
Contents
Depression as a Neuroendocrine Disorder: Emerging Neuropsychopharmacological Approaches beyond Monoamines
www.ncbi.nlm.nih.gov/pmc/articles/PMC6335777/
オンラインで2019年1月3日に公開
要旨
うつ病の有病率は増加の一途をたどっており,障害,罹患率,死亡率も増加していることから,うつ病は日常臨床において重要な問題であると認識されている。現在使用されている抗うつ薬は、寛解率が低く、副作用が問題となっている。
抗うつ薬の作用機序は、脳内のセロトニン、ノルアドレナリン、ドーパミンなどの神経伝達の障害を中心としたモノアミン仮説に基づいている。しかし、うつ病の病態に対する考え方は大きく変化しており、うつ病が全身に重要な影響を及ぼす複雑な神経内分泌疾患であると理解されるようになってからは、数多くの新しい神経精神薬理学的アプローチが注目されるようになっている。
モノアミン以外の革新的な薬理学的標的としては、グルタミン酸系やGABA系の神経伝達、脳由来向神経性因子、さまざまな内分泌軸、さらにはいくつかの神経ステロイド、神経ペプチド、オピオイド、エンドカンナビノイド、エンドバニロイドなどが挙げられる。
この総説では、これらの薬理学的標的に関する現在の知識と、うつ病の臨床管理におけるそれらの有用性の可能性についてまとめている。
1. はじめに
うつ病は,日常の臨床現場で最も頻繁に見られる精神疾患の1つであり,現在,世界中で障害の主な原因とされている[1]。大うつ病性障害(MDD)は、深刻な衰弱状態にあることに加えて、医学的併存疾患のリスクが高く[2]、直接的・間接的な経済的コストが非常に高い[3]ことから、公衆衛生上の重要な問題となっている。
このような見通しにもかかわらず、MDDに対する薬物療法の選択肢はまだ十分ではない。現在使用されている抗うつ薬(AD)は、4段階の治療を経て、約56%の寛解率しか得られていない[4]。さらに、現在入手可能な抗うつ薬の大部分は、問題のある副作用プロファイルと作用発現の遅れを示し、この疾患の管理をさらに複雑にしている[5]。神経精神薬理学の分野では、より効果的で忍容性の高い新薬の開発が急務となっているが、ここ数十年の間にMDDに対して承認された新薬は比較的少ないのが現状である[6]。
既存の抗うつ剤の効果が限られていることと、新しい選択肢が少ないことは、かつて画期的であったにもかかわらず、振り返ってみると、脳内のセロトニン(5-ヒドロキシトリプタミン、5HT)ノルアドレナリン(NA)ドーパミン(DA)の神経伝達障害を中心とした、うつ病の病態に対するモノアミン仮説に過剰で誤った方向に焦点が当てられていたことに起因していると考えられる[7]。実際、三環系抗うつ薬の偶然の発見は、この仮説の「リバースエンジニアリング」を推進し、その結果、歴史上すべての抗うつ薬の開発の多くを導いていた[8]。しかし、モノアミン仮説は、その妥当性や構成要素の相対的な重要性について、激しい論争を繰り広げてきた[9, 10]。現在、分子精神医学の進歩により、神経細胞のモノアミン調節不全は、いくつかの非モノアミン神経伝達物質といくつかの内分泌代謝成分が関与する病態生理学的経路の複雑な相互作用の最終状態であると再認識されている[11]。
このように、MDDの病態をより包括的に理解することで、モノアミン調節不全の最終状態以外の活性を有する、新規の有望な抗うつ薬候補薬を設計・検討することが可能となり、介入の余地が生まれていた[12]。このレビューでは,うつ病の神経生物学に関する現在の見解をまとめ,モノアミン神経伝達以外の新たな薬理学的標的に焦点を当てることを目的としている。
2. うつ病の神経生物学に関する見解の拡大
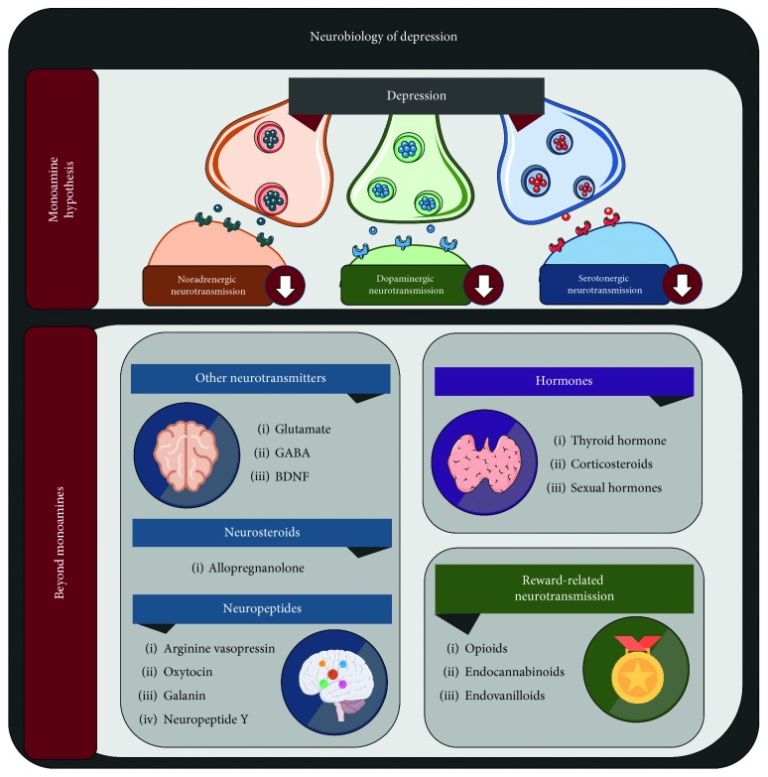
うつ病の臨床的な理解は,ヒポクラテスのメランコリアをはじめとするクレペリン以前の原始的な概念による初期の記述から,20世紀のさまざまな心理学的潮流から得られた豊かな記述,そして最近の精神薬理学と神経生物学の革命的な貢献まで,劇的に発展してきた[13]。後者の分野における研究の進歩は,特にうつ病や精神疾患の医学的モデルを推進し,これらの診断に対する理解が,どちらかというと無形でとらえどころのない概念から,特にモノアミン仮説を中心としたより具体的な生物学的用語へと移行したことを示している[14]。しかし、新しいアプローチは、このモノアミン神経伝達の中心的な機能障害を超えて、他の神経、内分泌、代謝の病態生理学的要素を巻き込んで絡み合っている(図1)。まず、古典的な3つのモノアミン以外の神経伝達物質については、以下の段落で説明する。
図1 うつ病の神経生物学に関する見解の拡大
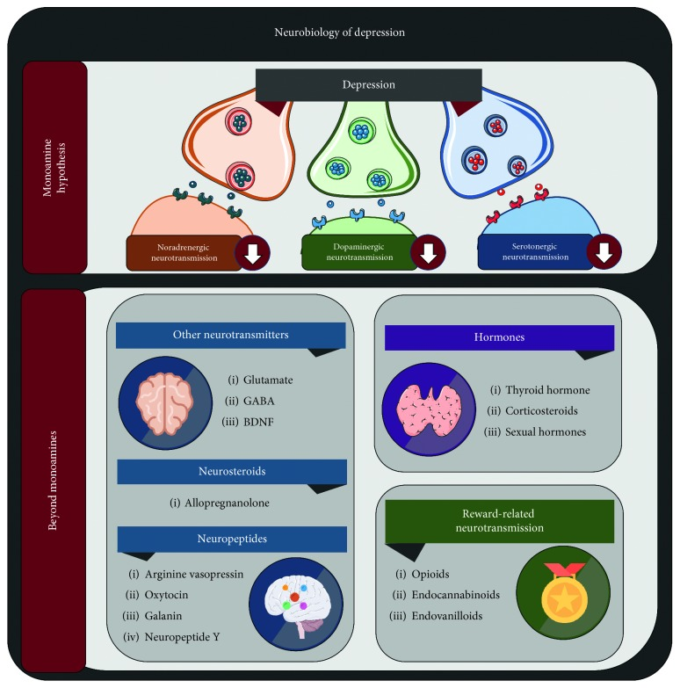
GABA:γ-アミノ酪酸、BDNF:脳由来向神経性因子。現在、うつ病に対する神経精神薬理学的アプローチは、モノアミン仮説を中心に行われている。しかし、現在使用されている抗うつ薬の臨床結果が不完全であることから、ノルアドレナリン、ドーパミン、セロトニン以外の様々な薬理学的標的が発見されている。
2.1. グルタミン酸。多才な調節因子
グルタミン酸(Glu)は,哺乳類の脳における主要な興奮性神経伝達物質である[15]。正常な状態では,グルタミン酸は,シナプス可塑性,学習,記憶に重要な役割を果たしている。しかし、病的な状態では、Gluは、急激な、あるいは遅延した神経毒性の強力な引き金となることが知られている[16]。グルタミン酸系薬剤に抗うつ効果があるという新たな知見が得られたことから,気分障害の病態生理におけるGluの役割についての憶測が生まれている[17]。特に,うつ病患者では,基礎的なグルタミン酸受容体の活動が亢進していると考えられている。そのため,グルタミン酸系の神経伝達を直接標的とする薬剤を用いた前臨床試験および臨床試験は,抗うつ薬治療の新たな可能性を提示している[18]。
