
Contents
www.ncbi.nlm.nih.gov/pmc/articles/PMC7016185/
Front Physiol. 2020; 11: 63.
2020年2月6日オンライン公開
要旨
筋萎縮性側索硬化症(ALS)は、脊髄、脳幹および運動皮質の運動ニューロンが選択的に失われる神経変性疾患である。ミトコンドリア機能障害や電子輸送チェーンの劣化に伴う酸化ストレス(OS)は、神経変性の一因であることが示されており、ALSの発症に関与している可能性がある。中枢神経系が冒された領域では、活性酸素種(ROS)が多量に発生し、抗酸化防御能が低下している。科学的研究では、特徴的な酸化ストレスに対抗するための抗酸化剤による治療と、前駆体の使用によるニコチンアミドアデニンジヌクレオチド(NAD+)レベルの再生が提案されている。本総説では、ALSの治療戦略としてのニコチンアミドリボシドとプテロスチルベンの役割の可能性について検討する。
キーワード:筋萎縮性側索硬化症、神経変性疾患、酸化ストレス、ミトコンドリア機能障害、ニコチンアミドリボシド、プテロスチルベン
酸化ストレス
ほとんどの真核生物は、細胞のエネルギー代謝が正常に機能するために酸素を必要としており、これは進化上の利点であり、エネルギー生産の高効率な形態でもある(Jie et al.、2013)。電子伝達系(ETC)において、酸素は電子の取り込みによって部分的に還元され、二次生成物としてフリーラジカルを生成する(Falkowski and Godfrey, 2008; Venditti et al.) フリーラジカルは、最終軌道層に1個以上の不対電子を持つ原子または分子であり(Chandrasekaran et al.、2017)、連鎖反応を行うことができる強い反応性を持ち、細胞や組織の酸化的損傷の原因となる(Soleiro-Villavicencio and Rivas-Arancibia, 2018)。これらは、ROSと反応性窒素種(RNS)に分類される(De Gara and Foyer, 2017)。また、反応性鉄種(RIS)(Dixon and Stockwell, 2014)、銅種(RCS)(Gyulkhandanyan et al, 2003)なども含まれる。主なROSは:スーパーオキシドアニオンラジカル(O・2)、ヒドロキシルラジカル(OH・)、過酸化水素(H2O2)(Popa-Wagner et al.、2013)である。RNSには、一酸化窒素ラジカル(NO・)、ニトロキシルアニオン(NO・)、ニトロソニウムカチオン(NO+)、ペルオキシナイトライトアニオン(ONOO・)などがある(Haliwell、2012; Rogers他、2014;Zuo他、2015)。
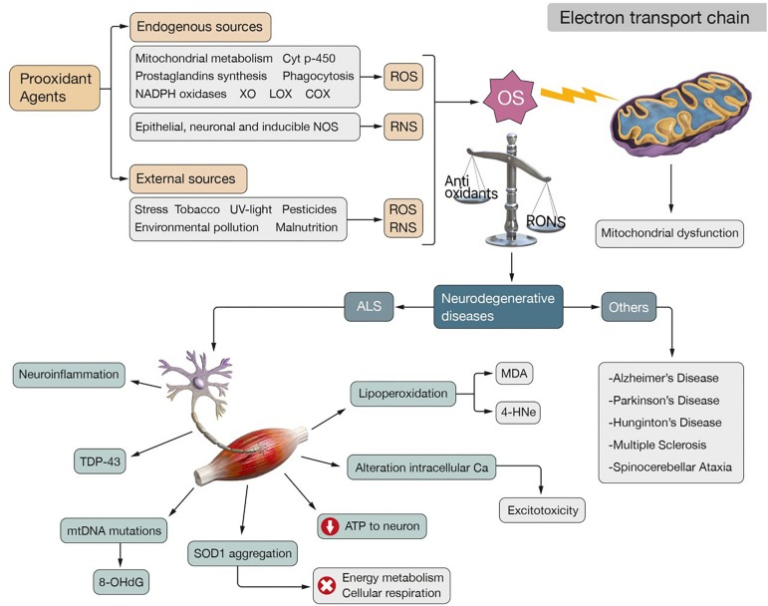
活性酸素の生成は、主に呼吸鎖、チトクロームp-450の活性、プロスタグランジンの合成、食作用に関わる酵素反応に二次的に生じる(Pizzino et al.、2017)。哺乳類細胞では、ミトコンドリア活性とシトクロムp-450の代謝が最も寄与している(Jha et al., 2017)(図1)。
図1 ALSの生理学的発症機構
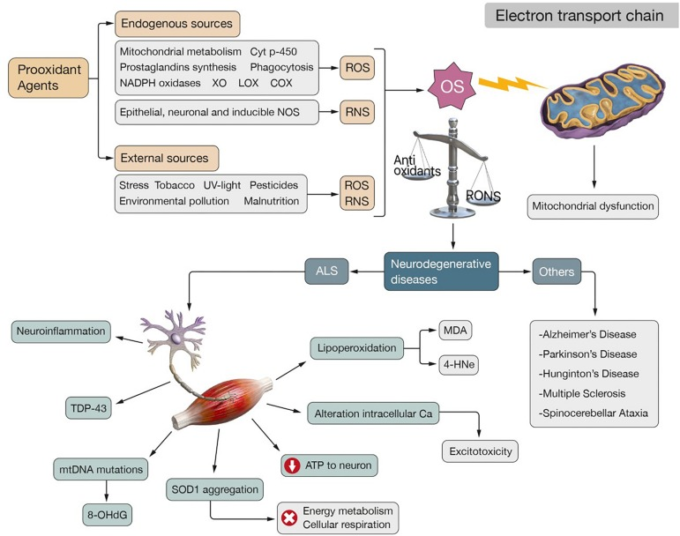
酸化ストレスは、抗酸化防御とRONSのバランスが崩れ、これらの第2のものが有利になることで引き起こされる。ROSの内因性産生は、ミトコンドリアおよびチトクロームp-450代謝、プロスタグランジン合成、食作用、NADPH酸化酵素、XO、LOXおよびCOXといった異なるプロオキシダント作用により、二次的に発生する。RNSの内因性供給源としては、上皮性、神経性、誘導性NOSの活性を挙げることができる。外因性物質もまた、ストレス、タバコ、紫外線、農薬、環境汚染、栄養失調など、RONSを生成する可能性がある。酸化ストレスはETCの正常な機能に影響を与え、ミトコンドリアの機能不全を引き起こし、さらに活性酸素を発生させ、代謝ストレスを増大させる「悪循環」をもたらす。このような状況は、次のような神経変性疾患を特徴づける。ALS、AD、PD、MS、HD、SAなどの神経変性疾患の特徴である。ALSの場合、酸化ストレスとミトコンドリア機能不全がその病因に関与する2つのメカニズムとして同定されている。これらは、神経炎症、TDP-43の凝集、8-OHdGなどの酸化ストレス特有のバイオマーカーを産生するmtDNAの変異、エネルギー代謝や細胞呼吸を損なうSOD1の凝集、ニューロンへのATP供給の影響、興奮毒性の原因となる細胞内カルシウムの恒常性障害に関連している。また、神経細胞膜の脂質酸化に伴うMDAや4-HNEなどの高反応性最終生成物の生成も観察される
ミトコンドリアによる活性酸素の生成は、生理的および病理的な条件下で、外膜、内膜、またはミトコンドリアマトリックスで起こることがある(Pizzino et al.、2017)。O∙-2の生成は、ニコチンアミドアデニンジヌクレオチドリン酸(NADPH)の蓄積がある場合、またはミトコンドリア内に還元型CoQプールがある場合に起こる(Murphy、2009年)。O∙-2はまた、アラキドン酸代謝の過程で、内皮細胞や炎症細胞によるリポキシゲナーゼ(LOX)やシクロオキシゲナーゼ(COX)の酵素活性(Valkoら、2007)により、二次的に生じる可能性がある(Al-Guboryら、2012年)。O・2は、H2O2やOH・を生成する反応に関与している可能性がある(Kumar and Pandey, 2015)(図1)。
肝臓に存在するチトクロームp-450酵素は重要な活性酸素生成源であり、その機能はNADPH依存的なメカニズムによってO∙2生成反応を触媒することだ(Liochev, 2013)。ここには遷移イオン、酸素、電子移動過程が含まれるため、ROS生成のリスクは高い(Liochev, 2013)。また、多形核細胞、マクロファージ、内皮細胞の細胞膜上には、NOX(NADPH oxidase)酵素群が存在し、NADPHを電子供与体として生体膜上で酸素をスーパーオキシドに変換し、副産物として活性酸素を放出することを促進する(アタシら、2015)(図1). 内皮のキサンチンデヒドロゲナーゼはキサンチンオキシダーゼ(XO)と相互作用してO・2とH2O2を生成し、その結果、別のフリーラジカルの発生源となる(Turrens, 2003)(図1)。
非酵素的な反応は、有機化合物と酸素の反応や、電離放射線に細胞がさらされた後の活性酸素の生成にも関与していると考えられる (Valko et al., 2007) (図1)。
一酸化窒素(NO∙)のようなRNSの内因性放出は、上皮性NOS、神経性NOS、誘導性NOSの3種類の一酸化窒素合成酵素の触媒反応によってl-アルギニンから生成され、これらは種々のエンドトキシンやサイトカインのシグナルに応答して活性化する(FörstermannとSessa、2012年)。したがって、酸素はこのNO・と反応し、ONOO-などの反応性の高い分子を形成することができる(Salisbury and Bronas, 2015; Sharina and Martin, 2017)(図1)。
活性酸素・窒素種(RONS)の内因性産生は、外因性プロオキシダントファクター:環境・大気汚染、水質汚染、農薬や工業溶剤などの化学物質、重金属や遷移金属、異なる種類の異生物、紫外線・X線・ガンマ線による照射、ストレス、タバコ、燻製肉、廃油の使用、栄養失調によって条件付けられうる(Phaniendra et al, 2015; Niedzielska et al., 2016; Solleiro-Villavicencio and Rivas-Arancibia, 2018; Zewen et al., 2018)(図1)。
