の構成がPEG4000と比較して高い透過性をもたらすことが示唆された[107]。治療薬を用いた試験管内試験の結果では、Zotによる透過率の上昇は20~300%の範囲であった。パクリタキセルとシクロスポリンAはそれぞれ3.13と1.2のPapp増強比を示した[107]。
5. 肺への投与経路
図4 強化された肺薬物送達のためのバイオエンハンサーの主な作用機序の図示
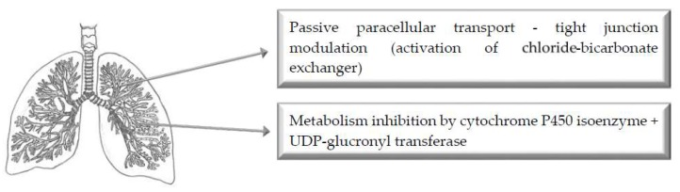
5.1. 胆汁酸塩
研究では、チロトロピン放出ホルモン(TRH、MW:362 Da)とインスリン(MW:5814 Da)の気管透過性がグリココール酸ナトリウムによって有意に増加することが示された[113]。摘出したウサギの気管の非倒立嚢セグメントを用いた。輸送試験に加えて、気管と空腸におけるペプチダーゼ活性を測定し、比較することで、ウサギの気管上皮を横断するTRHとインスリンの透過性のための酵素障壁を調べた[113]。その結果、TRH(5 mM)とインスリン(10 IU/mL)の両方の透過性は、10 mMのグリココラートの存在下で有意に増加することが示された。TRH(5および10mM)のPapp値は、それぞれ2.51±0.31×10-7および3.54±0.87×10-7 cm/sであった。グリココール酸ナトリウム(10 mM)はTRHの気管透過性を~3倍に増加させた[113]。インスリン(10 IU/mL)のPapp値は6.66±0.11×10-9 cm/sであった。しかし、インスリンの半減期は約14時間であり、実験期間(150分)の間は腔内分泌酵素によるわずかな分解を示したに過ぎなかった。気管および空腸ペプチダーゼは、以下のような活性の低下順を示した。DPP IV>ロイアミノペプチダーゼ>カテプシンB>トリプシン[113]。これら4つのペプチダーゼは空腸上皮細胞と比較して、気管上皮細胞において有意に低い活性を示した。TRHは気管透過中に代謝物を持たなかったので、タイトジャンクション調節の潜在的な作用機序、したがって強化された傍細胞透過性は、グリココラートナトリウム[113]のために示唆されている。グリココラートは、リューアミノペプチダーゼ阻害剤であることが知られている。しかし、インスリンは気管透過中にわずかに分解された。したがって、インスリン(MW:5814 Da)は、主に傍細胞拡散を介して輸送され、単離されたウサギの気管上皮を介してトリプシン様酵素やアミノペプチダーゼ様酵素などのタンパク質分解酵素によってわずかに代謝されることが示唆されている[113]。
同様に、以前の試験管内試験および生体内試験の研究では、吸入インスリンの吸収が胆汁酸塩タウロコリン酸ナトリウムによって促進されることが示されている[114]。生体内試験研究では、ビーグル犬を少なくとも16時間飢餓状態にした後、挿管してインスリンまたはインスリン-タウロコリン酸ナトリウムエアロゾルに時間を変えて曝露した。静脈内基準として,0.9%NaCl中のインスリンを右前脚静脈に5分間(0.2 U/kg,0.5 U/mL,0.08 mL/kg/分)注入した。インスリン-ナトリウムタウロコレート溶液をPARI LCジェットネブライザでエアロゾル化した。ネブライザーからの流量は3.2 L/min(1 Bar)で、インスリンの目標吸入量は1 U/kgであった。投与前および吸入開始(t=0)から5,10,15,25,35,50,65,95,125および245分後に血液サンプルを採取した[1144]。純粋なインスリンのバイオアベイラビリティーは2.6±3%であったが、ネブレーションしたインスリン溶液のバイオアベイラビリティーは32mMタウロコール酸ナトリウムの添加により23.2±4.4%に増加した。一方、エアゾール化した粉末を肺に投与した場合のインスリンのバイオアベイラビリティは3.81±1.12%であった。試験管内試験での結果は、25〜30mMの間のタウロコール酸ナトリウム濃度で、減少したTEERを伴うインスリン輸送の増加を示した。また、細胞層の生存率は、32 mM を超えるタウロコリン酸ナトリウム濃度では、残念ながら約ゼロであった [114]。
5.2. キトサンとその誘導体