グルタミン酸受容体は,イオン刺激性グルタミン酸受容体とメタボトロピックグルタミン酸受容体(mGluR)の2つの主要なファミリーに分けられる。ionotropic groupには、NMDA受容体、AMPA受容体、カイネイト受容体が含まれる[19]。安静時には,NMDA受容体は,膜が脱分極するまでマグネシウムによってブロックされている。このとき,2つのGlu分子と2つのグリシンまたはD-セリンが結合することで,カルシウムの流入が可能となり,興奮性入力の収束の機能的マーカーとしての役割を果たし,最終的にはより長い時間にわたって興奮を引き起こす[20]。AMPA受容体は,ほとんどのシナプスにおいて,急速に脱感作される速い興奮を媒介し,シナプスにおけるGluに対する最初の反応を担っている。AMPA受容体が活性化されると,ナトリウムが流入し,神経細胞膜が脱分極される。カイン酸受容体は,AMPA受容体と同様,電位依存性チャネルと関連しており,主にナトリウムイオンの流入を可能にし,高速の興奮性神経伝達を媒介するが,あまり広く普及しておらず,明確な分布を持っているようである[21]。一方,メタボトロピックファミリーは,シナプス前のグルタミン酸放出とシナプス後のNMDA電流の両方を増強するグループI受容体(mGluR1とmGluR5)と,グルタミン酸の機能を抑制する傾向のあるグループII(mGluR2とmGluR3),グループIII受容体(mGluR4,mGluR6,mGluR7,mGluR8)から構成されている[19, 22]。
現在までに,様々な前臨床モデルにおいて,NMDA受容体拮抗薬,グループIメタボトロピックグルタミン酸受容体(mGluR1およびmGluR5)拮抗薬,AMPA受容体の陽性調節薬が抗うつ様作用を有することを示す証拠が出てきている[23]。歴史的には、NMDA受容体拮抗薬であるAP-7およびMK-801の前臨床における抗うつ様作用が、Gluシグナルが潜在的な治療アプローチであることを最初に示唆した[24]。
この研究により、非競合的なNMDA受容体拮抗薬であるケタミンが実験的に使用されるようになった。ケタミンは、これまで麻酔学や神経学の分野で安全かつ良好な忍容性をもって使用されてきたことから、精神医学の分野においても妥当な候補であると考えられた[25]。ケタミンは,様々な盲検パイロット臨床試験において,麻酔下用量での使用後24時間以内に迅速な抗うつ効果を誘発し,1回の注入後少なくとも数日間は持続することが示されており,これを受けて「迅速作用型抗うつ薬」(RAA)という言葉が作られた[26-28]。この効果は、最初の精神刺激作用、解離作用、および多幸感が収まった後に始まることが報告されており、抗うつ作用が単に急性の気分の高揚の結果ではないことが示唆されている[29]。経口製剤や筋肉内製剤など、静脈注射以外のケタミン製剤も抗うつ効果を示している[30, 31]。中枢神経系への浸透性が高く、投与が容易であることから、経鼻ケタミンが最も有望視されている[32]。この迅速な作用により、従来のモノアミン系薬剤の作用発現の遅れを回避するための重要な薬理学的標的として、グルタミン酸神経伝達が挙げられると推測されている。しかし,強力な精神刺激薬であるフェンシクリジン(PCP)との薬理学的類似性や,幻覚を誘発するクラブドラッグとしての乱用性などから,抗うつ薬としてのケタミンの使用には懸念が残っている[33]。
ケタミンは,μ-オピオイド受容体アゴニストでありながら,NMDA受容体に対して優れた親和性を持つという興味深い薬理学的プロファイルを示している[33]。ケタミンは,GABA作動性介在ニューロンおよびシナプス後ニューロンのNMDA受容体に拮抗し,前者によって大脳皮質のグルタミン酸ニューロンの抑制が解除され[34],後者によって細胞内成長因子の合成が促進される[22]。また,ケタミンは,NMDA受容体を介した自発的な興奮性シナプス後電流の抑制を促進する。これにより,伸長因子2キナーゼ活性(eEF2K)が抑制され,神経可塑性や神経保護に重要な役割を果たす脳由来向神経性因子(BDNF)の翻訳が急速に増加する[35]。また、ケタミンは、一酸化窒素によるシグナル伝達を阻害し、細胞内のシグナル伝達を促進する低分子Gタンパク質であるニトラージックRhebの安定化を可能にすることが指摘されている[36]。実際,ケタミンは,哺乳類ラパマイシン標的(mTOR)経路をはじめとする多くの細胞内カスケードを活性化するようで,ラットの前頭前野(PFC)において,新しいシナプスの数と機能を増加させ,ストレスにさらされた結果生じるシナプスの欠損を回復させることが観察されている[22, 37]。さらに、ケタミンは、シナプス形成の調節に関与するp70 s6キナーゼ(p70s6K)および4E結合タンパク質のリン酸化を増加させるようである(図2)[37]。
図2 グルタミン酸シナプスに対するケタミンの効果
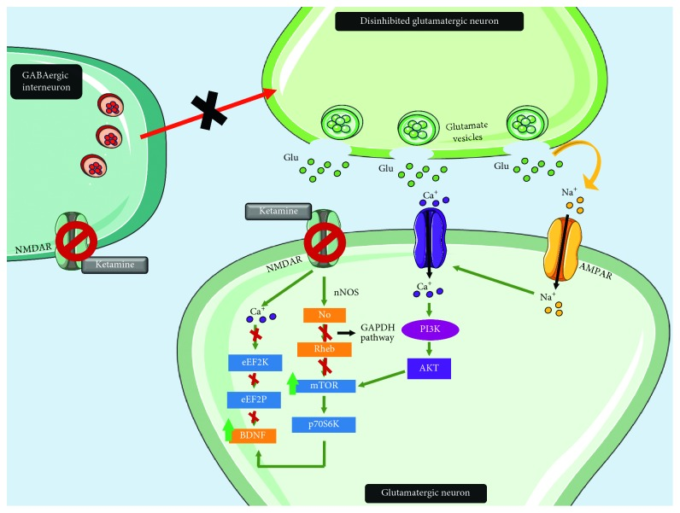
ケタミンは、GABA作動性介在ニューロンだけでなく、シナプス後のグルタミン酸作動性ニューロンにもNMDAアンタゴニストとして作用する。前者の拮抗作用により、シナプス前グルタミン酸ニューロンの抑制が解除され、シナプス後グルタミン酸ニューロンのAMPARの活性化が促進される。その結果、電位依存性カルシウムチャネルの活性化とともに、PI3K経路が活性化され、mTORの活性化につながる。さらに、NMDARに拮抗することで一酸化窒素経路が阻害され、Rhebの安定化とmTOR経路の増強につながる。mTORはp70s6K活性を増加させ、BDNFのシグナル伝達を促進する。また、NMDARに拮抗することでeEf2Kが不活性化されると、BDNFの活性化が促進される。
ケタミンの有望な結果を受けて、他の速効性抗うつ剤の研究が盛んに行われるようになった[38]。ケタミンの(S)および(R)異性体であるエスケタミンとアルケタミンも,前臨床および臨床試験で評価されている。特に、動物モデルにおいて、エスケタミンは、精神刺激作用を伴わずに、エスケタミンよりも強力で長期的な抗うつ作用を誘発すると考えられている[39]。一方,エスケタミンは,ケタミンよりも作用が弱く,副作用も同様であるが,抑うつ症状の急性改善に有効であると考えられている[40]。最後に、もうひとつのケタミン誘導体である(S)-ノルケタミンは、げっ歯類モデルにおいて、エスケタミンよりも低い効力で、迅速かつ長期的な抗うつ効果を誘発することが報告されているが、精神刺激性やその他の有害な生化学的および神経生理学的効果はない[41]。このように,ケタミン関連分子は,今後の研究によって,臨床効果と忍容性のバランスをとることができるだろう。
