
Contents
www.nature.com/articles/s41392-020-00233-4
要旨
ヒートショックプロテイン(HSP)、RNAシャペロン、ER関連ストレスタンパク質を含むストレスタンパク質(SP)は、細胞の恒常性維持に欠かせない分子シャペロンであり、その主な機能は、誤って折り畳まれたり、折り畳まれていないポリペプチドのシャペロンである。HSPの主な機能としては、ポリペプチドの折り返しや折り返していない状態のシャペロン、毒性ストレスから細胞を保護する機能、免疫・炎症性サイトカインを提示する機能などが挙げられる。
両刃の剣として考えられ、HSPはまた、多くのウイルスやがん細胞と協力してその生存を促進している。RNAシャペロンは、RNAポリメラーゼIIによって転写されたプレmRNA/hnRNAの機能と代謝の両方を操作するために不可欠な因子である不均一核内リボ核タンパク質(hnRNP)のグループである。
ストレスタンパク質の制御異常は、ヒト癌、心血管疾患、神経変性疾患(パーキンソン病、アルツハイマー病など)、脳卒中、感染症など多くのヒト疾患と関連している。本総説では、ストレスタンパク質の生物学的機能と、ウイルスの繁殖やウイルス感染によって引き起こされる疾患に関連したそのメカニズムに関する現在の進展についてまとめた。
また、SPは潜在的な抗ウイルス標的(COVID-19など)としても大きな関心を集めていることから、HSPをベースとした創薬開発のこの分野における現在の進捗状況と課題、および既に臨床評価中の化合物についても論じている。
ストレスタンパク質の概要
ストレスタンパク質(SP)は、細胞が細胞内または細胞外のストレス刺激にさらされたときに合成される多様なタンパク質群である。これらのタンパク質は、ストレスに対する保護効果を発揮する。
ストレスタンパク質には、ヒートショックプロテイン(HSP)、RNAシャペロン蛋白質(RNP)、小胞体(ER)で主に機能する蛋白質であるペプチジルプロピルイソメラーゼ、プロテインジスルフィドイソメラーゼ(PDI)、レクチン結合シャペロン系などがある1。
SPs はあらゆる種類の細胞に遍在的に発現しており、細胞内(病原体の侵入など)と細胞外(飢餓、サイトカイン/ケモカインやホルモンによる刺激など)の両方で発生するストレスを中和し、根絶するためのシグナルカスケードを誘発する。
SPによって誘発された応答は、細胞の生存を促進するための経路を活性化するか、または所定の条件下で特定の器官/組織を保護するために損傷を受けた細胞を排除するために細胞死(すなわち、アポトーシス、壊死、ピロプトーシス、またはオートファジー細胞死)を開始することができる。
ストレスタンパク質の制御異常は、心血管疾患、神経変性疾患(パーキンソン病、アルツハイマー病など)、脳卒中、ヒトの癌、感染症などの様々な疾患と関連していることが広く知られている。本総説では、その機能に焦点を当て、感染症、特にウイルス感染症に関連する知見を更新している。
ヒートショックプロテイン
1962 年、イタリアの遺伝学者リトッサは、ショウジョウバエの幼虫の培養温度を不用意に上昇させ、未知のタンパク質の遺伝子転写が増加することを発見した。さらに研究を進めていくと、大きなファミリーを形成し、細胞内でユビキタスに発現しているHSPが多数存在することが明らかになっていた。
HSPは分子量に基づいて、HSP100s、HSP90s、HSP70s、HSP60s、HSP40s、そして一部の小さなHSP(15-40 kDa)に分類されている3,4,5。HSPの発現は、細胞が飢餓、高温、低酸素、高酸素、病原体の侵入、栄養失調、化学物質や紫外線への曝露などの生理的または環境的な攻撃を受けたときに急速に誘導される。
HSP は、基質タンパク質の正しい折り畳みを促進したり、安定化させたりするためのネットワークを形成し、その機能的/活性的なコンフォメーションを獲得している(図 1)。HSPは、細胞の生存、分化、細胞死を制御する重要な因子であり、いくつかのHSPが自然免疫だけでなく、適応免疫応答における抗原提示にも関与していることを示すエビデンスが蓄積されている7,8。血漿中のHsp70の増加は心不全と関連していることが示されている9 。
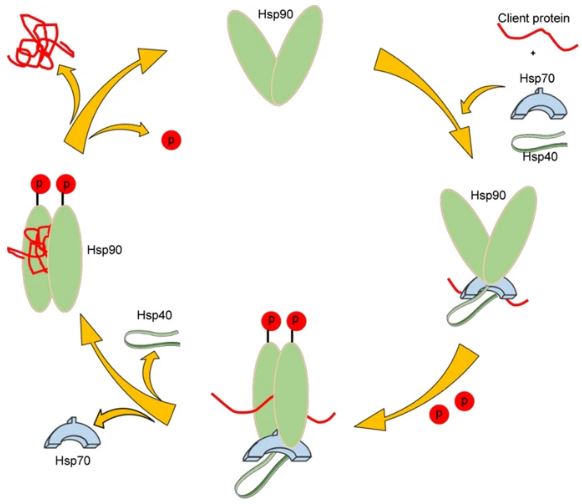
図1
図1
ヒートショックプロテインの一般的なシャペロンサイクル。最初は、HSP70-HSP40シャペロンに結合したアンフォールドされたクライアントタンパク質がHSP90と相互作用する。HSP90へのATP結合は、クライアントタンパク質のHSP70からHSP90への移行を誘導する。その後、HSP70-HSP40シャペロンのコンフォメーションが解除される。最後に、ATPの加水分解は、クライアントタンパク質の放出につながる追加のコンフォメーション変化を誘導する
HSP90s
5,6,11 Hsp90 は ATP 依存性のシャペロンであり、異なるアイソフォームを持つ
1) Hsp90α(HSP90AA1、または HSPC1)、Hsp90α-A2(HSPAA2、または HSPC2)、Hsp90β(HSPAB1、または HSPC3)が細胞質に位置する。
2) グルコース調節タンパク質Grp94(HSPC4またはGP96)は小胞体に存在する。このうち、ヒトではHsp90αとHsp90βが最も多くを占めている。Hsp90は、N末端領域にATPase依存性加水分解ドメイン、中間リンカー領域、C末端領域に二量化ドメインの3つの領域を有している。Hsp90がATPを結合すると、N末端ドメインの一過性の二量化により、基質ペプチドがHsp90に結合する。また、Hsp90はテロメアの維持、アポトーシス、細胞周期の進行などにも関与している6,13。Hsp90 は、RNA ポリメラーゼの組み立てや一部の(-) ssRNA ウイルス(インフルエンザウイルスの PB2 など)の核インポートに関与しているだけでなく、ウイルスのキャプシドタンパク質やウイルスのアセンブリの折り畳み過程でも重要な役割を果たしていることが知られている14。
Hsp90 のコシャペロン
Hsp90のコシャペロンは、Hsp90の機能を多面的に制御している。CDC37(p50とも呼ばれる)はHsp90にキナーゼを送達し、そのATPase活性を阻害している。
Hsp70s
HSP70は、分子量70 kDaのHSPスーパーファミリーのサブファミリーであり、細胞内の分子シャペロンの大部分を占めている。細胞内の分子シャペロンの大部分を占めている。
(1) Hsp72 (HSPA1A)、Hsp70-2 (HSPA2)、Hsp70B’ (HSPA6) および Hsc70 (HSPA8) は一般的に細胞質に位置し、(2) Grp75 (HSPA9) はミトコンドリアに位置し、(3) Grp78 (HSPA5) は ER に関連している16。Hsp70は2つのドメインから構成されている:N末端領域には4つのサブドメイン(IA、IB、IIA、IIB)に分割可能な44kDaのヌクレオチド結合ドメイン(NBD)と、C末端領域にはC末端αへリックス(SBDα)とN末端βシート(SBDβ)のサブドメインからなる28kDaの基質結合ドメイン(SBD)から構成されている17,18。細胞内タンパク質監視の重要な構成要素として、ATP依存性分子シャペロンはストレスによる損傷から細胞を保護し、新たに合成されたポリペプチドの折り畳み、誤って折り畳まれたまたは凝集したタンパク質の認識とリフォールディング、タンパク質の可溶化または分解、タンパク質の輸送、オリゴマータンパク質複合体の組立または分解、および特定のネイティブに折り畳まれたタンパク質の制御など、多くの折り畳みプロセスに関与している5,13,19,20。
Hsp70の機能は宿主のタンパク質フォールディングに限定されない。その機能は、ウイルス感染時にはかなりのものである。Hsp70のメンバーは、ウイルスのライフサイクルの過程で全く異なる役割を示す。
例えば、Hsp72、Hsp70B’およびHsc70は、HCVウイルスの侵入、ウイルスの組み立ておよびウイルスゲノムの翻訳に関与している。ERのGrp78は、ウイルスタンパク質の恒常性に関連し、宿主細胞におけるウイルスタンパク質の過負荷を防止する。21 Grp75 はミトコンドリアで HCV の NS5A タンパク質と相互作用している22 。
Hsp70のシャペロンサイクル
シャペロンサイクルは、N末端のNBDが媒介しており、2つの状態を切り替えることでHsp70と基質の結合を制御している。第一の状態は、基質結合に対する親和性が低いATP結合状態、すなわち基質ペプチドのSBDへの高い会合・解離率を持つ状態である。
第2の状態は、ATP加水分解によりADP結合状態とヌクレオチドフリー状態に切り替わる状態である。この状態では、基質交換率は低いが、基質への親和性は高い。Hsp70のシャペロン活性は、ほとんどがATP加水分解に依存している。
Hsp70の基底ATPアーゼは、基質ペプチド自体によって刺激されない限り、通常は低い。30℃でHsp70の1分子あたりのATPが加水分解されるのに20〜30分かかる。その結果、ATP加水分解を誘導し、基質ペプチドに対する親和性の増加を助けるために、いくつかのコシャペロンがHsp70 ATPと遭遇する必要がある。
Hsp70のコシャペロン
Hsp70の最も重要なコシャペロンは、Jドメインタンパク質(JDPs)ファミリーとヌクレオチド交換因子(NEFs)ファミリーのメンバーである。これまでの研究では、Hsp70機構の機能に焦点を当てており、その作用様式の「正準モデル」の開発につながっている。
このモデルは2つのステップを含んでいる。まず、展開されたペプチド基質は、Hsp40ファミリーのJDPと結合し、その後、基質はHsp70に送達され、Hsp70のATPアーゼ活性を刺激する。同時に、JDPは、アンフォールドされていないタンパク質の凝集を防ぐ。
第二に、NEFは、基質が正しい活性なコンフォメーションに折りたたまれることを保証する基質放出因子として働く。このようにして、コシャペロンはHsp70の機能を強力に促進する。したがって、Hsp70は一般的に個別に働くのではなく、コシャペロンと協力して働くことになる23,24。
Hsp60s
Hsp60sは、2つのリングが背中合わせになっている大きな円筒形のオリゴマーである。19 各リングの中央の空洞の非ネイティブタンパク質は、ATP依存性のプロセスを経てネイティブタンパク質に折り畳まれる25,26。25,26 Hsp60sは2つのサブファミリーに分類され、第1グループは主に原核生物に存在し、第2グループは真核生物の細胞質や一部の古細菌に存在する。
第一グループで最もよく研究されているのは、細菌の細胞質に存在するGroEL-GroESシャペロニン系である。GroELは約57 kDaのタンパク質で、2つのリングが背中合わせに配置され、7つのサブユニットがテトラデカマー構造を形成している。GroESはGroELのコシャペロンである11。2つの環-四量体構造は、非対称(1GroEL:1GroES)と対称(1GroEL:2GroES)の2つの形態で発現し、それぞれ「弾丸」14,29,30と「アメリカンフットボール」31の形をしている。
GroEL-GroESシャペロニンは、ATPに依存してアロステリックな制御を受け、タンパク質のフォールディング機能を完成させる。このポリペプチドはGroELの7つのサブユニットのうちの1つの疎水性部位に結合し、ATPの結合や加水分解によって構造を変化させる。GroEL-GroES システムとは対照的に、哺乳類のホモログ Hsp60/Hsp10 システムはあまり研究されていない。Hsp60 はミトコンドリアに取り込まれ、58 kDa の分子サイズを持つ成熟型に変換されると考えられている33,34,35 。
グループIIのシャペロニンには、古細菌のサーモソームと真核生物のCCT(シャペロニン含有TCP1、またはTriCと呼ばれる)があり、各環に分子量57~61 kDaの8~9個のサブユニットを持つオリゴマーである。I群と比較して、II群のシャペロニンはATP結合による異なるアロステリックな動きを示する。
HSP60ファミリーは、ウイルスの付着からウイルスゲノムの複製までの様々な段階でウイルスのライフサイクルに関与していることが知られている。Hsp60は宿主細胞の免疫調節に必須である。いくつかのウイルスタンパク質は、宿主細胞内での転座にHsp60を必要とする。PB2 はインフルエンザ A ウイルス RNA ポリメラーゼのサブユニットであり、主に核内に位置するが、ミトコンドリアにも現れる38 。
PB2はミトコンドリア抗ウイルスシグナル伝達タンパク質(MAVS)と相互作用し、侵入したウイルスが宿主細胞の防御から容易に逃れることができるようにIFNβのレベルを低下させることで細胞内免疫応答をダウンレギュレートする39。また、Hsp60はTNF-a、IL-6、IL-1bなどの炎症性サイトカインの放出を誘導することで、自然免疫の制御に大きな役割を果たしている40。
スモールヒートショックプロテイン
スモールヒートショックプロテイン(sHSP)は、~15~40 kDa の低分子量を持つ小型蛋白質のグループである。sHSPファミリーには10のメンバーが存在し、Hsp27 (HSPB1)、HSPB5 (αB-クリスタリン、またはαBC)、Hsp20 (HSPB6)、Hsp22 (HSPB8)などのユビキタスなものもある。他のものは組織特異的であり、HSPB2(筋萎縮性ジストロフィープロテインキナーゼ、MKBP)、HSPB3、HSPB4(αA-クリスタリン、αAC)、HSPB7(cvHsp)、HSPB9、およびHSPB10(精子外緻密繊維蛋白質、ODF)などがある。 41
sHSPは、細胞内ではモノマー、二量体、あるいは大きな多量体の複合体として存在する可能性がある42。sHSPの構造は、そのあまり保存されていない配列のため、他のHSPとは異なる。sHSPsの基本構造は、保存されたα-クリスタリンドメイン(ACD)を挟んで、N末端配列(NTS)とC末端配列(CTS)を含む2つの保存されていないドメインが配置されている。これらのドメインのうち、ACDが異なるsHSPの特徴となっている43,44。
sHSP は、ストレス耐性、アポトーシス、老化、長寿に関するいくつかの生理学的プロセスにおいて重要な役割を果たしている45,46,47,48 。リン酸化と N 末端 WDPF モチーフは、sHSP のホモまたはヘテロオリゴマーの形成を助けている49,50 。
オリゴマー化のダイナミクスは、シャペロンの活性にとって非常に重要であり、それは、幅広い基質をシャペロンするために、それぞれが異なる結合特性を持つ異なるホモおよびヘテロオリゴマーを形成する可能性をもたらすからである。小さなリン酸化二量体/四量体は、F-アクチンと結合してアクチン重合を調節する。
様々なsHSPの中で、Hsp27は広く研究されている。Hsp27 はすべての組織に存在し、主に心臓、骨格筋、平滑筋で発現していることが知られているが54 、その重要性は細胞分化、細胞生存、細胞の宿命免疫、ウイルスタンパク質の翻訳、細胞内ウイルス輸送などで実証されている55,56,57 。Hsp27 のリン酸化は可逆的なプロセスである。60,61
Hsp27 は、最大 800 kDa の大きなホモオリゴマーを形成するだけでなく、他の sHSP(例:Hsp20)と協力してヘテロマー構造を形成することもできる56,58 Hsp27 は、感染症の際に上昇し活性化される62,63 。67,68,69 Hsp27 はまた、発生、分化、細胞増殖に関与する様々なシグナル伝達経路にも関連している70,71 。
様々なストレス(HBV や EBV 感染など)によって刺激された Hsp27 の長期的かつ高レベルの発現は、発がん、細胞生存、がん細胞の幹細胞性、がん転移、腫瘍形成、薬剤耐性を増強する。
HSPの転写制御
ヒートショック因子(HSF)は、hsp遺伝子の転写活性化に大きく貢献している。すべての無脊椎動物では HSF1 のみが転写活性化に関与している。脊椎動物では、HSFファミリー(HSF1-4)の4つのメンバーがHSPの発現を調節している72が、その中で最も重要なのはHSF1である。
ストレス条件下では、細胞質内の元々単量体であった HSF1 が三量体化して核内に移動し、プロモーター領域のヒートショックエレメント(HSE)に結合して hsp 発現を促進することが知られている74。
タンパク質ジスルフィドイソメラーゼ
タンパク質ジスルフィド異性化酵素(PDI)は、小胞体(ER)におけるジスルフィド結合の形成、異性化、還元を触媒する多機能酸化還元酵素およびシャペロンである。ジスルフィド結合形成の際に、PDIのCGHC活性部位のシステイン残基がポリペプチド基質のシステイン残基から2個の電子を受け入れ、PDIの還元と基質の酸化につながる。
チオールジスルフィド異性化酵素としての PDI の触媒機能に加えて、グリコシル化タンパク質の品質管理のための分子シャペロンとしての特性も発揮する。ERp57(PDIA3, Grp58)は、おそらく最もよく研究されているPDIファミリーメンバーであり、4つのドメイン(すなわちa-b-b’-a’)からなる類似の構造を共有し、2つの局在化配列-ERリテンションシグナル(QDEL)と核局在化シグナル(KPKKKK)-を持っている。
還元酵素または異性化酵素活性のために基質に直接結合する他のPDIファミリーメンバーとは異なり、ERp57のbドメインはカルレティキュリン(CRT)およびカルネキシン(CNX)と高い親和性を有しており、これは糖タンパク質のポリペプチドセグメントを認識してリクルートするのに役立つであろう77。
タンパク質が正しく折りたたまれない場合、UDP-グルコース:糖タンパク質グルコシルトランスフェラーゼ(UGGT)がタンパク質を再糖化するためにリクルートされ、それらがERp57/CRT/CNX複合体によって認識され、再結合されることを可能にする76,78,79。
RNAシャペロン
RNA と非特異的に相互作用し、非機能的な阻害構造を解消するタンパク質は、通常、RNA シャペロンと呼ばれ、共通の配列やモチーフを持たずに明確な役割を持っている80,81 。RNA分子は、その機能を果たすために、ほとんどの場合、よく定義された立体構造に依存している。82
多数の可能性のあるRNAの塩基対合と二重構造の高い安定性により、機能的なネイティブ構造と同様に熱力学的に安定な代替的な二次構造や三次構造が多数存在すると考えられている。RNAシャペロンは、キネティックフォールディングトラップからの脱出を促進することにより、RNAのフォールディングを促進し、RNAが非機能的な構造にトラップされるのを防ぐ。
HnRNPsは不均一核内リボ核タンパク質の一群である。HnRNPは、RNAポリメラーゼIIによって転写されたpre-mRNA/hnRNAの機能と代謝の両方を操作するために不可欠な因子である。hnRNP は、アルギニン・グリシンボックス(RGG ボックス)、RNA 認識モチーフ(RRM)、hnRNP K 相同性(KH)ドメイン、ジンクフィンガー(ZF)ドメイン(KHZF ドメイン)などの共通の RNA 結合モチーフを含んでいる。 87
