
Contents
Can magnesium reduce central neurodegeneration in Alzheimer’s disease?
Basic evidences and research needs
pubmed.ncbi.nlm.nih.gov/30905744/
2019年3月22日
ハイライト
- 生理学的には、マグネシウムは興奮性、細胞内の代謝、細胞の生存に主要な役割を果たしている。
- マグネシウムは、アミロイドタンパク前駆体のβプロセッシングを切り替え、タウタンパクのリン酸化を抑制する。
- マグネシウムは炎症性サイトカインの発現や影響を抑え、神経炎症を抑制する。
- 神経変性の血液脳関門の障害は、マグネシウムによって弱められ、逆転することもある。
- マグネシウムはNMDA受容体の異常の原因と結果に対して抗興奮性の特性を示す。
概要
マグネシウム(Mg)は、300以上の細胞機能を持つ重要な2価の陽イオンである。マグネシウムは、300以上の細胞機能を持つ重要な2価の陽イオンであり、いくつかの神経疾患において治療効果を発揮する。マグネシウムが神経細胞の変性に関与する病理学的プロセスを抑制することを示す多くの基礎的証拠があるにもかかわらず、この低コストの選択肢は、今のところ臨床研究や実践において十分に考慮されていない。しかし、現在、古典的なガイドラインで推奨されている高価な薬剤(生理学的なリハビリテーションに加えて)には、特別な効果を示すものはなかった。ここでは、アルツハイマー病に焦点を当て、神経新生と神経保護のためのMgの使用をサポートする治療経路を分析する。これまでに報告されている実験結果によると、Mgは、毒素排出の促進、神経炎症の抑制、アミロイドタンパク質前駆体(APP)の病理学的処理やタウタンパク質の異常なリン酸化の抑制、N-メチル-D-アスパラギン酸受容体の異常を回復させるなどの興味深い能力を有している。現在のところ、これらの証明された効果に関わるメカニズムの重要な詳細はまだ不明であり、臨床的背景も乏しい。そのため、Mgの薬力学的標的をよりよく理解し、Mgの臨床利用のための最適な薬理学的戦略を見出すためには、さらなる研究が必要である。
1. はじめに
マグネシウム(Mg)は、その存在量からして、細胞内では2番目、人体では4番目の陽イオンである(Allgrove, 2009)。このイオンは、300以上の細胞質の酵素反応(ミトコンドリアのエネルギー生産、タンパク質合成、生存シグナル、タンパク質の空間構成、核酸の活性調節など)に大きな役割を果たしている(Allgrove, 2009; de Baaij, 2015)。その複数の機能と特性から、Mgはいくつかの疾患、特に過剰なアポトーシスや興奮性の異常を伴う神経系の病態において治療に用いることが検討されている(Kirkland er al 2018)。さらに、臨床医にとっては、Mgは大きな治療濃度域を持つ低コストの治療オプションを提供する。しかし、このイオンの使用は、その使用可能な病態のほとんどについて、十分に成文化されていない。実際、いくつかの神経病理学的プロセスにおいてMgが有効であることを示す多くの基礎的な証拠があるものの、ガイドラインを編集するための十分な翻訳的・臨床的研究はない。そのため、従来の治療法の補助としてMgが有効であるにもかかわらず、臨床現場ではMgの使用があまり考慮されていない。現在、神経変性疾患では、緩和的な内科的・外科的治療(通常、高価な薬剤や深部神経刺激)やリハビリテーション(運動療法や認知トレーニング)を行い、致命的な症状の進行をできるだけ遅らせることが求められている(Bond er al)。 ここでは、アルツハイマー病を例にとり、神経変性疾患の管理においてMgをより考慮すべきであることを示する。そこで、Mgの関連する生物学的特性に基づいて、Mgをアルツハイマー病に使用する際に期待される薬理学的・力学的ターゲットを示した。
2. アルツハイマー病の概要
アルツハイマー病は、最も頻度が高く(認知症の60-70%)死亡率の高い神経変性疾患である(Burns and Iliffe, 2009)。早期進行型の認知症で、主に側頭葉、頭頂葉、帯状回、前外側前頭皮質などの脳の萎縮、βアミロイド(アミロイドβ)が凝集した細胞外のプラーク、神経原線維内のタングル(NFT)を特徴としている(Burns and Iliffe, 2009)。プラークは、大きなタンパク質(図1)である全長のアミロイド前駆体タンパク質(APP)が断片化されて合成された39~43アミノ酸の小さなペプチドの組み合わせであるアミロイドβ′sポリマーが蓄積された結果、不溶性の堆積物となる(Burns and Iliffe, 2009; Chen er al)。 APPは、神経細胞の可塑性と生存に重要な膜貫通型タンパク質である。このタンパク質は、大きな細胞外ドメイン、短い膜貫通ドメイン、および細胞質尾部から構成されている(Chen er al 2017)。APPのプロセシングは、細胞内の異なるゾーンに位置する3つのプロテアーゼに依存している。αセクレターゼ(α-セクレターゼ)は細胞膜に固定されているが、βおよびγセクレターゼ(β-セクレターゼおよびγ-セクレターゼ)はエンドサイトコンパートメント内にある(Chen et al 2017,Seegar et al 2017)。