キトサンとN-トリメチルキトサン(TMC)による気管支オクトレオチドの吸収促進を試験管内試験/生体内試験相関法を用いて評価した[109]。キトサンをアルカリメチル化することにより、20%および60%のQDを有するTMC誘導体(TMC20およびTMC60)を合成した。キトサンを1.5%(w/v)でHBSSに溶解し、30mM HEPESで緩衝し、pH5.5に調整した。TMC20およびTMC60は、pH7.4でHBSS/HEPESに1.5%(w/v)で溶解した。TEER試験の2時間前に、培地を除去し、Calu-3細胞を基底側チャンバ内のpH7.4で1mlのHBSS/HEPES、および先端コンパートメント内のpH5.5またはpH7.4で200μLのHBSS/HEPES中で平衡化した。t = 0において、アピカルコンパートメントの培地を200μLキトサン、TMC20またはTMC60製剤、またはコントロールとして機能したpH5.5またはpH7.4での200μLHBSS/HEPESで置換した。TEERは、投与前のt = 120および60,ならびに投与後のt = 0,30,60,90,120,150,210および240分で測定した。
オクトレオチドは、1.5%キトサン(pH5.5)、TMC20(pH7.4)またはTMC60(pH7.4)を含む0.9%生理食塩水に0.97mMの末端濃度になるように溶解した。コントロール溶液は、pH5.5および7.4の0.9%生理食塩水中のオクトレオチドを含んでいた。これらの溶液から 200μLをラットの気管内に注入した。ラットの気管支への投与量を確保するために、気管内注入を採用した。6匹のラットのグループには、オクトレオチドの絶対的なバイオアベイラビリティーを決定するために、190μMのオクトレオチドを0.9%生理食塩水に溶解した200μLの静脈内ボーラスを投与した[109]。
キトサン、TMC20およびTMC60の存在下では、TEERの有意な、しかし可逆的な減少が観察され、それぞれ21倍、16倍および30倍のオクトレオチド透過率の向上を伴った。バイオアベイラビリティーはそれぞれ2.4倍、2.5倍、3.9倍に増強された。TMC60はTEERの強い低下を誘導したが、これはpH、溶解度、およびカチオン性電荷密度が傍細胞バリアの調節に重要な因子であることを示唆している[109]。計算された吸収率の間には、試験管内試験/生体内試験で直線的な相関が観察された(R2 = 0.93)。これは、類似のメカニズムにより、試験管内試験と生体内試験の両方で、ポリマーによる浸透促進が起こることを示唆している。浸透研究ではゼロオーダーの速度論的プロファイルが示されており、ポリマーがタイトジャンクションの構造的完全性を変化させ、オクトレオチドの受動的な傍細胞拡散を促進していることを示唆している[109]。
5.3. クエン酸(キレート剤)
溶液および乾燥粉末からのインスリンの肺吸収に対する添加物の効果を、雄性のSprague-Dawleyラットで検討した[110]。この研究の結果、クエン酸の非存在下ではわずかに血糖降下作用があることが示された[110]。クエン酸の存在下では、インスリン投与後に有意で拡張した用量依存性の血糖降下効果が観察された。クエン酸緩衝液(0.1mL中のクエン酸0.19mg/用量)を用いた場合、pH5及び3でのバイオアベイラビリティはそれぞれ43%及び57%であった。しかし,乾燥粉末のクエン酸塩では,溶液よりも大きな血糖降下効果が得られた。クエン酸0.2%(0.036 mg/dose)製剤では、インスリン乾燥粉末投与後に血糖値が急速に低下し、その効果は実験期間中継続した[110]。50%以上の絶対的なバイオアベイラビリティーが達成された。肺細胞への急性毒性の敏感な指標であるLDH活性は,0.2%クエン酸ではPBS投与時と同様に低かった。これは、クエン酸が安全な吸収促進添加物であることを示唆した。クエン酸のために提案された可能性のある作用機序は、肺の上皮膜の完全性を変更し、酵素活性と肺胞マクロファージの貪食活性を抑制することにより、インスリン分解を減少させた[110]が含まれてた。
5.4. シクロデキストリン(CD)