このファミリーのよく定義された機能には、転写調節、プレmRNAスプライシング、3′末端形成、mRNAパッケージング、RNA輸送、翻訳調節、RNAサイレンシング、DNA修復、およびテロメア生合成が含まれる。また、これらのタンパク質は核と細胞質の間をシャトルする能力を持っているため、核内でのRNP複合体の形成を一時的に助け、細胞質でのRNA代謝にも関与している可能性がある88 。
ストレスタンパク質の重要性
ストレスタンパク質の主な機能の一つは、細胞の恒常性を維持することである。圧力がかかると、ストレスタンパク質は圧力を解放するためにハイパーアクテ ィブになる。Hsp27、Hsp70、およびhsp90は、かなりの数の種類の癌細胞において非常に高いレベルで蓄積されている90,91,92。腫瘍の進行に伴い、蓄積されたがん原性タンパク質は強力なタンパク質フォールディング能力を必要とする。
長期ストレス下では、HSP は発がん、細胞生存、抗アポトーシス、血管新生、がん細胞の幹細胞化、浸潤、転移に関与したり、促進したりする。しかしながら、アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症、ハンチントン病、脊髄小脳失調症94,95などのポリグルタミン疾患を含むほぼすべての加齢性神経変性疾患では、HSPsとRNAシャペロンがダウンレギュレーションされている(図2)。
減調したRNAシャペロンは、RNA代謝の障害につながる;94一方、減衰したHSPは、神経疾患におけるタンパク質の折り畳みやタイムリーな分解を適切に処理するためのタンパク質品質管理(PQC)システムの障害につながる95。このレビューでは、これらのトピックにはあまり注目せず、病原体感染、特にウイルス感染によって引き起こされるヒトの疾患におけるストレスタンパク質の主な生物学的機能と標的値のみに焦点を当てることにする。
図2
図2
ストレスタンパク質の機能障害とウイルス感染による制御異常が原因の疾患 左図は、がんや神経変性疾患を含む多様なヒト疾患におけるストレスタンパク質の発現レベルを示している。ストレスタンパク質の発現量は、がん細胞では有意に上昇しているが、神経変性疾患では劇的に低下している。右図は、がんや神経変性疾患など、ウイルス感染が原因となる疾患を軽度から重度まで示したものである。ストレスタンパク質がこれらの疾患に寄与していることがわかる。
ウイルス感染は、ストレスタンパク質の調節障害と関連性の高い様々な疾患を引き起こす(図2)。例えば、呼吸器症状96,97,97 胃腸炎98,99,100,101 出血性発熱102,103 などである。例えば、HIV感染は神経認知障害を引き起こす可能性がある。今日までに、全身性のウイルス感染が、アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症、多発性硬化症、自閉症スペクトラム障害を含むいくつかの神経変性疾患としばしば関連していることを示す証拠が増加している104,105。
がん症例の20%以上が世界中のウイルス感染に起因している。がん症例の最大数に関連するウイルスは、ヒトパピローマウイルス(HPV)および肝炎ウイルス(HBVおよびHCV)である。HPV感染は子宮頸がんおよび他のいくつかの上皮性悪性腫瘍を引き起こし、HBV/HCV感染は肝細胞がんの大部分を引き起こする。
その他のがんウイルスとしては、エプスタインバーウイルス(EBV)、カポジ肉腫関連ヘルペスウイルス(KSHV)、ヒトT細胞白血病ウイルス(HTLV-I)、メルケル細胞ポリオマウイルス(MCPyV)、HIVなどがある106 。本総説では、ウイルス感染に応答するストレスタンパク質が、ウイルスの繁殖と疾患の発症に与える影響に注目している。
ストレスタンパク質は、ウイルスの侵入、コーティング解除、複製、遺伝子発現、ウイルスの組み立て・放出など、ウイルス感染過程の多くのステップに関与している(図3)。ストレスタンパク質とウイルス感染、宿主細胞応答の関係を表1にまとめた。
図3
図3
ストレスタンパク質は、ウイルスのライフサイクルの多様なステップに関与している。ウイルスのライフサイクルの5つの主要なステップは、ステップ1-5としてラベル付けされている:ウイルスの侵入とコーティング解除、ウイルスの複製、遺伝子発現、組み立てと放出である。ほとんどのウイルスはこれらのステップを細胞質内でのみ行うが、特にEV-A71、DENV、JEVなどのようなRNAウイルスはそうである。
一方、一部のウイルス(インフルエンザ、SV40、HBVなど)は宿主細胞の核内にも侵入する。これらのウイルスは、そのライフサイクルの中で、a-cとしてラベル付けされた他のステップを経ることがある:ウイルス核の輸入、核の輸出、およびウイルスRNAの処理。ウイルス感染の過程において、Hsp90、Hsp70、Hsp60、Hsp40、Hsp27、およびPDIは、ウイルスの侵入およびコーティング解除のステップに参加する。
Hsp90、Hsp70、Hsp60、Hsp40、Hsp27およびRNAシャペロンは、ウイルス複製ステップに参加する。Hsp70、Hsp40およびRNAシャペロンは、ウイルス遺伝子発現ステップで必要とされる。Hsp90、Hsp70、Hsp40はウイルスの組み立てを助ける。
Hsp70とRNAシャペロンはウイルスの放出に寄与する。一方、Hsp90はウイルス核のインポートとエクスポートにも重要である。Hsp70はウイルス核のインポートで役割を果たしている。そして、RNAシャペロンは、複製、翻訳開始、安定化、崩壊を含むウイルスRNAの処理に大きな役割を果たす
表1 多様なウイルスの複製過程のさまざまな段階でストレスタンパク質が関与している
フルサイズテーブル
ウイルス感染におけるHSP90ファミリーの機能
本節では、RNAウイルス、DNAウイルス、レトロウイルスを含む様々なウイルスのライフサイクルにおけるHSP90ファミリータンパク質に関する新たな知見を紹介する。また、ウイルス誘導細胞応答におけるHSP90ファミリーの機能についても考察する。
RNAウイルス感染におけるHSP90ファミリーの機能
ウイルスの侵入
RNAウイルスの場合、Hsp90はエンテロウイルスA71(EV-A71)、107 日本脳炎ウイルス(JEV)、デングウイルス(DENV)の侵入に重要である108 。DENV ウイルスと JEV の両方がフラビウイルスに属しているため、これらの 2 つのウイルスの侵入は、神経芽腫細胞と微小グリア細胞の両方で Hsp70s のサポートを受けて Hsp90 を異なる形で利用している。111
さらに、Hsp90 と Hsp70 は共に DENV 感染に反応して膜脂質ラフトと関連している。110 しかし、DENV 感染肝細胞(HepG2)では、Hsp90 も Hsp70 も DENV の内部化を可能にする受容体として機能していない。Hsp90とHsp70の受容体機能は、他の未知の分子で置き換えられている可能性が高い。
ウイルス複製
Hsp90タンパク質は、いくつかの側面でウイルスの複製を促進する。第一に、Hsp90は、ウイルスタンパク質を安定化させる古典的なシャペロンタンパク質として機能する。Hsp90は、パラミクソウイルスのポリメラーゼやLタンパク質を安定化させ、ウイルスの複製を助けている。
Hsp90の阻害は、ウイルスの複製を妨げ、水胞性口内炎ウイルス(VSV)、ヒトパラインフルエンザウイルス-2(HPIV-2)、ヒトパラインフルエンザウイルス-3(HPIV-3)、シミアンウイルス41(SV41)、または呼吸器性合胞体ウイルス(RSV)感染細胞におけるLタンパク質の半減期を短縮する可能性がある112,113 同様に、Hsp90は、nsP3(RNA合成に必須のタンパク質)およびnsP4(RNA依存性RNAポリメラーゼ、RdRp)を含むチクングニヤウイルス(CHIKV)非構造タンパク質(nsPs);114、およびHCV非構造タンパク質NS3のプロテアーゼの安定性を維持することが示されている115.2番目に、Hsp90はウイルス複製を促進するためにウイルスポリメラーゼ活性を調節する。
HCV を例にとると、Hsp90 は NS5 リン酸化キナーゼ PRK2 の上流キナーゼであるキナーゼホスホイノシチド依存性キナーゼ l (PDK1) の安定性を維持することで、HCV ポリメラーゼ NS5 活性を間接的に調節している116 。Hsp90 阻害剤は、Hsp90-NS5 複合体の形成を阻害することにより、ウイルスの複製を抑制する。
インフルエンザウイルス感染中、Hsp90はウイルスRdRpサブユニットであるポリメラーゼ塩基性タンパク質-1(PB1)および-2(PB2)と相互作用して複合体を形成し、核内に共移動する14,118。Hsp90は核内に侵入した後、Hsp90/PB1/PB2複合体から解離し、ポリメラーゼ酸性タンパク質(PA)と新たな機能性複合体を形成する。HDAC6/8阻害剤は、ポリメラーゼの核輸入を効率的に制限し、ウイルスの複製を抑制する。
最近の研究では、Hsp90もPIM1シグナルを介してEV-A71の複製に重要な役割を果たしていることが示されている(未発表)。122 上昇したPIMはEV-A71の複製を促進し、PIM1のノックダウンはEV-A71の複製を減少させることが示されている。Hsp90βをノックダウンすると、感染12時間後(p.i.)にはウイルス複製の60%が減少し、分泌されるウイルスは約80%減少することから、ウイルスの複製と分泌の両方においてHsp90βが重要な役割を果たしていることが示された(図4a-c)。
他の研究者は、Hsp90βがEV-A71の組み立てに関与していることを報告しており、これが本研究でEV-A71ウイルスの分泌を抑制している理由かもしれない。また、Hsp90βをノックダウンまたはノックアウトすると、EV-A71構造タンパク質の発現レベルが低下することが示された(図4d,e)。
また、Hsp90阻害剤である17-AAG、ゲルダナマイシン(GA)、VER50588は、いずれもEV-A71タンパク質の発現を劇的に阻害することがわかった(図4f)。その中でもVER50588は、これまでに報告されていない最も強い阻害効果を示した。このことから、Hsp90βがPIM1の標的となる可能性があることが予想された(図4g)。
この仮説に対処するために、PIM1を過剰発現させた実験とPIM1をノックダウンした実験を行った。その結果、Hsp90βのリン酸化状態が増加したり、減少したりした(図4h,i)。さらに重要なことに、CRISPR/Cas9媒介遺伝子編集によるHsp90βのノックアウトでは、EV-A71複製に対するPIM1シグナル伝達の効果はほぼ完全に消失した(図4j,k)。
図4
図4
Pim1シグナルはHsp90βのリン酸化を介してEV-A71複製を促進する(未発表データ).a RD細胞をスクランブル/Hsp90β siRNAで24h処理し、Hsp90βノックダウンの効果をRT-qPCRアッセイで測定した。結果は平均値を示し、±誤差は標準偏差(SD)を示す。データは3回の実験から得た。*b, c RD細胞をスクランブル/Hsp90β siRNAで4時間処理した後、指示された時間だけ1のMOIでEV-A71に感染させた。細胞内(b)および細胞外(c)のウイルスRNAレベルをRT-qPCRアッセイにより検出した。結果は平均値を示し、±誤差は標準偏差(SD)を示す。データは3回の実験から得た。*d、e siRNAによるHsp90βのノックダウンまたはCRIPSR/Cas9媒介遺伝子編集によるノックアウトをRD細胞で行い、その後、指示された時間の間、細胞を1のMOIでEV-A71に感染させた。EV-A71のタンパク質レベルはウエスタンブロットによって決定された。f RD細胞を異なる濃度のHsp90阻害剤で処理し、0.01のMOIで48時間EV-A71に感染させた。g オンラインGENEMANIAプログラムを用いてPim1-タンパク質相互作用ネットワークを予測した。Hsp90 のリン酸化状態をウエスタンブロットで検出した。そして、細胞溶解物を採取し、ネイティブページ解析を行った。 j WT RD細胞およびHsp90βノックアウト(Hsp90β-KO)細胞を、2μM PIM1阻害剤SGI-1776で2時間前処理し、その後、EV-A71をMOI 0.01で48時間感染させた。細胞内ウイルスRNAレベルをRT-qPCRにより決定した。結果は平均値を示し、±errorは標準偏差(SD)を示す。データは3回の実験から得た。*k Pim1をWT/Hsp90βノックアウトRD細胞で過剰発現させ、1のMOIで12時間EV-A71に感染させた。
ウイルスタンパク質の成熟、ウイルスの集合、放出
ウイルスタンパク質の発現と成熟の間、Hsp90は古典的なシャペロンとして機能し、ウイルスタンパク質の適切なフォールディングを監視する。Hsp90は、HCV非構造タンパク質2/3(NS2/3)キナーゼの成熟を調節する。123 Hsp90とそのコシャペロンp23は複合体を形成し、ポリオウイルス、ライノウイルス、およびコックスサッキーウイルスのキャプシド前駆体ポリタンパク質P1の適切な折り畳みを支援する。ウイルスの組み立ての間、Hsp90はノロウイルスのキャプシドVP1タンパク質およびマウスノロウイルス1ゲノムの末端と相互作用する。124 Hsp90は、ウイルスの放出に関与する主要な表面糖タンパク質であるインフルエンザノイラミニダーゼ(NA)と相互作用し、安定化させる。
DNAウイルス感染におけるHSP90ファミリーの機能
ウイルスの侵入
DNA ウイルスの侵入には、細胞膜を越えての侵入と核内への侵入がある。Hsp90 は主にウイルスの核内輸送を補助する能力を示している。多くのウイルスの核内輸送は、微小管(MT)とMT依存性分子モーターダイニン/ダイアクチン複合体に依存している。Hsp90の阻害剤は、Hsp90のアセチル化チューブリンへの結合を阻害し、それによってHSVキャプシドタンパク質の核輸送を阻害する。
ウイルスの複製
レトロウイルスのDNA複製の際、Hsp90は主に逆転写酵素(RT)活性の調節・維持に寄与している。B型肝炎ウイルス(HBV)を例にとると、逆転写は、VIII型ウイルスにおいてウイルスゲノムDNAを生成するために不可欠なステップである。逆転写の開始は、事前ゲノムRNA上のRNAシグナル(パッケージングシグナルε)とRTの認識と相互作用である134 。Hsp90は、ウイルスのRTと相互作用することでアヒル型B型肝炎ウイルス(DHBV)の複製を促進する必須の宿主因子であることが明らかになった135 。ポリメラーゼの末端タンパク質(TP)およびRTドメイン内の2つの独立した領域は、N末端およびC末端フラグメントでHsp90と別々に結合しており、両ドメインはリボヌクレオタンパク(RNP)およびタンパク質のプライミングに不可欠である137,138。第一に、Hsp90シャペロンまたはHsp70シャペロンがRT-ε相互作用に必須であるかどうかである。Stahlらは、Hsp70シャペロンがRT-ε相互作用にとってより重要であると考えていた。また、Hsp90/Hop複合体はRT活性の質ではなく量に影響を与えることを提案している。最後に、別の研究では、Hsp90はHBV RT/ε RNA複合体を維持するのではなく、HBV RTのプライミングを助けることが示されている142 。
ほとんどのDNAウイルスの複製は、ウイルスが複製センターで形成される核で行われる。したがって、ウイルスタンパク質の適切な位置は、ウイルスの複製に非常に重要である。Hsp90はまた、ウイルス感染細胞におけるウイルスDNAポリメラーゼの位置を調節することがわかっている。Hsp90阻害剤で処理すると、HSVポリメラーゼは核内から細胞質に誤局在化し、その後プロテアソーム依存的に分解される。Hsp90阻害剤は、ウイルスDNAポリメラーゼのトランスロケーションを効果的にブロックする。
ウイルス遺伝子発現
Hsp90は、転写と翻訳の両方のレベルでウイルス遺伝子の発現に重要である。HSV immediate-early α(IEα)遺伝子の転写は、八量体結合転写因子1(Oct-1)、宿主細胞因子1(HCF-1)、およびウイルスタンパク質16(VP16)からなる転写因子複合体によって開始される148,149。転写過程において、Hsp90αは、マクロオートファジーを介した方法でVP16を分解から守ることで、VP16の安定性を維持することが示されている150 。同様に、Hsp90はまた、主要な即時初期遺伝子の転写に重要なAktとNF-κBシグナル伝達経路を活性化することで、ヒトサイトメガロウイルス(HCMV)即時初期遺伝子の転写を制御している151,152。
翻訳レベルでは、Hsp90は、単純ヘルペスウイルス1型および2型(HSV-1、HSV-2)、水痘帯状疱疹ウイルス(VZV)、EBV、KSHVを含む保存されたヘルペスウイルスプロテインキナーゼ(CHPK)の翻訳を促進する。EBV核抗原1(EBNA1)タンパク質の翻訳もまた、Hsp90によって操作されている163,164 EBNA1は、細胞の形質転換、腫瘍形成、およびウイルスエピソームの維持に重要である165,166,167 EBNA1の翻訳は、Gly-Alaリピートドメインを介してHsp90によって厳密に制御され、EBNA1を比較的低いレベルに維持している168。163,164 Hsp90はEBNA1と直接相互作用しないため、ブリッジタンパク質がこのプロセスに関与している可能性がある。EBNA1の発現を阻害すると、試験管内試験(in vitro)でのEBV誘発性原発性B細胞形質転換と生体内試験(in vivo)でのSCIDマウスのリンパ増殖性疾患の両方が強く抑制される。
ウイルスの組み立て
DNAウイルスの組み立てにおけるHsp90の機能を報告している論文はごくわずかである。活性化されたHsp90はHBVの組み立てに必要である。169,170 Hsp90は、キャプシド形成のためのコアタンパク質二量体の親和性を高め、キャプシドの解離を防止する。
ウイルス誘発性腫瘍形成
前述のように、EBVはバーキットリンパ腫171 や上咽頭癌(NPC)などのいくつかの腫瘍の原因となる前駆体である。 172 潜在膜タンパク質1(LMP1)は、マトリックスメタロプロテアーゼ9(MMP9)の発現を誘導し、腫瘍壊死因子受容体(TNFR)スーパーファミリータンパク質を模倣し、NF-kB、MAPK、PI3K/Akt、JAK/STATシグナル伝達経路を活性化することにより、腫瘍の転移や浸潤を促進するオンコプロテインと考えられている173,174。175,176 Hsp90阻害剤であるAT13387およびBIIB021は、上述の下流のシグナル伝達経路を活性化することでLMP1の機能を阻害し、細胞増殖とアポトーシスを強力に阻害すると考えられている。
免疫の変調