生理的濃度の活性酸素・窒素種は、数多くの細胞機能の調節因子である:細胞シグナル伝達経路、細胞生存の制御、血管緊張の調節、細胞膜受容体によるシグナル伝達、膜更新、ホルモンの合成・放出、炎症性サイトカイン転写調節の増加(Robberecht、2000; Ray et al, 2012)、タンパク質のリン酸化、イオンチャンネルや転写因子への作用、甲状腺ホルモンの産生、細胞外マトリックスへの架橋(Brieger et al.)
生体は、RONSの産生と抗酸化系による除去能力の間でレドックスホメオスタシスを維持しようとし(Zuo et al., 2015)、これにより、高濃度のRONSに一時的にさらされた後にレドックス状態を再確立し、酸化ストレスと呼ばれる不均衡状態である酸化還元恒常性が悪化するリスクを最小化する(Sies, 1986; Coyle and Puttfarcken, 1993; Liguori et al, 2018) (図1)。しかし、生体のほとんどの反応は酸化還元状態に依存しているため(Tan et al., 2018)、恒常的な曝露は細胞内シグナルや遺伝子発現に深刻な影響を与え、不可逆的な病理的結果をもたらすことがある(Rhee et al., 2003)ため、酸化還元恒常性はフリーラジカルへの曝露の大きさと期間によって条件付けされる。
神経変性疾患などの酸化ストレスに関連する疾患は、老化(Liguoriら、2018)、組織および器官機能の進行性損失(Flatt、2012)、調節プロセスの変化、生体の抗酸化能の低下、酸化還元バランスの達成を損なうRONSによる不可逆的組織損傷を伴う生理学的段階(Romanoら、2010)と関連している。酸化による損傷は、酸化還元シグナル伝達経路に関与する酵素の欠陥に依存する(Tan et al.、2018)。
フリーラジカルは、細胞を横断し、主要な高分子(脂質、タンパク質、核酸)に修飾を引き起こし、その構造を損傷し、正常な機能を変化させる(Salisbury and Bronas, 2015)。過酸化脂質は、さまざまな疾患状態と関連しており、不安定な細胞膜、低密度リポタンパク質(LDL)および多価不飽和脂肪酸(PUFA)の酸化の原因となっている。リジン、アルギニン、プロリン、スレオニンなどの様々なアミノ酸が酸化されることにより、タンパク質の機能不全が引き起こされる。DNAの酸化的損傷は、深刻な変異や転写への悪影響を生じさせ、酸化に対してより脆弱なRNAを生成する。このような状況が慢性的に続くと、細胞死を引き起こす可能性がある(Huiyong et al, 2011; Dizdaroglu and Jaruga, 2012)。
酸化ストレスは、慢性的な炎症性病態(Kehrer and Klotz, 2015)、進行性の脳損傷、および以下のような神経変性疾患の発症において重要な役割を担っている。筋萎縮性側索硬化症(ALS)、アルツハイマー病(AD)、パーキンソン病(PD)、多発性硬化症(MS)、ハンチントン病(HD)、脊髄小脳失調症(SA)(Diasら、2013; Islam、2017; Zewenら、2018) (図1)。
酸化ストレスと神経変性疾患
神経変性疾患の正確な病因は依然として不明だが、遺伝的素因、特定の内因性因子、環境因子への曝露などの側面が関与する複雑かつ多因子性の起源が確立されている(Correia and Moreira, 2010; Correia et al.) 活性酸素は、これらの病態の病因の重要な要因であり(Bhat et al., 2015)、神経変性疾患の患者では、高レベルの酸化ストレスバイオマーカーが観察されている(St-Pierre et al., 2006; Niedzielska et al., 2016)。しかし、生理的濃度のフリーラジカルは、正常な脳機能において重要な役割を果たす(De Silva and Miller, 2016; Scialò et al.) したがって、ROSは以下のことに寄与し得る:血管形成、脳灌流、細胞シグナル伝達、シナプス可塑性、神経伝達物質分泌および脳血管拡張(Masaad and Klann, 2011; Grochowski et al, 2018; Neal and Richardson, 2018)。ミトコンドリア活性に続発するROSの適度な増加は、有害な薬剤に対する神経保護機能をもたらし、大量のROS形成に対して予防的でさえあるプレコンディショニングを生み出す(Dirnagl and Meisel, 2008; Jou, 2008; Dirnagl et al, 2009)。問題の起源は、酸化還元ホメオスタシスの障害にあり、細胞膜に損傷を与え、中枢神経系(CNS)の生存能力と完全性を損なう(Van Horssenら、2011; Payne and Chinnery、2015)。活性酸素の過剰は、脳の微小循環の変化と関連している(Freeman and Keller, 2012; Staiculescu et al, 2014)。O・2やH2O2は脳血管の収縮を引き起こすことができる(Allen and Bayraktunan, 2009)。H2O2レベルの増加は、脳血管細胞におけるプロアポトーシス剤の増加と関連しており(Li et al., 2003)、酸化ストレスはNO・シグナルの中断、ひいてはその血管拡張能と抗炎症能を介して脳血管機能を変化させることができる(Miller et al., 2010; De Silva and Miller, 2016)。さらに、活性酸素は、サイトカインおよびケモカインの分泌に続いて、炎症性状態の維持に寄与する(Hsieh and Yang, 2013; Kehrer and Klotz, 2015)。
CNSは特に酸化的な損傷を受けやすい(Cordeiro, 2014)。脳は大部分が過酸化脂質に高い感受性を持つPUFAによって形成されており(Nunomura et al, 2006)、運動神経細胞は酸化ストレスに高い感受性を持ち(Simpkins and Dykens, 2008)、CNSはグルタチオンペルオキシダーゼ(GPx)、カタラーゼ(CAT)またはスーパーオキシドディスムターゼ(SOD)などの保護酵素の活性が低く、細胞再生の能力が低く、抗酸化能力が低い(Fischer and Maier, 2015; Kim et al, 2015; Formella et al, 2018)。脳は体重の2%しか占めておらず(Mergenthaler et al., 2013)、ミトコンドリア密度は神経細胞よりも筋細胞で高い(Gandhi and Abramov, 2012)ものの、体の総酸素の20%まで消費できる高い代謝率を持つ器官である(Moreira et al., 2009;Ronnett et al.) これは、他の組織よりも10倍高いエネルギーとグルコースの消費量と、ミトコンドリアのエネルギー生産への依存度が高く、ROSの増加の原因となることを意味する(Carvalho and Moreira, 2018)。脳の高い需要率とエネルギー消費量を考慮すると、ミトコンドリア変異の大部分はCNSの機能に影響を与えることができ、神経変性疾患と関連している(Angelova and Abramov, 2018)。したがって、ミトコンドリア機能不全は現在、神経変性における「収束点」とみなされている(Correia et al., 2010; Bennett et al., 2014; Arun et al., 2016; Kurtishi et al., 2018)。