アミロイドβを生成するタンパク質分解反応は、β-セクレターゼ、次いでγ-セクレターゼの2つのプロテアーゼによって触媒される(Chen er al 2017)。アミロイドβのプラークがアルツハイマー病の主な神経病理学的特徴であるが、いくつかの研究により、アミロイドβの可溶性凝集体がより早く発症に関与し、臨床的重症度とより相関している可能性が高いことが明らかになったことに注目したい(Cline et al 2018)。これらの可溶性凝集体は、アミロイドβのオリゴマー(oアミロイドβ)であり、アルツハイマー病の神経膠障害においては、アミロイドβ二量体とアミロイドβ*56の2種類がよく研究されている(Amar et al 2017,Klyubin et al 2008,Lesné et al 2013,Shankar et al 2008)。アルツハイマー病のもう一つの発症現象であるNFTは、タウタンパク質の異常な凝集によるものである。生理的条件下では、この分子はリン酸化されると微小管を安定化させるため、細胞の輸送に不可欠である。アルツハイマー病では、タウの過剰なリン酸化(主にoアミロイドβ′sの毒性が関与)によりクロスペアリングが起こり、神経細胞の輸送システムの変化を伴うもつれの構成が生じる(Amar et al 2017,Lesné et al 2013,Sherman et al 2016)。これらの病理学的プロセスの最終的な結果は、脳の連結性の地域的損失と神経細胞の死である。
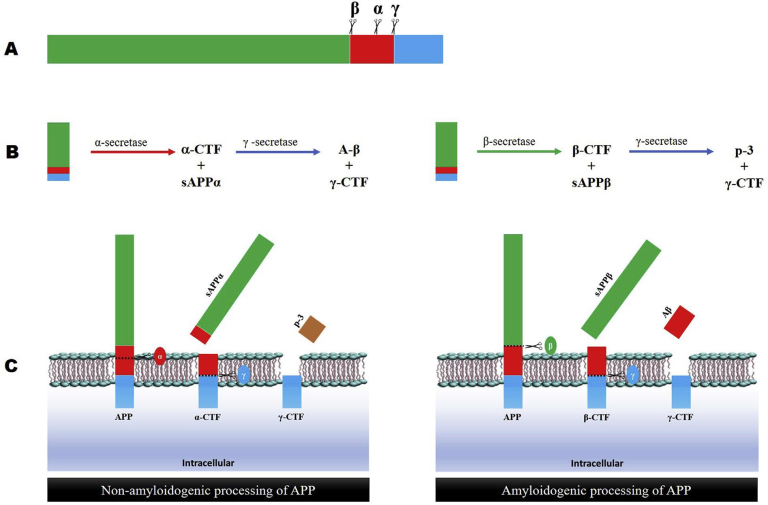
図1 アミロイド前駆体タンパク質(APP)のプロセシング|A)
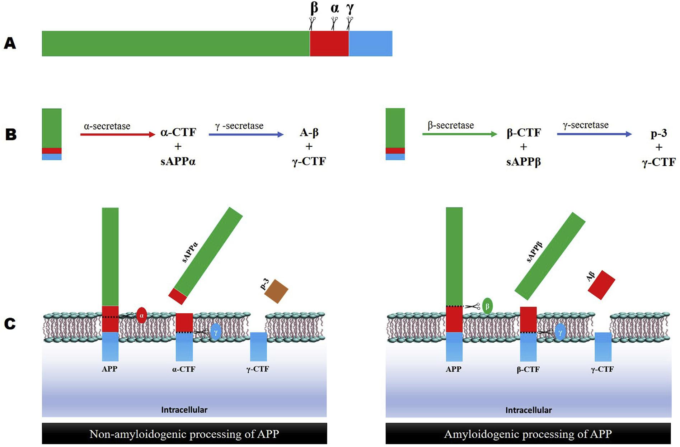
A) α-β-およびγ-セクレターゼによるAPPの切断部位。B-C) APPのα-β処理:α-セクレターゼはAPPをアミロイドβタンパク質ドメイン内で切断し、可溶性APP-α(sAPPα)を放出するが、β-セクレターゼの作用はsAPPβを放出する。どちらのプロセスでも、残留する鎖(C末端フラグメント、CTF)は膜に結合したままである。αプロセスではCTFαが、βプロセスではCTFβが膜に結合したままとなる。そして、γセクレターゼの作用により、CTFαからは細胞外のP3が、CTFβからはアミロイドβタンパク質が放出される。
3. Mgの生理機能
形質細胞内のMg濃度(0.75-1.4mmol/L)は、吸収量の変化に応じて、尿中排泄量の変化によって細かくバランスをとっている(de Baaij, 2015)。血漿中では、マグネシウムは主に遊離イオン化形態(55%)で存在する(Sztark and Cochard, 1998)。約34%はプロテインに結合しており、残りの11%はリン酸塩やクエン酸塩などの分子と複合体を形成している(Sztark and Cochard, 1998)。細胞内では、遊離イオン化した状態で存在するのは10%以下である(Sztark and Cochard, 1998)。
Mgは、興奮しやすい細胞の代謝や、膜貫通部や細胞内のイオン伝達に影響を与えている(Allgrove, 2009)。これらの機能により、膜の電気的安定性、神経膠の情報伝達、ひいては神経膠の生存に重要な役割を果たしている(Trapani et al 2011)。さらに、細胞内の遊離MgとMg-ATP複合体は、タンパク質合成と細胞増殖を協調的に制御している(Rubin, 2011)。また、Mgイオンは神経伝達、主にN-methyl-D-aspartate(NMDA)グルタミン酸伝達にも関与している(Spasov et al 2009)。この活動において、Mgはグルタミン酸に対するNMDA受容体チャネルの感度を調節することで、シナプス後の活性化を制御している(Nikolaev er al)。 このイオンは、シナプス領域外のNMDA受容体の制御にも関与している(Nikolaev et al 2012)。シナプス後の活性とは別に、Mgはグルタミン酸のシナプス前放出の確率や、アストロサイトによるこの神経伝達物質の放出/取り込みも調節する(Furukawa er al)。 全体として、Mgは、その細胞内での利用可能性に応じて、細胞の生存だけでなく、細胞死に関連する細胞経路に関与している(Trapani et al 2011,Wolf and Trapani 2008)。この二重の役割は、細胞内のMgが、その細胞質レベルに応じて、生存カスケードとアポトーシスカスケードのいずれかを活性化することを意味する(Wolf and Trapani, 2008)。
4. Mg の濃度と神経変性を関連づける主な臨床的・実験的観察結果
ヒトおよび動物モデルを対象としたいくつかの研究により、Mgの低濃度または高濃度が神経変性疾患(Kieboom et al 2017,Kirkland et al 2018年)てんかん(Arjona et al 2014,Gandhi et al 2012年)頭痛(Lodi et al 2001年)およびその他の神経疾患につながることが示された。さらに、Mgの補給は、ディフォルトの病態生理学的な脳の状態で治療効果を示する。
4.1. 脳神経変性症におけるマグネシウムの代謝異常