Hsp90がDNAウイルスによってハイジャックされ、ウイルスの複製を促進することについては、上記で述べた。特定の条件下では、Hsp90は細胞免疫を促進することで抗ウイルス活性を発揮する。急性感染段階では、Hsp90はEBV感染B細胞の細胞表面に発現するように誘導される。γδT細胞は、宿主がHIV、インフルエンザ、センダイ、コクサッキー、ワクシニア、VSV、またはHSV-1に感染した急性期のファージにおいて、強力な抗ウイルス能力を有する。Hsp90は免疫センサーとして働き、抗原提示を助けるので、EBV感染においても同じように機能する可能性がある。
レトロウイルス感染時の細胞形質転換におけるHSP90ファミリーの機能
いくつかのHSPは、細胞形質転換を促進するためのオンコプロテインとして機能している。Hsp90 は HTLV-1 誘導性細胞形質転換に関与している。HTLV-1 の tax タンパク質は、ウイルスの複製を制御し、T リンパ球の形質転換を誘導する。 153,185 Hsp90 阻害剤 17-DMAG を経口投与すると、ATL マウスの多臓器への攻撃的な浸潤が有意に抑制される。
ウイルス感染におけるHSP70ファミリーの機能
RNAウイルス感染におけるHSP70ファミリーの機能
ウイルスの侵入
異なるファミリー(例えば、Picornaviridae、Flaviviridae、およびReoviridae)のウイルスは、宿主細胞への侵入のためにHSP70ファミリータンパク質を利用する。ピコルナビル科の場合、例えば、コックスサッキーウイルスA9(CAV-9)は、その侵入のためにHsp70ホモログGrp78を利用する186。187 細胞をGrp78とインテグリンαvβ3抗体で同時に処理すると、ウイルス結合は完全にブロックされる。
そのため、Grp78はCAV-9の共受容体として機能する。さらに、Grp78は、CAV-9の感染後、宿主細胞膜上の主要組織適合性複合体(MHC)I分子と相互作用することができる。186 EV-A71感染の過程で、Hsp70は劇的にアップレギュレートされ、細胞表面でEV-A71と相互作用する。エンテロウイルスの他に、フラビウイルス科の多くのウイルスもまた、宿主細胞への侵入にHsp70を必要とする。
110,189 Hsp70はDNEVエンベロープタンパク質(Eタンパク質)と相互作用し、ウイルスの付着に重要な役割を果たしている。190 同様に、Hsp70とHsp90に対する抗体はDENV感染を有意に阻害する。191 JEV 感染した Huh7 細胞では、Hsp70 は脂質ラフトに濃縮され、E タンパク質とコロケーションしている。
これらの結果から、Hsp70 は JEV の受容体として機能し、脂質ラフトは JEV の侵入を促進する組織化センターとして機能していることが示唆されている。JEV 侵入の後期には、JEV 感染時に C6/36 細胞で Hsc70(アイソフォーム D)がアップレギュレートされる。しかし、Hsc70 はウイルスの細胞膜への付着には必要ではなく、ウイルスの宿主細胞への侵入に必要とされているようである。
最近では、Grp78 もウイルスの細胞膜への付着と侵入の両方に必要であることが報告されている。Grp78 のノックダウンは JEV の内部化も阻害する。興味深いことに、Grp78 は JEV 感染後に宿主細胞外に分泌され、分泌された Grp78 は JEV と協力してウイルス感染を促進する。最近の研究では、Grp78がSARS-COV、MERS-CoV、およびSARS-CoV-2ウイルスの受容体であることが示されている。198 Hsc70特異的モノクローナル抗体は、ウイルスの付着に影響を与えることなく、ウイルスの内部化と感染を阻害する。199 さらに、ウイルス粒子全体とVP5のC末端領域の短いドメイン(またはペプチド)がHsc70に結合するのに十分であることが示されている。
ウイルスの複製
HSP70ファミリータンパク質は、様々なメカニズムでウイルス複製に関与している。第一に、HSP70ファミリータンパク質は、ウイルス複製複合体の形成を促進し、または複合体タンパク質の安定性を維持する。いくつかのケースでは、HSP70ファミリータンパク質は、ウイルスポリメラーゼと直接相互作用して、ウイルス複製を増強する。例えば、ムンプスウイルス(MuV)感染時には、Hsp72の発現レベルが上昇する。Hsp72のC末端領域は、RdRp複合体の必須構成要素であるPタンパク質のN末端領域と相互作用する。Hsp72 のノックダウンにより、ユビキチン化された P タンパク質が蓄積され、細胞のアポトーシスが増加することが報告されている。Hsp70はHsp90と協力してLタンパク質レベルを調節している。Hsp90 阻害剤である 17-AAG は、Hsp70-interacting protein (CHIP) の C 末端を介したプロテアソーム経路での分解を促進し、L タンパク質レベルを低下させる。Hsp70阻害剤VER155008は17-AAGとともにLタンパク質の分解を促進する。202 犬ジステンパーウイルス(CDV)感染の場合、Hsp70 の増加は、ポリメラーゼ活性を示す軽核カプシド(NC-L)バリアントの発現上昇をもたらする。さらに、Hsp70は、Hsp70 ATP活性に依存してNCポリメラーゼ活性を調節し、Hsp70抗体がNCポリメラーゼ活性を有意に阻害し、精製組換えHsp70を補充することで、基底およびストレス誘発性のNCポリメラーゼ活性が増強されることが示されている205。
Hsp70sの他のメンバーもまた、VRCの形成を調節する。Hsp72は、NS5A、NS3、NS5B(RdRp)を含むフラビウイルスのいくつかの複製タンパク質と物理的に相互作用する。206 Hsp72のダウンレギュレーションはHCV感染細胞におけるVRCの数の減少をもたらし、一方、Hsp72の過剰発現はVRCの数を増加させる。Hsc70 は HCV RNA ゲノムの 3′ polyU/UC モチーフに結合することで VRC と関連している。209 N タンパク質はウイルス RNA との相互作用に関与し、P タンパク質は N タンパク質および RdRp L と相互作用してヌクレオカプシドを形成する。Hsp70 阻害剤は RSV ポリメラーゼ活性を抑制するが、Hsp70 はウイルス遺伝子の発現を阻害するだけで、RNA の重合には影響を及ぼさない。
エボラウイルス(EBOV)の複製に関しては、Hsp70の関与メカニズムはより複雑である。免疫沈降法と質量分析法を用いて、Nタンパク質はHsp70、NEF、BAG2、およびHsp70のコ・シャペロンであるDNAJA2と相互作用することが明らかになっている。この複合体はさらにウイルスタンパク質VP30、VP35、RdRp Lと相互作用し、最終的にVRCを形成する。212,213 さらに、Hsp70 は昆虫細胞内で L ポリメラーゼと共精製されている。また、Hscs70 は EBOV ゲノムの末端非コード領域と相互作用し、結合部位を変異させることで相互作用を阻害することで EBOV のミニゲノム複製を強力に阻害する。
Hsp70がウイルスの複製をサポートするために用いるもう一つのメカニズムは、ポリメラーゼまたは核内カプシドの核内輸入を調節することである。いくつかのRNAウイルスもまた、CDVやインフルエンザウイルスのように核内で複製する。そのため、核内輸送はウイルスの複製に重要なステップとなる。CDV感染時には、Hsp70は細胞質から核内への核カプシド粒子の移動を促進することで、ウイルスの複製に強く寄与することが示されている216。同様に、インフルエンザ感染時には、Hsp70はHeLa細胞およびHEK293T細胞において、PB2またはPB1モノマーおよびPB2/PB1ヘテロダイマーと相互作用し、PB2モノマーまたはPB2/PB1ヘテロダイマーとともに核内に順次トランスロケーションする。核内コンパートメントと細胞質コンパートメントの間でのHsp70のシャトリングは、インフルエンザウイルスの複製に対するHsp70の調節効果の根底にある。
ウイルス遺伝子発現
ウイルスの侵入および複製に加えて、Hsp70は、ウイルスのタンパク質翻訳にも寄与する。陽性一本鎖RNAウイルス(例えば、SARS-CoV-2、HCV、ZIKA、EV-A71など)は、内部リボソームエントリーサイト(IRES)を利用して自身のタンパク質の翻訳を開始するが、真核生物の翻訳開始/伸長因子(EIFs/EEF)の調節または切断を介して宿主細胞キャップ依存性の翻訳を阻害する。218 翻訳開始段階では、Hsp70 は IRES-acting factor lupus autoantigen タンパク質の発現をアップレギュレートし、キャップ依存性翻訳抑制因子である eIF4E binding protein 1 (EIF4EBP1) を活性化する。伸長段階では、Hsp70 は Akt-哺乳類ラパマイシン標的複合体 1 (mTORC1)シグナルカスケードを活性化し、キナーゼ p70S6K と Cdc2 介在性リン酸化と EEF2 キナーゼ (EF2K) の不活性化を介して EEF2 の活性化につながる218 。Hsc70 は EV-A71 の 2A プロテアーゼと相互作用して EIF4G の切断を促進し、宿主細胞のキャップ依存性の翻訳を損なうが、ウイルスの IRES 介在性の翻訳を促進する。ウイルスの中にはIRES配列を持たないものもあり、ウイルスの複製は多くのdsRNAを産生し、これがプロテインキナーゼ-RNA活性化(PKR)-EIF2aシグナル伝達カスケードの活性化の引き金となり、細胞内でのグローバルな翻訳をシャットダウンし、ストレスを放出する220,221。222,223,224,225,226 感染していない細胞やストレスを受けていない細胞では、p58IPKの活性は複合体を形成してHdj1と閉塞している。これらの知見は、p58IPKの活性化は、単量体化したp58IPKがPKRを阻害することを可能にするp58IPK-Hdj1複合体からのHdj1のHsp70介在性の放出の後続であるように見えることを示唆している。
ウイルスの集合体
Hsp70は、いくつかのウイルスの組み立てを助けることが報告されている。227 シグマ1タンパク質のN末端セグメントはHsp70に依存しない方法で共翻訳的に折り畳んで三量化するが、C末端グロビュラードメインの翻訳後の折り畳みはHsp70に依存している。この過程で、Hsp70はN末端のαへリックスコイルドコイルの下流領域に共翻訳的に結合し、不要な相互作用やミスフォールディングを抑制するのに役立つと考えられている。シグマ1タンパク質のC末端ドメインの三量化は、リボソームからのHsp70のATP依存性の放出と結合している。Hsp70-P1複合体は、ピコルナウイルスの集合体中間体と考えられている。
興味深いことに、いくつかの研究者は、Hsp70がウイルスリボ核タンパク複合体(vRNP)の核輸出を阻害することによってインフルエンザウイルスの複製を阻害し、その後のウイルスの形態形成をvRNPからM1を解離させることによって阻害することを実証している。
ウイルスリリース
ウイルス放出に関するHSPの証拠は限られている。Hsp70とHsc70はどちらもNS5Aタンパク質と相互作用することができ、HCV感染においては異なる役割を果たしているが、Hsp70のサイレンシングはウイルスタンパク質の発現を低下させるが、ウイルスタンパク質レベルには影響を与えない232,233。さらに、Hsc70はウイルスキャプシドに埋め込まれており、Hsc70とHCVのコアおよびE2構造タンパク質との共局在化が脂質液滴中で発見されている。したがって、Hsp70とHsc70は、異なる段階でHCV感染放出を調節している可能性がある。
DNAウイルス感染におけるHSP70ファミリータンパク質の機能
ウイルスの侵入とゲノムの放出
ここでは、ウイルスが宿主細胞に付着して侵入するのではなく、ERからの細胞質へのウイルス侵入の話をした。ERからの細胞質への侵入は、SV40感染において重要なステップである。このステップにはHsc70が必須であることが報告されている。234 さらに、HSP70 スーパーファミリーメンバーである Hsp105 は、Hsc70-SGTA 複合体のサブユニットを形成し、SV40 の細胞質侵入を促進することが報告されている。Grp78 は DNAJB11 に依存した方法で SV40 のキャプシドタンパク質と相互作用し、SV40 の分解と細胞質への侵入を助けている。
Hsp70はまた、いくつかのDNAウイルスのゲノム放出に必要である。このようなプロセスは、アデノウイルス感染症に記載されている。ウイルスが細胞内小胞から細胞質に放出された後、Hsp70とHsc70は直ちにアデノウイルスの主要なコートタンパク質の一つであるヘキソンタンパク質に付着する。無傷のヌクレオカプシドは、典型的な NLS 依存性の核内導入機構を介して核内に輸送される240 。ヌクレオカプシドは、そのヘキソンタンパク質を介して、細孔複合体の構成要素と相互作用して核内細孔に固定される。240 ウイルスDNAはHsp70に依存した方法で核内に転送されるが、精製されたヘキソンはウイルスDNAの代わりにHsp70に依存しない方法で核内に入るため、ヘキソンは核外に残される。しかしながら、このような説明にはより強固な証拠が必要である。238 宿主の核内へのウイルスゲノム放出におけるHsc70の寄与の他の例としては、HSVとポリオマウイルスがある。HSV感染細胞では、細胞質から核へのHsc70の移動は、即時初期ウイルスタンパク質ICP0によって引き起こされる。Hsc70は、26SプロテアソームおよびウイルスUL6ポータルタンパク質の構成要素とコロケーションしており、これはキャプシドからのDNAの出入りのための導管を提供する。UL6 は核内で高度にユビキチン化されており、UL6 が Hsc70 の基質であるという直接的な証拠はなかったが、Hsc70 がユビキチン-プロテアソーム経路で UL6 を正しく折りたたんだり分解したりすることに関与している可能性が示唆されている241 。Hsc70 は、ポリオマウイルスに感染すると、キャプシド蛋白質の転座に伴い、細胞質から核へと転座すると考えられている。
遺伝子発現とタンパク質の成熟
ほとんどのウイルスは、細胞内の転写・翻訳機械を操作し、宿主タンパク質合成を遮断することで、これらの機械を利用して、ウイルスタンパク質の発現のために開始因子や伸長因子をリクルートすることができる。ウイルスによって利用されるいくつかの宿主因子は、Hsp70複合体の構成要素と密接に相互作用する。したがって、シャペロンシステムは、ウイルスの遺伝子発現にとって非常に重要である。いくつかの転写開始因子は、試験管内試験(in vitro)でHsp70コシャペロンBag1と物理的に相互作用することがよく知られている。Bag1はHsp70に依存した方法で全般的な転写活性を刺激する。242,243,244 グローバルな転写活性の刺激は、ヒトポリオマウイルス、ジョンカニンガムウイルス(JCV)またはHCMVのいずれかに感染した細胞で検出される。
ウイルスタンパク質の成熟を制御する Hsp70 システムの典型的な例は、HBV ラージエンベロープタンパク質(LHBsAg)に示されている(247,248,249)。最初は、LHBsAgのC末端は共翻訳的にERに存在し、N末端は細胞質に常駐し、後にERにトランスロケーションされて、Grp78の発現が刺激されていることがポスト翻訳で報告されている。正しいトポロジーを確保するために、Hsp70システムはLHBsAgのN末端のポストトランスロケーションを厳密に制御している。248,249 Hsc70 のコシャペロンである Hip と Bag1 もまた、Hsc70 の活性を拮抗的に制御している。過剰発現した Hip は Hsc70 活性を促進し、LHBsAg の N 末端の細胞質保持量を増加させる。翻訳後のプロセスでは、Grp78はLHBsAgおよびHsc70と結合し、HBV大表面抗原のERトランスロケーションを促進する。Grp78の機能は、正のレギュレーターであるER局在化DnaJドメイン含有タンパク質4(ERdj4)と負のレギュレーターであるBiP関連タンパク質(BAP)の両方によって制御されている(253 BAPの増加はLHBsAg/BiP複合体を不安定化させる)。
ウイルスの組み立て
多くのDNAウイルスは、通常、感染した細胞の核で集合する。ポリオマウイルスを例にとると、すべてのキャプシドタンパクは細胞質で合成されるのに対し、その後のウイルスの集合は核内でしか行われない。ポリオマウイルス感染時には、Hsp70とHsc70の構成型は、3つのウイルスキャプシドタンパク質(VP1、VP2、VP3)と共沈する。Hsp70 は感染後期に細胞質から核へと移動し、ウイルスキャプシドタンパク質の局在変化と一致している。原核生物の Hsp70 シャペロンである DnaK もまた、組み換え VP1 と C 末端ドメインで相互作用しており、ここでは組み立てられたキャプシドのペンタマーが連結されている。しかし、DnaK、DnaJ、およびGRpEを含むHsp70シャペロン系とVP1を一緒に組み合わせることは、カルシウムなしでATP単独の存在下でVP1を均一なカプシドに組み立てるのに十分である255。
レトロウイルス感染におけるHSP70ファミリーの機能
ヒトTリンパ原性ウイルス1型(HTLV-1)は、Hsp70タンパク質と相互作用するレトロウイルスのよく研究された例である。HTLV-1感染の間、シンシンチウム形成は、細胞間ウイルス移動の重要な因子である。256,257 Hsc70特異的モノクローナル抗体がシンキュウム形成とHTLV-1感染性を排除するため、細胞膜常駐型のHsc70はこのプロセスに必要である。興味深いことに、Hsc70 はシンチチュウム形成を促進するが、ウイルスの侵入には影響を与えません。
Hsp70もまた、ウイルス核内侵入後の段階で重要な役割を果たしている。ヒト免疫不全ウイルス1型(HIV-1)感染時には、ウイルス蛋白質R(Vpr)は、ウイルス前統合複合体とカリオフェリンαとの相互作用を刺激して、ウイルスの核内侵入を促進する。興味深いことに、Hsp70とVprは同じ基質を共有しているため、Hsp70の抗ウイルス的役割を主張する研究者もいる。Hsp70が競合してVprの機能を阻害すると思われる。HIV-1 は細胞周期やアポトーシスを操作するために Vpr を必要としているため、HSP70 は HIV-1 感染における Vpr の機能を中和すると考えられている。Hsp70 ATPase活性を阻害するとHsp70-ビリオンコアの結合が阻害され、ウイルスの感染性が低下するため、Hsp70-ビリオンに組み込まれたHsp70 ATPase活性とHsp70-ビリオンの正しい構造はHIV感染に不可欠である。
DNAウイルス感染におけるHSP70sの宿主細胞への影響
細胞変換
ゲノム内にポリメラーゼをコードしないDNAウイルスは、宿主のDNA複製機構に依存している。休止細胞で複製するためには、ウイルスは細胞周期を再活性化して宿主細胞を変化させなければならない。ウイルスが細胞周期の制限点を克服できるようにするために、いくつかのメカニズムが進化していた。最もよく研究されている例は、SV40である。大T抗原と小T抗原(TAg)は、SV40の形質転換能力の中心となるものである。両タイプのTAgのN末端には、Hsp70コシャペロンのシグネチャーモチーフであるJドメインが含まれている。