レパスチネルは,ケタミンとは無関係のもう1つのRAAであり,B6B21モノクローナル抗体の軽鎖に由来するテトラペプチドで,NMDA受容体モジュレーターおよびグリシンサイト部分作動薬として作用する[41]。動物モデルにおいて,レパスチネルは,NMDA受容体依存性のシグナル伝達を増強することにより,海馬およびPFCにおける電気生理学的活動の長期増強を促進すると考えられている[42]。レパスチネルの臨床研究はまだ初期段階にあるが,レパスチネルは迅速かつ持続的な抗うつ効果を誘発し,精神刺激作用を伴わずに忍容性が高いと考えられている[43]。
これらのどちらかといえば選択的なグルタミン酸系薬剤とは対照的に、非選択的なムスカリン受容体拮抗薬であるスコポラミンもGlu調節活性を示し、これが抗うつ効果と相関している可能性がある[44]。動物モデルでは、スコポラミンはケタミンと同様にRAAであり、PFCにおいてグルタミン酸系神経伝達を増加させ、mTORC1シグナルを活性化することが示されている[22, 45]。この増強は、ケタミンの作用の基礎となる最初の細胞標的と同様に、PFC内側のGABA作動性介在ニューロンに存在するムスカリン受容体の遮断を介して起こると考えられている[46]。いくつかの臨床研究では,低用量のスコポラミンを静脈内に注入すると,迅速な抗うつ効果が得られることが報告されており,反復投与(5~7日間で3回)すると,最長2週間にわたって気分の改善が持続した [47, 48].このシナリオでは、スコポラミンは、治療抵抗性の患者が依然として抑うつ症状の有意な減少を示すものの、治療を受けていない患者においてより多くの有効性を示している[49]。
ケタミン、その誘導体、およびケタミンとその関連作用機序とは無関係でありながら、迅速な臨床効果と忍容性および精神模倣作用という臨床上のジレンマを抱えるシロシビンなどの新興RAAには、現在限界があるため[50-52]、より少ない副作用で抗うつ効果を再現できる、より忍容性の高いNMDA受容体拮抗薬を求める研究が行われている。現在、アルツハイマー病の治療薬として承認されているNMDA受容体拮抗薬であるメマンチンは、気分障害を対象として研究されている。ネズミのうつ病モデルを用いた前臨床の報告では、抗うつ剤のような効果が認められている[53]。しかし,現在実施されているプラセボ対照臨床試験では,ヒトでの抑うつ症状に対する有効性を示すことができなかった[54, 55]。
筋萎縮性側索硬化症の治療薬としてFDAに承認されているリルゾールも、いくつかのケタミン延長試験で使用されている[56, 57]。この薬はGlu受容体モジュレーター[58]であり、単剤療法[52]および治療抵抗性大うつ病性障害(MDD)患者の併用療法[59]の両方で明らかな有効性を示している。この抗うつ効果は、治療抵抗性の患者において、特に4週間で顕著に現れることが研究で明らかにされているが、プラセボ対照試験では有意差が認められない傾向にある[60]。したがって、このような状況におけるリルゾールの臨床効果をより明確にするためには、さらなる調査が必要である。薬力学的研究によると、リルゾールはNMDA受容体のオープンチャネルアンタゴニストではなく、培養海馬ニューロンにおけるAMPA受容体サブユニットの表面発現を促進することで作用し、この点ではラモトリギンの作用に似ていることが示唆されている[61]。リルゾールは,BDNFの発現を促進し[62],アストロサイトによるGluの再取り込みを促進することで,シナプス間隙からのGluのクリアランスを促進することも報告されている[63]。これらの作用はいずれも,前臨床のげっ歯類モデルにおいて,抗うつ薬のような効果と関連している[64]。
現在、うつ病に対する初期の実験的研究が行われているGlu調節分子は他にもいくつかある。NR2Bサブユニットに選択性をもつNMDA受容体アンタゴニストであるRo(25)-6981は,おそらくmTOR経路などのシナプス後カスケードの発現増加を介して,抗うつ様作用を示している[21]。同様の候補であるMK-801(ジゾシルピン)は,NMDA受容体の高親和性非競合アンタゴニストであるが,抗うつ効果は比較的安定していない[65]。どちらも短期的には有意な抗うつ効果があるようだが、ケタミンのように長時間作用するものではないようだ[21, 66]。
メタボトロピックGlu受容体もまた、うつ病の潜在的な治療標的として検討されている[67]。mGlu2/3受容体のアンタゴニストであるLY341495は、この受容体群のアゴニストであるLY379268ではなく、抗うつ効果と関連している[68, 69]。これは、ケタミンと同様に、別のmGlu2/3受容体拮抗薬であるMGS0039と同様に、mTORC1経路の活性化が関与しているようである。LY341495はまた、動物モデルで慢性的なストレスにさらされることで生じる快感消失を迅速に回復させる。これは、抗うつ薬の迅速な作用を示す厳密なげっ歯類実験である[70]。
実際、mTORカスケードの刺激、およびPFCにおけるAMPA受容体の同時活性化は、Gluモジュレーターに関連した抗うつ効果に不可欠であるかもしれない[71]。抗うつ作用のメディエーターとしてのAMPA受容体の重要性は、Altらによって強調されている[72]。AMPA受容体の特定の構造上の変異が、AMPA受容体を介した電流の調節に関連している可能性がある。その結果、BDNFのシグナル伝達を促進することで、モノアミン系の神経伝達が促進されると考えられる。
一方、Palucha-Poniewieraらのラットを用いた研究では、mGlu5受容体拮抗薬であるMTEPとmGlu7受容体作動薬であるAMN802の抗うつ作用が短時間であることが報告されている。両者の作用発現は、投与後60分以内に始まり、投与後23時間程度で消失したという。その結果、MTEPはmTORC1シグナルを促進せず、AMN802はこの経路を刺激するが、同時にAMPA受容体を活性化することはできなかった[66]。このクラスの分子に関する洞察は、まだ実験の初期段階にあり、さらなる研究が必要である。
最後に、亜鉛とマグネシウムは、前臨床試験[73-75]と臨床試験[76-78]で抗うつ作用を示しており、うつ病の薬物療法における貴重な補助剤となりうる。亜鉛とマグネシウムは、BDNFとグリコーゲン合成酵素キナーゼ-3(GSK-3)のシグナル伝達の増加に関連して、NMDA受容体の活性を低下させるようである。