ミトコンドリアの機能不全は、神経細胞へのATPのエネルギー供給、カルシウムの恒常性を損ない、ミトコンドリアDNA(mtDNA)の変異率や神経細胞膜の脂質過酸化を加速させる大量の活性酸素をもたらし、PUFAの分解や反応性の高い最終生成物の生成を引き起こす。マロンジアルデヒド(MDA)および4-ヒドロキシ-2-ノネナール(HNE)(細胞損傷およびアポトーシスに関連する最も毒性の高い化学種)(Petrozzi et al. , 2007; Albarracín et al., 2012; Fritz and Petersen, 2013) (図1)。mtDNAに変異が蓄積すると、酸化的ダメージの増加、エネルギー速度の低下、活性酸素の増加が引き起こされる。このように、ミトコンドリア機能障害は、神経細胞の損傷、遺伝子変異、代謝ストレスを担う「悪循環」を発生させ、アポトーシスにつながる状況である(Van Houten et al., 2006; Selvaraji et al., 2019; Wang et al., 2019)。
ミトコンドリア機能障害は、神経変性疾患の病態(ALS、PD、AD、MS、HD)の主な原因と考えられている(Lin and Beal, 2006; Federico et al, 2012)(図1)。これらの疾患の患者の脳における死後の研究により、ミトコンドリア機能不全が神経細胞の変性と死につながる共通の事象であることが判明している(Schon and Manfredi, 2003; Bao and Swaab, 2018; Du et al, 2018)。
脳の老化は、酸化ストレスに対する感受性を高め、抗酸化防御の有効性を低下させ、機能不全、炎症、弾力性の低下、神経変性疾患の発症に関与する病因に対する感受性を高める(Federicoら、2012; Guoら、2013)。また、mtDNAの変異の蓄積、酸化的リン酸化経路の機能不全、レドックスホメオスタシスの障害にも寄与している(Sarasija and Norman, 2018)。
現在、活性酸素は、神経変性疾患の発症および進行の主な要因の1つとして提唱されている(Morató et al.、2014)。RONS濃度の上昇は、組織損傷、ミクログリアやアストロサイトの活性化、神経細胞機能障害、神経変性、細胞死の誘発因子であることを示す証拠がある(Federico et al, 2012; Morató et al, 2014; Schieber and Chandel, 2014)。このように、酸化ストレス、ミトコンドリア機能不全、老化、神経炎症の間の「悪循環」が実証されている(Witteら、2010;Van Giauら、2018)。しかしながら、酸化ストレスが神経変性疾患の開始を誘発する主要な原因であるか、または神経細胞への損傷の広がりに関連する副作用であるかは不明であるため、関与する生理学的メカニズムを理解するためにさらなる研究が必要である(Schettersら、2018)。
筋萎縮性側索硬化症(Amyotrophic Lateral Sclerosis)
ALSは、シャルコー病またはルー・ゲーリッグ病としても知られ、もともとJean-Martin CharcotとJoffreyによって記述され(Chenら、2013;Niedzielskaら、2016)、最も一般的な運動ニューロン疾患である(Rechtmannら、2015)。運動皮質の上部運動ニューロン(UMN)と脳幹および脊髄の下部運動ニューロン(LMN)の選択的な喪失を引き起こす(Brown and Al-Chalabi, 2017; Valko and Ciesla, 2019)。
ALSの患者は、全筋( bulbarと呼吸器)の脱力、筋脱神経、萎縮と進行性の麻痺、嚥下障害(Al-Chalabi and Hardiman, 2013; D’Amico et al., 2013; Gowland et al., 2019)、呼吸筋力の低下を起こし、呼吸不全を起こして死に至る(Sferrazza-Papa et al., 2018)。生存率は発症後3~5年である(Andersen, 2006; Gordon, 2013; Coan and Mitchell, 2015; Wier et al.、2019)。
ALSの発症率および有病率は、研究の地理的およびデザインによって異なり(Marin et al., 2017; Kaye et al., 2018)、アフリカ系アメリカ人およびヒスパニック系集団の間で違いがある(Gordon et al., 2013)。発症率は西ヨーロッパ諸国では1.000.000人/年で2~3例、有病率は100.000人/年で4.6~5例である(Chiò et al., 2013; Talbot, 2016)。ALSの全有病率は、2020年までに人口10.000人あたり8.58件、2116年までに人口10.000人あたり9.67件と大幅に増加すると予想されている。英国では2010年の有病率は1415例で、2020年には1701例、2116年には2635例に増加すると予測されている(Gowland et al.、2019年)。
ALSは一般的に50~60歳の白人成人に発症し(Chiò et al., 2013; Mehta et al., 2018)、小児での発症は非常に稀である(Martin, 2010; Bäumer et al., 2014; Ingre et al., 2015)。発症年齢は女性より男性の方が早く、男性は脊髄病変を起こしやすいのに対し、女性では嵩上げ病変が多く見られる(McCombe and Henderson, 2010)。
ALSは、家族性ALS(fALS)と散発性ALS(sALS)の2種類に分類される。fALSは症例の約5~10%を占め、常染色体優性遺伝パターンを持つ一連の遺伝子の不均一な変異に関連している(Mitchell and Borasio, 2007)。fALSの最大20%は、Cu-Znスーパーオキシドジスムターゼ(SOD1)をコードする遺伝子の変異に起因する。sALSは最大90-95%の症例で発生するが、その起源はまだ不明である(Martin, 2010; Andersen and Al-Chalabi, 2011; Pan et al, fALSとsALSはあらゆる随意筋を侵す可能性があるが、その提示、表現型、進行は多様であり(Wei et al., 2019)、鑑別診断の妨げとなっている(Swinen and Robberecht, 2014; Ghasemi, 2016; Van Es et al., 2017)。ALSは、四肢の病変(80%)または嵩上げ病変(20%)という二重発症を示すことがある(Parakhら、2013年)。
運動ニューロンの死は、痙性、筋力低下と萎縮、反射亢進、けいれん、筋収縮、バビンスキー徴候、協調性の喪失、随意筋群の麻痺、嚥下障害、嚥下、言語、呼吸の困難といった特徴的な臨床的特徴をもたらす(Weiduschatら、2014;Zufiríaら、2016;Soleiro-Villavicencio and Rivas-Arancibia、2018)。体重減少、代謝亢進、高脂血症などの代謝障害と関連している(Dupuis et al., 2011)。fALSとsALSは感覚(視覚、嗅覚、味覚、聴覚、触覚)に影響を与えない(Fiala et al., 2013)。
ALSの病態に関連するいくつかの因子が同定されている。酸化ストレス、ミトコンドリア機能障害、神経炎症、神経伝達物質グルタミン酸の増加による興奮毒性、軸索伝達およびRNAの代謝の欠陥、アポトーシス、細胞骨格異常、膜輸送の乱れ、小胞体ストレス、タンパク質ミスフォールドおよび凝集(Papadimitriou et al, 2010; Blokhuis et al., 2013; Peters et al., 2015; Bond et al., 2018; Yusuf et al., 2018)、およびシステイン残基プロテオスタシスの一般的な異常に寄与する酸化またはパルミトイル化などのシステイン修飾(Valle and Carrì, 2017)などが挙げられる。環境条件とALSの疫学との間には、アルコール、タバコ、座りがちな生活、真菌およびウイルス感染、または電磁波への曝露などの関係が確立されている(Yu et al.)