J-ドメインの変異または欠失は、TAg と Hsp70s との機能的な結合を阻害する。また、TAg は網膜芽細胞腫ファミリータンパク質(pRb、p107、p130)を隔離し、Hsc70-ATP 加水分解依存性の方法で転写因子の E2F ファミリーのメンバーを解放する。まず、大きなTAgがpRB-E2F複合体と結合する。第一に、大きな TAg が pRB-E2F-Ag 複合体と結合し、ATP の存在下で Hsc70 と結合し、Hsc70 の ATP 結合型は TAg-Hsc70 複合体形成を促進するために高い基質結合率を示す。
第三のステップは、TAg-Hsc70複合体のTAgのJドメインがATP加水分解を刺激することである。このプロセスは、Hsc70の活性部位に依存しており、T-抗原結合部位では発生しない。一方、Hsc70は、基質タンパク質pRBまたはE2Fのトラップを可能にする高親和性コンフォメーションに移行する。最後のステップでは、Hsc70は複合体中の基質タンパク質のコンフォメーションを誘導し、これによりpRBとE2Fの解離が促進される。
ADP解離とATPのHsc70への再結合の後、E2FとpRB-Ag複合体はHsc70-基質複合体から放出される。271,272,273,274 TAg は Bag1 と同様に、Hsc70 の存在下でそれぞれのプロモーター上で転写開始複合体のアセンブリーを補助するように機能する。また、TAgはHsc70を誘導して抑制性サイレンサー複合体を分解したり、クロマチンのリモデリングを助けたりすることも可能である(図5)。
図5
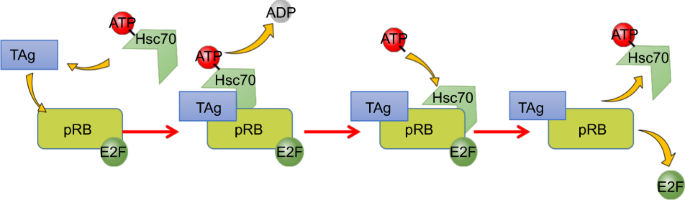
図5 TAgが誘導するpRBとE2Fの分解にHsc70が関与するモデル。
まず、TAgはpRB-E2F複合体と結合し、T-抗原-Hsc70複合体の形成を促進する。その後、TAg-Hsc70複合体はATPの加水分解を刺激し、Hsc70は高親和性コンフォメーションに移行し、基質タンパク質であるpRBやE2Fのトラップを可能にする。最後に、Hsc70は基質タンパク質のコンフォメーションを誘導し、細胞周期の再プログラムを開始するためにpRBとE2Fの解離を助ける複合体を形成する。
HPV とアデノウイルスは、pRB-E2F 複合体を破壊することで、同様の形質転換活性を持つ。E7(HPV)とE1A(アデノウイルス)のどちらのタンパク質もJドメインを含まないが、どちらのタンパク質もSV40 TAgで説明されているのと同様の方法で細胞を形質転換する可能性がある。275 hTid-1との相互作用を媒介するE7のC末端は、pRBとの直接的な相互作用には必要ではないが、pRB-E2F複合体の物理的な破壊には不可欠である。これらの観察から、hTid-1との相互作用がpRB-E2F複合体の崩壊に関与していることが示唆され、E7はSV40 TAgの機能に類似した形で複合体の解離のためにHsc70をリクルートするために必要なJドメインを提供している。あるいは、E7のhTid-1への結合は、hTid-1の想定される腫瘍抑制機能を阻害することによって細胞を形質転換し得る。E1AはHsc70と直接相互作用してpRB-E2F複合体を解離させる。
結論として、ほとんどの二本鎖DNAウイルスは、宿主細胞が細胞周期に再突入するように再プログラムするために、Hsp70シャペロンに依存している。
細胞の生存とアポトーシス
Hsp70系はストレス条件下での細胞生存に不可欠な調節因子であるため、Hsp70タンパク質の誘導は、成熟したウイルスが退出する準備が整うまで細胞を生存させることにより、ウイルス感染を促進する。279,280,281 ウイルスのライフサイクルの初期段階では、ウイルスの繁殖は細胞死に対して単純に脆弱である。当然のことながら、ウイルスは細胞のアポトーシスを操作するように進化している。EBV感染細胞では、核内オンコプロテインEBNA3Aは、アポトーシスを阻害してB細胞を不死化するために、Hsp70核内転座とHsp70シャペロン複合体の形成を助けている。291,292 一方、Hsp70 mRNAレベルの低下は、アデノウイルス感染の後期段階でのアポトーシスの適時誘導につながる可能性がある。
自然免疫
Hsp70は自然免疫や炎症反応に影響を与えることが以前に報告されている(293)が、肝細胞は自然免疫を欠いていると考えられてきた。Maらは、HBVの複製によってGrp78の発現が刺激されることを報告している。さらに、Hsp70はウイルスや細菌の感染に応答して細胞の自然免疫に大きく寄与していることが明らかになった。細菌感染の過程では、Hsp70の細胞内レベルの上昇は、LPSが媒介する炎症から細胞を保護する。TNF受容体関連因子6(TRAF6)との相互作用により、Hsp70はそのユビキチン化を阻害し、それによって転写因子NF-kBの活性化をブロックすることができる。Hsp70 は IKK と結合し、NF-κB/IkBα/IKK 複合体の機能と安定性を阻害し、さらに IkBα のリン酸化を阻害する。
さらに最近の研究では、Hsp70 が炎症を制御するための別のアプローチを提供している。299 細胞内Hsp70の抗炎症機能とは別に、分泌された細胞外Hsp70は、その結合要素によって認識される前に、樹状細胞やマクロファージに結合し、その大部分は自然免疫受容体である293,300。
Toll様受容体(TLR)は、ウイルスの侵入を検出し、細胞内の自然免疫応答を直ちに誘発する。Hsp70 は TLR2 と TLR4 の両方を利用して CD14 依存的に炎症性シグナルを誘導し、MyD88/IRAK/NF-κB 軸シグナル伝達カスケードを介して炎症性サイトカイン産生を促進している8,301。
リガンド結合後、TLR は二量体化し、アダプター分子である骨髄分化一次応答タンパク質 88 (MyD88)、IL-1R-associated kinases (IRAKs)、TGFβ-activated kinase (TAK1)、TAK1-binding protein 1 (TAB1)、TAB2 および TNF-receptor-associated factor 6 (TRAF6) を含む下流のシグナル伝達分子をリクルートするために必要な構造変化を受けます。 304,305,306,307 他の多くの自然免疫関連シグナル伝達経路は、その後、例えば、TAK1/IKK活性化を介したNF-κBのリン酸化など、活性化されるであろう。MAPK p38、JNK、および ERK 経路もまた活性化され、その後、CREB および AP-1 転写因子を活性化する。AP-1 と NF-κB の両方が TNF-α、IL-6、IL-1β、および多数の他のサイトカインやケモカインを含む炎症性サイトカインの発現を活性化する。
ウイルス感染におけるHSP60ファミリーの機能
RNAウイルス感染時のHSP60sの宿主細胞への影響
免疫の変調
RNA ウイルスのライフサイクルにおける Hsp60 の機能を示す研究はほとんどないが、宿主免疫の調節における Hsp60 の機能は広く研究されている。JEV 感染の場合、Hsp60 は NLRP3 イン フラマソーム活性と NFκB リン酸化を介して IL-1β 産生を促進することでウイルス誘発性炎症を促進する。ゲノムワイド RNA 干渉(RNAi)スクリーンは、インフルエンザウイルスの PB2 と Hsp60 の相互作用を同定している。このように、Hsp60はミトコンドリアの安定性とIFN-β産生レベルの両方に対するPB2の効果を決定している38,39。Hsp60をサイレンシングすると、マクロファージでのIFN-α産生が増加し、ウイルスの繁殖が減少する。
アポトーシス制御
ウイルスはアポトーシスを調節するために異なるメカニズムを使用する。HCV感染の場合、活性酸素産生は細胞のアポトーシスも促進するが、HCCの主な原因と考えられている。320 研究によると、HCVコアタンパク質のN末端ドメインはHsp60と相互作用することで活性酸素産生を誘導し、タンパク質ストレスを放出するHsp60の正常な機能を阻害することができることが示されている321,322 一方、別のRNAウイルスであるロタウイルスSA11は、Hsp60の安定性を調節することで初期のアポトーシスを遅らせることに最善を尽くそうとしている323。323 ウイルスがHsp60を介してアポトーシスを延期したとしても、Hsp60の主な機能はミトコンドリアでのタンパク質のリフォールディングである。
DNAウイルス感染におけるHSP60ファミリーの機能
ウイルスの複製
これまでのセクションでは、HBV感染におけるRT-ε RNA複合体形成にHsp90が重要であることを述べてきた。しかし、Hsp60がRT-ε RNA複合体形成の前にRT活性を直接調節することが示されている325,326。さらに詳細な研究によると、2つのRTフラグメントのうちの少なくとも1つ、末端タンパク質(TP)ドメインの残基1-1-99とRnase H(RH)の残基680-842がHsp60の結合に必要であることが示されている。
アポトーシス制御
HBV感染と同様に、HBV感染も強いアポトーシスを誘導するが、これは主にHBxタンパク質が寄与していると考えられている328。Hsp60はHBxの小さなドメイン(残基88-117)に結合している329 。さらに、Hsp60はミトコンドリア内でHBxやHsp70と複合体を形成している330 。
免疫調節
ここでは、HBVが宿主免疫応答を回避するためにHsp60をどのように利用するかに焦点を当てる。331 これらの戦略のうち、HBVの感染はCD4+CD25+ T調節細胞(Tregulation cells: Tregs)の集団を増加させ、これはIL-10とTGF-βを大量に産生することができる。334 HBV感染は血清sHsp60レベルを上昇させ、Hsp60を利用してTLR2/MyD88/IL-10シグナリングを介してCD4+CD25+調節性Tを活性化させる。
レトロウイルス感染におけるHSP60ファミリーの機能
DNA/RNA ウイルス感染過程における Hsp60 の役割は広く研究されているが、レトロウイルス感染における知見は限られている。Hsp60はHIV粒子にカプセル化されているが、その機能については不明である。Hsp60-Hsp10複合体はインテグラーゼを活性型に維持し、ATP依存的に活性を刺激する。
ウイルス感染におけるHSP40(Hsp70コシャペロン)の機能
RNAウイルス感染におけるHSP40ファミリーの機能
ウイルスの複製
Hsp40s は、ポリメラーゼ活性、複製複合体、核輸送を調節することにより、RNA ウイルスの複製を制御している。JEV感染細胞では、Hsp40/DNAJホモログHdj2がウイルスRNAゲノム複製に必須のRdRpであるNS5タンパク質と相互作用し、コロケーションしている。339 インフルエンザウイルスもまた、Hsp40 を利用して VRC が核内に再配置されるのを助けることで複製を促進している。インフルエンザウイルスの複製は宿主細胞の核内で起こるため、ウイルスリボ核タンパク質(vRNP)複合体の核内移動が必要である。このvRNPは、ウイルスRNA(vRNA)、ポリメラーゼヘテロ三量体(PA、PB1、PB2)、核タンパク質(NP)で構成されている340 。第一に、Hsp40/DnaJB1は感染の初期段階でNPと相互作用し、NPとインプリンピンαの間の効率的な関連付けを確実にしている。344 DnaJB1 とは異なり、DnaJA1 は主に C 末端の基質結合ドメインに依存してウイルス RNA 合成を管理している。
ウイルス遺伝子発現
ウイルスのRNA複製では、二本鎖RNA(dsRNA)分子が大量に生成される。宿主細胞はこれらの二本鎖RNAを検出し、インターフェロン誘導プロテインキナーゼ(PKR)を活性化して、真核生物開始因子eIF2αをリン酸化し、タンパク質合成を阻害することでウイルスの複製を制限している220,221,345。インフルエンザウイルスNS1はPKRのN末端に直接結合し、PKRの活性化を阻害している。Hsp40 が p58IPK の阻害剤であるのに対し、III 型 Hsp40/p58IPK は PKR の阻害剤 222,223,224,226,347 であることが報告されている225,348 。その後、p58IPKはPKR/eIF-2αの活性を阻害する。その結果、インフルエンザウイルスはタンパク質合成の阻害を解除する。しかし、感染の後期には、インフルエンザウイルスM2、Hsp40およびp58IPKは、PKRの活性化、ER-ストレス誘発細胞死およびウイルス放出につながるであろう安定な複合体を形成している。
タンパク質の成熟
フラビウイルスゲノムは大きなポリタンパク質をコードしており、後にいくつかの成熟構造体と非構造体タンパク質に切断される。成熟したタンパク質はVRCを形成する。フラビウイルスのVRC形成には、最初にHSP40ファミリータンパク質DNAJC14が参加している。黄熱病ウイルス(YFV)感染時には、DNAJC14はNS3とNS5との非構造タンパク質クラスタリング部位にリクルートされ、VRCを形成する。DNAJC14の過剰発現はNS3/4AとNS4A/2Kの切断部位を変化させ、NS3とNS3-4Aの比率に異常をきたすことから、DNAJC14のシャペロン活性がNS3/4A/2Kの切断を調節し、NS3とNS4Aの適切な発現レベルを確保していることが示唆された。DNAJC14を異所性に発現させた場合のVRC形成の阻害は、シャペロンの調節障害によるものである。
免疫の変調
Hsp40は、ウイルスの宿主免疫の回避を助ける働きをすることもあれば、特定の条件下で宿主免疫応答を増加させることで抗ウイルス活性を示すこともある。Hsp40 は Hsp70 と協働して MDA5 と相互作用し、MDA5 多量体形成を阻害しながら MDA5/MAVS 経路を抑制することが報告されている。HFDV 感染時には、VP1 は IRF3 のリン酸化、二量体化、核内転座の抑制を介して I 型インターフェロンシグナルを抑制することができるが、DNAJA3 は VP1 タンパク質のリソソーム分解を誘導する。したがって、DNAJA3は間接的に宿主細胞の免疫応答を刺激することになる。
DNAウイルス感染におけるHSP40ファミリーの機能
ウイルスの複製
証拠は、Hsp40sがDNAウイルス複製の開始を制御していることを示唆している。HPVの場合、複製はオリジン(Ori)配列上のタンパク質E2の認識と複製開始因子E1のリクルートから始まり、ATPaseとヘリカーゼ活性を発揮する。355,356,357 Hsp40 (Hdj1,Hdj2) と Hsp70 は独立して、相加的に E1 のオリへの結合を促進するが、Hsp40 は E1 と直接結合し、E1-オリ複合体に留まる。同様に、Hsp40(hTid1)もまた、独立したシャペロン活性を有するHdj1およびHdj2と同様の機能を有する。Hsp40はHSV-1の複製開始タンパク質およびヘリカーゼタンパク質UL9と相互作用し、それによってそれらの複製起源への結合を促進する。
HBVの複製を促進する以外に、Hsp40の負の役割の可能性も報告されている。HBxは多機能性ウイルス性因子であり、ウイルスの複製やヒトの肝がん化に関与している。361,362 複製競合性のあるHBV構築物をトランスフェクトした肝細胞における各Hsp40の個別発現は、ウイルスの複製とキャプシド形成の両方に抑制効果を示している。さらに研究を進めると、HBVのコアタンパク質とHBxタンパク質は、Hdj1またはhTid1との共発現によって不安定化することが明らかになった;その理由は、それらが強化されたタンパク質分解の標的となっているからである。HBVの複製を阻害する以外に、Hdj1はHBxのプロテアソーム媒介分解を促進する。
ウイルス誘導細胞変換
上述したように、ウイルスの中には細胞形質転換や腫瘍形成を誘導するものがある。ここでは、ウイルス誘導性細胞形質転換におけるHsp40タンパク質の役割について議論する。Hsp40は、HBV誘導細胞形質転換に負の役割を有する可能性がある。HBxが肝細胞形質転換の誘導の主要な因子であることが実証されている.363,364,365,366 それにもかかわらず、Hsp40s(Hdj1およびhTid1)の過剰発現は、HBxのプロテアソーム媒介分解を有意に増強することが示されている。別の研究では、E7の結合配列がSV40のTAgオンコプロテインと高い類似性を共有していることから、hTid1がE7オンコプロテインと相互作用し、HPV16;275による細胞形質転換を促進することが示唆されている。
レトロウイルス感染におけるHSP40ファミリーの機能
ウイルス前統合複合体の核内侵入
このためには、核膜を破壊することなく、ウイルス前統合複合体(PIC)を核内に積極的に輸送することが必要である。核内インポートの制御に関与するPICの構成要素には、HIV-1(またはHIV-2のVpx)の中央DNAフラップとウイルスタンパク質IN、MA、Vprが含まれている。同様に、DnaJB6はHIV-1感染時にもVpr核局在化を促進するが、374,375、特にDnaJB6のロングアイソフォームはこの過程において極めて重要である。CstF64 が高レベルであれば DNAJB6-S イソフォームの産生が促進され、低レベルであれば DNAJB6-L イソフォームの産生が促進される。
ウイルス遺伝子の発現制御
レトロウイルス遺伝子発現調節におけるHsp40の関与の注目すべき例として、HIV-1感染時の遺伝子発現増強における重要性が挙げられる。Hsp40とNefは核内に共移入し、そこでCDK9と結合した転写複合体の一部となり、長末端リピート(LTR)を媒介とする遺伝子発現を増強する。Hsp40とは異なり、Hsp70はウイルスの複製と遺伝子発現を抑制するが、Hsp40はHsp70の抑制されたウイルス遺伝子発現を救済する。Hsp70 は CDK9 リン酸化を阻害するが、これは HIV-1 のトランザクティベーターである転写陽性転写伸長因子 b 複合体が応答 RNA をトランザクティベートするために高親和性で結合するために必要不可欠な事象である。Hsp40A1、B1、B6、C5(C3は除く)はHIV-1の産生を制限することができるが、アデノウイルス、HSV-1、ワクシニアウイルス感染時のウイルス遺伝子発現には影響を与えない。保存されたDNAJドメインは、HIV-1の産生を抑制する役割を担っていることが示唆されている。Hsp70/Hsp40複合体は、Rev翻訳を特異的に認識して阻害するか、またはその分解を促進し、ウイルス遺伝子発現の阻害につながる。
ウイルス感染におけるsHSPの機能