低マグネシウム血症は、脳の神経変性に最も関与する代謝異常である。まず、重度の先天性低マグネシウム血症では、低マグネシウム血症誘発性の発作がよく知られている(Chen er al 2016)。興味深いことに、これらの発作は、適切な補充が早期に開始されない場合、通常、重度の認知障害や脳の萎縮と関連している(Ndiaye et al 2013)。その他の臨床研究では、原発性神経変性疾患におけるMg濃度の異常の役割が示唆されている。脱認知症については、Kieboom et al 2017)が、ベースラインで認知機能が正常な9569人の患者(追跡調査期間中央値7.8)を対象に、この障害の発生率を調査した。その結果、血清Mg濃度の低値(≦0.79mmol/L)だけでなく、高値(≧0.90mmol/L)も認知症のリスク上昇と関連することがわかった。(Barbagallo et al 2011)は、101人の患者を対象とした別の研究において、アルツハイマー病群では、Mg-ion(総Mgではない)が、年齢をマッチさせた対照群のアルツハイマー病でない成人と比べて有意に低いことを明らかにした(0.50±0.01 mmol/L vs 0.53±0.01 mmol/L; p < 0.01)。さらに、Veronese et al 2016)は、13件の研究(アルツハイマー病患者559人、健常対照者381人、他の医学的疾患を有する患者126人を含む)を分析したシステマティックレビューにおいて、アルツハイマー病患者の脳脊髄液(脳脊髄液)中のMgは低いが、血清中のMgは低いことを報告している。パーキンソン病(PD)についても、脳脊髄液中のMg濃度が低いことが報告されているが、パーキンソン病患者対対照者では血清中のMg濃度は明確に低下していなかった(Kirkland et al 2018)。さらに、動物モデルでは、Oyanagi et al 2005, 2006)が、2世代にわたってMgを継続的に低摂取することで、ラットのドーパミン神経細胞の選択的な損失を誘導することを報告している。神経細胞の変化以外にも、細胞外のMg濃度が低いと、過酸化水素の暴露によって内皮細胞に発生する酸化ストレスが増大する(Wolf er al 2008)。このような代謝異常は、血液脳関門(BBB)の機能不全や脳の微小血流の変化につながり、変性的な結果をもたらす可能性がある(Burns and Ilffe, 2009; Horgusluoglu et al, 2017)。
4.2. 脳神経変性に対するマグネシウムの補給
数多くの研究が、神経変性におけるMgの興味深い治療特性を示している。しかし、これらの研究のほとんどは、試験管内試験の調製物や動物モデルを用いた経験的なものであった。中側頭葉てんかんのカイネイトマウスモデルでは、Mgの補給が空間記憶障害の重症度を軽減する(Toffa er al)。 別の研究では、Hashimoto et al 2008)は、1-methyl-4-phenylpyridinium(MPP+)が関与するPD疾患の試験管内試験ラットモデルにおいて、予防と改善の両方のEfficacyを報告した。MPP+の治療効果を評価するために,ドーパミン神経細胞の生存率とドーパミン神経細胞の神経突起の長さを評価した。治療効果は用量依存的であり,Mg濃度1.2mMでMPP+の毒性が有意に抑制されたのに対し,4.0mMでは神経細胞の損失を完全に防ぐことができた。
5. アルツハイマー病の神経変性プロセスに対するマグネシウムの働き:基礎サポート
アルツハイマー病に関与する様々な病理学的現象がMgの標的となることが期待されている。
5.1. 神経炎症
神経炎症は、主要な神経変性疾患における神経細胞の損失および神経膠機能障害の重要な要因と考えられている(Ilievski et al 2018,Rakic et al 2018,Walker et al 2015,Wenzel et Klegeris 2018)。アルツハイマー病では、初期段階ではアミロイドβオリゴマーによって炎症が引き起こされ、その後、アミロイドβ′sの沈着や血液脳関門からの毒性漏出によって炎症が引き起こされる(Wenzel et Klegeris, 2018; Yamazaki et Kanekiyo, 2017)。過剰な炎症性サイトカインの放出、特にガンマインターフェロン(IFN-γ)インターロイキン1β(IL-1β)腫瘍壊死因子α(TNF-α)の放出と、それに続く結果が主なプロセスである(Ilievski et al 2018;Rakic et al 2018;Wenzelら、Klegeris 2018)。アストロサイト、次いでミクログリア(後に血液免疫細胞が関与)が、この炎症内プロセスにおいて主要な役割を果たしている(Chun et al 2018;Perez-Nievas and Serrano-Pozo 2018)。Park et al 2018)は、γ-インターフェロン(IFN-γ)(反応性アストロサイトから放出される)と過剰なアミロイドβ(ニューロンから放出される)の両方が、IFN-γ受容体とToll Like受容体4(TLR4)を介してそれぞれミクログリアの活性化を誘導することを示した。ミクログリアは、誘導性一酸化窒素合成酵素(iNOS)や核内因子カッパB(NF-κB)の発現を促進し、TNF-αや細胞傷害性のNOを放出して、神経細胞の変性を引き起こすとしている。興味深いことに、Mgを投与すると、ラットの重症子癇前症モデルにおいて、生体内試験でミクログリアの活性化が抑制されること(Johnson et al 2014)や、試験管内試験でカルシウムがリポポリサッカライド暴露に対するマクロファージ様細胞の炎症反応を抑制すること(Lin et al 2010)などが報告されている。