2.2. GABA 抗痙攣薬から気分安定薬、抗うつ薬へ?
γ-アミノ酪酸(GABA)は、哺乳類の脳における主要な抑制性神経伝達物質であり、その様々な受容体は中枢神経系に広く存在している[79]。GABA-Aファミリーでは,受容体はリガンド依存性イオンチャネル複合体の一部であり,各アイソフォームはGABAによってゲートされる中央の塩化物イオン選択チャネルを囲む5つの相同または同一のサブユニットから構成されている。GABA-Bファミリーは,シナプス後膜に存在し,神経細胞を即時的に抑制する一方で,シナプス外膜に存在し,周囲のGABAに反応して長期的な抑制をもたらす。対照的に,GABA-Bファミリーには,セカンドメッセンジャーシステムを介してイオンチャネルを開閉するメタボトロピックGタンパク質共役型受容体が含まれている[81]。抗けいれん薬(ACD)などのGABA調節薬は,うつ病の管理に有効であることが広く知られている[82]。この効果は、ACDによってGlu-GABAバランスがより良好になることによるものと提唱されている。しかし、各ACDの気分調整パターンは明確であり、また、一部のACDでは気分調整が行われないことから、GABA作動性薬物の抗うつ薬理作用は、単にGlu-GABA平衡の変化だけではなく、より複雑であることが示唆されている[83]。
いくつかの抗うつ薬を慢性的に投与した後、ネズミの脳でGABA-B受容体の結合が増加することが報告されており[84]、抗うつ薬の作用のGABA作動性仮説を支持している。それにもかかわらず、GABA-B受容体結合による抗うつ作用と称されるものについては、さらなる研究で矛盾が生じている[85]。現在、気分障害におけるACDの臨床効果が証明されていることから、この文脈でのGABA作動性調節の研究が続けられている[86]。
事実上,GABAおよびGluシステムの既知の分子成分のすべてが,潜在的な治療標的として考えられている[87]。特に、α5サブユニットを含むGABA-A受容体のネガティブアロステリックモジュレーター(α5 GABA-NAM)は、新しいクラスの即効性抗うつ薬として提案されている。これは、前脳においてケタミンと同様の正味の効果をもたらすが、前脳ではα5サブユニットの発現が比較的少ないため、副作用が軽減されるというものである。オスのマウスを用いた研究では,α5 GABA-NAMであるMRK-016が,ロタ棒パフォーマンス,条件付場所嗜好性,運動量などの副作用モニタリングパラメータに変化を与えることなく,抗うつ薬様の反応を示した[88].この有望な分子のヒトにおける有用性と適合性をより明確にするためには,さらなる研究が必要である.
2.3. 脳由来の向神経性因子
BDNFの海馬での発現低下は、うつ病、不安、ストレスの神経生物学的構成要素としてよく知られており、抗うつ薬治療によってこのメッセンジャーのレベルが上昇することが観察されている[89]。BDNFは、トロポミオシン関連キナーゼ受容体(TrkB)を介したシグナル伝達を活性化することで、神経発生や神経細胞の生存・成長に中心的な役割を果たしている[90, 91]。BDNFは当初、うつ病の代替治療薬として提案されたが、組換えBDNFを用いた臨床試験では、有意な抗うつ効果を得ることができなかった[92, 93]。
最近では、BDNFシグナルの有益な効果を利用する別の方法を見つけるために、特にTrkBリガンドの研究が行われている[94]。特に,TrkBアゴニストである7,8-ジヒドロキシフラボンは,マウスモデルにおいて抗うつ作用を示すとともに,神経新生を促進することが明らかになっている[95, 96]。一方,ANA-12は,選択的なTrkBアンタゴニストであり,神経細胞の生存を損なうことなく,側坐核での向神経性活性を阻害することができる.逆説的に、このアンタゴニストは、マウスモデルにおいて、抗うつ作用や抗不安作用とも関連している[99, 100]。したがって,うつ病に対するTrkBリガンドの使用の薬理学的および臨床的相関関係を明らかにするためには,さらなる研究が必要である。
3. うつ病の内分泌薬理学的ターゲット
精神医学と内分泌学の分野は、古くから大きく関連していることが知られており、様々な精神疾患と内分泌疾患はしばしば双方向の関係を示する[101]。ホルモン異常を有する被験者には、抑うつ症状や不安症状がよく見られ、臨床家にとっての課題となっている[102]。うつ病患者は必ずしも明らかな、あるいは重度の内分泌疾患を有しているわけではないが、この集団ではホルモンの変化が頻繁に見られ、うつ病の病態生理学的役割を支持する証拠が蓄積されており、ひいては内分泌障害がこの疾患の治療標的となる可能性がある[103]。
甲状腺ホルモンは、脳の発達と機能に関与していることはよく知られており、精神神経学的な症状は甲状腺疾患の特徴である[104]。逆に、精神疾患はしばしば視床下部-下垂体-甲状腺軸(HPTA)の乱れを特徴とする。うつ病患者では、甲状腺刺激ホルモン(TSH)とチロトロピン放出ホルモン(TRH)に対する反応が異常になり、脳脊髄液中のTRH濃度が上昇し、抗甲状腺抗体の有病率が高くなることがわかっている[105]。現在までの利用可能なデータによると、うつ病の被験者では明らかな甲状腺病変はまれであることがわかっている[106]。それとは逆に、潜在的な甲状腺病変は精神疾患の重要な危険因子であるようであり[105]、複数の研究では、潜在的な甲状腺機能低下症の患者では、年齢と性別をマッチさせた対照群に比べて鬱症状の頻度や程度が有意に高いことが示されている[107-109]。このような見通しは、うつ病の病態生理におけるHPTAの関与の複雑さを示している。
実際、うつ病におけるHPTAの役割は、総体的な甲状腺疾患よりも微妙である可能性があり、それはすべての組織に甲状腺ホルモン受容体が遍在していることと、このシステムがストレスに弱いことによって明確になっている。HPTAのダイナミックな調整は、激しい身体活動や妊娠などのストレス状態における生理的反応を表している[110]。しかし、うつ病では、低悪性度の全身性炎症の慢性的な状態が、HPTAやその他多くの内分泌・免疫軸を混乱させるようである[111]。In vitroおよび生体内試験の研究では、T3がミクログリアの移動と活性化を刺激することが示されており、これは、うつ病、統合失調症、自閉症スペクトラム障害で見られる顕著な現象である[112, 113]。このような状況では、T3シグナルは、PI3K、MAPK/ERKカスケードの障害、および一酸化窒素を介したミクログリアの遊走と活性化の増強をもたらすと考えられる[114-116]。これらの事象は、うつ病に見られる向神経性、神経可塑性、および神経伝達の機能障害に寄与する可能性がある[117]。
甲状腺ホルモンの投与は、たとえ甲状腺機能亢進症の患者であっても、難治性うつ病の管理における補強戦略として十分に支持されている[118-120]。しかし、ホルモン補充だけでは、甲状腺機能低下症患者のうつ病を治療するには不十分である可能性があり、この介入から最も恩恵を受ける患者のプロファイルをより明確にするために、さらなる研究が必要である[121]。