スモールヒートショックプロテインは、例えば、細胞が高い活性酸素レベル、異常に高い温度、または病原体の侵入にさらされた場合など、ストレス条件下で宿主細胞で同定された最もアップレギュレーションされた蛋白質である。Hsp27 は、骨格筋、平滑筋、心筋で最も高いレベルで発現している最もユビキタスに発現している sHSP の一つである59,385 他のすべての sHSP と同様に、Hsp27 は高度に保存されたα-クリスタリンドメイン(いわゆる C 末端ドメイン)を共有している。Hsp27は6-8個のβ鎖を含み、分子間相互作用部位として2つのβシートを形成している41,59。
Hsp27はウイルス感染において特に重要であることが示されている。Rajaiya らは、Hsp27 と p38 や NFκB/p65 との結合がウイルス感染細胞における炎症促進メディエーターの発現制御に重要な役割を果たしていることを示唆している。
RNA ウイルス感染における Hsp27 の機能
プロテオミクス解析により、Hsp27はEV-A71感染時にアップレギュレートされる。Hsp27 をノックアウトすると、ウイルスの複製とタンパク質発現レベルが抑制され、Hsp27 が回復すると回復することがわかった。また、Hsp27は2Aproを介したEIF4Gの切断を促進することで、ウイルスIRES活性を高めている。383 Hsp27 はコロナウイルス感染の初期段階(感染後 4 時間)で急速に発現が亢進しており、ウイルスの初期複製に重要な役割を果たしており、SARS-CoV-2 感染症の治療薬としても有効である可能性が示唆されている。豚熱ウイルス(脳脊髄液V)はフラビウイルス科に属するウイルスである。Hsp27は、ウイルスの複製およびアセンブリに応答して非構造タンパク質であるNS5Aと結合することがわかっている。Hsp27 が欠乏するとウイルスの複製が促進され、PK-15 細胞では NF-κB シグナルを活性化することで Hsp27 の異所性発現が抑制される。しかし、Hsp27 の異所性発現によりウイルス力価は低下する。Hsp27 は NF-κB シグナルを活性化し、IFNβや下流のインターフェロン刺激遺伝子(ISG)の転写を促進する可能性がある。
図6
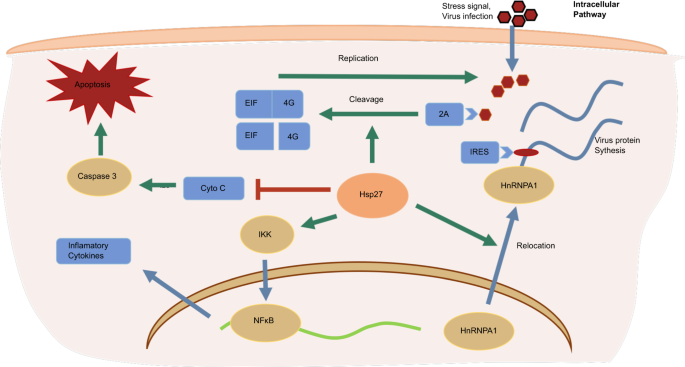
図6
ウイルス感染時のHsp27の機能については議論の余地がある。Hsp27は2Aタンパク質を介したeIF4Gの切断を促進し、IRESの機能とウイルスゲノムの複製を促進する。Hsp27は、宿主細胞核から細胞質へのhnRNPA1の転座、およびその結果として生じるウイルスタンパク質の翻訳と相関している。また、Hsp27はカスパーゼ3の阻害を介してアポトーシスを抑制することができる。一方、Hsp27 は IKK 複合体を活性化し、NF-κB や AP-1 を調節して炎症性サイトカインの発現を誘導することが期待されている。
DNAウイルス感染におけるHsp27の機能
プロ炎症性サイトカインは、初期の抗ウイルス感染において重要な役割を果たしている。390 アデノウイルスの内部化は ERK1/2 の活性化につながり、これにより NF-κB と AP-1 がトランス活性化され、異なる実験系でプロ炎症性サイトカイン IL-8 発現を誘導する可能性がある。IL-8 の増加は NF-κB インヒビターによって抑制され、Hsp27 が低下した細胞における IκB-α 分解およびリン酸化された IκB-α 蓄積の亢進と相関している。さらに、Hsp27は、IκBキナーゼ(IKK)複合体と関連していることが示されている。プロスタノイド PGE2 と IL-8 の合成は NF-κB によって制御されていることから、炎症性サイトカイン産生を調節する Hsp27 の機序と考えられる。
Hsp27 は、グルタチオン還元酵素やグルコース-6-リン酸デヒドロゲナーゼなどの還元型グルタチオンを保持することで酵素活性を制御していることが一般的に知られている。395,396 しかしながら、ヘルペスウイルスの複製に利益をもたらす酸化タンパク質の蓄積に Hsp27 が関与していることを示す証拠も議論の的となっている。実験モデルは、2つの遠縁のヘルペスウイルスであるアカゲザルラディノウイルス(RRV)とHSV-1を用いて設定されている。これらはカポジ肉腫関連ヘルペスウイルス(KSHV)の近親者である。酸化されたタンパク質は、これらのウイルス感染の間に蓄積される。その結果、プロテアソーム依存的な方法で酸化タンパク質の一部だけが除去され、他の一部は分解に抵抗することがわかった。さらに、酸化されたタンパク質の蓄積は、Hsp27 が欠損した細胞でより顕著である。Hsp27の役割は、ウイルス感染時には相互に排他的なものではない可能性が高い。
Hsp27はウイルスの複製にも寄与している。ポルシン・サーコウイルス2型(PCV2)は、豚の離乳後多系統性消耗症候群(PMWS)を引き起こす一本鎖DNAウイルスである。PCV2に感染したPK-15細胞では、リン酸化されたHsp27が核内でアップレギュレートされている。SB203580のようなHsp27阻害剤はPCV2の複製を抑制する。同じ結果は、Hsp27をノックダウンした場合にも現れる。399 さらに、Hsp27のリン酸化は、リン酸化された(活性化された)Aktレベルと同様に、EBV陽性細胞でもアップレギュレーションされている。EBV陽性細胞をPI3K阻害剤で処理すると、リン酸化されたHsp27のレベルは低下し、EBV感染時にPI3K/Aktシグナル伝達経路を介してHsp27のリン酸化がアップレギュレーションされることが示唆されている。Tongらは、Hsp27が肝細胞におけるIFN産生を促進することにより、HBV複製に対する抗ウイルスタンパク質として働くことを報告している。Hsp27の発現レベルは、HBV感染ヒト肝組織およびHBV産生HepG2.2.15細胞の両方で増加している。
ウイルス感染におけるPDIの機能
ウイルスの侵入とコーティング解除におけるPDIの機能
いくつかのウイルスの真核細胞への侵入は、酸化還元反応によって制御されている。その一例がニューカッスル病ウイルス(NDV)であり、パラミクソウイルス科の鳥類ウイルスである。PDIとERdJ5(余分なJドメインを持つPDIファミリー還元酵素)の過剰発現は、ウイルス膜融合の増加をもたらし、ウイルスがPDIファミリーを利用して宿主細胞に侵入する経路を示している。また、チオールブロッカーやPDI阻害剤はMA104細胞におけるロタウイルスの侵入を減少させ、感染性にチオールが関与していることを示唆している。
HIVの侵入はPDIによって規制されている。HIVのエンベロープは侵入のためにPDIによって解離される。Ryserらは、HIV-1エンベロープのgp120表面成分の2つのジスルフィド結合の開裂が、CD4+細胞へのウイルスの侵入に必要であることを最初に報告した。407 現在、エンベロープが宿主細胞にどのように結合するかについては、多くの研究が行われている。現在、エンベロープがどのようにして宿主細胞に結合するのかについては、多くの研究が行われているが、様々なグループの研究結果では、gp120が膜表面に沿って横方向に移動し、典型的な脂質ラフトとは異なる膜のドメインにあるPDIのパッチと衝突することが示されている。PDIはgp120の2つのジスルフィド結合を減少させ、おそらくCD4へのgp120の結合を安定化させ、その後のケモカイン受容体への結合のためにV3ループを露出させるコンフォメーション変化を生じさせる。これに続いて、gp41はその発熱性中間体への再配列を受け、侵入が起こる。
他の例としては、非開 発型ポリオマウイルス(Py)がある。Py粒子は、コートタンパク質VP1の層を含んでいる。この単一のタンパク質は、72個のペンタマーとして配置され、ウイルスゲノムを取り囲む殻を形成している。例えば、SV40への侵入は、洞質/脂質ラフトを介したエンドサイトーシス、小胞輸送、細胞質への移動を伴う。ERp57はVP1をつなぐ鎖間ジスルフィドを異性化してウイルスのコーティングを解除する。PDIとERp72はPyを減少させ、ERp57は主に試験管内試験(in vitro)でウイルスを異性化させる。突然変異誘発研究により、VP1の残基C11とC15が感染に重要であることが確認され、異性化中のこれらの残基の役割が示唆された。
PDIはウイルスタンパク質の翻訳を制御する
PDIがウイルスタンパク質翻訳に関与していることを示す証拠は少ない。いくつかの陽性一本鎖RNA(+ssRNA)ウイルスは、ウイルスタンパク質合成のためにIRES媒介翻訳に依存している。EV-A71の感染は、ハーブGarcinia oblongifoliaから単離された活性化合物Oblongifolin M(OM)によって強力に阻害される。また、ERp57の異所性発現がIRES活性を増加させ、OM処理によるウイルス複製の減少を部分的に救済することが示されている415。ERp57がIRES活性をダウンレギュレートする詳細なメカニズムについては、さらに研究が進められる。
PDIは酸化ストレスとERストレスに影響を与えてウイルス活性を調節する
いくつかの研究では、ウイルス感染の確立とウイルス誘発性疾患の進行に酸化還元バランスの乱れが関与していることが実証されている。416,417 ウイルスによる酸化ストレスは、HIV,418 インフルエンザウイルス,419 HBV,420 HCV,421 脳筋炎ウイルス(EMCV),422 呼吸器同期ウイルス(RSV),423 JEV424 感染時に報告されている。
ERp57は、そのチオレドキシン様部位を考慮すると、細胞内のどの位置にあっても酸化ストレスに対する細胞保護機構の主役であると考えられてきた。HPV陽性組織のレドックスプロテオミクス解析では、ERp57とGSTの発現レベルが組織のレドックス状態と正の相関を示しており、ウイルス誘発性の酸化的DNAおよびタンパク質損傷との関連性が示唆されている。
ERストレスは、ウイルスの活動によって引き起こされるもう一つの結果である。ウイルス感染は、ER膜を悪用し、誤ったタンパク質の蓄積、カルシウム濃度の不均衡などを引き起こする。インフルエンザAウイルス(IAV)、HBV、JEV、DENV、ZIKAウイルスは、すべて宿主細胞のプロセスをハイジャックして、ウイルスのフォールディングとトラフィッキングの促進、受容体の相互作用への影響、宿主免疫応答の調節など、ウイルスの発病を促進している(426,427,428)。
図7
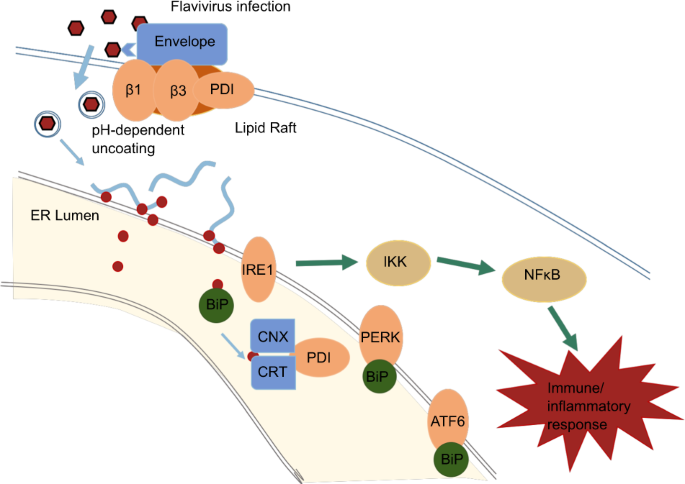
図7
フラビウイルス感染時のPDI機能とERストレス応答の一例。フラビウイルスの内皮細胞への侵入はPDIのサイレンシングによって抑制される可能性がある。研究はまた、PDIがフラビウイルスエンベロープタンパク質とともに細胞表面脂質ラフトと共局在化し、細胞表面インテグリン(b1およびb3)の活性化につながり、これがウイルスの宿主細胞への侵入を助けることに直接的な意味を持つことを示している。ウイルスRNAはその後、放出され、翻訳のためにERの周りに配置される。タンパク質合成の増加は、ERの恒常性を乱す可能性があり、PERK、ATF6およびIRE1を活性化するためのPERK、ATF6およびATF6とGRp78/BiPの解離を介してアンフォールドされたタンパク質応答を導く。下流の応答は、NF-κBおよび他のシグナル経路の活性化を含み、免疫応答および炎症応答に続く
ウイルス感染におけるRNAシャペロンの機能
RNAウイルス感染におけるRNAシャペロンの機能
ほとんどのRNAウイルスのライフサイクルは、宿主細胞の細胞質で完了する。ライフサイクルを完了するために、ウイルスはしばしば多くの宿主細胞タンパク質の再分配を誘導することができる;特に、RNA代謝およびウイルスRNAの機能的調節に関与するタンパク質。
ウイルスは、hnRNPを再分配するために様々な手段を採用する。例えば、EBOVはhnRNP C1/C2の核内インポートを阻害する。VP24は、C末端領域(アミノ酸424〜457)でNPI-1サブファミリーのカリオフェリン-α核内輸入タンパク質に結合し、チロシンリン酸化STAT1(pSTAT1)およびhnRNP C1/C2との相互作用を阻害する。阻害の結果、pSTAT1およびhnRNP C1/C2.430の細胞質保持が得られる。 あるいは、ウイルスは、核輸出を促進することによってhnRNPを再分配することができる。Rae1はmRNA輸出因子である。VSV 感染は Rae1 依存的な方法で hnRNP A1 の細胞質再分配を誘導し、再分配された hnRNPA1 は VSV 誘導性アポトーシスに関与している。HnRNP A2 は JEV の負鎖 RNA に結合し、ウイルス複製を促進する。
ウイルス複製
hnRNPがウイルスの複製に大きく貢献していることはよく知られている。例えば、hnRNP I/PTBおよびhnRNP Cは、コロナウイルス436 HCV437およびポリオウイルスなどの様々なウイルスの複製に関与している。
ウイルスRNA合成におけるhnRNP I/PTBの役割は、異なるウイルス間で異なるようである。PTBは、IRESおよび3′-UTRにおけるUCUAAペンタヌクレオチドリピートに結合する。PTBはコロナウイルスRNA合成を調節する436。しかしながら、hnRNP I/PTBの機能はHCV RNA複製においてある程度複雑である。それはHCVゲノムRNAの3′末端と選択的に相互作用する。上流のSL2およびSL3ステムループ構造はhnRNP-I/PTB I結合に必須であるが、最も3′末端のステムループSL1は不要である。HnRNP I/PTB IおよびhnRNP Cは、HCV RNAゲノムの3′NTRのピリミジンに富んだ領域に特異的に結合する。おそらく、hnRNP I/PTB Iは、ウイルスのネガティブ鎖(-)RNAゲノム複製の開始、RNAゲノムの安定化、および/またはゲノムRNAのカプセル化を調節するためにリクルートされているのではないかと考えられる439,440。いくつかの研究では、PTBがHCV RNAゲノムの合成を阻害する効果があることが示されており441 、一方で相崎らは、PTBが効率的なHCV RNA複製に必要であることを報告している442 。HuRは、ウイルスRNAの3′UTR上のhnRNP I/PTB I結合と競合してLaの3′UTRへの結合を促進することができることが示されている;一方、Laタンパク質はHCVゲノム複製に不可欠である。
hnRNP CはhnRNP Iと多くの類似点を有し、両者ともHCV RNAゲノムの3′UTRに結合してウイルスRNAの複製を促進するが;437、より詳細にはhnRNP Cのみが負極性と正極性の両方でウイルスRNAの3′末端に結合していることが示されている446。さらに、hnRNP Cはまた、ポリオウイルスのネガティブストランドRNAの5′末端に結合し、ポジティブストランドRNAの合成を促進する;438;一方、miR-555はhnRNP Cの発現を減少させ、それによってポリオウイルスの複製を阻害する。
ウイルスRNAスプライシング
hnRNPの主な機能は、スプライシング、安定化、輸送を含むRNAのプロセスと代謝を調節することである。hnRNP KはIAVのRNAスプライシングを助けることが報告されている。IAVは、8つの負鎖RNAセグメントからなるゲノムを持つ主要なヒト病原体である。つのウイルスRNAセグメント、NS1およびMは、代替スプライシングを受け、NS1、NS2、M1およびM2タンパク質を含むいくつかのタンパク質を産生する。インフルエンザウイルスRNAセグメントのうちの2つは、スプライスされた産物を生成する。NSセグメントは非構造タンパク質NS1と核輸出タンパク質NEP/NS2をコードし、Mセグメントはマトリックスタンパク質(M1)とイオンチャネル(M2)をコードしている。NS1-BPは、M4 mRNAやNS mRNAセグメントのスプライシングに影響を与えることなく、ウイルスM1 mRNAセグメントを適切にスプライシングしてM2 mRNAを生成する。このプロセスにおいて、hnRNP Kは、NS1-BP(結合タンパク質)とM mRNAの相互作用を橋渡しするメディエーターとして機能する。NS1-BPもhnRNP Kも欠乏すると、M mRNAの適切なスプライシングが保証される。NS1-BPは5′ssの最も近位に結合し、U1 snRNP結合部位と部分的に重なり、hnRNP Kはさらに下流に結合し、U1 snRNPリクルートを促進する。hnRNP KおよびNS1-BP結合部位のいずれかまたは両方の変異は、MセグメントのミススプライシングおよびIAV複製の減衰をもたらする。
ウイルス翻訳
ウイルスRNAの翻訳は、しばしば宿主細胞タンパク質を利用する。ほとんどのhnRNPは、ウイルス翻訳をポジティブに調節する。陽性センス一本鎖RNAウイルスは、通常、細胞キャップ依存性翻訳のウイルスタンパク質翻訳独立性を駆動するIRES配列を含む。 hnRNPは、異なるウイルスのIRESと結合して翻訳を補助する。HCVウイルス感染中、hnRNP I、hnRNP L、hnRNP D、452,453,454 hnRNP A1およびhnRNP K455はすべて、5′UTR配列と結合することによりHCVウイルスタンパク質の翻訳に参加している。 hnRNP I(PTB3)は、ウイルスRNAと再帰的に結合することが報告されているポリピリミジントラクト結合タンパク質(PTB)の1つである。PTBはHCVのUTR領域441カリシウイルスRNA457およびコロナウイルスRNAにも結合している。ピコルナウイルスおよびHCVでは、PTBがIRESの翻訳可能な構造への折り畳みを助ける可能性が提案されている458 。RNAシャペロンが共有する定型的な一過性の相互作用とは異なり、IRESが適切に折り畳まれた後にPTBが排除されるかどうかは明らかではない459 。翻訳アッセイでは、PTB抗体は試験管内試験(in vitro)で効率的にIRES媒介翻訳をブロックする460.PTBモノマーの3つのRRMモチーフがFMDVのIRESに直接結合してIRES構造を安定化し、IRESへのeIF4Gのエントリーを増強する459。461,462,463 hnRNP Lは、一本鎖RNAをmiR-122でプレアニーリングする際に、一本鎖HCV RNAと結合する463 hnRNP Dは、AU-rich element RNA-binding protein 1(AUF1)とも呼ばれ、細胞質と核の間をシャトルする。hnRNP Dはまた、HCV 5′UTRのステムループIIと相互作用し、その過剰発現はHCV IRES依存性翻訳を増強することがわかっている。同様に、hnRNP KはHCVゲノムの5′-UTRに位置するSL1と相互作用し、そこでmiR-122結合部位と結合する。464,465 miR-122はHCV複製に必要である。それは5′-UTRの保存された配列に結合し、HCV RNAの安定性を増加させる466,467,468 HCVコアタンパク質のN末端付近の親水性領域である残基25-91がhnRNP Kのプロリンに富んだドメインに結合し、ヒトチミジンキナーゼ転写への影響をネガティブに調節することも報告されている469 しかし、ウイルス複製に対するその機能については十分に検討されていない。