さらに、Mgは、神経細胞のγ-secretaseやミクログリアのTNF-αに対する抑制効果を示した(Yu er al 2018)。なお、γ-セクレターゼは、アミロイドβの合成に重要なプロテアーゼ複合体である。その亢進は、この基質の神経細胞への蓄積と、ミクログリアのTNF-αの過剰発現をもたらし、その後の神経変性を引き起こす。Yu et al 2018)は、試験管内試験のヒトおよびマウス由来の細胞を用いて、Mgが神経細胞のγセクレターゼ活性を低下させるとともに、ミクログリアのTNF-α発現を抑制することで作用することを強調した。また、これらのMgの神経保護作用には、ホスホイノシチド-3-キナーゼ/プロテインキナーゼB(PI3-K/Akt)とNF-κBのシグナルが重要であることを実証した。また、Mgの補給によりIL-1βの抑制が認められている(Wang et al 2017)。実際、Wang et al 2017)は、MgがIL-1βおよびアミロイドβ誘発性の炎症をブロックすることを示した。さらに、この抑制は、細胞外シグナル制御プロテインキナーゼ1および2(ERK1/2)とペルオキシソーム増殖剤活性化受容体γ(PPARγ)の刺激に起因することを実証した(Wang et al 2017)。
5.2. APPの切断とタウタンパク質の代謝
アルツハイマー病の神経ネットワーク機能障害では、アミロイドβおよびタウタンパク質の蓄積が重要な役割を果たしている(Crimins et al 2013;Kamat et al 2016)。これらの現象は、部分的に可逆的である(Pooler et al 2014)。したがって、このような毒性蓄積をもたらす酵素反応は、興味深い治療標的となる。このような反応の一部は、Mgによって調節可能であることが、いくつかの実験結果から明らかになっている。Yu et al 2010)は、様々な濃度の細胞外MgがAPPの処理に及ぼす影響を評価した。Yu et al 2010)は、細胞外のMg濃度を変化させて、APPのプロセッシングに及ぼす影響を評価した。この研究では、生理学的濃度(0.8 mM、通常のコントロールとして)低濃度(0〜0.4 mM)高濃度(1.2〜4.0 mM)を用った。この研究では、低濃度のMgはアミロイドβの分泌を促進し、細胞の生存率を低下させるのに対し、高濃度のMgはαセクレターゼによるAPPの切断経路を促進する(αセクレターゼによるAPPの切断の結果、sAPPα+αCTFでsAPPαの合成が増加する)。Mg濃度が高いと、βセクレターゼからαセクレターゼへの切断が切り替わるという効果があるが、これはAPPの膜内保持が一部関係していると考えられている(Yu er al 2010)。対照的に、Mgレベルが低いと、APPの膜内保持率が低くなる。さらに興味深いことに、sAPPαの割合が高いと、外傷性脳損傷(Plummer et al 2016年)プロテアソームストレス(Copanaki et al 2010年)BAG3によって誘発されるプロテオトキシックストレス(Renziehausen et al 2015)などのダイオキシンストレス状態において、神経保護特性を示する。また、sAPPαは、加齢に伴う神経前駆細胞の増殖(NPC)の低下を回復させるとともに(Demars et al 2013)成体海馬NPCの増殖やグリア分化を促進することで、向神経性学的な効果を示した(Baratchi et al 2012)。そのメカニズムについて、Ryanらは、ラット海馬スライスを用いて、sAPPαの向神経性特性には、即時型初期遺伝子転写因子(AP-1,Egr1)のアップレギュレーション、クロマチン環境の調節、構成的転写因子CREBおよびNF-κBの明らかな活性化など、複数のレベルの転写反応が関与していると結論づけている(Ryan et al 2013)。
図2 アルツハイマー病における血液脳関門(BBB)の機能不全とMgの潜在的標的
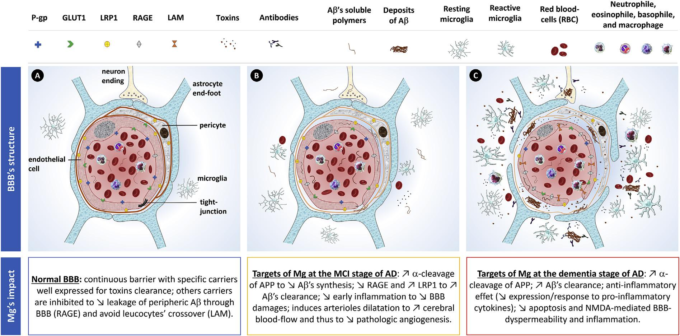
A)正常なBBB:内腔側の内皮細胞、中間部の周皮細胞、基底膜からなる連続したバリアであり、その上には外腔側のアストロサイトが存在する。内腔側のアストロサイトのエンドフットからなる連続したバリアである。血管周囲の空間は狭く、パラセルラー・ジオフュージョンは制限されている。特異的なトランスポーターの中には、毒素の除去や神経膠に送られる血清物質のろ過のためによく発現しているものがある。その他の輸送体は,末梢のアミロイドβが関門を通過して漏出するのを抑制したり(RAGE輸送体),白血球のクロスオーバーを回避するために抑制される(LAM)。B) MCIの段階。アミロイドβの代謝異常と脳血流の変化が引き金となり、BBBの初期障害が生じる。アミロイドβの代謝異常と脳血流の変化が相まって、BBBの初期障害が引き起こされる。主な変化は、毒素クリアランスの担い手の減少と基底膜の障害であり、微小な異常漏出とミクログリアの最小限の炎症反応を引き起こす。Mgは、局所での過剰なアミロイドβ合成を防ぎ、そのクリアランスを改善することが期待されている。また、初期の炎症過程や病的な血管新生促進シグナルを抑制することも必要である。C) 認知症段階:BBBの主要構成要素に深刻な変化が見られる:タイトジャンクションや基底膜の破壊、内皮細胞の異形化、アストロサイトの末端足の後退、GLUT1,P-gp、LRP1の低下、RAGEやLAMの上昇などが見られる。これらの障害は、血液物質の毒性漏出を引き起こし、血球の実質的なクロスオーバーの結果、神経膠コンパートメントにおける主要な神経炎症および酸化プロセスを引き起こす。血液、内皮細胞、神経膠に由来するアミロイドβの沈着は、このような主要なプロセスにおいて重要な役割を果たしている。Mgは、アミロイドβの内皮・神経膠での合成を抑制するとともに、すべての毒素による炎症・酸化の影響を軽減することが期待される。