ヒドロキシプロピル-β-シクロデキストリン(HPβCD)ヒドロキシプロピル-γ-シクロデキストリン(HPγCD)ランダムメチル化β-シクロデキストリン(Rameb)および2-0-メチル化β-シクロデキストリン(Crysmeb)の化学修飾シクロデキストリン(CD)の試験管内試験透過性試験をCalu-3細胞を用いて実施した[111]。この研究からの結果は、それぞれのβCD誘導体の存在下でCalu-3層を横断する14C-マンニトールフラックスの濃度依存的な増加を示したが、HPγCDによる透過性の変化は見られなかった。Ramebは、最低濃度(10mM)ではマンニトール輸送を増強し、最高濃度(50mM)ではマンニトールフラックスを10倍に増加させる唯一のCDであった。CrysmebとHPβCDは25mMと50mMでマンニトール透過性を増加させることができた。TEERの減少は、Calu-3細胞層を横断するマンニトール輸送のすべての増加に対して観察された。共焦点顕微鏡は、Crysmeb(25 mM)は可逆的にタイトジャンクション複合体を混乱させることができることを明らかにした[111]。
同様に、以前の研究では、テトラデシル-β-マルトシド(TDM)とジメチル-β-シクロデキストリン(DMβCD)は、主に薬物と相互作用するのではなく、膜に作用することによって、試験管内試験および生体内試験の両方で低分子量ヘパリン(LMWH)の肺吸収を高めることができることが実証された[115]。Calu-3細胞を用いて、3H-エノキサパリンおよび14C-マンニトールをTDMまたはDMβCDの異なる濃度(0.0625%,0.125%、および0.25%(w/v))の有無にかかわらず、pH7.4で輸送試験を行った。TEERは輸送実験中に測定した。生体内肺吸収試験では、TDMおよびDMβCDを生理食塩水に溶解し、それぞれ1%のストック溶液を調製した。エノキサパリン、ダルテパリン(MW:5000 Da)および未分割ヘパリン(MW:15,000-20,000 Da)をそれぞれ生理食塩水またはTDMおよびDMβCD(0.0625%,0.125%および0.25%(w/v))のいずれかと混合し、100μLの製剤あたり15 Uの抗因子Xa活性の最終濃度を得た。これらの製剤(50 U/kg)をラットに気管内投与または皮下投与し、それぞれ吸収試験およびバイオアベイラビリティー試験を行った。その後,0, 15, 30, 60, 120, 240, 360, 480 分後に血液サンプル(300μL)を採取した。エノキサパリンの吸収は、比色法を用いて血漿中の抗因子 Xa レベルを測定し、その後、標準的な薬物動態解析を行うことで決定した [115]。
結果は、先端液中のTDMまたはDMCD(0.0625~0.25%)の存在下でのエノキサパリンの用量依存的な増加を実証した。TDM濃度を0.0625%から0.25%に増加させると、エノキサパリンのPappは統計的に有意に4倍増加することが観察された。マンニトールの輸送についても同様の結果が得られた。TDMまたはDMβCDの存在下でのマンニトールの全体的な透過性の増加は、作用機序がタイトジャンクションの緩みであることを示唆しており、したがって、傍細胞輸送が強化されている。これは、2時間後に0.25%TDMおよび0.25%DMβCDの存在下でそれぞれ初期値の51.9%および70.7%まで低下したTEER値で確認されたが、TEER低下効果は可逆的であり、これらのバイオエンハンサーが呼吸器上皮細胞に広範な損傷または細胞毒性を引き起こす可能性がないことを示している。TDMおよびDMβCDの濃度を増加させると、エノキサパリンについてはCmaxが増加し、Tmaxが減少した。全体として、薬物動態解析の結果から、TDMはエノキサパリンの肺吸収を促進することが示唆された。バイオアベイラビリティのデータは、DMβCDと比較してTDMがLMWHの肺吸収を促進する効果が高いことを示した。興味深いことに、肺投与されたエノキサパリンの薬物動態プロファイルは、皮下投与されたエノキサパリンと比較して、抗因子Xa活性の迅速な発現を示した。ダルテパリンはまた,0.125%TDMまたはDMβCDの存在下で抗因子Xa活性を迅速かつ実質的に増加させたが、未分画ヘパリンは抗因子Xa活性のわずかな増加しかもたらさなかった。未分画ヘパリンの吸収率の低下が観察されたが、これはLMWH(エノキサパリンおよびダルテパリン)よりも大きくてかさばるという事実に起因している可能性がある。本研究の結果は、LMWHの経鼻投与に関する先行研究[217]と一致している。さらに、比較分析により、エノキサパリンの相対的なバイオアベイラビリティは、経鼻投与されたエノキサパリンと比較して肺投与で4~6倍高く、肺投与されたLMWHsから治療用抗因子Xa活性を産生するのに必要なバイオエンハンサーの量は2~5倍少ないことが実証された[115]。