hnRNP A1はHCV RNAの5′-UTRおよび3′-UTRの両方と結合するが、それらはNS5bおよびセプチン6と複合体を形成し、ウイルスタンパク質の翻訳を補助する。hnRNP A1のC末端およびセプチン6のN末端は、翻訳過程で必要とされる。hnRNP A1はhnRNP A/B、hnRNP A2/B1、およびhnRNP A2のような多くの相同性を有しているため、それらのすべてがIRES媒介翻訳の調節においてhnRNP A1の代わりとなる可能性がある470 。エンテロウイルスのIRES配列もまたhnRNPと相互作用する。アピゲニンは、食餌性フラボノイドであり、hnRNPsと相互作用し、そのRNA編集活性を阻害する。この実験では、hnRNP A1の再分配は影響を受けないことに留意されたい。最近の研究では、hnRNP A1の細胞質への移動は、MINK/p38 MAPK経路473やHsp27などのシグナル伝達やストレスタンパク質によって厳密に制御されていることが示されている。
ウイルスIRES配列と結合してウイルスタンパク質の翻訳を促進する以外は、hnRNPはウイルスタンパク質と直接結合してウイルス複製を促進することができる。CVB3感染時には、3CproはhnRNP Mと結合して切断し、ウイルスの複製を促進する。DNEV のコアタンパク質は hnRNP K と相互作用して転写活性化因子 C/EBPb に対する hnRNP K の抑制効果を放出することが示されている。hnRNP Dはエンテロウイルスの5′-UTRと3′-UTRの両方に結合し、RNAの崩壊に影響を与えることなく翻訳を阻害する。434 一方、ウイルスはプロテアーゼ3CDの切断を介してhnRNP Dの阻害効果を放出する戦略を適用している。hnRNP Dの細胞質への移動は、2Aproの発現およびウイルスRNAの複製に依存している。
ウイルスRNA輸出
hnRNP A2/B1はインフルエンザウイルスのNS1と相互作用し、ウイルスNS1 RNA/タンパク質レベルの低下とNS1 RNA核輸出を導く。
RNAポリアデニル化
ルース肉腫ウイルスのgag遺伝子には、シス作用型ネガティブ・レギュレーター・オブ・スプライシング(NRS)要素が含まれている。NRSの一般的な機能は、通常、セリン/アルギニンリッチ(SR)タンパク質hnRNP HとU1/U11 snRNPとの結合により発現し、その結果、5′スプライスサイトのデコイとして作用してスプライシングを阻害する。試験管内試験(in vitro)でのポリアデニル化解析により、3′LTRポリアデニル化に必要とされるNRS要素の新たな機能が明らかになった。この過程で、hnRNP HはNRS要素に結合してポリアデニル化を促進するが、U1およびU11 snRNPのNRSへの結合部位の変異はポリアデニル化に影響しないことが明らかになった。
DNAウイルスのウイルス活性を調節するRNAシャペロンの機能
ウイルスの複製
HCMVの一過性のリチン化DNA複製は、シス作用を持つオリサイト、ウイルスがコードする6つのコアタンパク質、提案されているDNA複製開始タンパク質UL84、IE2、IRS1、そしてUL112/113遺伝子座の遺伝子産物に依存している。ここでは、hnRNP KがHCMVのUL84タンパク質と十分に相互作用し、それによってウイルスの複製を促進することが報告されている。この相互作用は、別の2つのウイルスタンパク質であるUL44とIE2.488によってさらに増強されている。
遺伝子発現
hnRNP は、転写レベルと転写後レベルの両方で DNA ウイルス遺伝子の発現を正または負のいずれかのレベルで制御する。
転写レベルでは、hnRNP D0BおよびhnRNP A/Bは、HPV18後期プロモーターのシス作用型AAGTATGCAコアエレメントと結合して、ウイルスキャプシドタンパク質の構成要素である後期遺伝子L1およびL2を抑制することができる。hnRNP Kの異所性発現はHBV複製を増大させるが、hnRNP KのノックダウンはHBVウイルス負荷を有意に減少させる。490 さらに研究を進めると、APOBEC3BはhnRNP Kと超複合体を形成し、それが構造変化を介してENII結合活性を変化させ、その結果、S遺伝子プロモーターの活性を抑制することが明らかになった。CK2はhnRNP KとICP27をリン酸化することができる。リン酸化されたICP27は、その核細胞質への移動とhnRNP Kとの相互作用に関与している。
転写後のレベルでは、hnRNPs は DNA ウイルス感染時のポリアデニル化、スプライシング、翻訳を制御している。HPVゲノムは初期領域と後期領域に分けられ、それぞれ近位初期(pAE)と遠位後期(pAL)のポリアデニル化シグナルに従う。hnRNP Hは、初期ポリアデニル化シグナルpAEの下流174ヌクレオチドに位置する複数のGGGモチーフと相互作用することにより、後期遺伝子L2を早期にダウンレギュレーションすることが示されている。hnRNP Hの結合は、初期ポリアデニル化シグナルでのポリアデニル化を促進し、L2 mRNAの産生を阻害することがわかっている。ポリアデニル化を調節するhnRNPの別の例としては、EBV初期タンパク質のSMポリメラーゼ(pol)mRNAが切断され、非効率的にポリアデニル化されることが挙げられる。
hnRNP A1は、RNAスプライシングに影響を与えることでHPV後期遺伝子の発現をネガティブに制御している。502 hnRNP A1はスプライシングサイレンサーエレメントと直接結合して、AUGのすぐ上流に位置する3′スプライスサイトの後期1(L1)mRNAでの使用を抑制する。一方、スプライスサイトのすぐ下流には、さらに17ヌクレオチドが存在しており、これがhnRNP A1結合型スプライシングサイレンサーの効果を打ち消している。
翻訳レベルでは、hnRNP IとhnRNP Kは、L2 mRNAの3′末端にある特定のシス作用要素に結合することにより、HPV後期遺伝子L2の翻訳を阻害する;508 一方、L2翻訳の阻害は、複数のチロシン残基でのhnRNP Kのc-Src媒介のリン酸化により阻害される可能性がある509。
細胞変換
hnRNPはまた、ウイルス感染中にいくつかの他の機能を示す。AUF1は、Cプロモーター結合因子2(CBF2)の主要な構成要素として働き、EBNA2と相互作用し、EBNA2をEBVの潜伏Cプロモーター(Cp)にターゲティングすることを仲介し、それによってヒトのB細胞の不死化とウイルス潜伏を誘導する510。EBV陽性細胞では、EBER1は豊富に存在するため、宿主細胞内でAUF1と相互作用する標的と競合する可能性がある。
免疫調節
ウイルスのライフサイクルに直接影響を与える以外に、hnRNPは免疫応答を調節することによってウイルスの活動を調節することができる。HSV-1感染中、hnRNP A/BはウイルスDNAと複合体を形成し、その後ホモ二量化と脱メチル化が起こる。これらの事象は、複合体を細胞質に移動させ、I型インターフェロンシグナルを介して自然免疫を活性化させる。さらに、この複合体は、DNAウイルス感染時にN6-メチルアデノシン修飾およびcGAS-STING関連mRNAの転座を促進し、ウイルス排除のための免疫応答をさらに強化する。
レトロウイルスのウイルス活性調節におけるRNAシャペロンの機能
上述したように、hnRNP A1はRNA/タンパク質に結合し、真核生物ではmRNAのオルタナティブスプライシングだけでなく、細胞内核細胞質輸送にも関与している513。レトロウイルス感染時には、hnRNPはウイルスRNAの転写、514 スプライシング、515、516、517 転座516、翻訳に関与している。
転写
518 Nef、Eed、キナーゼLck、nPKCサブファミリー(PKCδ/θ)が複合体NAKC(Nef-associated kinase complex)を形成し、ウイルスの複製を促進することが報告されている519 。Nef、Eed、キナーゼの相互作用の橋渡しをしている。hnRNP K 核化複合体は ERK1/2 を活性化し、結果として HIV プロモーターを抑制し、転写開始のための最適量以下の Tat と転写因子(例えば NF-kB)を可能にする。
転写後
HIV-1は、代替スプライシングを利用して、さまざまなウイルスタンパク質をコードするメッセンジャーRNAを大量に生成する。1つのプレmRNAから代替スプライシングによって40以上のメッセージRNAが生成されることが知られている520,521,521 スプライシングパターンの変化は、HIV-1の感染性と病原性に劇的に影響する521,522
HIV mRNAのスプライシングは、主にプレmRNA上でのスプライスソームの組み立ての初期段階で、小核リボヌクレオタンパク粒子(snRNP)U1、U2、およびU5-U4/U6の段階的な結合によって制御されている。523 アルギニンとセリンを豊富に含むドメイン(RSドメイン)を含むRSスプライシング因子は、これらのステップを支援する。U2AF65とU2AF35の2つのサブユニットからなるスプライシング因子の1つであるU2AFは、それぞれポリピリミジングルートおよび3′スプライスサイトと相互作用する524。SR スプライシング因子は、スプライスサイトの早期認識、プレmRNAへの基本的なスプライシング因子のリクルート、および他のRSドメイン含有スプライシング因子とのブリッジングコンタクトの形成を含むスプライスソームアセンブリの実質的にすべてのステップに不可欠である。
529 プレ-mRNA は、スプリソソームを形成する前に hnRNPs で満たされている。構成的スプライシングにおける hnRNPs の直接的な役割は観察されていないが、hnRNPs の結合は非特異的な性質を示すようである。hnRNPは、プレmRNAの構造をモデル化し、スプライス部位の認識を開始するという重要な機能を担っていると広く信じられている。
代替スプライシングを受ける細胞やウイルスのプレmRNAのシス作用配列要素は、そのプロセスをポジティブまたはネガティブに制御している。これらは、トランススプライシング因子の機械を一緒に結合し、スプライシング複合体を形成する。トランススプライシング因子の大部分はhnRNPまたはSRファミリー蛋白質である。また、これらのタンパク質はオルタナティブRNAのスプライシングを正または負に制御している。SRタンパク質はスプライシングをポジティブに制御することが多いであるが、hnRNPの中には主にネガティブに制御するものもある。
HIV-1ゲノムmRNAの代替的不完全スプライシングは、シスエキソンスプライシングサイレンスエレメント(ESS)とトランスhnRNPによって高度に正確に制御され、HIV-1感染細胞内で40種類以上のユニークなウイルスmRNAを産生する。Tat、RevおよびVprタンパク質の産生は、これらがHIV-1の増殖に重要な役割を果たすため、高度に制御されている。ESS2は、エクソン4内の3′ ss A3の下流に位置し、tat mRNAスプライシングを特異的に抑制する、HIV-1ゲノム内で最初に同定されたESSである531。ESS3はエクソン7に位置し、3′ ss A7でスプライシングを抑制している。vifコーディング配列内のエクソン3の3′ ss A2の下流に位置する第3のESS(AUAGUUAGUCCUAGG、ESSV)は、3′ ss A2.535でスプライシングを抑制する。
Tatタンパク質の発現を調節するために、いくつかのhnRNPは、これらのESS、イントロンスプライシングサイレンサー(ISS)要素と直接相互作用するか、またはエンハンサースプライシング要素(ESE)活性を妨害することによってスプライシングプロセスに参加している。例えば、hnRNP A1とhnRNP KはESS2上で相乗的に結合し、A3スプライシングサイトの利用を阻害する。hnRNP A1のC末端Glyドメインは結合に重要である。サイトA3の強力な活性化因子であるSRタンパク質SC35およびSRp40は、hnRNP A1と同様の結合部位を有する。hnRNP HはESS2に結合し、3′スプライスサイトA3に隣接するエクソン配列に結合するためにU2AF35と競合する。この結合は、3′-スプライスサイトA3でのスプライシングの阻害をもたらす。538 hnRNP A1はまた、エクソンスプライシングサイレンサー(ESS2)とは独立しており、U2 snRNPの侵入をブロックするが、U2AF65ではないISSと結合することにより、tatのスプライシングを阻害する。 539,540,541 hnRNP A1のUP1ドメインはISSとの結合に関与しているが、hnRNP A1のRGGドメインはオルタナティブスプライシング活性には不要である542,543 hnRNP A1によって制御されるESS2およびISS要素に加えて、ESSへのSRタンパク質SC35の結合をブロックすることは、ウイルスが感染初期の段階でTat発現を阻害するための別の戦略である。 hnRNP A/Bタンパク質は、Tatエクソン2に位置するESSに結合してスプライシングを抑制するのに対し、SC35はESSに結合してスプライシングを活性化する。hnRNP A1とSC35の結合部位は、並置されたESE/ESS9の中で重複している。hnRNP A1はESSに結合し、SC35の結合部位を直接マスキングすることで、上流イントロンのスプライシングを阻害していると考えられる544,545。したがって、ASF/SF2とhnRNP A1の比率は、サイトA7での活性化または抑制のためのESE3/(GAA)3の利用を決定する515,541,546
hnRNP A/Bタンパク質は、vpr mRNAを生成するために使用される3′スプライスサイトA2でのスプライシングを阻害する。ESSV内の3つのPyUAGモチーフが変異すると、スプライスサイトA2でのスプライシングが増加し、vpr mRNAのレベルが上昇し、A2とD3に挟まれたノンコーディングエクソンのスキップが減少することにつながる547。ESSVにおけるhnRNP A/Bタンパク質の結合は、抑制された3′スプライス部位のPPTへのU2AF65の結合をブロックし、3′スプライス部位のスプライシング効率を阻害する。
興味深いことに、hnRNP H は HIV mRNA のエクソン 6D スプライシングを積極的に制御していることがわかっている。hnRNP H ファミリーは、配列 CGGA と相互作用し、エクソン 6D への U1 snRNP 組立を可能にしている。
RNA転座
HIVのRNAは転写・スプライシングされた後、核内にはスプライシングされたものとされていないものがたくさん存在している。RNAの細胞質への移動は、Revタンパク質に依存している。一方、いくつかのhnRNPはRNAの細胞質への移動を促進する。HIV-1ゲノムにおいて、hnRNP A2応答エレメント(A2RE)に類似した2つの配列は、トランス作用性輸送因子hnRNP A2に結合するシス作用性RNA輸送配列として機能する。この結合は、オリゴデンドロサイトで広範囲に特徴づけられた特異的なRNA輸送経路を媒介している。A2RE-1はgag遺伝子の主要な相同性領域内に位置し、A2RE-2はvprとtatのコーディング配列の間に重なる領域に位置している。A2REを介したRNA輸送は、微小管とhnRNP A2の両方を必要とする。A2RE-1およびA2RE-2をそれぞれ含むgagおよびvpr RNAをそれぞれ異なる標識をすると、それらが同じRNA輸送顆粒に共集合し、細胞の周縁部に共輸送されることが観察される。一方、hnRNP A2B1、hnRNP C、hnRNP U は核内に HIV-1 ゲノム RNA を保持することができる。hnRNP A2B1の欠乏は、revが存在しない場合にウイルスRNAゲノムの細胞質的再分配をもたらす557,558 hnRNP UのN末端フラグメントは、3′長末端リピート(3′LTR)を特異的に標的とし、mRNAの細胞質的蓄積をブロックし、それによってHIV遺伝子の発現に影響を与える559。
CK2はORF57をリン酸化し、ORF57-hnRNP K相互作用を促進する。ORF57 はウイルスの mRNA の核内輸送に関与しているので、ORF57-hnRNPK-CK2 複合体は KSHV の RNA 輸送に重要である可能性がある560,561 hnRNP A1 は Rex の rex-response element (XRE) への結合を阻害する。HTLV-1 の Rex タンパク質は、スプライスされていない、あるいはスプライスされていないウイルス mRNA の核輸出を媒介している。このプロセスは部分的に Rex の rex-regulatory sequence XRE.562 への結合に依存している。
ウイルスタンパク質の翻訳
HnRNP A1はHIV感染後期に細胞質への再分配を誘導し、IRESを介した翻訳を促進する。hnRNP DはHIV mRNAの細胞質への再分配を助け、p45とp42のアイソフォームはウイルスGagタンパク質の合成を増加させ、p40とp37はこのプロセスを抑制した。
抗ウイルス治療のためのストレスタンパク質の標的化への挑戦
ウイルス感染によるHSP、ERストレスとヒト疾患
HSPとそのコ・シャペロンのネットワークは、細胞がタンパク質の恒常性を維持するために不可欠であり、初期のタンパク質フォールディング、膜を越えたタンパク質の転座、タンパク質複合体の形成などが含まれる。熱ストレス、栄養飢餓、化学的毒性、酸化ストレス、低酸素、炎症、ウイルス感染など、多くのストレス刺激がタンパク質の恒常性を乱す可能性がある565,566,567,568 ストレスは、細胞や組織の損傷を防ぐために迅速に解決する必要があるタンパク質のミスフォールディングや凝集を引き起こす可能性がある。
多くの証拠のラインは、ウイルス感染から宿主細胞を保護するためのタンパク質サーベイランスの主要な因子としてHSPを示唆している。Hsp70/Hsp90の発現は、HSV感染細胞において劇的に増加することが観察された。この相互作用は自然感染ではまだ調査されていないが、試験管内試験(in vitro)での知見から、Hsp70はウイルスキャプシドによって誘導される細胞毒性効果を廃止することによってWNV感染から宿主細胞を保護するために、ウイルスキャプシドタンパク質のネガティブレギュレーターとして作用する可能性が示唆されている570 我々のグループや他のグループの研究は、ほぼすべてのHSPサブファミリーメンバー(Hsp90s、Hsc70、Hsp70、Grp78、Erp57、Hsp27)がエンテロウイルス感染に高度に関与し、ウイルスのライフサイクルのすべての段階で重要な役割を果たしていることを示している。驚くことではないが、全てのHSP(Hsp90s, Hsp70s, Hsp60s, Hsp40s, Hsp27)がコロナウイルス感染に関与していることが実証されている145,196,387,571は、抗COVID-19薬の開発のための良いターゲットであることを示唆している。
572,573,574 HSPは細胞内の自然免疫だけでなく、細胞外の自然免疫や適応免疫も調節することができる。腫瘍細胞から放出された HSP は、抗原提示細胞(APC)の表面受容体に結合し、抗原の交差提示によって腫瘍特異的なキラーを誘発することができる。PEDV感染は、宿主の抗ウイルス監視から逃れるためにHsp27の発現をダウンレギュレートする。