コルチコステロイドもまた、うつ病にみられる神経内分泌系の障害に一役買っていることが広く認識されている。コルチゾールは、視床下部-下垂体-副腎軸(HPAA)の最終産物であり、「ストレスホルモン」として広く認識されており、急性および慢性のストレス条件下で、すべての器官系における無数の必須機能を媒介する [122] 。しかし,コルチゾールは,うつ病に見られるように,慢性的なストレス関連疾患の病態生理にも深く関わっていることが知られている[123]。中枢性コルチコトロピン放出ホルモン(CRH)の過剰刺激は、慢性ストレスとうつ病の重要な永続因子であると考えられ、CRH活性の抑制が、MDDの安定した寛解に必要な抗うつ作用の最終的かつ共通のステップである可能性が示唆されている[124]。中枢神経系では、神経解剖学的な発現パターンにより、2つのCRH受容体サブタイプ-CRHR1とCRHR2-が報告されており、CRHR1は、うつ病や不安症におけるCRH誘発作用の媒介に重要な役割を果たしていると考えられている[125]。特定のCRHR1ハプロタイプは、MDDの発症と関連している[126]。したがって、CRHR1遺伝子は、抗うつ薬の薬理遺伝学の候補遺伝子と考えられる[127]。
CRH活性の増加を示す長年の証拠により、当初はうつ病の推定治療法としてCRH受容体拮抗薬の開発が行われた[123]。R121919は、ラットおよび霊長類において不安を軽減し、MDDと診断された20人の被験者を対象とした小規模な研究では、良好な忍容性でうつ病のスコアを低下させることが示されている[128]。また、もう一つのCRH1受容体拮抗薬であるNBI-30775/R121919は、パロキセチンと同等の臨床プロファイルと有効性を有することが報告されている[129]。一方、選択的な非ペプチド性CHRH1アンタゴニストであるCP-316,311は、忍容性が高いにもかかわらず、MDDの治療に有効性を示すことができなかった[130]。もう一つのCRH1受容体拮抗薬であるNBI-34041は、全身性の副作用を伴わずに、投与時にストレスによるコルチゾールの分泌を減少させることが報告されている[131]。このように、CRH1受容体拮抗薬は、将来的にうつ病や不安症の新たな治療法として有望である。このような進歩は、十分なバイオアベイラビリティーと血液脳関門の通過を保証するために、これらの化合物の薬学的処方と薬物動態に関して現在認識されているいくつかの困難を回避する必要があるだろう[132]。
ステロイドホルモンもまた、うつ病の神経生物学に関与しており、特に、ライフサイクルを通しての非常に特徴的な変動や、男女ともに気分障害との相関関係が指摘されている[133]。気分障害のエピソードは、思春期、更年期、産後など、ホルモンが大きく変化する人生の重要な時期に多く見られることがわかっている[133-135]。実際、女性は、男性に比べて、気分障害のリスクが2倍以上であることと一致するように、より劇的な結果をもたらすさまざまな変動にさらされている [136, 137]。この格差は、気分障害の病因における性腺ホルモンの役割の可能性を浮き彫りにしている。エストロゲン受容体は、脳内に広く分布しており [138]、これらのホルモンは、発達中のニューロンの組織化や成熟したニューロンの活性化に関与している。エストロゲンは、神経突起の成長とシナプス形成を促進し、BDNF活性を増強し、5HT、NA、DA、Glu、Achの神経伝達を調節する[139]。特に、エストロゲンは、5HTの代謝と5HT受容体の発現を調節することにより、セロトニン作動性を増加させ、また、瀬状核のセロトニン作動性ニューロンの自発的な発火を調節する[139]。
臨床的には、うつ病におけるエストロゲンの役割は、更年期においてより徹底的に研究されている。いくつかの研究では、うつ病を軽減する方法としてエストロゲン補充療法(ERT)に焦点が当てられ、興味深い結果が得られている。Schmidtら[140]による更年期女性を対象としたプラセボ対照試験では、ERTは治療開始後3週間という早い時期に、鬱症状の部分的または完全な寛解と関連していたが、Klaiberら[141]は、MDDを有する更年期女性を対象に、高用量のERTを3ヶ月間投与することで同様の効果が得られることを報告した。より大規模な研究では、661名の更年期女性が、エストロゲンとプロゲステロンの経口投与を受ける群、エストラジオールとプロゲステロンの経皮投与を受ける群、プラセボ群の様々なグループに分けられた。48ヵ月の追跡調査の結果、経口ERTを受けた女性は、プラセボ群と比較して、抑うつ症状の有意な軽減を示した[142]。
しかしながら、1,208人の更年期女性を対象とした10件の研究のメタアナリシスでは、血管運動症状の管理には有効であるにもかかわらず、プロゲステロンを併用した場合でも、バイオアイデンティカルエストロゲンの補充は、抑うつ症状に対して有意な効果がないことが確認された[143]。さらに、KNHANES研究では、長期の経口避妊薬の使用とERTの使用は、うつ病のリスクが有意に高いと判断されている[144]。したがって、うつ病に対するエストロゲンの臨床使用については、依然として議論の余地があり、さらなる研究が必要である。
更年期に起こることと同様に、出産後に見られるエストラジオールレベルの急激な低下は、産後うつ病(PPD)に顕著に寄与するという仮説が立てられている[138]。さらに、黄体ホルモン濃度の変動も大きく介在する可能性がある。プロゲステロンやその他の黄体ホルモンは、おそらくGABA神経伝達を阻害することで、ネガティブな気分状態に関係していると考えられている[145]。プロゲステロンを投与された閉経後の女性やアロプレグナノロン(APG)を投与された動物では、血清APG濃度と気分の悪さとの間に逆U字型の曲線に似た二峰性の関連性が見られる。さらに、ヒトでは、ネガティブな気分を生じさせるAPGの最大有効濃度は、生理的な黄体期の血清濃度の範囲内にある[146]。
PPDにおけるエストロゲンまたはプロゲストーゲンの使用に関する臨床データは、現在のところ乏しいが、利用可能な結果では、エストラジオールの舌下投与が抑うつ症状の迅速な軽減につながることが示唆されている[147]。経皮的なE2の投与は、この方法では肝代謝がバイパスされ、静脈血栓塞栓症のリスクが減少することを考慮すると、可能な治療経路としても考えられている。しかし、最近のパイロット研究では、まだ成功していない[148]。産後うつにおける有望なターゲットと考えられているのは、前述のAPGである。Kanesらは、無作為化比較試験において、APGの静脈内投与を研究し、有害事象が少なく、症状が大幅に改善したことを明らかにした[149]。PPDは、治療が著しく困難であると認識されているため、PPDにおけるホルモン療法の役割を明らかにするために、さらなる研究が必要である。
最後に、副腎および性腺由来のアンドロゲンは、血液脳関門を通過することができ、脳に様々な影響を与え、テストステロンを含む様々なアンドロゲンが脳内で新たに合成されることがある [150, 151]。アンドロゲンは、GABA-A受容体のアロステリックモジュレーターとして作用し、受容体に付随するクロライドチャネルの開口部の持続時間と頻度を増加させ [152]、様々な神経伝達システムとニューロンの興奮性を調節し、気分障害の神経生物学において重要な意味を持っている [152-155]。このプロファイルは、臨床的にも重要な相関関係があるようである。テストステロンレベルは、加齢とともに徐々に低下し、陰性気分、疲労、イライラ、性欲低下など、うつ病に見られる症状と興味深いほど類似している [156]。さらに、抗アンドロゲン薬で治療を受けた男性は、MDDを発症するリスクが高いことがわかっている[157]。