現在、ALSに有効な治療法はなく、治療は対症療法と緩和治療に限られている(Bhattacharya et al.) リルゾール(Millerら、2012年)とエダラボン(Rothstein、2017年)は、生存率を2-3ヶ月増加させる唯一の承認された薬理療法ですが(Dorstら、2017年、Hodgkinsonら、2018年)、エダラボンは早期のALSの人々のサブセットにのみ有益である(Abeら、2017年)と言われている。他の治療ルートは、以下を含む集学的治療を示している:看護師、栄養士、作業療法士、理学療法士、心理学者、ソーシャルワーカー、言語療法士(Gordon, 2013)。
酸化ストレスと筋萎縮性側索硬化症
酸化ストレスバイオマーカーの存在と高レベルのROSは、特にALS患者のCNS領域で決定されており(Goleniaら、2014)、これは、酸化還元恒常性の障害が関連因子であり、fALSおよびsALSにおける神経変性の発生および進行に関連することを示す(Carrìら、2015;Petrovら、2017;Smithら、2017)。患部細胞では、ある種の活性酸素(H2O2およびO・2)の高レベルが観察されている(Bealら、2005年)。
酸化制御の喪失と過剰なROS生成は、特にSOD1の変異型(Ferri et al., 2006; Hooten et al., 2015; Angelova and Abramov, 2018)と関連しており、最大で150種類の変異がある(Gamez et al., 2006; Al-Chalabi et al., 2013)。SOD1は、O・2のH2O2およびO2への変換を触媒する抗酸化酵素であり、酸化ストレス、細胞損傷(Mattiazzi et al., 2002; Li et al., 2011; Pehar et al., 2014)、エネルギー代謝および細胞呼吸(Saccon et al., 2013)の調節において重要である。SOD1の酸化および/またはミスフォールディングは、上記プロセスの制御の悪化、ミトコンドリア機能不全、およびスーパーオキシドの産生の増加をもたらす(Israelsonら、2010; Picklesら、2013、2016; Richardsonら、2013)。その変異は、細胞小器官が酸化的損傷を受けやすくなり(Dengら、2006;Ahtoniemiら、2008)、アストロサイトを攻撃する過剰なレベルのROSが発生する(Islam、2017)。酸化ストレスは、SOD1の凝集(Brujinら、2004年)ミトコンドリア機能不全(Liuら、2004年;Vijayvergiyaら、2005年)を増強することに寄与する。
ALSのトランスジェニックマウス(SOD1G93A)により、発症メカニズムの同定と「悪循環」の確立が可能になった。酸化ストレス、ミトコンドリア機能不全、ETCの劣化(Jungら、2002年、Menziesら、2002年、Pfohlら、2015年、Xiaoら、2018年)。CNSで形成される酸化種のほとんどは、酸化的リン酸化の二次的なものであり(Brookesら、2004;Balabanら、2005;Karamら、2017)、ミトコンドリア機能不全はROS産生の最大の源なので、脊髄および運動皮質の運動ニューロンの影響の明確な兆候である(Bendottiら、2001;Kawaharaら、2004;Loizzoら、2010;CozzolinoおよびCarrìら、2012)。ミトコンドリア損傷は、細胞内カルシウムレベルおよびETCの正常な機能の変化を引き起こし(Bond et al., 2018)、それがROSの増加および酸化的損傷の拡大につながる連鎖反応の活性化を助長し、「悪循環」を強化する(Harraz et al, 2008; Federico et al., 2012; Rojas et al., 2015; Loeffler et al., 2016; Van Horssen et al., 2017)、アポトーシスに対する細胞の感受性を高める(Guegan et al., 2001; Iverson and Orrenius, 2004)。
ALS患者において、抗酸化防御マーカーの変化が観察されている(Covaら、2010)。しかし、この疾患の異質性、発症形態、発症メカニズムの多様性が広いため、結果はランダムである(Johnson et al., 2012)。ALS患者の赤血球(Babuら、2008)および運動皮質(Ikawaら、2015)における還元型グルタチオン(GSH)の低レベル、全身の炎症性状態(IL-6およびIL-8のレベル上昇)、抗酸化系の障害(Erhartら、2015)が強調されている。ある研究では、11人のALS患者の運動皮質におけるGSHのin vivoレベルを、年齢をマッチさせた11人の健常ボランティアと比較し、ALS患者のGSHレベルは健常ボランティアよりも31%低いことが明らかになった(Weiduschat et al.) また、グルタチオンレベルの低下は、hSOD1wt過剰発現マウスモデル(野生型hSOD1を中程度のレベルで過剰発現させたヘミ接合体マウス)において運動ニューロン変性を引き起こし(Killoyら、2018)、SOD1G93Aマウスでは運動ニューロン死を加速した(Peharら、2014)。カタラーゼ(CAT)活性は、sALS患者の赤血球で低下し(Nikolic-Kokicら、2006;Babuら、2008)、神経学的に健康な対照群と比較して有意に低下している(Goleniaら、2014)。
試験管内で行われた研究では、神経細胞(アストロサイトおよびオリゴデンドロサイト)の酸化的損傷が神経変性に関係することが示された(Pollari et al.) 酸化的不均衡は、グリア細胞の数を減少させ、軸索信号の伝達能力を低下させる(Park et al.、2007)。
酸化ストレスのマーカーは、ALS患者の脳脊髄液(CSF)、組織、血液、尿で測定されている(Blasco et al.、2017年)。sALSおよびfALS患者の神経細胞組織を死後分析した結果、タンパク質、脂質、DNAにおいて酸化ストレスバイオマーカーの増加が認められた(Beal, 2002; Agar and Durham, 2003; Kim et al, 2003; Turner et al, 2013; Bozzo et al., 2016)。最も頻繁に研究されているバイオマーカーは、カルボニル化および/またはグリコシル化タンパク質、脂質過酸化およびDNA損傷である(Shibata et al.、2002)。
ALS患者の神経組織では、運動皮質で高レベルのカルボニル化タンパク質が測定されており(Sasaki et al., 2000; Guareschi et al., 2012; Tan et al., 2014)、sALS患者の赤血球の血漿では高レベルのタンパク質酸化の高度生成物(LoGerfo et al., 2014)である。ALSマウスモデルでは、ペルオキシナイトライトによるアミノ酸残基の酸化に二次的に起因する非機能性タンパク質が観察されている(Yoshino and Kimura, 2006)。また、酸化損傷により、ALSの病態に関わる主要な疾患関連タンパク質であるTDP-43の凝集が起こり(Moujalled et al., 2017; Oberstad et al., 2018)、神経細胞における酸化ストレスを促進する(Duan et al., 2010; Cohen et al., 2012, 2015; Magrané et al., 2014)(図1)。
CNS脂質は酸化的損傷を受けやすく(Niki, 2014)、脂質過酸化は高レベルのHNE、MDA(Radakら, 2011)およびF2-イソプロスタン(F2-IsoPS)(Milneら, 2007; Milatovicら, 2011; Van’t Erveら, 2017) を生成する (図 1)。ALS患者のCSFでは、高レベルのHNEと3-ニトロチロシン(3-NT)が得られている(Simpson et al., 2004; Duffy et al., 2011; Perluigi et al., 2012; Ayala et al., 2014; Santa-Cruz and Tapia, 2014)。