さらに、細胞外HSPは樹状細胞(DC)によるサイトカイン産生を調節することができ、HSPによって調節される自然免疫応答と適応免疫応答との間に関連性があることを強調している577。ヒト単球および樹状細胞への細胞外Hsp72の結合は、TNF-α、IL-1β、IL-6、IL-12およびIFN-γを含むプロ炎症性サイトカインの産生を誘導することができる。上記の HSP 誘導性サイトカイン産生の過程において、HSP は、さらに NF-κB および MAPKs シグナルを活性化する CD14/TLR (TLR2 および TLR4) 複合体シグナル伝達経路の内部刺激として作用する。
HSPの主な機能はストレスから細胞を保護することであるが、多くのウイルスに乗っ取られて感染を成功させることが多い。最近の研究では、DENV NS3タンパク質がHsc70と相互作用してRNAサイレンシングのウイルス抑制因子として作用し、それによってRNAi経路による宿主の抗ウイルスシステムを阻害し、その後DENVの複製を促進することが実証されている。
ウイルスはまた、感染後に宿主細胞にERストレスを発生させる。ウイルスは炎症誘導、オートファジー、アポトーシスではなく、細胞の生存につながるUPR活性化を操作しなければならない。DENVは、感染によって誘発されたERストレスに対処するための細胞適応を可能にすることで、ウイルスのライフサイクルを延ばすために、UPR活性化を逐次的に調節している。UPRはPERK経路によって一過性に誘導され、その結果、eIF2αのリン酸化とその後の感染初期段階でのトランスレーショナルな減衰をもたらす。この一過性のイベントは、ウイルスタンパク質の合成と蓄積を可能にし、最終的には感染中期のIRE1-XBP1(X-box binding protein 1)軸によるUPRの引き金となる。その結果、タンパク質の折り畳みを容易にするためのGrp78の発現が増加し、また、eIF2ɑを脱リン酸化するGADD34(growth arrest and DNA damage 34)の発現が増加し、タンパク質の翻訳を継続することができるようになる。また、増加したGrp78は、CHOP(ストレス持続時のプロアポトーシスタンパク質)によって媒介される細胞のアポトーシスを阻止する。最後に、増加したウイルスタンパク質は、UPRのATF6アームを一過性にトリガーし、後期段階でのUPR活性化を維持するための活性スプライスされたXBP1を提供する。
195 細胞表面のHsp70はJEVエンベロープタンパク質と直接相互作用する。192 Hsp70または90に対する抗体はウイルスの侵入を有意にブロックする。さらに、Hsp70 は NS3、NS5、ウイルス dsRNA と相互作用し、JEV 感染時の VRC の形成を安定化させる。
ERストレスと抗ウイルス
哺乳類細胞では、ERストレスは3つのER膜貫通受容体:膵臓ERキナーゼ(PKR)様ERキナーゼ(PERK)、イノシトール要求酵素1(IRE1)、活性化転写因子6(ATF6)によって感知され、媒介されている。安静時の細胞では、これら3つのセンサーは、ER常駐シャペロンGrp78との相互作用を介して不活性状態に維持されている。アンフォールドまたはミスフォールドされたタンパク質がER内腔に蓄積すると、Grp78はこれら3つのトランスデューサーから解離し、活性化とUPRの開始をもたらする。
ウイルスはまた、感染の特定の段階でそのライフサイクルを有利にするために、異なる手段でERストレス応答を利用したり、反乱させたりしている。ERストレス応答は、ER内のタンパク質フォールディングを阻害する様々な条件によって引き起こされる細胞内プロセスを構成している。真核細胞は進化的に保存された適応機構、すなわち、ERストレスとUPRを開発し、折り畳まれていない/折り畳まれていない、過剰なタンパク質産生を排除し、ERの恒常性を回復させることを目指していた。ERストレスが解消されない場合には、UPRが誘発されて細胞死に至る。ERストレスとUPRは、ほとんどのウイルスがウイルスのタンパク質合成と修飾に宿主のER機械を必要とするため、大量のウイルス感染で観察される可能性がある。ウイルス感染は通常、ERストレスを引き起こす。多量の誤ったタンパク質や過剰に発現したウイルスタンパク質がER内腔内に蓄積されると、ERシャペロンタンパク質(Grp78/Bip、Grp94、カルネキシン、カルレティキュリンなど)の発現が高速に上昇することで示されるように、ERストレス応答が誘発される。ERストレス応答は、インフルエンザウイルス、感染性膵壊死ウイルス(IPNV)、トラウイルス(TULV)、PEDV、ウシウイルス性下痢ウイルス(BVDV)、DENV、JEV、HBV、HCV、E型肝炎ウイルス(HEV)、HSV-1、イヌジステンパーウイルス(CDV)、RSV、シミアンウイルス5(SV5)、CVB3、およびHIV-1などを含む多くのウイルスによって誘発されることがよく知られている。
いくつかのケースでは、ERストレスがウイルスの発病に役割を果たしている。例えば、JEV、BVDV、TULV、重症急性呼吸器症候群コロナウイルス(SARS-CoV)およびWNVはすべて、UPRを介してアポトーシスを誘導することが示されている585,586,587,588,589 酸化ストレスは、HCV感染中にERストレスによって媒介される21 。HCV感染はATF6経路を活性化する一方、IRE1経路をブロックする。代わりに、HBV感染はATF6とIRE1の両方のシグナル伝達を刺激するが、PERKシグナル伝達には影響を及ぼさない(どちらのウイルスも同じ種類の肝細胞に感染するが)。
UPRのいくつかの結果は、ウイルスのライフサイクルに有益である。例えば、ATF6誘導されたシャペロンタンパク質の発現は、ウイルスのタンパク質フォールディングを助け、タンパク質の凝集を防ぐことができるかもしれない。PERK-eIF2αで活性化されたATF4は、細胞代謝を再確立し、タンパク質翻訳を再開するのに役立つかもしれない。594 活性化された ATF6 は、ASFV とリンパ球性絨毛膜炎ウイルス(LCMV)の複製を促進する。この複合体は、DENV E タンパク質の適切な折り畳みと安定性を大幅に改善し、それによってウイルス産生の増加につながる。597 DENV 以外にも、Grup78 は HCMV、JEV、RGNNV の感染を促進することも実証されている。PERK-eIF2α媒介のグローバル翻訳減衰は、DENVやWNVによる感染など、ウイルスの複製を制限する抗ウイルス応答として知られている。活性化されたPKRはリボソーム界面でeIF2αをリン酸化し、その結果、タンパク質合成の全般的な阻害とVSV複製の阻害を引き起こす。
ウイルスの発病に寄与するERストレスのもう一つのアプローチは、宿主細胞の免疫応答の調節を介して行われる。VSV、HCVおよびSARS-CoVは、PERKシグナル伝達を活性化することでタイプIのIFNシグナル伝達経路を阻害することができ、これはIFNAR1のリン酸化依存性ユビキチン化およびそれに続く分解をもたらし、それによって免疫回避およびウイルスの発病を促進する。WNVはまた、ERストレスを誘導し、宿主免疫応答から逃れるためのタイプIFNシグナル伝達経路を阻害することも報告されている。601 HCMVのUS11タンパク質は、UPRを活性化してクラスI主要組織適合性複合体(MHC1)の分解を促進し、免疫逃避につながる。例えば、HCVはIRE1を活性化することで炎症性応答を誘導し、これはTRAF2と相互作用してJNKをリン酸化し、炎症性メディエーターの活性化につながる。HCV の NS4B および NS5A タンパク質は、ER ストレス誘発性カルシウム枯渇と活性酸素産生を介して NF-κB を活性化する。
ウイルス感染によるRNAシャペロンとヒトの病気
hnRNPs は、転写産物の合成、処理、分解、翻訳を含む mRNA の代謝に影響を与える。したがって、核-細胞質間のシャトルリングを制御するメカニズムは非常に重要である。ほとんどのhnRNPは、通常の核局在化シグナル(NLS)を持ち、定常状態では主に核内に存在している。609 メチル化、リン酸化、ユビキチン化、スモイル化などの翻訳後修飾は、hnRNPsの生物学的活性や細胞内局在に影響を与えることが報告されている。
hnRNPsの発現異常がどのようにしてヒトの病気を引き起こすのかが注目されている。hnRNPs の発現レベルは、ヒトの様々な癌や神経変性疾患(脊髄筋萎縮症(SMA)、筋萎縮性側索硬化症(ALS)、アルツハイマー病(AD)、前頭側頭葉認知症など)を含む多くの疾患で変化している。ここでは、主にhnRNPとウイルス感染による疾患との関係を中心に解説する。
エンテロウイルスは、世界中でヒトの病気の原因となる一般的な病原体である。エンテロウイルス感染症の多くは不顕性であるが、軽度の上気道疾患(感冒)、熱性発疹(手足口病やヘルパンギーナ)、無菌性髄膜炎、心不全、胸膜症、脳炎、急性弛緩性麻痺(麻痺性ポリオ骨髄炎)、新生児敗血症様疾患など、幅広い疾患を引き起こす可能性がある。さらに、いくつかの研究では、60-88%の筋萎縮性側索硬化症(ALS)患者の脊髄の神経細胞体からエンテロウイルス配列が検出されることが示されており、エンテロウイルスの持続感染がALSと関係している可能性が示唆されている295,297,299。ポリオウイルス、EV-A71、コックスサッキーウイルスの複製と翻訳は、細胞質に多数のhnRNP(hnRNP A1など)を再分配するため、エンテロウイルス感染はALSのリスクを引き起こすか、または増加させると考えられているが、ALSの発症時にも同じ局在性を示する。例えば、hnRNP K、hnRNP A1、hnRNP M、およびhnRNP Dは、ウイルスの複製や翻訳を助けるために細胞質にシャトルする。
302 HSV 感染はしばしば、口、唇、鼻、または性器の皮膚または粘膜に水疱を生じる615。神経栄養性および神経侵入性のウイルスとして、HSV-1 および-2 は、免疫監視から神経細胞に潜伏して感染者に持続する。保菌者の免疫力が低下すると、HSVは再活性化され、新たなただれを引き起こする。さらに深刻なことに、HSV感染はアルツハイマー病の発症と関連していることが明らかにされている105,616,617。
HBV、HCV、EBV、HPVおよびHIVは、ヒトのがんの20%以上を占める発がん性ウイルスである。HCVまたはHBVの感染は、急性肝炎(劇症肝炎を含む)から慢性肝炎、肝硬変、肝細胞がん(HCC)に至るまで、幅広い範囲の肝疾患を引き起こす619,620。HDVとHBVの共感染は、肝硬変および肝がんのリスクを増加させる。HBV感染者の10%は、肝臓にダメージを与える以外に、血清病様症候群、膜性糸球体腎炎、急性壊死性血管炎(結節性多発動脈炎)、小児の丘疹性肢端皮膚炎(ジャイアノッティ-クロスティ症候群)などの肝臓以外の症状も有している623 。HCVのRNA複製にはhnRNP I、hnRNP C;437,442,446の存在が必要であるが、ウイルスのRNA翻訳にはhnRNP A1、hnRNP D、hnRNP I、hnRNP K、およびhnRNP Lが必要である452,460,462,463,464,465。8,304,305,624
EBVは、感染性単核球症、625 バーキットリンパ腫、626 ホジキンリンパ腫、627 上咽頭癌、628 多発性硬化症629 およびリンパ腫様肉芽腫症など、いくつかの疾患に関与している。EBV感染に関連する他の疾患には、ジャイアンティック・クロスティ症候群、 多形性紅斑、急性性器潰瘍、口腔毛様白板症631、α-シヌクレイン凝集関連疾患(例:多発性萎縮症、レビー小体型認知症、パーキンソン病)などがある632。632 EBV感染中、hnRNP A1とhnRNP CはEBV RNAのポリアデニル化を助ける。
HPV は、皮膚および粘膜上皮に感染する 150 以上のメンバーを持つ DNA ウイルス群である。HPVの急性感染は、性器以外の性器イボや性器コンジロームなどの良性の皮膚病変、または扁平な子宮頸部コンジロームを引き起こす633。これらのがん関連HPVは高リスク型に分類され、子宮頸がんとは関連しないHPVは低リスク型とみなされる。HPV16およびHPV18は、子宮頸がん、扁平上皮がん、腺がんの割合が全地域で最も多い発がん性型である。hnRNP A1はHPV RNAのスプライシングに関与しており、hnRNP HはHPVのポリアデニル化に関与している。
後天性免疫不全症候群(AIDS)以外にも、HIV感染は免疫系の機能を損なうため、ヒトのがんを引き起こす可能性がある。AIDS患者は、細菌、ウイルス、真菌、寄生虫に感染しやすいだけでなく、バーキットリンパ腫、カポジ肉腫、原発性中枢神経系リンパ腫、子宮頸がんなど、さまざまなウイルス性がんを発症するリスクが高い638。hnRNP A1, hnRNP D, hnRNP HはHIV RNAのスプライシングを制御している。また、hnRNP A2、HNRNP A/B、HNRNP A2B1、HNRNP U、HNRNP C、HNRNP IおよびHNRNP Kを含む、他のいくつかのHNRNPがHIVのライフサイクルに関与している。 515,532,533,534,535,536,537,544,545,546,547,548,549,550,551,552,553,554,555,556,557,558,559,564
ストレスタンパク質を標的とした治療効果の可能性
ストレスタンパク質を標的とした様々な疾患への関心は年々高まっている。本パートでは、ストレスタンパク質を標的とすることで、どのような治療効果が期待できるのかを議論する。
まず、ストレスタンパク質を標的とすることで、抗ウイルス能力は多岐にわたる。例えば、抗がん剤として開発されたHsp90阻害剤は、ポリオウイルス、ライノウイルス、EV-A71,107に対する抗ウイルス活性を有することが培養細胞で実証されている。 109 コックスサッキーウイルス、124 RSV、112、113 VSV、パラミクソウイルス(HPIV2、HPIV3、SV5、SV41)、インフルエンザウイルス、126 CHIKV、114 HCV、115、124およびHSV、131、144、145、148、149 HBV、135、169 EBV、146、147、163、164 HCMV151、152およびHTLV。 153,185 siRNAによるHsp60の枯渇は、インフルエンザウイルス38,39 DENV,319 HBV,325,329,331 HCV,321,322 ロタウイルス,323およびHIVによる感染を機能的に抑制する。hnRNP A1を標的とすると、HIV,539,540,541,563 HPV,503,504,505 HCV,471 EV-A71472およびHTLV-1の複製を阻害することができる。HnRNP Dを標的とすると、HCVとHIVの複製がブロックされる。PTB/hnRNP Iを標的とすると、HCV,442,460ピコルナウイルス,89,456,459HPV507およびHIV557,558の生殖を阻害することができる。インフルエンザウイルス、449,450 EV-A71,455 DENV,476 VSV,479,480,481,482 HCMV,488 HBV,490 HSV,492,493 HPV,508 HIV514,536,537 および KSHV を含む。 560,561 hnRNP LおよびhnRNP Mは、HCVおよびエンテロウイルスの複製に重要である461,462,463,475 したがって、ストレスタンパク質は、治療法を欠いている患者や新たに出現したウイルス性疾患の抗ウイルス標的として特に魅力的である。
第二に、疾患状況下では、ストレスタンパク質の細胞の必要性は通常、ストレスタンパク質を標的とする薬剤の特異的選択性を可能にする通常の状況よりも強い。例えば、変異体p53は、野生型p53よりもはるかに多くのHsp90機能に依存している。Hsp90 阻害剤 GM は、野生型 p53 には影響を与えないが、変異型 p53 と Hsp90 の関連性を容易に阻害し、変異型 p53 の分解をもたらすことができる。腫瘍細胞由来のHsp90と活性化コシャペロンp23およびHOPとの複合体は、ATPアーゼ活性の増加を示し、Hsp90阻害剤に対する高い親和性を有するため、腫瘍細胞由来のHsp90は、正常細胞由来のHsp90よりも100倍高い17-AAGに対する結合親和性を示する。294 さらに研究を進めると、がん細胞では、Hsp90 の特徴的な部分ががん原性パートナーと複合体を形成しており、例えば、SkMel28 メラノーマ細胞の変異型 B-RAF-Hsp90 の Bcr-Abl-Hsp90 複合体、K562 慢性骨髄性白血病細胞、MDA-MB-468 乳がん細胞の Her3-Hsp90 および Raf1-Hsp90 複合体などがその原因であることが明らかになった。Hsp90 阻害剤 PU-H71 は、これらの癌細胞における特定の Hsp90-オンコタンパク質ネットワークに選択的に結合する。
同様に、ストレスタンパク質の阻害剤は抗ウイルス効果が期待できる。Hsp70 阻害剤 JC40 は、MDDCs におけるパンフラビウイルス(DENV2 および DENV4)の複製を阻害し、宿主細胞への毒性は無視できる程度であった。
第三に、抗ウイルス剤としてHSP阻害剤を用いても薬剤耐性は観察されていない。例えば、Hsp90阻害剤は薬剤耐性を発現しにくい。これはRSV感染症で明らかに証明されている113。RSVをHsp90阻害剤で繰り返し治療した場合、培養細胞内でのウイルスの広範な通過でも、Hsp90阻害剤での長期治療を受けたマウスでも、薬剤耐性は観察されないであった。同様の結果は、DENV感染症でも観察されている。640 Hsp70阻害剤JC40で10回までの継代でDENVを処理しても、薬剤耐性は生じません。Hsp90およびHsp70阻害剤に対するウイルスの薬剤耐性の欠如は、このような抗ウイルスアプローチが、薬剤耐性が最も頻繁に観察される慢性ウイルス感染症の治療に特に有用である可能性を示唆している。
ストレスタンパク質阻害剤の3つの特徴は、それらを非常に強力な抗ウイルス剤にしており、COVID-19のようなウイルス感染症によって引き起こされる疾患の治療に大きな応用の可能性を示唆している。
課題と展望:ターゲットベースの創薬開発
Hsp90を標的とした抗ウイルス薬開発
Hsp90 は、最も豊富で進化的に保存されているヒートショックタンパク質であると考えられている。Hsp90 の N 末端領域にはユニークなポケットがあり、ATP やコ・シャペロンとの結合に必要とされている。Hsp90の中間ドメインは、アンフォールドされたクライアントタンパク質をリクルートして集合を促進する傾向がある。Hsp90 の C 末端ドメインは、コシャペロンと相互作用するための高度に保存されたペプチド配列を有している。したがって、ほとんどのHsp90阻害剤は、ATPase活性を抑制するか、またはHsp90とそのコシャペロンとの間の相互作用を阻害することによって、その阻害効果を達成する。