興味深いことに、臨床研究では、男性のうつ病管理における増強戦略としてのテストステロン投与の有効性は示されていないが [158] 、治療抵抗性のMDDを有する女性に低用量のテストステロンを投与すると、プラセボと比較して抑うつ症状が有意に改善することが観察されている [159] 。これらの逆説的な知見は、女性のアンドロゲンに対する感受性の高さと一致しており、アンドロゲンへの曝露が高いほど、女性の気分に明確な悪影響を及ぼすことが指摘されている[160, 161]。今後の研究では、このような男女間の違いとその臨床的な関連性について検討する必要がある。
4. うつ病における神経ステロイド薬理学的標的
多くの神経細胞、特にグルタミン酸系やGABA系の神経細胞では、神経の興奮性を調節するステロイド系のメッセンジャーが新たに合成されている[162]。アロプレグナノロンは、これらの分子の中で現在最もよく知られている分子であり、鎮静作用や麻酔作用のほか、気分のコントロールにも影響を与えることが指摘されている[163]。他のステロイドと同様に、APGの合成には、神経細胞でいくつかのCYP酵素の発現が必要である[164]。
APGは、GABA-A受容体のアロステリックモジュレーターとして、また、HPAAのネガティブフィードバックシグナルとして作用するようである。慢性的なストレス状態では、APGレベルが低いと、HPAAの活性化が進み、ホメオスタシスへの回復が遅くなることが示唆されている[165]。この仮説は、MDDを含む様々な感情障害の患者の脳脊髄液(脳脊髄液)および末梢血中に見られるAPGレベルの低下と一致している[166]。選択的セロトニン再取り込み阻害剤、三環系抗うつ剤、ミルタザピンは、APGレベルを上昇させることが観察されているが、電気けいれん療法、反復経頭蓋磁気刺激、睡眠遮断はそのような影響を及ぼさないようである[167]。APGの合成誘導体であるSAGE-217は、現在、第2相臨床試験が行われており、予備的な知見では、短期使用後にうつ病の臨床スコアがわずかに改善したことが示されている[168]。MDDにおけるAPGまたはその合成類縁体の使用については、さらなる研究が必要である。
うつ病における他のニューロステロイドの役割は、まだはっきりとはわかっていない。アンドロステンジオンは、HPAAおよびアンドロゲン代謝の調節因子として特に関連があると考えられるが、現在の知見は矛盾している。アンドロステンジオンと抑うつ症状の間には、直接的な関係と逆の関係の両方が記述されている[169, 170]。今後の研究により、これらの矛盾が解消され、神経ステロイドを用いた治療戦略の可能性が示されるかもしれない。
5. うつ病における神経ペプチド薬理学的標的
5.1. オキシトシンとアルギニンバソプレシン
ニューロステロイドの研究と並行して,将来の治療法を模索する現代の精神医学研究では,神経ペプチド(図3),特にオキシトシン(OXT)とアルギニンバソプレシン(AVP)に大きな関心が寄せられている[171]。中枢のオキシトシンシグナルは抗不安作用や抗うつ作用を示すのに対し、バソプレシンは不安や抑うつ行動を促進する傾向がある。このような相反する作用は、情動調節においてこれらの神経ペプチドのバランスのとれた活性が重要であることを示していると考えられる。ポジティブな社会的刺激や心理薬物療法によって、この均衡をオキシトシンにシフトさせることが、うつ病の管理に役立つ可能性がある[172]。
図3 うつ病における神経ペプチド薬理学的標的
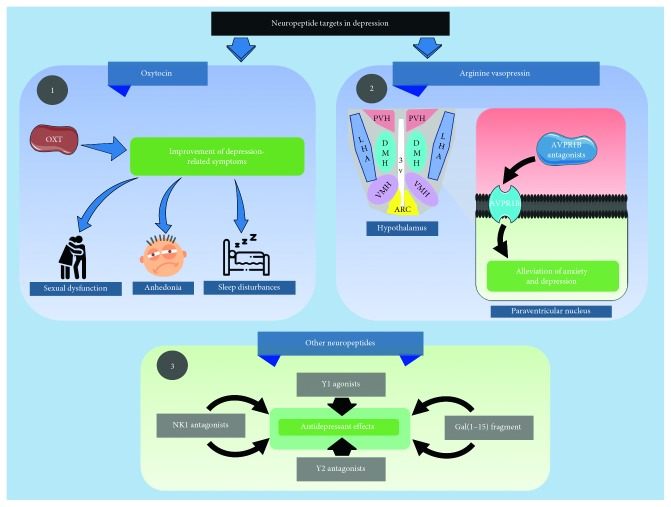
OXT:オキシトシン、LHA:側坐核、PVH:室傍核、DMH:背内側核、VMH:腹内側核、ARC:弧状核、AVPR1B:アルギニンバソプレシン受容体1B、NK1:ニューロキニン1。 うつ病の神経精神薬理学における神経ペプチドに関する現在の知見について、主なものは以下の通りである。1)オキシトシンが、性機能障害、快感消失、睡眠障害などのうつ病関連症状の改善に大きく寄与することを示唆する前臨床および臨床のエビデンスが豊富であること。2)AVPR1Bアンタゴニストは、動物モデルとヒトモデルの両方において、不安や抑うつの症状を軽減すると考えられている。(3)神経ペプチドのシグナル伝達を調節するいくつかの薬が抗うつ作用を示しているが、その意義や有用性を明らかにするにはさらなる研究が必要である。
実際、げっ歯類の研究では、OXTはポジティブな社会的相互作用と明確に関連しており[173, 174]、合成OXTは、中枢および末梢の両方に投与すると、げっ歯類のストレス反応をより能動的な対処スタイルにシフトさせることが示されている[172]。さらに,最近,成人ラットの海馬において,AVPではなくOXTが神経細胞の成長を促進し,グルココルチコイドやストレスによる神経新生の抑制を救済することが示された[175]。ヒトでは、精神病性および非精神病性のうつ病[176]や双極性うつ病[177]では、OXTのレベルが有意に低いことが観察されている。さらに、MeynenらのmRNA発現研究で明らかになったように、OXTは、うつ病のメランコリック表現型を持つ被験者で特に低いようである[178]。
さらに,OXTは,性機能障害を含む他のうつ病関連症状の改善にも寄与する可能性があることが前臨床および臨床で示されている。オスのラットにOXTを腹腔内注射した研究では,室傍DA受容体を刺激することで陰茎の勃起が誘発されるだけでなく,オキシトシンニューロンを活性化することで中脳辺縁系DA神経伝達が増加することが示された。これらの知見は、これらのメディエーターが、性行動の消費的側面と動機・報酬的側面の両方に強力に影響を与えることを示唆している[179]。[180]、さらには睡眠障害にも影響を及ぼす可能性がある。さらに,最近,成人ラットの海馬において,AVPではなくOXTが神経細胞の成長を促進し,グルココルチコイドやストレスによる神経細胞新生の抑制を救済することが示された[175]。
その一方で,AVPは不安を煽る作用があるようである[182].AVPの受容体としては,AVPR1A,AVPR1B,AVPR2が古くから知られているが [183],AVPは構造的に関連のあるOXT受容体(OXTR)にも高い親和性で結合する.AVPR1A受容体は血管に広く分布しており,脳室傍核などの中枢神経系にも見られるが,AVPR2受容体は主に腎集合系の主要な細胞に存在している[172].AVP受容体ファミリーは,Gタンパク質共役型の受容体です.AVPR1AとAVPR1Bは共にGq/11に結合しており,ホスホリパーゼCを介してシグナルを送る [184, 185].AVPR2はGsに結合しており,活性化されるとアデニル酸シクラーゼを誘引してcAMPレベルを上昇させる [186].