DNAに含まれるグアニンは酸化されやすく、活性酸素の標的として働く(Lee et al., 2007; Chang et al., 2008)。8-オキソデオキシグアノシン(8-OHdG)は、ALSにおける運動野の酸化の特異的バイオマーカーと考えることができる(Cooke et al, 2000, 2002; Ihara et al, 2005; Mitsumoto et al, 2008)(図1)。本疾患に罹患した被験者の尿中には、対照群と比較して8-OHdGとイソプロスタノイドの増加が観察されている(Miller et al., 2014)。
ALS患者におけるGPx活性、グルタチオン還元酵素(GR)、SOD、血清総抗酸化状態(TAS)、MDA、8-OHdGの評価では、TASレベルの有意な低下と8-OHdGおよびMDAレベルの上昇、ならびに酸化/還元グルタチオン(GSSG/GSH)比およびIL-6、IL-8の有意な上昇が認められた(Blasco et al.,2017)。
抗酸化物質と筋萎縮性側索硬化症
ALSの病因に関連する因子として、フリーラジカルと酸化ストレスが関与していることが証明されている。この疾患の管理における有用な薬剤として、以下のような抗酸化能を有する物質に関心が集まっている:ビタミンE、カロテン、フラボノイド、レスベラトロール、エピガロカテキンガレート、クルクミン、コエンザイムQ10、メラトニンおよびエダラボン(Table 1)。
表1 ALSの治療に用いられる抗酸化化合物
| 酸化防止剤 | 特徴 | 分子メカニズム | 治療効果と治療対象 | 参考文献 |
| ビタミンE | -親油性抗酸化剤 -細胞膜を通過する能力 |
-脂質過酸化に対する保護-ROS およびRNSに対する保護 |
-疾患の臨床的発症の遅延- 血漿中のGSHレベルの上昇-ALS による死亡のリスクの低下- 疾患のリスクの有意な低下 -ALS率の低下 |
Gurney et al。、1996 ; Desnuelle et al。、2001 ; Ascherio et al。、2005 ; Veldink et al。、2007 ; Wang H. et al。、2011 |
| カロチン | -天然色素 -さまざまな種類:β-カロテン、ルテイン、アスタキサンチン、リコピン |
ROSに対する抗酸化および中和特性 | -ALSの予防および/または発症の遅延を支援し ます-ALSのリスクを低減する -神経炎症およびアポトーシスの治療 |
Fitzgerald et al。、2013 ; Krishnaraj et al。、2016 |
| フラボノイド | -果物と野菜の天然物質 -さまざまな種類:7,8-DHF、フィセチン、ケルセテイン |
-ROSに対する保護 -代謝経路を調節する |
-運動障害を改善し、ニューロンの生存率を低下させる- 細胞内ROSレベルを低下させる -運動ニューロンの喪失を低減する- 運動活動と生存率を改善する -SOD1の凝集と誤った折り畳みの抑制 |
Korkmaz et al。、2014 ; Ip et al。、2017 ; Wang et al。、2018 |
| レスベラトロール | -天然ポリフェノール化合物 -ブドウ、ピーナッツ、ベリー -機械的損傷、真菌感染、紫外線に反応して植物で 生成-フリーラジカルのスカベンジャー |
-変異SOD1(G93A)タンパク質と相互作用する-SIRT1を アップレギュレートし ます-骨髄間葉系幹細胞におけるAMPK/SIRT1シグナル伝達のダウンレギュレーション |
-ALSの発症を遅らせる -脊髄運動ニューロンの生存を増加 させる-下部および上部運動ニューロンの機能を維持する-運動ニューロン の喪失を軽減する -筋萎縮 を改善する-筋線維のミトコンドリア機能を改善する |
Mancuso et al。、2014a、b ; Song et al。、2014 |
| エピガロカテキン ガレート |
-緑茶に含まれるカテキン -血液脳関門を通過する |
-酸化ストレスに対するミトコンドリアの応答を調節する-脂質過酸化に対する保護-細胞内シグナルを変化させる -NF -kBカスパーゼ-3とiNOSの濃度を低下させる |
-酸化ストレスによる死を防ぐ-ALS の発生または進行を遅らせる- 病気の発症を遅らせる- 耐用年数を延ばする -運動ニューロンの数を増やする- ミクログリアの活性化を減らす |
Koh et al。、2004、2006 ; Xu et al。、2006 |
| クルクミン | -天然および脂溶性染料 -ターメリックから得られる -化学的不安定性 -低い経口バイオアベイラビリティ -低い水溶性率 -さまざまなタイプ:DMC |
-Nrf2を活性化する- 細胞内ROSレベルを低下させる -TDP-43によって誘発される興奮性を排除する-DMC はミトコンドリア機能障害を低下させる |
-生存率を改善 する-ALS進行の減少と酸化的損傷の減少 |
Ahmadi et al。、2018 ; Chico et al。、2018 |
| コエンザイムQ10 | -内因性抗酸化物質 | -ETCの補因子 -レドックスバランスの作用 -ミトコンドリア機能障害を改善する |
-生存率を高める | Matthews et al。、1998 ; ビール、2002年 |
| メラトニン | -両親媒性分子 -強力な抗酸化剤 |
-抗酸化剤 -ミトコンドリアの生体エネルギー機能の調節因子 |
-病気の進行を遅らせる -生存率を高める |
Zhang et al。、2013 Weishaupt et al。、2006 |
| エダラボン | -低分子量抗酸化剤 -静脈内投与 -フリーラジカルスカベンジャー -安全 -血液脳関門を容易に通過する -高い脳浸透能力 -両親媒性能力 -脂質および水溶性ペルオキシラジカルと鎖を運ぶ脂質ペルオキシラジカルを除去する |
-プロスタサイクリングの生成を強化する -ヒドロキシルラジカルをトラップし、活性酸素をクエンチする |
-脳虚血時に過酸化脂質とヒドロキシルラジカルを削除し ます-虚血領域内または周辺の神経細胞をフリーラジカル損傷から保護する -酸化ストレスを改善し、脊髄運動ニューロンの変性を抑制する -ニューロン、ミクログリア、星状細胞、希突起膠細胞に対する抗炎症作用と保護作用 -機能的な運動障害の進行を遅らせる |
池田と岩崎、2015年; Banno et al。、2005 ; 吉野と木村、2006 ; 宮本ほか、2013 ; 阿部ほか、2014 ; バイリー、2019 |
ビタミンE
ビタミンE(α-トコフェロール)は、細胞膜を通過する能力と、脂質過酸化(Butterfieldら、2002;Di Matteo and Esposito、2003)、ROSおよびRNS(Barber and Shaw、2010)に対する保護のために最も研究された親油性抗酸化物質の1つである(表1)。このビタミンの栄養補給に関する研究では、相反する結果が得られている。
この抗酸化物質は、SOD1をコードする遺伝子の変異コピーを発現するトランスジェニックマウス(ALSの動物モデル)において、病気の臨床的発症の遅延(表1)と関連している(Gurneyら、1996)。3つの主要なラジカル消去酵素(SOD1、CAT、GPx)の赤血球活性を測定したところ、14人のALS患者にビタミンEを補給(300〜3000U/日)しても3つの酵素の活性には影響がなかった(Przedborski et al.、1996年)。ある研究では、非トランスジェニックマウスと比較して、脊髄にビタミンEが蓄積し、マウスの生涯にわたってMDAレベルが上昇することが示された (Hall et al., 1998)。
二重盲検プラセボ対照無作為臨床試験において、ビタミンEはALSの生存率と運動機能に影響を与えないようであった。しかし、ビタミンEとリルゾールを3ヶ月間投与したところ、ALS患者の血漿中で典型的に変化するマーカーである血漿GSHレベル(表1)の上昇と血漿チオバルビツール酸活性種(TBARS)レベルの低下が観察された(Desnuelle et al.、2001)。ビタミンEはALSの進行を遅らせる効果があり(Orrellら、2004)、ある研究では、ビタミンEサプリメントの常用はALSによる死亡リスクの低下と関連していることが観察された(Ascherioら、2005)(表1)。