5.5. ランタニド
ラット肺からのランタノイド(Ln3+)すなわちランタン、セリウムおよびガドリニウムを事前投与または共投与したインスリンの吸収を、in situ 肺吸収実験 [112] を用いて調査した。ランタニドイオン溶液は、酸化物を5mol/Lの塩酸水溶液に溶解し、加熱して過剰な酸を除去することにより調製した。残渣を生理食塩水で希釈し、Ln3+濃度を測定した。事前に投与された気管内薬物送達のために、最初に25μLの生理食塩水(pH7)中のLn3+(0.2 mg/kg)を注射し、次いで30分後にインスリン(40μLの生理食塩水(pH7)中の1 IU/kg)を投与した。他の実験では、インスリンとLn3+を共投与した。インスリン投与後、所定の時間間隔で血液サンプル(0.5 mL)を採取し、血清インスリン濃度を測定した[112]。
その結果、血清インスリン濃度はCeCl3とGdCl3の存在下で有意に上昇し、相対的なバイオアベイラビリティはそれぞれ57.9%と59.5%であった。さらに、ガドリニウムのアニオン性形態は、そのカチオン性形態(Gd3+)に比べて、肺インスリン吸収のより大きな増強を示した。また、GdCl3を併用投与した場合と投与前に投与した場合の相対的なバイオアベイラビリティーは、それぞれ80.1%と59.5%であった。対照的に、LaCl3はインスリンの肺吸収の弱い増強効果を示し、相対的なバイオアベイラビリティは30.9%であったが、LuCl3は増強効果を示さなかった[112]。Gd3+(0.2 mg/kg)とインスリンの共投与(Fr = 80.1%)は、肺からの最大のインスリン吸収促進を示した。興味深いことに、Gd3+(0.6 mg/kg)の高濃度投与ではインスリン吸収が低下したが、これはおそらく肺の入り口でGd3+が部分的に加水分解されるためであると考えられる。Ln3+の作用機序として提案されているのは、ランタノイドが膜脂質やタンパク質に結合して構造変化や細孔形成を誘導し、細胞内や外因性物質の透過性を高めることで、局所的な粘膜組織の変調である[218,219]。もう一つの作用機序は、Ln3+が亜鉛(II)結合部位に結合することでインスリンの構造変化が誘導され、肺からのインスリンの良好な吸収が促進されることである。さらに、血清中のインスリン結合Ln3+により、細胞膜中の受容体とインスリンの相互作用が増加する可能性がある[112]。
5.6. プロテアーゼ阻害剤
ラットにおける組換えヒトコロニー刺激因子(rhG-CSF)の肺吸収促進剤として、プロテアーゼ阻害剤の効果を評価した[108]。プロテアーゼ阻害剤としては、(p-アミジノフェニル)メタンスルホニルフルオリド-HCl(p-APMSF)アプロチニン、ベスタチンが挙げられた。それぞれのプロテアーゼ阻害剤(10mMのp-APMSF、500IU/mLのアプロチニン、1mMのベスタチン)をrhG-CSF溶液(pH6.5)に添加し、最終的なrhG-CSF濃度を250μg/mLに調整した。試験溶液(プロテアーゼ阻害剤を含む)および対照溶液(rhG-CSFのみ)を静脈内、皮下および気管内投与した後、8時間定期的に血液サンプルを採取し、酵素免疫測定法によりrhG-CSFの血漿中濃度を測定した。
血漿中のrhG-CSF濃度は、プロテアーゼ阻害剤の存在下で大幅に増強され、p-APMSFは、気管内投与30分後に3.9から11.7 ng/mLまで血漿中のrhG-CSF濃度が約3倍に増加し、最大の増強を示した[108]。吸収促進のメカニズムを調べるために、界面活性剤であるポリオキシエチレン9-ラウリルエーテル(ラウレス-9)とプロテアーゼ阻害剤であるp-APMSFをrhG-CSFと一緒に投与した。その結果、rhG-CSF濃度は3.9±1.4 ng/mLから481.5±96.7 ng/mLへと123倍以上に上昇した。その結果、提案されている作用機序は、膜透過性を高め、rhG-CSFの酵素分解を阻害することであった[108]。