Hsp90およびそのクライアントタンパク質の発現は、ウイルス感染中およびほとんどの癌細胞において増加する。Hsp90は有効な抗がん剤のターゲットとしてすでに注目されており、研究室、前臨床、臨床の各場面でHsp90阻害剤が集中的に研究されている。Hsp90阻害剤のがん治療への応用が成功したことで、ウイルス感染症の治療への応用が容易になった。
以上のように、ウイルスの多様な生殖過程におけるHsp90とそのクライアントタンパク質の機能を包括的にまとめた。Hsp90阻害剤の可能性は、EV-A71,107,109ポリオウイルス、ライノウイルス、コキサッキーウイルスによる感染に対して培養細胞を保護することが十分に示されている。 124 パラミクソウイルス(HPIV2、HPIV3、SV5、SV41)、VSV、RSV、112、113インフルエンザウイルス、126 CHIKV、114 HCV、115、124 HSV、131、144、145、148、149 HBV、135、169 EBV、146、147、163、164 HCMV、151、152およびHTLV。 153,185 注目すべきことに、Hsp90阻害剤を感染動物に投与すると、毒性はほとんどなく、ポリオウイルス、107,109 EBV,163,164 HBV,135 CHIKV114およびHCVの複製が強力に抑制される。
ここでは、現在のHsp90阻害剤とその抗ウイルス療法への可能性を簡単に列挙する。ほとんどのHsp90阻害剤は、HSP90のATP結合部位に競合的に結合することにより、HSP90の機能を阻害したり、コシャペロンとの相互作用を阻害したり、アセチル化を調節したりする。このようにして、Hsp90阻害薬を抗ウイルス薬候補として再利用する可能性にも取り組んでいる(表2)。
表2 Hsp90阻害剤 原文参照
Hsp90 ATPase活性を標的とした阻害剤
一部のHsp90阻害剤は、Hsp90のN末端のATPポケットに競合的に結合し、ATP加水分解を阻害し、Hsp90二量体のN末端を閉鎖することが報告されている。さらに、別のATP結合部位がHsp90のC末端に発見されている。648 最近では、いくつかの天然物やその誘導体がHsp90のC末端のATPポケットに競合的に結合することが報告されている。
一般的にHsp90 ATPaseの阻害剤は3つのタイプに分類される。(i)アンサマイシン、(ii)非アンサマイシン、(iii)Hsp90のC末端ATPase活性を阻害するものの3種類に分類される。
アンサマイシン類は、ベンゾキノンを構造核とする抗生物質である。これらの抗生物質としては、ゲルダナマイシン(GA)、ヘルビマイシンA、マクビシンなどが挙げられる。649 GAはN末端のATPポケットに競合的に結合し、ATPアーゼ活性を阻害する。タネスピマイシン(17-アリルアミノ-17-デメトキシゲルダナマイシン、17-AAG)は、GAのアナログである650,651 アルベスピマイシン(17-ジメチルアミノエチルアミノ-17-デメトキシゲルダナマイシン、17-DMAG)は、17-AAGのアナログであり、水への溶解度が高い。さらに、レタスピマイシン塩酸塩(IPI-504)は、17-AAGのより水溶性の誘導体である652 Hsp90の非アンサマイシン阻害剤としては、Luminespib,653 BIIB021,654 Ganetespib,655 Onalespib,656 SNX-5422,657などが挙げられる。
別のタイプのHsp90阻害剤は、N末端ATPポケットではなくC末端ATPポケットに結合する。ノボビオシンは、C末端ATPase活性をブロックする658,659 試験管内試験(in vitro)および生体内試験(in vivo)アッセイでは、ノボビオシンによる治療は、Raf-1およびp60v-srcなどのHsp90依存性クライアントタンパク質の発現を強く減少させる。天然のロテノイドであるデグエリンは、N末端のATPポケットに影響を与えることなく、C末端のATPポケットに結合することが示唆されている。Hsp90のN末端ATPポケットを阻害する場合と比較して、C末端ATPポケットを標的とする阻害剤は、Hsp70の安定性とHsp90のクライアントタンパク質の負の不安定性をもたらする。しかし、これらのC末端阻害剤の可能性とその分子機構は未だ解明されていない。Hsp90のC末端ドメインの構造的特徴をさらに解析する必要がある。このような解析は、HSRを誘導することなく効果的なHsp90阻害剤を設計するための重要なヒントを提供する可能性がある。
Hsp90のコシャペロンの阻害剤
Hsp90シャペロン機構の機能は、関連するコシャペロンに大きく依存しているため、特定のタンパク質-タンパク質相互作用を阻害することにより、Hsp90の下流シグナル伝達を選択的に阻害することが可能である。この相互作用を標的とした阻害剤は、他のHsp90阻害剤による毒性を防ぐための代替的なアプローチとなる可能性がある。その結果、Hsp90とそのコシャペロン間の相互作用を阻害することに多大な努力が払われ、この分野では最近になって報われるようになった。ここでは、Hsp90-Cdc37複合体とHsp90-Hop-Hsp70三元系複合体について簡単に紹介する。
ヒトCdc37はHsp90のコ・シャペロンとしてよく研究されている。Cdc37の中間ドメインとC末端は、Hsp90との相互作用に重要である。
現在、Hsp90とCdc37の相互作用を操作するために報告されている薬剤のほとんどは、天然物およびその誘導体である。
これらの薬剤には、セラストロール、アフェリンA、スルフォラファン、コンゲンシンA、プラチコジンDなどがあり、これらはHsp90-Cdc37複合体を解離させることができる。
さらに、ペプチド(Pep-1、Ac-KHFGMLRRWDD-NH2)が開発され、Hsp90-Cdc37複合体の形成に強い阻害効果を示すようになっている668 別のよく知られたHsp90/コシャペロン複合体は、Hsp90-Hop-Hsp70三元複合体であり、Hsp70からHsp90へのアンフォールドされたクライアントタンパク質の移行を促進する。注目すべきことに、NCGC化合物ライブラリーをスクリーニングした後、6つの活性化合物が同定されている669 これらの化合物は、類似の構造コアを有しており、Hsp70発現に影響を及ぼさない。
Hsp90-コシャペロン阻害剤に関する研究は限られている。Hsp90-co-シャペロン阻害剤の研究は限られており、今後の創薬研究や臨床研究のためにも、有効性や毒性については十分な検討が必要である。
Hsp90の脱アセチル化を阻害するHsp90阻害剤
Hsp90 のシャペロン機能は、翻訳後修飾(PTM)によっても制御されている。Hsp90の翻訳後修飾は、Hsp90のMドメインにおけるアセチル化と脱アセチル化である。Vorinostat (suberoylanilide hydroxamic acid, SAHA)は、Her2/ErbB2の分解につながるHsp90のアセチル化を介してHsp90からHer2/ErbB2を解離させる.670 LAQ824は、Hsp90のアセチル化を誘導し、Hsp90クライアントタンパク質のレベルを低下させる.671 ロミデプシンは、Hsp90 のアセチル化を介して変異型 p53 と Raf-1 の Hsp90 からの解離に関与している672 。さらに、IAV ポリメラーゼの核内導入には、ヒストン脱アセチル化酵素 6/8(HDAC6/8)によって厳密に制御されている Hsp90 の脱アセチル化が必要である120 。他のウイルス(SARS-CoV-2など)と戦うためのHsp90阻害剤の潜在的な使用法については、広範囲に検討する必要がある。
Hsp90開裂を阻害する新規クラスのHsp90阻害剤
近年、紫外線照射、673 アスコルビン酸/メナジオン、674 HDAC 阻害剤、675 プロテアソーム阻害剤676 など、様々な刺激下で Hsp90 の開裂が観察されている。そのため、Hsp90 開裂は別のメカニズムとして考えられている。これらの阻害剤によって誘導されるHsp90開裂は、酵素的開裂と非酵素的開裂の2つのタイプに分類される674,676。さらに、いくつかの物質がHsp90の開裂活性を示すことが報告されているが、そのメカニズムは不明である。
医薬品開発のためのHsp70sの標的化
現在、抗ウイルス薬の一種である抗ウイルス薬は、HSPの異なる特性に基づいて設計されている。Hsp70およびHsc70がHCVの潜在的標的であることを考慮すると、複数のエピトープに対する抗原および腫瘍特異的T細胞応答を長期化するためにDCにHsp70を負荷する677 Hsp70阻害剤(ケルセチン、VER155008およびJC40など)もまた、フラビウイルスおよびエンテロウイルス感染症の治療に大きな可能性を示している202,208,640。Grp78とコロナウイルスのスパイク蛋白質との相互作用を阻害する阻害剤は、SARS-CoV、MERS-CoV、SARS-CoV-2感染症対策に有用なアプローチであると考えられている。
薬物開発のためのHsp60sの標的化
上述のように、Hsp60s は自己免疫疾患、ヒト癌、ウイルス感染誘発性疾患を含む様々な疾患において重要な役割を果たしている。研究は、siRNAによるHsp60sのサイレンシングは、免疫応答を活性化することにより、インフルエンザウイルス38,39 DENV319およびHBV325,331の生殖を有意に減少させ;そしてHCV321,322、ロタウイルス323 HBV329およびHIV338の感染誘発性病因を放出することが可能であることを示している。
したがって、Hsp60sを標的とした低分子再生剤は、これらの疾患の治療薬として有用である可能性がある。メカニズム的には、これらの阻害剤は2つのタイプに分類される。タイプIの阻害剤は、ATPの結合と加水分解を阻害する。タイプIIの阻害剤は、Hsp60の特定のシステイン残基と共有結合的に相互作用する。しかし、詳細な結合部位は同定されていない。
Hsp60阻害剤の天然物としては、ミゾリビン、エポラクタエン、ミルトゥコムロンA、ステファシジンBが挙げられる。 ミゾリビンは、Eupenicillium brefeldianum.680から単離されたイミダゾールヌクレオシド系抗生物質である。エポラクタエンは、Hsp60 に直接結合し、Hsp60-Hsp10 複合体のシャペロン活性を阻害する。683,684 興味深いことに、ETBはHsp60のATPase活性を阻害しない。
686 MCは非プレニル化アシルフロログシノールであり、抗菌性、687,688,688 抗酸化性、689 抗炎症性、690,691 抗腫瘍性を含む複数の生理活性が報告されている692,693 これらの天然物に加えて、いくつかの他の天然物も、直接的な証拠がないままHsp60と相互作用することが報告されている。694,695 興味深いことに、二量体のステファシジンBは組織培養液中で単量体のアブラインビルアミドに変換され、アブラインビルアミドが実際の活性種であることを示唆している。
表3 Hsp60阻害剤 原文参照
Hsp60 阻害剤として上記で同定された天然物の他にも、かなりの数の合成分子が Hsp60 活性を調節できることが発見されている。697 後に、O-carboranylphenoxyacetanilide が Hsp60 に選択的かつ直接結合し、Hsp60-Hsp10 の ATPase 活性とリフォールディング活性を阻害することが明らかになっている。Hsp60はHIF-1αと相互作用することができ、カルボラニルフェノキシアセトアニリドによるHIF-1α活性の阻害はHsp60を標的としたものである可能性が示唆されている。
いくつかのHsp60阻害剤がウイルスの生殖を強力に抑制しているが、ウイルス感染によって引き起こされる疾患の治療薬として機能するかどうかについては、まだ多くの研究がなされていない。
hnRNPを標的とした抗ウイルス薬開発
RNAシャペロンを標的とした薬剤は、広範囲の抗ウイルス能力を有する可能性がある。 hnRNP A1は、HIV,539,540,541,563 HPV,503,504,505 HCV,471 EV-A71472およびHTLV-1の複製に関与している。 562 hnRNP Cの阻害はHCV,438 ポリオウイルス,447,448 DENV,479,480,481,482 EBV,501およびHIVの複製を抑制する。hnRNP Dは、HCV452,453,454およびHIV548,556の複製に必要である。HnRNP Hは、EBV,499,500 HIV,538,550,551,552およびRSV485,486,487感染の増殖に関与している。PTB/hnRNP Iの枯渇は、HCV,442,460ピコルナウイルス、89,456,459HPV507およびHIV.557,558のライフサイクルを損なうことができる。hnRNP K の奪取はインフルエンザの生殖を減少させる,449,450 EV-A71,455 DENV476 VSV,479,480,481,482 HCMV,488 HBV,490 HSV,492,493 HPV,508 HIV,514,536,537 および KSHV.560,561 HnRNP L および hnRNP M は HCV およびエンテロウイルスの複製に参加している461,462,463,475 特筆すべきは、HIV と RNA シャペロンの関係が非常に強いことである。ほとんどのhnRNPはHIVのライフサイクルに参加しており、hnRNPA1,563 hnRNPA2,553,554,555 hnRNP A/B,544,545を含む。 547 hnRNP A2B1,557,558 hnRNP U,559 hnRNP C,hnRNP D,548,556 hnRNP H,538,550,551 hnRNP I557,558,564およびhnRNP K. 514,536,537
しかし、hnRNPを標的とした抗ウイルス薬として開発されたレジェントは限られている。現在までにhnRNPを標的とした薬剤は3種類しか報告されていない。アピゲニンは、果物や野菜に多く含まれるフラボノイドである。アピゲニンは hnRNP A2 を標的とし、hnRNP A2 の二量体化を阻害し、主要な mRNA のオルタナティブスプライシングに影響を与える。アピゲニンは hnRNP A1 の C 末端領域に結合し、核と細胞質の間をシャトルする hnRNP A1 の能力を損ない、最終的にはその細胞質保持をもたらする。Quercetin の抗ウイルス能は、インフルエンザの侵入抑制 702 EV-A71,703 や HCV,208 JEV,DENV の生殖抑制など、いくつかのウイルスで確認されている704 が、これらの研究では、hnRNP A1 の機能に言及することなく、 Quercetin の抗ウイルス機序は Hsp70 発現208 やウイルスプロテアーゼ703 の阻害によるものであると説明されている。別の化合物 VPC-80051 は hnRNP A1 の RNA 結合モチーフを標的とし、hnRNP A1 のスプライシング活性を変化させている705 。
ERストレス経路を標的とした創薬開発
ここ数年の間に、ERストレスを媒介とする経路の役割の拡大は、広範な抗ウイルス治療、特にウイルスの生殖・病原性の分野において、非常に魅力的なトピックとなってきており、潜在的な抗ウイルス剤の開発にも大きな進展が見られている。
ERストレスに関与する経路の中では、3つのUPR経路が最もよく研究されており、これらの経路に関連した抗ウイルスターゲットは第一級のものと考えられている。抗ウイルス剤開発のためには、PERK-eIF2α経路の研究が盛んに行われている。
Boyceらによって同定されたサルブリナルという小さな化合物は、PP1/GADD34複合体の形成を特異的に阻害するとともに、HSVγ134.5を介したeIF2αの脱リン酸化を抑制する。706 彼らの研究から、サルブリナルはPP1/GADD34依存性のeIF2αのリン酸化を減衰させることができ、それによってDENVの複製を抑制することが示唆された。この薬剤は抗ヘルペスウイルス薬の候補であり、臨床治療薬として試験されることが期待されている。
eIF2α以外にも、IRE1-XBP1経路を標的とした薬剤の開発にも多大な努力が払われている。3,5-ジブロモサリチルアデヒドはIRE1と特異的に相互作用し、下流のIAV複製を阻害するエンドリボヌクレアーゼである600 WP1130もIRE1-XBP1阻害薬であり、複数のウイルス(MNV-1、LCVなど)に対して幅広い抗ウイルス効果を示している709。
また、ERストレスを媒介としたアポトーシスシグナル伝達は、医薬品開発や選択のためのもう一つの注目すべき経路である。Rana catesbeiana ribonuclease(RC-RNase)は、カスパーゼ-3、カスパーゼ-8、カスパーゼ-9経路の活性化を介して、JEVの複製を効率的に抑制することが報告されている(710 ダブ ル鎖RNA活性化カスパーゼオリゴメライザー(DRACO)は、臨床治療において幅広い抗ウイルス効果があることが報告されている。DRACOはdsDNAウイルスに感染した細胞のアポトーシスを選択的に活性化するが、正常な健康な細胞には有害な影響を与えません。DRACOは、HIVやデングウイルスを含む15種類以上のウイルスを根絶することができる。711 また、JNK、JAK、Bcl-2、CHOPなどの他のシグナル伝達物質を標的とした薬剤も研究されている。有望な薬剤であるバチタノールBは、ERストレスによるアポトーシスから宿主細胞を保護することができる。
SFVの非エンベロープ糖タンパク質、LCMVの前駆体糖タンパク質、TULVの糖タンパク質、SARS-CoVのいくつかの非構造タンパク質、フラビウイルスファミリーの複数の非構造タンパク質、PEDVのエンベロープタンパク質、HCVのE1、E2および非構造タンパク質、MHVの表面タンパク質、HSV1のICP0糖タンパク質B、CHIKVの構造タンパク質4など、ERストレスを誘発する可能性のあるウイルスタンパク質に注目する研究者もいる。UPRの活性化とアポトーシス誘導に関与するウイルス蛋白質は、HCV(E1, E2, core)、HCMV(pUL37x1, pUL38)、SV5 V蛋白質など、特に注目されている。この分野では大きな進歩があり、現在の進歩には、HCV NS4Bのためのクレミゾール、HSV-1 γ134のためのワクチンベクター、およびIAVヘマグルチニンAのためのノーラキンが含まれている。
ストレスタンパク質を標的とした阻害剤は、正常な細胞機能に副作用を及ぼす可能性があるため、臨床現場での使用は非常に慎重であり、また、一部のウイルス(例えば、SARS-CoV)の感染様式が風化しているため、臨床試験の機会が限られており、コロナウイルスによる重症急性呼吸器症状(SARS)のような一般的な感染症や特殊な感染症に対するストレスタンパク質を標的とした薬剤の開発には、さらなる課題がある。