他の同様の研究では、プラセボとα-トコフェロールを投与したグループとの間に有意差は検出されず、ビタミンEの大量投与が病気の進行を遅らせる効果があると主張するには不十分な結果が示された(Grafら、2005年)。ビタミンE 600mg/dayの補給は、プラセボ群に対する差を示さなかった(Galbussera et al.) ALS患者132名を対象とした症例対照研究では、ビタミンEの摂取量が平均値より多い場合、病気のリスク(表1)が有意に減少することが確認された(Veldink et al.、2007)。ビタミンEの長期的な補給はALS発症率の低下と関連しており(Wang H. et al., 2011)、男性ALS患者50例を含む研究では、ベースラインの血清α-トコフェロールが平均値以下の男性において、α-トコフェロールの補給(50mg/日)を受けた者は受けていない者に比べてALSリスクが有意でなく低下すると結論付けている。ベースラインの血清α-トコフェロールが平均値以上であれば、α-トコフェロールの補給はALSのリスクに影響を与えなかった。この場合、α-トコフェロールの補給はALSリスクに対して有意な保護効果を示さなかった(Michal-Freedmanら、2013)。
カロテン類
カロテンは天然色素で、果物や野菜のオレンジ、赤(Krinsky and Johnson, 2005)、黄色または緑色(Guest and Grant, 2016)の原因となり、活性酸素に対する抗酸化および中和作用(Fiedor et al, 2012; Nisar et al, 2015)がある(表1)。
ALSとカロテン類の摂取との間には有益な関連がある(Okamoto et al, 2009; Nieves et al, 2016)。したがって、それらの摂取はALSの予防および/または発症の遅延に役立つ可能性がある(Fitzgeraldら、2013)(表1)しかし、ALSの診断を受けた77人の韓国人を対象とした症例対照研究では、カロテンの食事摂取はALSに負の関連があると判断された(Jinら、2014)。5つのコホートで行われた研究では、これらの色素の高い摂取量はALSのリスク低減と関連し、ß-カロテンとルテインの高い食事摂取量は、この病気にかかるリスクと逆相関することが決定された。アスタキサンチンとリコピンはALSに有益な効果を示したが(Longeckerら、2000;Isonakaら、2011)、ALSのSOD1G93Aモデルマウスを用いた研究では、トマト強化食品(リコピンが豊富)は病気の発症と生存に影響を与えなかった(Espositoら、2007)。β-カロテンは、ALS患者の神経炎症およびアポトーシスを治療するための潜在的な治療分子として機能する可能性がある(Krishnarajら、2016年)(表1)。
フラボノイド類
フラボノイドは、主に果物や野菜に存在する天然物質であり(He et al., 2017)、活性酸素に対する保護効果(Cao et al., 1997)、認知障害および神経機能障害の軽減(Vauzour et al., 2008)、神経炎症の抑制(Solanki et al., 2015)に関する異なる代謝経路の活性(Mansuri et al., 2014)(表1)であると言われている。
7,8-ジヒドロキシフラボン(7,8-DHF)は、神経筋伝達に対する神経保護および調節特性を有する(Mantilla and Ermilov,2012)。7,8-DHFの慢性投与は、トランスジェニックALSマウスモデルにおいて運動障害を有意に改善し、下位神経細胞生存率を向上させた(Korkmazら、2014年)(表1)。
フィセチンによる処理は、ALSの3つの異なるhSOD1関連変異モデル(hSOD1G85Rを発現するショウジョウバエ、hSOD1G93ANSC34細胞およびトランスジェニックマウスhSOD1G93A)において細胞内ROSレベル、運動ニューロン損失を低減し、運動活性および生存率を改善した(Wang et al.、2018)(Table 1)。
ケルセチンは、食事(タマネギ、リンゴ、ブロッコリー、ベリー類)に豊富に含まれるフラボノイドであり(Espositoら、2002)、酸化ストレスの様々な動物モデルにおいてミトコンドリア損傷を軽減することが示されてきた。ケルセチンによる治療は、ラット海馬におけるアルミニウム誘発性神経変性に対する神経細胞死を減衰させるための治療戦略となり得る(Sharmaら、2016年)。ケルセチンおよびその誘導体であるケルセチン3-β-d-グルコシド(Q3BDG)は、ALSに関連するSOD1の凝集およびミスフォールディングの治療的阻害剤となり得る(Ipら、2017年)(表1)。
レスベラトロール
レスベラトロール(RES)は、変異型SOD1(G93A)タンパク質(ALSの特徴)と相互作用でき(Julien、2007;Srinivasan and Rajasekaran、2018;Laudatiら、2019)、ALSの変異hSOD1-G93A保有運動ニューロン様細胞培養モデルにおいてサーチュイン1(SIRT1)の発現を上昇させることでプラスの効果を有する(Wang J. ら、2011)(Table 1)。ALSのSOD1G93Aマウスモデルにおいて、ALSの発症を遅らせ、脊髄運動ニューロンの生存率を高め、下部および上部運動ニューロンの機能を維持し(Mancusoら、2014a、b)、運動ニューロンの損失を減衰させ、筋萎縮および筋線維のミトコンドリア機能を改善させた(Songら、2014)(表1)。ある研究では、ALS患者の骨髄間葉系幹細胞がAMPK/SERT1シグナルのダウンレギュレーションを示し、RESによる処理で回復することが示された(Yun et al.、2019)(表1)。
エピガロカテキンガレート
エピガロカテキンガレート(EGCG)は、緑茶に存在するカテキンで(Chung et al., 2010)、血液脳関門を通過し酸化ストレスに対するミトコンドリア応答を調節する(Bedlack et al., 2015)ことから、特に運動ニューロンに対する抗神経変性および抗酸化効果(Nie et al., 2002; Panickar et al., 2009)に起因している(Table 1)(注:この文献では、EGCGは運動ニューロンに対する抗神経変性および抗酸化効果を有するとされている)(Koh et al. また、in vitroの研究では、細胞膜二重層のリン脂質に活性酸素を暴露することにより、脂質過酸化(表1)から保護する(Terao et al, 1994)。それは、細胞生存および死シグナルの変化により、変異型SOD1運動ニューロン細胞の酸化ストレス誘発死を防ぐ(Kohら、2004)。EGCGの投与は、細胞内シグナルの変化を通じてALSの発生または進行を遅らせ、生存シグナル(PI3-KおよびAktなど)を増加させ、死シグナル(GKS-3ß、細胞質チトクロームc、活性化カスペース3および切断型ポリADP-リボースポリメラーゼなど)を減少させ得る(Levitesら、2002; Mandelら、2003,2005;Kohら、2006)(表1)。ALSのトランスジェニックマウスモデルにおいて、運動ニューロン数の増加、ミクログリアの活性化の減少、NF-kB、カスパーゼ-3およびiNOSの濃度の減少に加えて(Xuら、2006)、前症候群の段階からEGCG10 mg/kgを経口投与すると病気の発症を遅らせ、有用寿命を延長させる(表1)。
クルクミン
クルクミンはウコンから得られる天然色素で脂溶性色素である(表1)。それは神経保護効果を有し、酸化ストレス(Kimら、2012)、ミトコンドリア機能障害、炎症およびタンパク質凝集に対する保護を提供する(Ullahら、2017;RakotoarisoaおよびAngelova、2018;Ghasemiら、2019)。