略語
グルコーストランスポーター1(GLUT1)P糖タンパク質(P-gp)低密度リポタンパク質受容体関連タンパク質1(LRP1)進行性糖化最終産物の受容体(RAGE)マトリックスメタロペプチダーゼ9(MMP-9)白血球接着分子(LAM)。
タウの蓄積については、ストレプトゾトシン誘発の散発性ADモデルラットにおいて、Xu et al 2014)が硫酸Mgの神経保護作用を見出している。彼らの研究では、MgがGlycogen synthase kinase 3-β(GSK-3β)の9番セリンでの抑制性リン酸化を増加させることで、タウの過リン酸化を減少させ、それによってAktの473番セリンでの活性とPI3Kの458/199番セリンでの活性をインクリメントし、さらにインスリン感受性を改善することが明らかになった。
5.3. 血液脳関門(BBB)を介した毒素のクリアランス
アルツハイマー病では、アミロイドβ′sプラークやタウのもつれが生じる前の初期の病態変化の中に、血管やBBBの機能障害が含まれることが多いと広く考えられている(Merlini et al 2011,Sweeney et al 2019,Thal et al 2003,Yamazaki and Kanekiyo 2017)。また、血管と脳のインターフェースの障害は、アルツハイマー病におけるAPPの代謝異常の原因および/または結果として、悪循環的に作用することが認められている(Anderson et al 2011;Jefferies et al 2013;Yamazaki and Kanekiyo 2017)。実際、ミ血管の変化は、アミロイドβ′s代謝異常に対する活性化されたアストロサイトとミクログリアの炎症反応から生じる可能性がある。この炎症は,主にトロンビンと血管内皮成長因子(VEGF)を媒介とする過剰な血管新生シグナルと関連している(Jefferiesら,2013)。これらのメディエーターは、二次的に内皮のアミロイドβ′sの過剰分泌を促進し、その後のプロオキシダントな代謝異常を引き起こし、さらに内皮/神経細胞にダメージを与える(Jefferies et al 2013)。また、病的な血管新生は、動脈の変化による脳血流(脳血流)の変化が引き金となり、microangiogenesisの不適切なアップレギュレーションを引き起こすと考えられている(Jefferies er al)。 そうでなければ、NMDA受容体を介したBBBの伝染性の過剰な増加が関与しており、アルツハイマー病の中期および後期において重要である可能性が高い(Xhima er al 2016)。ヒトでは、アルツハイマー病の初期/後期の微小血管障害の発生は、いくつかの画像研究(DCE-MRI、T2*およびSWI MRI、FDG-PETおよびベラパミル-PET)死後の神経病理学的所見、および生体流体マーカー(主に、アルブミン漏出)の動態分析によって裏付けられている(Sweeney et al 2019)。さらに、通常のアルツハイマー病の遺伝的動物モデル(PSEN1,APP、APOE4トランスジェニックマウスなど)では、BBBの機能不全がアミロイドβ/tauの蓄積や脳の萎縮に先行している(Sweeney er al)。 BBBの変化は、血管周囲のアストロサイト端部、周皮細胞、基底膜、内皮細胞をアホ化する(Sweeney et al 2019; Yamazaki and Kanekiyo, 2017)。その結果、BBBを介したパラセルラー/トランスセルラーの移動が異常になる(Jefferies et al 2013)。このような細胞間移動の異常は、特定のトランスポーターの規制解除に関連しており、グルコーストランスポーター1(GLUT1)P-グリコプロテイン(P-gp)低密度リポタンパク質受容体関連タンパク質1(LRP1)のダウンレギュレーションや、RAGE(receptor for advanced glyca-tion end products)MMP-9(matrix metallopeptidase 9)LAM(leucocyte adhesion molecule)のアップレギュレーションが示唆されている(Sweeney et al, 2019; Yamazaki and Kanekiyo, 2017)。) 重要なのは、超細胞伝染性の低下により、フィブリン、トロンビン、アルブミン、免疫グロブリンG、ヘモジデリンなどの血液由来物質の毒性のある周辺斑状蓄積を伴う微小出血が生じることである(山崎・金清 2017)。その結果、神経炎症、神経膠膜の過酸化、アポトーシスが起こる(Jefferies et al 2013,Sweeney et al 2019)。さらに、毛細血管へのアミロイドβポリマーの沈着は、内皮細胞の異形性/機能不全、毛細血管の閉塞、基底膜の肥厚/破壊、間質空間の構成の変化をもたらす:これが脳アミロイド血管症(脳アミロイド血管症)である(Anderson et al 2011,Jefferies et al 2013,Thal et al 2003)。要約すると、アルツハイマー病における微小血管の変化は、アミロイドβの代謝異常、アミロイドβの過剰合成(神経細胞および内皮細胞)に対応した炎症促進シグナル、内皮細胞の酸化ストレス、BBBを介した有害物質の漏出、そして最終的には神経細胞の酸化およびアポトーシスのカスケードの誘発を意味する(図2)。