同様に、先行研究では、ベスタチン(アミノペプチダーゼBおよびロイシンアミノペプチダーゼ阻害剤)およびアプロチニン(トリプシンおよびキモトリプシン阻害剤)により、インスリンの気管透過性が有意に増加したことが実証されている[113]。切除したウサギの気管の非倒立嚢(2cm)セグメントを使用した。簡潔に、HEPES緩衝液(0.2 mL、pH7.4)中のプロテアーゼ阻害剤(0.1および1 mMベスタチン;1000および10,000 KIU/mLアプロチニン)の有無にかかわらず、試験溶液を嚢粘膜側に注入し、酸素化(O2/CO2,95%:5%)HEPES(7 mL、pH7.4)からなる血清培地に入れた。サンプル(0.2 mL)を、あらかじめ決められた時間に150分間、血清液から除去し,0.2 mLの新鮮な液と交換した。別の実験では、RIA法を用いて気管粘膜液中のインスリンの分解速度を計算するために、10μLのサンプルを予め決められた時間で粘膜液から除去した。さらに、気管と空腸におけるペプチダーゼ活性を測定し、比較することで、ウサギの気管上皮における TRH とインスリンの透過性の酵素的障壁を調べた [113]。
その結果、インスリンの透過性(10 IU/mL)は、1 mMベスタチンと10,000 KIU/mLアプロチニンの存在下で有意に増加することが示された。しかし、インスリンの半減期は約14時間であり、実験の持続時間(150分)の間、腔内分泌酵素によるわずかな分解を示しただけであった[113]。気管および空腸ペプチダーゼは、以下のような活性の低下順を示した。DPP IV>ロイアミノペプチダーゼ>カテプシンB>トリプシンであった。これら4つのペプチダーゼは空腸上皮細胞に比べて気管上皮細胞では有意に活性が低かった。しかし、インスリンは気管透過時にわずかに分解された。このことから、インスリン(MW:5814 Da)は主に傍細胞拡散を介して輸送され、単離されたウサギの気管上皮を介してトリプシン様酵素やアミノペプチダーゼ様酵素などのタンパク質分解酵素によってわずかに代謝されていることが示唆された。まとめると、インシュリンなどのいくつかのペプチド薬物の気管内気管挿管による肺投与は、これらの薬物の全身吸収に寄与している可能性がある[113]。
5.7. 界面活性剤
ラットにおける組換えヒトコロニー刺激因子(rhG-CSF)の肺吸収促進剤として、天然由来の界面活性剤の効果を評価した[108]。界面活性剤としては、ポリオキシエチレン9-ラウリルエーテル(ラウレス-9)とグリコリン酸ナトリウム(SGC)を用いた。界面活性剤(1% w/v)をそれぞれrhG-CSF溶液(pH 6.5)に添加し、最終的なrhG-CSF濃度を250 µg/mLに調整した。試験液(界面活性剤を含む)および対照液(rhG-CSFのみ)を静脈内および気管内投与した後、8時間定期的に血液を採取し、酵素免疫測定法によりrhG-CSFの血漿中濃度を測定した。
血漿中のrhG-CSF濃度は、界面活性剤の存在下で大きく上昇した。ラウレス-9およびSGCの静脈内および気管内投与で達成されたrhG-CSFの相対的なバイオアベイラビリティは、それぞれ37%(静脈内)88%(気管内)84%(静脈内)および197%(気管内)であった[108]。吸収促進のメカニズムを調べるために、界面活性剤ラウレス-9とプロテアーゼ阻害剤(p-アミジノフェニル)メタンスルホニルフッ化物-HCl(p-APMSF)の両方をrhG-CSFと一緒に投与した。その結果、rhG-CSF濃度は3.9±1.4 ng/mLから481.5±96.7 ng/mLへと約123倍に上昇した。その結果、提案されている作用機序は、膜透過性の亢進とrhG-CSFの酵素分解の阻害を伴うことが明らかになった[108]。
6. 結論
天然由来のバイオエンハンサーの包括的なリストを、4つの特定の薬物投与経路(経鼻、頬、肺、経口)に対する作用機序とともに表1に示す。記載されているすべてのバイオエンハンサーの研究から、薬物投与の経口ルートでは、薬物のバイオアベイラビリティ向上の主要なメカニズムは、全身性前代謝の阻害だけでなく、排出トランスポーターおよびタイトジャンクションの変調であることが明らかになった。
経鼻、頬側、肺側の投与経路では、タイトジャンクション変調と膜破壊が薬物吸収促進の優勢なメカニズムとなっている。
様々な要因が異なる粘膜表面における薬物の溶解および吸収に影響を及ぼす可能性があるため、バイオアベイラビリティの向上が必要とされる特定の薬物ごとに、バイオエンハンサーの研究を実施する必要がある。多くの研究で吸収促進剤の可能性が示されているが、市販の薬剤にはほとんど含まれていない。