クルクミンは、初代脊髄アストロサイトの核因子赤血球2関連因子(Nrf2)標的遺伝子を活性化し、細胞内ROSのレベルを低下させ、酸化的損傷とミトコンドリア機能障害を減衰させる(Jiangら、2011年)(表1)。ALSの運動ニューロン様細胞モデルにおいてTDP-43(散発性ALSの主要な病理学的タンパク質)によって誘発される興奮性を排除する(Mackenzie and Rademakers, 2008; Dong et al, 2014)。ジメトキシクルクミン(DMC)は、変異TDP-43を安定的にトランスフェクトした細胞株におけるミトコンドリア機能不全を減少させることができた(Luら、2012)(表1)。SOD1のプレフィブリル凝集体に結合し、そのアミロイド形成経路を変化させ、細胞毒性を減少させる(Bhatiaら、2015)。クルクミンは、ALS患者、特に嵩上げ病変を有する患者の生存率を改善する可能性がある(Ahmadiら、2018年)。二重盲検治療試験において、クルクミンによる治療は、ALSの進行の減少および酸化的損傷の減少を示した(Chicoら、2018年)(表1)。
ある研究では、クルクミンに基づく薬物送達システムがALSの治療のために提案されることが示されているが(Tripodoら、2015)、他の研究では、ALSにおける治療としてのクルクミンの使用は、化学的不安定性と低い経口バイオアベイラビリティおよび水溶性率という欠点があると示されている(Rakotoarisoa and Angelova、2018)(表1)。したがって、これらの限界を克服するための新しい技術の開発が必要である(Liu et al.、2016)。
コエンザイムQ10
Co-enzyme Q10(CoQ10)は内因性の抗酸化物質であり、酸化還元バランスに関与するETC(Petrovら、2017)の補酵素である(Yamamoto、2016)(表1)。ALSのSOD1トランスジェニックマウスモデルでは、CoQ10の補充により生存期間が6日延長し、脳内ミトコンドリア濃度がコントロールと比較して増加した(Matthewsら、1998; Strong and Pattee, 2000; Beal, 2002) (Table 1)。20 名の sALS 患者と年齢・性別をマッチさせた健常対照者の血漿中 CoQ10 の酸化還元状態を比較した結果、sALS では CoQ10 の酸化型が有意に増加していた (Sohmiya et al., 2005)。31 名の被験者を対象とした研究では、8 ヶ月間にわたり 3000 mg/day の CoQ10 を大量に投与することにより、ALS のミトコンドリア機能障害が改善することが示された (Ferrante et al., 2005)。しかしながら、ある研究では、血清CoQ10濃度はALSのリスクとは無関係であることが示唆されている (Molina et al., 2000; Kaufmann et al., 2009)。
メラトニン
メラトニンは両親媒性分子であり、ミトコンドリア機能障害に関連する神経変性疾患(Polimeni et al, 2014)において強力な抗酸化治療薬として同定されている(Ganie et al, 2016)(表1)。メラトニンがALSの神経保護候補となり得ることを示した研究がある(Jacob et al, 2002)。ALSのSOD1G93Aトランスジェニックマウスモデルにおいて、高用量のメラトニンを経口投与すると、疾患の進行を遅らせ、生存率を増加させた(Weishauptら、2006;Zhangら、2013)(表1)。しかし、腹腔内メラトニンは神経保護作用がなく、神経変性を悪化させる可能性があることを示唆する証拠がある(Dardiotisら、2013年)。神経保護剤としての相対的な利点が決定的に確立される前に、50〜100mg/日の範囲のメラトニンを採用する臨床試験が必要である(Pandi-Perumalら、2013)。最近の研究では、メラトニンではなくリルゾールが急性運動ニューロン変性を改善し、ALSにおけるミトコンドリアカルシウム信号の乱れを誘発するSOD1媒介興奮毒性を適度に抑制することが示唆されている(Jaiswal, 2017)。
エダラボン
エダラボンは、フリーラジカルスカベンジャーとして作用する(Jackson et al., 2019)低分子抗酸化剤(Radicava®)、静脈内投与される(Petrov et al. 2015年、日本ではエダラボンがALS治療薬として承認され(Okada et al., 2018)、2017年には米国食品医薬品局(Food and Drug Administration of United States)より承認された(Watanabe et al., 2018)。
エダラボンは血液脳関門を容易に通過し、高い脳浸透能を示す(Jin et al, 2017)。その両親媒性能力により、エダラボンは脂質と水溶性のペルオキシルラジカルと鎖状脂質ペルオキシルラジカルの両方を消去する(Nagase et al.、2016)(表1)。
エダラボンの抗酸化機構は、プロスタサイクリン産生促進、ヒドロキシルラジカル捕捉、活性酸素のクエンチ(澤田、2017)(表1)である。エダラボンは脳虚血時の過酸化脂質やヒドロキシルラジカルを除去し、虚血領域内またはその周辺の神経細胞をフリーラジカル障害から保護し(阿部ら、2014)、脊髄運動ニューロンの酸化ストレス改善と変性抑制を行う(池田・岩崎、2015)。神経細胞、ミクログリア、アストロサイト、オリゴデンドロサイトにおける抗炎症作用(Bailly, 2019)および保護作用(Banno et al, 2005; Miyamoto et al, 2013)が帰属している(表1)。
この抗酸化剤を静脈内投与された20名のALS患者におけるエダラボンの安全性と有効性の調査により、この薬剤は安全であり、酸化ストレスを減少させることにより機能運動障害の進行を遅らせる可能性があることが示された(吉野と木村、2006)(表1)。
ニコチンアミドリボシドと神経変性疾患
NAD+の役割とレベル
NAD+は、ミトコンドリア代謝の動作に重要な酸化還元反応(その還元型はNADH)に関与する補酵素である(Bergerら、2004;Yoshinoら、2018)。生存に不可欠な多くの機能により重要な生物学的メディエーターである:酸化還元反応、シグナル伝達経路、エネルギー代謝、ミトコンドリア機能、カルシウム恒常性、DNA修復、遺伝子発現(Guarente, 2014; Yaku et al, 2018)、脳代謝、神経伝達、学習、記憶、軸索神経保護(Araki et al., 2004; Conforti et al., 2006; Gong et al., 2013)、老化に関連するNAD+/PARP/SERT1軸への参加(Mendelsohn and Larrick, 2017)(図2)などがある。
図2 ニコチンアミドリボシドの作用機序

NRは細胞内に入り、そこで2つの機構を経てNAD+に変換される。ひとつはNrk1とNrk2、もうひとつはPNPとNAMPTである。NAD+は活性酸素によるDNA損傷の修復に関係するPARPSの副基質である。NAD+ は、エネルギー代謝、炎症制御、DNA 修復、ミトコンドリア代謝に関連する SIRT の副基質でもある。SIRTの活性化は、SOD2やCatのような活性酸素を解毒する代謝経路の増加を通じて、酸化ストレスに対する抵抗性を増加させる。SIRT1はPGC-1αを活性化し、SOD2やGSHを介した抗酸化防御の増加に関与する。SIRT3は、SOD2およびCatを活性化する。このように、SIRTはミトコンドリア機能と老化プロセスを制御し、活性酸素の解毒に関与している
NAD+は、サーチュイン(SIRT)(Verdin, 2015)、活性がNAD+レベルに依存するクラスIIIヒストン脱アセチル化酵素(Tang, 2016; Jęśko et al., 2017)や、ROSによって損傷を受けたDNAの修復に必須なタンパク質ファミリーであるポリ(ADP-リボース)ポリメラーゼ(PARPs)に対する共基質として作用し得る(Shen et al, 2015)(図2)。
サーチュインは細胞の代謝状態に関係し、細胞代謝、特定の遺伝子の発現調節(O’Callaghan and Vassilopoulos, 2017)、エネルギーおよびミトコンドリア代謝(Covington and Bajpeyi, 2016)、炎症およびDNA修復(Tang, 2017)で重要な役割を担っている。細胞質、核、ミトコンドリアに存在する(Kupis et al.、2016)。神経変性疾患や老化において、酸化ストレスやDNAの損傷に対する反応を組織化し、異なる組織における呼吸器官の機能性やROSの生成に関係している(Huang et al.)