AVPR1Bアンタゴニストの研究では、動物やヒトのモデルにおいて、不安や抑うつを軽減するなど、良好な結果が得られている[187, 188]。ラットモデルでは、AVP遺伝子は形質不安と強い相関があるとされている[189]。さらに、臨床試験では、AVPR1Bアンタゴニストの使用は、MDDの被験者におけるHPAAの変調と臨床症状の改善に関連している[190]。
5.2. その他の神経ペプチド
ニューロキニン1(NK1)拮抗薬は、うつ病の非モノアミン関連生物学的治療法として提案された最初の選択肢の一つであり、これらの分子の一つであるMK-869の慢性投与がうつ病の症状の改善と関連するという知見が得られたためである[91, 191]。これを受けて、別のNK1アンタゴニストであるアプレピタントの臨床試験が行われた。初期の報告は好意的なものであったが、第III相臨床試験では有効性を示すことができず、この問題に対するさらなる科学的関心は失われた[192]。
しかし、より最近の研究では、うつ病の治療で効果を得るためには、NK1受容体のほぼ完全な中枢ブロックが必要であることが示唆されている[193, 194]。カソピタントとオルベピタントという2つのNK1アンタゴニストは、はるかに大きな遮断能力を持っており、様々な単離した無作為化試験で抗うつ効果を示している[193-195]。この有望なデータにより、うつ病に対するNK1拮抗薬やその他の神経ペプチド関連の代替薬への関心が高まっている。
ニューロペプチドY(NPY)は、中枢神経系に非常に広く存在する神経伝達物質であり、様々な受容体を通して作用する[196-198]。近年、NPYは、うつ病、不安、ストレスにおいて、血漿[199]と髄液[200]の両方で減少することが報告されている。逆に、抗うつ薬の投与は、NPYレベルの上昇と関連している[201]。
これらの知見を踏まえて、NPYに関連する治療的介入が注目されている。マウスモデルからのデータは豊富である。NPYの中枢投与は、無動状態の減少や強制水泳テストでの水泳時間の延長[202]やその他の同様の相関関係[203-205]と関連しているが、Y1受容体ノックアウトマウスは逆の結果を示す傾向がある[206]。一方、Y2およびY4受容体のノックアウトマウスは、これらの試験でより回復力のある表現を示し[206, 207]、Y1アゴニストと同様にY2アンタゴニストを注入することで抗うつ効果が得られることがわかっている[202]。このことは、異なるタイプのNPY受容体が異なる役割を果たしていることを示唆しており、今後の研究が期待される仮説である。
ガラニンもまた、うつ病の神経生物学に関与していると提案されている[208, 209]。ガラニンシステムには、3つの主要なGタンパク質共役型受容体(GALR1,GALR2,GALR3)があり、いずれも中枢神経系に広く分布しており、モノアミン受容体と共役してヘテロ受容体複合体を形成する傾向がある[210]。このように、ガラニンシグナルは、神経伝達の重要な調節因子である。ガラニンの過剰発現は、うつ病やストレスで記述されており[211]、血清ガラニンレベルは、うつ病のバイオマーカーとして示唆されている[212]。siRNAによるGALR1およびGALR2のノックダウンラットモデルでは、フルオキセチンとGal(1-16)フラグメントを併用することで、より大きな抗うつ効果が得られた[213]。他のいくつかの研究でも、様々なガラニンリガンドで同様の結果が得られている[214-217]。とはいえ、ガラニンの神経生物学への理解は、特に気分の調節に関してはまだ始まったばかりである。
6. 報酬系神経回路におけるうつ病の薬理学的標的
報酬系は,外部および内部からの刺激に反応して,意欲的な行動や学習を媒介するさまざまな神経回路を含んでいる[218]。解剖学的には、このシステムは腹側被蓋領域(VTA)に由来し、側坐核(NAcc)、外側視床下部、外側中隔、海馬、扁桃体、PFC、前帯状皮質(ACC)に投影される[219]。前臨床および臨床における神経画像診断の結果、うつ病に見られる快感消失や意欲喪失は、モノアミン仮説の柱の一つであるドーパミン神経伝達の低下とともに、報酬系のいくつかの核、特にNAccとACCのサイズと機能の低下と密接に関連していることが明らかになっている[220, 221]。
機能的には、報酬処理には、相互に関連する2つの要素が含まれている。1つは、報酬を与える刺激に注意と行動を集中させる動機付け処理で、基本的にドーパミン神経伝達が関与している。もう1つは、これらの刺激に対する快感反応を仲介する快感処理で、NAcc、腹側淡蒼球、島皮質、眼窩前頭皮質全体のGABA、オピオイド、エンドカンナビノイド(EC)エンドバニロイド(EV)のシグナル伝達が関与している[224, 225]。これらのシステム間のクロストークは複雑で、まだ完全には解明されていない[226, 227]。しかし,これらの回路の特定の構成要素は,うつ病の神経精神薬理学的アプローチにおける潜在的な標的としてすでに登場している(図4)。
図4 報酬神経回路におけるうつ病の薬理学的治療目標
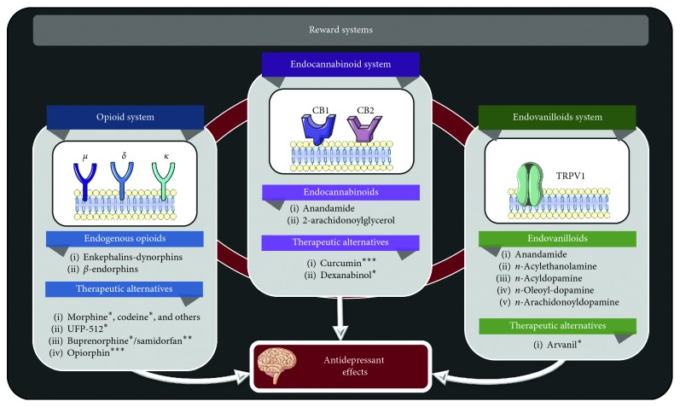
∗アルゴニスト。∗∗アンタゴニスト ∗∗∗μ:μ-オピオイド受容体 δ:δ-オピオイド受容体 κ:κ-オピオイド受容体 ∗:モジュレーター δ:δ-オピオイド受容体 κ:κ-オピオイド受容体 CB1:カンナビノイド受容体1。CB2:カンナビノイド受容体2。TRPV1: transient receptor potential cation channel V1. 報酬系うつ病の薬物療法のターゲットは、まだほとんど前臨床段階にある。現時点では、これらの物質のうつ病治療における臨床的有用性が示唆されているが、その有効性の程度や薬理学的プロファイルは様々である。
オピオイドシグナルの調節障害は、長い間、うつ病と関連しており、実際、現代の抗うつ薬が導入されるずっと前から、うつ病の管理にオピオイドが試行されてきた[228-230]。オピオイド受容体は、Gタンパク質共役型受容体スーパーファミリーに属し、異なる生理的役割を持つ3つの異なるタイプ、すなわち、ミュー(μ)、デルタ(δ)、カッパ(κ)を含んでいる。内因性オピオイドは、そのネイティブリガンドであり、特定のNH-末端配列(Tyr-Gly-Gly-Phe)を共有することを特徴とするペプチド群である。内因性オピオイドは,その構造と異なる受容体タイプへの親和性によって,エンケファリン,ダイノルフィン,β-エンドルフィンに分類されている[231]。