これらのプロセスの発症経路に対して,MgによるAPPのα-leavageの促進は,微小血管や神経膠の保全のための抗アミロイドβ予防の鍵となる効果である(Yu er al)。 また、微小血管平滑筋の弛緩を誘導するMgの能力(Gröber et al 2015)は、脳血流を大幅に改善し、病的な反応性血管新生を防ぐことができる。さらに、Mgの神経膠/内皮の抗炎症作用は、上に引用したいくつかの研究で報告されている(Wang et al 2017,Wolf et al 2008,Yu et al 2018)。BBBを介して漏出したアミロイドβや毒素によって引き起こされる炎症を軽減するこの能力は、アルツハイマー病の後期段階においてMgの補充が有用な選択肢であり続けることを示唆している。このような潜在的な薬理作用に加えて、Mgは内皮機能を改善することもできる。この点について、(Zhu et al 2018)は、Mgの補給がBBBの伝染性とその機能を改善し、神経変性を抑制することを示した。これらの著者によると、このプロパティーは、主にアミロイドβタンパク質のバリアーを介したクリアランスに対するそのeffectsに基づいている。実際,Mgは,低密度リポタンパク質受容体関連タンパク質(LRP)やホスファチジルイノシトール結合クラスリン集合タンパク質(PI-CALM)を介して,BBBを介して脳から血液側へのアミロイドβトランスサイトーシスを促進する。逆に、RAGE(receptor for advanced glycation end products)やカベオラへの作用により、脳側へのアミロイドβの流入を減少させる(Zhu er al 2018)。
5.4. シナプスの活動とエネルギー代謝
アルツハイマー病では、特に初期段階において、シナプス機能不全とシナプス消失が病態生理に重要な役割を果たしている(Pozueta et al 2013;Spires-Jones and Knafo 2012)。これらの段階では、シナプスの変化は、樹状突起の減少とシナプス接点の機能低下からなる。この変化は主に、プラークが形成される前の可溶性oアミロイドβ(アミロイドβのオリゴマー)の蓄積によるものである(Cline et al 2018,Price et al 2014)。oアミロイドβの病原的役割は、Lambertらの観察によって初めて裏付けられた(Lambert et al 1998)。彼らは、合成oアミロイドβ(フィブリルなし)がシナプス伝達増強の急速な阻害を引き起こし、その後、神経細胞の損失を引き起こすことを指摘した。その後、多くのex vivoおよび生体内試験の研究により、この仮説が確認された(Klyubin et al 2008,Müller-Schiffmann et al 2016,Shankar et al 2008)。アルツハイマー病におけるoアミロイドβの毒性作用は、細胞性プリオンタンパク質(PrPC)とメタボトロピックグルタミン酸受容体5(mGluR5)を介したシグナリングに基づいている(Beraldo et al 2016; Brody and Strittmatter, 2018)。oアミロイドβはまた、タウのリン酸化やインスリン受容体基質の不活性化を誘導することによっても作用する(Ma and al., 2009)。Mgが完全長APPの毒性のあるβフラグメンテーションを有意に減少させることを考慮すると(Yu et al 2018)その予防的使用は、軽度認知障害(MCI)が診断された時点で神経保護のための興味深い視点である。この点、Li et al 2014)は、APPswe/PS1dE9マウス(ADモデル)を用いて、早期のMg-L-threonate投与により、シナプスの消失とAPPのβプロセッシングを事前に防ぐことができることを明らかにした。MCIが必ずしも認知症に発展するわけではないが、アルツハイマー病発症のリスクは最大で15倍も高いとされている(Burns and Iliffe, 2009)。したがって、Mgを早期に補給することで、認知症になるのを防ぐことができると考えられる。
アルツハイマー病のもう一つの病因は、アミロイドβ複合体によるNMDA再受容体(NMDAR)の制御異常である(Deng et al 2014,Dewachter et al 2009,Mota et al 2014,Snyder et al 2005)。NMDARは、高いカルシウム伝染性とMgによる電圧依存的なチャネルブロックを有するグルタミン酸系の興奮性チャネルである(Calabresi er al)1992)。神経細胞へのカルシウム流入は、静止状態に近い膜電圧ではMgによって減少するが、集中的な膜の脱分極によってロックが外れる(Hansen er al)。 NMDARは、7種類のサブユニット(GluN1,GluN2A-D、GluN3A-B)が4量体の複合体に結合して可変的に会合することで形成される(Hansen er al)。 これらの受容体、特にシナプスに存在する受容体のほとんどは、d-セリンを共役とし、主に大量のカルシウム流入シグナルを介して、シナプスの円滑化や網目構造の発達のための神経可塑性に重要な役割を果たしている(Volianskis er al 2015)。神経可塑性の最も重要なプロセスは、シナプスの増強であり、最初の短期増強(STP)に続いて長期増強(LTP)があり、ネットワークの円滑化の究極の安定した段階である。STPには2つの要素があり、速い方のメカニズムはGluN2AとGluN2Bサブユニットの活性化に関係し、遅い方のメカニズムはGluN2BとGluN2Dサブユニットの活性化に関係する。その後のLTPでは、主にGluN2AとGluN2Bサブユニットを持つトリヘテロメリックなNMDARの活性化が必要である。GluN2Aサブユニットを持つNMDA再受容体のシナプスでの活性化は、αセクレターゼによるAPPのアミロイド前駆体の非アミロイド化に関与する(Hoey et al 2009)一方、GluN2Bサブユニットはβセクレターゼによるアミロイドβ合成を促進する(Hardingham and Bading, 2010)。対照的に、シナプス領域外のNMDA受容体(GluN2Bサブユニットを含む)は、グリシンをコナゴニストとして、長期抑圧(LTD)や、急性脳損傷や神経変性に関連した毒性プロセスに主に関与している(Bordji er al 2010; Hardingham et Bading, 2010)。シナプス領域外の活性化は、興奮毒性または非興奮毒性のアポトーシスに至るまでの複雑な反応のカスケードを開始する(Papouin and Oliet, 2014)。多くの研究が、シナプス領域外受容体の慢性的な活性化により、アミロイドβが過剰に合成されることを示している(Bordji et al 2010; Rush and Buisson, 2014)。GluN2B-サブユニット型の一部のシナプスNMDA受容体もこのプロセスに関与している(Hardingham and Bading, 2010)。アルツハイマー病では、様々な異常経路により、シナプス外NMDARの過剰発現/過剰刺激、およびシナプスNMDARの興奮毒性促進活性が生じる(Kamenetz et al 2003)(Dewachter et al 2009,Mota et al 2014,Pozueta et al 2013,Wang and Reddy 2017,Zhang et al 2016)。実際、アミロイドβの蓄積は、過酸化水素やヒドロキシルラジカルの産生が増加することにより、膜のプロオキシデーション効果がある(Butterfield et al 2010,Mota et al 2014,Zhang et al 2016)。これらの変化は、膜の過興奮性とNMDAの高活性化をもたらし、その後の興奮毒性を引き起こす。アルツハイマー病におけるタウの興奮毒性の役割は、シナプス外のNMDARを過剰に刺激することでタウのリン酸化が進み、樹状突起が蓄積されることを示唆している(Zhang et al 2016)。
そのため、いくつかの薬理学的研究がNMDAを標的としているが、開発された薬剤のEfficacyは、回避効果、一過性の反応とその後のリバウンド効果、そして狭い治療窓によって制限されている。興味深いことに、いくつかの関連研究では、シナプスおよびシナプス領域外のNMDA受容体が、細胞外のMgによるチャネルブロックに対して非常に敏感であることが示された(Burnashev et al 1992年、Calabresi et al 1992年、Nikolaev et al 2012,Wang and MacDonald、1995)。なお、NMDAチャネル遮断のためのMgの深い結合部位は、メマンチンのものと同じである(Glasgow et al 2018)。メマンチンは、アルツハイマー病管理に一定の効果を示し、中等度から重度の段階で適応となる(Bond er al 2012)。Mgはメマンチンと同様に、NMDARを介した興奮毒性を抑え、さらにAPPのβプロセッシングを抑制することが期待されている。Nikolaev et al 2012)は、高濃度のMgを用いて、ラットの海馬スライスにおけるニューロンのNMDA活性を阻害し、NMDA-Rブロック仮説を確認した。この阻害は、APP-processingの変調と相まって、アミロイドβ合成の大幅な減少をもたらすことができる(Yu et al 2010;Yu et al 2018)。Li et al 2014)の研究では、NMDARシグナルの有意なダウンレギュレーションとβセクレターゼの発現の安定化が報告されている。さらに、彼らの研究結果は、高い細胞外レベルのMgが、カルシニューリンの過剰な活性化を防ぐことで、アミロイドβによるシナプスのNMDARの過剰な減少を防ぐことを示唆していた(Li er al)。