オピオイドペプチドとその受容体は、中枢および末梢神経系に十分に分布しており、侵害受容、鎮痛、内分泌および免疫学的調節、気分の調節、快楽および動機付けの処理、中毒行動の調節などに関与している[229]。中枢神経系では、オピオイド受容体は、主に脳幹、辺縁系核、大脳皮質に発現しており、後者2つは特に気分障害に関連している[232]。実際に、うつ病や自殺行為のある被験者を対象とした神経画像診断や病理学的研究では、特にPFC、NAcc、ACCにおいて、オピオイドシグナル伝達の構造的・機能的変化が示されている[233-237]。
オピオイドの誤用と慢性的な高用量治療は、うつ病の発症、再発、再燃の増加に一貫して関連しているが [238-241]、この点に関しては、より微妙な使い方が有益であろう。前臨床研究では、モルヒネ、コデイン、レボルファノール、メタドン、トラマドールなどの様々なオピオイドアゴニストが、うつ病行動を評価するマウスモデルにおいて、一般的に使用されている抗うつ剤よりも良い結果をもたらすことが観察されており、これらの効果はオピオイドアンタゴニストであるナロキソンによって可逆的であった[242]。δ選択的オピオイドアゴニストであるUFP-512をマウスおよびラットに脳室内および腹腔内投与した結果、オピオイドの抗うつ効果は、δ受容体アゴニストに特異的に依存している可能性が認められた[243]。非選択的なδアゴニストには、耐性、依存性、乱用の可能性があるため[244]、このような状況でのオピオイドの使用は、オピオイドアゴニスト/アンタゴニストの併用に関心が移っている。特に、δ受容体の部分作動薬であり、δおよびκ受容体の拮抗薬であるブプレノルフィンと、δ選択的拮抗薬であるサミドルファンを併用することで、迅速な抗うつ効果が得られるとされている[245]。
うつ病に対する別のオピオイド関連の潜在的薬理学的標的は、エンケファリンの迅速な不活性化に介在するZn-アクトペプチダーゼ、ニュートラルエンドペプチダーゼ、アミノペプチダーゼNの阻害剤であるオピオラルフィンに見出されている[246]。前臨床モデルでは、オピオイドを投与すると、抑うつ反応が弱まり、この効果は、δ選択的アンタゴニストであるナルドリンドール[247]やナロキソン[248]を投与することで可逆的であった。これらの有望な結果にもかかわらず、ヒトを対象とした研究はまだ少なく、これらの物質の誤用に関する懸念を解消するにはさらなる研究が必要である。
エンドカンナビノイド系もまた、気分の制御に重要な役割を果たしており、脳脊髄液中のEC代謝物のレベルは、うつ病の重症度と相関している[237]。ECは、報酬回路内の様々な核で必要に応じて合成される親油性の物質であり、主な分子は、バニロイド受容体にも親和性を示すアナンダミドと2-アラキドノイルグリセロールである [249, 250]。カンナビノイド受容体1は、基底核全体のグルタミン酸系およびGABA系のシナプスに局在し、カンナビノイド受容体2は、中枢神経系と免疫細胞の両方に見られることから、うつ病と全身の健康との間には、刺激的な関連性があると考えられている[251]。これらの受容体をコードする遺伝子の多型は、特異的な治療抵抗性 [252]、重度のうつ病症状 [253]、不安感、自殺行動 [254]と相関している。さらに、脳脊髄液中のカンナビノイド受容体リガンドのレベルは、電気けいれん療法を受けた後のうつ病患者で増加していることが判明している[255]。
うつ病におけるECモジュレーションに関する臨床研究はまだ少ないが、前臨床での知見は有望である。最近、Xiaolieらは、カンナビノイド受容体モジュレーターであるクルクミンと、デキサナビノールを担持した固体脂質ナノ粒子が、マウスおよび培養細胞におけるうつ病治療に果たす役割を調査した。その結果、この化合物はECの発現を増加させ、カンナビノイド受容体の活性化を増強し、多くの抗うつ薬の活性に重要なカスケードであるERK1/2経路の活性を増加させることがわかった[256]。
最後に、エンドバニロイドは、バニリル基を持ち、TRPV1受容体に親和性を示す内因性物質で、このカテゴリーの主な分子は、アナンダミド、n-アシルエタノールアミン、n-アシルドパミン、n-オレオレオイルドーパミン、n-アラキドノイルドーパミン、さらには12-ヒドロペルオキシエイコサテトラエン酸のようなアラキドン酸のリポキシゲナーゼ誘導体である[257]。TRPV1受容体を薬理学的に調節することで、痛み、不安、うつ病、様々な神経疾患の治療に役立つと考えられている[258]。TRPV1受容体は、気分、ストレス反応、および記憶処理の制御に関与する重要な領域である青斑部への入力を調節することが示されている[259]。
前臨床試験において、TRPV1ノックアウトマウスは、無動時間が短く、新奇性を抑制した摂食パラダイムにおける潜伏時間が短くなる傾向があり、これは抑うつ反応の減少と一致している[260]。TRPV1受容体の調節は、抗うつ薬の効果を高めることが観察されており[261]、TRPV1およびカンナビノイド受容体の合成アゴニストであるアルバニルは、マウスにおいて有意な抗うつ効果を誘発するようである[262]。今後、この分野でのヒトを対象とした研究により、うつ病における報酬系の神経精神薬理学的調節の意義と有用性が明らかになるはずである。
7. おわりに
うつ病が神経内分泌疾患であるという理解が広まったことで、神経精神薬理学の分野では大いに期待されている。特に、グルタミン酸系の代替薬は、近い将来、最も実現性の高いものになるかもしれない。現在、有望で活発な臨床試験が行われており、ケタミンの静脈内投与と経口投与が評価されており、他のいくつかの関連分子も使用されている[263, 264]。同様に、非薬理学的介入も、生活習慣の改善や電気けいれん療法などの確立されたものから、脳深部刺激、経頭蓋磁気刺激、精神外科手術などの新しいものまで、うつ病の治療におけるそれらの位置づけが近年より明確になってきていることから、軽視すべきではない。
さらに、うつ病の病態生理に関する見解の進展は、免疫学的、代謝学的、循環器系的側面を中心とした薬理学的および非薬理学的介入が、将来的に精神科治療の分野で新たな地平を切り開く可能性を示唆している。したがって、現在の治療成果は早急に改善する必要があるが、うつ病の治療法の選択肢という難問に対して楽観的でいるためには、これから十分な革新があるかもしれない。