シナプス制御以外にも、Mgは慢性的なグルタミン酸刺激によるエネルギー代謝異常(アルツハイマー病発症に関与)に対して神経保護作用を示す。この性質は、生理的濃度のMgを含むラット大脳皮質神経細胞標本に、興奮毒性レベル(100μM)のグルタミン酸を作用させたときに明らかになっている(Clerc er al)。 このような条件下で硫酸Mgを添加すると、神経細胞の予備呼吸能力が改善された。ミトコンドリア内のアミロイドβ蓄積は、呼吸鎖の複合体IIIおよびIVの機能低下→細胞質カルシウムレベルの上昇と酸化ストレスおよびATPの産生低下→最終的にカルシウムレベルと酸化ストレスの悪化という有害なサイクルで呼吸鎖を障害することが知られているので、この効果はさらに興味深いものである(Moreira er al)。
6. 現在の限界と研究の必要性
上記の研究では、アルツハイマー病患者では脳脊髄液のMg濃度が低いことが報告されているが、このイオンの有効性に関する臨床的背景は弱いものである。今のところ、実験結果は心強いものであるが、そのような結果は、臨床研究のものとして強い価値を提供するものではない。第一に、実験において、動物モデルはアルツハイマー病の人間の状態を正確に再現することができない(Chun er al 2018)。この限界は、実際の人間の認知的影響の推定や、有効な臨床用量に関連する可能性のある回避的効果の予測を損なう。さらに、試験管内試験の神経化学/神経組織学的変化や動物の記憶の改善を期待される臨床結果と相関させることは難しい課題である。実際、ヒトの相対的な治療反応は、侵入した特定の要因(遺伝や環境の決定要因)や神経細胞の特異性(ニューロンネットワークや神経伝達を含む)に左右される。とはいえ、Mgは内因性物質であり、これまでにアルツハイマー病に対して薬剤的に開発された薬剤よりもはるかに多くの神経膠機能を標的としている可能性があるため、現在入手可能なデータは臨床研究チームがもっと関心を持つべきである(Bond et al 2012,Kamat et al 2016,Wenzel and Klegeris 2018)。さらに興味深いことに、アルツハイマー病へのMgの使用は今のところヒトのガイドラインでは考慮されていないが、様々な神経学的病理におけるMgの特性は、臨床的に良好な忍容性と、神経興奮性および神経膠の生存に対する強固な影響を示している(Trapani et al 2011)。今後の課題は、このイオンの最適な薬理作用を明らかにし、大規模なヒトコホートでの使用を評価することである。
とりわけ、Mgの潜在的な神経保護効果は、生体内でのバイオアベイラビリティ、主に神経膠のアバイラビリティによって制限される。実際、全身のMgは、腸管吸収、糸球体濾過、尿細管再吸収、尿中排泄のバランスによって厳密に制御されている(Allgrove, 2009; Schmitz er al)。 多くの循環副成分と同様に、MgのBBBや血液-脳脊髄液関門を通過する際の移動も細かく制御されており、主にTRPM6型とTRPM7型の一過性受容体電位メラスタチン(TRPM)が必要とされている(Ghabriel et Vink, 2011)。そのため、高用量を使用しない限り、サプリメントの神経細胞への影響は限定的である。例えば、抗けいれん薬の予防のためには、血清Mg濃度が4〜7mEq/Lの範囲が必要である(正常範囲は1.5〜2.5mEq/L)(Hunter and Gibbins, 2011)。Mgの治療域の広さを考慮すると、アルツハイマー病に必要と思われる高用量を投与しても、ほとんどの患者は十分に耐えられると考えられる。静脈内投与は、血清中での存在感を高め、脳関門を通過させるための興味深い選択肢であるが、この方法は長期治療には適していない。もう一つの方法である髄腔内投与は、最適なバイオアベイラビリティを得るための理想的な選択肢であったかもしれないが、このようなアプローチは通常、感染のリスクが高い複雑な移植手術を必要とする。したがって、BBBの脳側で高濃度のMgを得ることができる最適な方法を特定することが重要である。その他
また、Mgのアジュバントとしての利用も検討すべきである。実際、ほとんどの抗アルツハイマー病薬は、実質的な抗神経変性作用を示しているが、標的とする病態生理経路は限られている。そのため、高用量の投与が必要となるが、治療の幅が狭くなってしまう。このような観点から、Mgは、合成薬の主要な作用を潜在化させるだけでなく、合成薬によって明らかにされた薬力学的標的に作用するという興味深い選択肢を提供する。いずれにしても、神経細胞環境でのMgの利用可能性に関連した治療濃度域を慎重に検討する必要がある。実際、YangとKsiezak-Reding(1999)は、Mgとカルシウムの局所的な上昇が、一対のらせん状フィラメントであるタウ(PHF-tau)の病的な蓄積を引き起こすことを示唆している。この研究は試験管内試験で行われたものであるが、神経細胞環境におけるMgの高濃度と中濃度の上昇がもたらす回避的な慢性効果に注意を払う必要があることを指摘している。今後の専門的な研究の方法論としては、それぞれの選択肢の長所と短所を明確にすることが必要である。
7. 結論
Mg は、MCI の場合には アルツハイマー病 の予防に、アルツハイマー病 の後期段階ではアジュバントの選択肢として考慮できることを裏付ける確かな証拠がある。実際、いくつかの実験結果から、Mg は アルツハイマー病 や他の神経変性疾患の主な病態生理学的プロセスを阻害することがわかっている。しかし、アルツハイマー病におけるマグネシウムの有効性の臨床的背景はまだ乏しいため、より大規模な臨床研究が必要である。さらに、最適な薬物動態と薬力学のための最良の戦略を特定するためには、さらなる基礎研究が必要である。