SIRTの活性化は、スーパーオキシドジスムターゼ2(SOD2)、イソクエン酸デヒドロゲナーゼ2(IDH2)およびCAT(TennenおよびChua、2011)などのROSの解毒(Salvatori et al、2017)を担う代謝経路の増加により酸化ストレスに対する抵抗性を高める(Haigis and Sinclair、2010)。これらは、代謝能力を向上させ、ミトコンドリアの酸化的代謝を促進し、DNA損傷の修復を促進する(Haigis and Sinclair, 2010)(図2)。
SIRT1は、エネルギー代謝の基本的な調節因子であるペルオキシソーム増殖剤活性化受容体γコアクチベーター1α(PGC-1α)の経路に作用する(Alquilanoら、2013;Tang、2016)(Bayerら、, 2017; Thirupathi and de Souza, 2017)およびミトコンドリア動態(Singh et al., 2016)SOD2およびGSH(Liang and Ward, 2006; Zhong and Xu, 2008; Wang et al., 2015)による抗酸化防御の上昇と関連する(図2)。SIRT1活性の低下は、酸化的代謝と抗酸化力を低下させ、ETCの複合体I、ミトコンドリア機能および生合成に影響を与える(Rodgersら、2005年)。この効果は、老化やさまざまな病態:神経変性疾患、代謝障害、がんにおいて観察されている(Rass et al., 2007; Hou et al., 2017b; Jęśko et al., 2017)。ALSでは、患者の死後組織(Körnerら、2013)およびALSのマウスモデル(Hanら、2012)において、SIRT1レベルの変化が決定されている。
サーチュイン3(SIRT3)は、活性酸素の解毒に関与するSOD2やCAT(Salvatori et al., 2017)を活性化することにより、ミトコンドリア機能および老化プロセスを制御する(Qiu et al., 2010; Kida and Goligorsky, 2016) (図2)。
NAD+量の減少は、その生合成の欠陥または枯渇に続発する可能性があり(Imai and Guarente, 2014)、核およびミトコンドリアにおけるSIRTの活性の欠損につながり(Chang and Guarente, 2013; Gomes et al, 2016)、ミトコンドリアの unfolded protein response を低下させ(Mendelsohn and Larrick, 2017)、ATP生合成を乱し、細胞内勾配に対するカルシウムのポンプ能力を低下させ(Camandola and Mattson, 2011)、カルシウムの恒常性を乱し、興奮毒性の増加(Rzheshevsky, 2014)、筋肉の健康に対して有害な影響(Goody and Henry, 2018) があると言われている。
NAD+レベルの生物学的利用能の低下は、神経変性病態(Essaら、2013;JohnsonおよびImai、2018)などの多くの疾患に関与し、エネルギーの生産に影響を与え、ATPのレベルを下げ、SIRT1の保護を制限し(Houtkooperら、2010;Cantóら、2012;今井およびGuarente、2014)、これらの疾患ではその進行性から悪化し得る側面である(Cantóら、2015)。NAD+レベルは、加齢とともに減少し、脳内の低レベルにつながることが示されている(Zhu et al.、2015)。いくつかの研究により、NAD+代謝がニューロン機能および神経変性疾患の病態生理に関与すること(Hikosakaら、2019)、および組織におけるNAD+レベルがこの種の疾患において有益な治療効果をもたらすことが示されている(Amanら、2018; Kulikovaら、2018)。
NAD+およびSIRT1を介したミトコンドリアおよびエネルギー生産に関連する代謝経路の活性化は、現在、治療戦略として示唆されている(YangおよびSauve、2016;Tang、2017;Rajmanら、2018;ZhangおよびSauve、2018)。
NAD+レベルの回復および/または増加は、骨格筋を進行性の劣化から保護し(Fletcherら、2017)、脳エネルギー代謝の損傷を逆転し、酸化ストレスに対する保護力を高めることができる(López-Otínら、2013;Brownら、, 2014; Verdin, 2015)、SIRTおよびミトコンドリア機能に関与するタンパク質の活性を促進し(Bonkowski and Sinclair, 2016)、過酸化物毒性に対する耐性を与え、ミトコンドリアROSを減少させ(Harlan et al, 2016)、神経変性に対する保護(Sasaki et al, 2006)およびシナプス可塑性および神経細胞ストレス耐性に関係する依存酵素のアップキープ(Lautrup et al, 2019)。
したがって、前駆体を用いたNAD+レベルの補充は、この加齢に伴う機能的欠陥を改善し(Imai and Guarente, 2014)、神経変性疾患に特徴的な発症プロセスの逆転に役立つ(Zhang et al.、2016)。
NAD+の前駆体
NAD+の前駆体には、ニコチンアミドモノヌクレオチド(NMN)、ニコチンアミド(NAM)、ニコチン酸(NA)、ニコチンアミドリボシド(NR)などがある(Harlanら、2016年)。NAD+前駆体の多くは、NAD+よりも安定であり、神経細胞への侵入能力が高い(Sasaki et al.) NMNとNRは最も使用されており、どちらも細胞内のNAD+レベルの増加を示している(Nikiforov et al.、2011)(表2)。ほとんどの証拠は、NRがNAD+レベルの大幅な増加を刺激する能力が高く(Cantóら、2012)、他の前駆体の使用よりも有利であると結論づけている(表2)。
表2 異なるNAD+前駆体間の比較
| NAD +前駆体 | 利点 | 短所 | 参考文献 |
| ニコチン(NA) | -ペラグラを防ぐ -脂質レベルの調節(総コレステロール、トリグリセリド、LDL) |
-皮膚の紅潮を引き起こする-大部分の細胞では NAD +前駆体ではない |
デビッドソン、2008 ; Guille et al。、2008 ; Prousky et al。、2011 |
| ニコチンアミドモノヌクレオチド(NMN) | -最も使用されているものの1つ- セル内の NAD +レベルの増加を示している |
-細胞への輸送方法に関するコンセンサスはない | Nikiforov et al。、2011 ; Grozio et al。、2019 ; シュミットとブレナー、2019年 |
| ニコチンアミド(NAM) | 細胞の刺激剤になることができる | NRと比較して NAD +の増加が小さい | Cantóetal。、2012 |
| ニコチンアミドリボシド(NR) | -哺乳類細胞への作用 -最小限の毒性 -高いバイオアベイラビリティ -血液脳関門を通過する高い能力 -ニューロンのNAD +合成をサポート-NAD +レベルと中間前駆体 の有意な増加を刺激する優れた能力 |
証拠がない | ヤンH.他、2007年; ボーガンとブレナー、2008年; Cantóetal。、2012 ; Trammell et al。、2016 ; Airhart et al。、2017 ; Martens et al。、2018 |
NAはペラグラを予防し(Prousky et al., 2011)、脂質(総コレステロール、トリグリセリド、LDL)の調節に良い影響を与える(Guille et al., 2008)(Tabla 2)。しかし、皮膚紅潮を生じ、大部分の細胞でNAD+前駆体として働かない(Davidson, 2008)(表2)。NRのNAに対する最も重要な利点の1つは、神経細胞のNAD+合成をサポートし(Bogan and Brenner, 2008)、マウスやヒトの細胞を含むいくつかのタイプの培養哺乳類細胞に対して作用を有することだ(Yang H. et al, 2007)(表2)。
NMNがどのように細胞に運ばれるのかについては、コンセンサスが得られていない(Grozioら、2019;SchmidtとBrenner、2019)(表2)。しかし、NRは、骨格筋の細胞内NAD+レベルを増加させるのにNMNよりも5倍効果的である(Oakeyら、2018年)(表2)。
マウスでNRとNAおよびNAMを比較した結果、NRはNAD+濃度のピークが有意に高く、NMN、NAモノヌクレオチドおよびNAアデニンジヌクレオチドを含む中間NAD+前駆体が有意に増加した(Trammellら、2016)(表2)。NRは、NAMおよびNAと比較して異なる時間経過を有し、同等の用量で他の前駆体よりも多くのNAD+を生成する(Trammell et al.、2016)。NRは、NMN、NAMおよびNAと比較して、NAD+/NADHの比をより顕著に増加させ、これは、ミトコンドリアの酸化的能力の向上(Cantó et al.、2012)およびDNAおよびミトコンドリアストレスに対する酸化的損傷率の減少(Ramamoorthy et al.、2012;Srivastava、2016;Dropikou et al.、2019)(表2)に対して貢献し得る(表2)。
NRは、ニコチンアミドとリボース(Belenky et al., 2007; Chi and Sauve, 2013)基を組み込んだヌクレオシドである。ビタミンB3(Lanska, 2012)として知られる微量栄養素で、特定の食品(乳製品、魚、卵、野菜)(Mintoら, 2017)、栄養補助食品、強化食品(Colbourneら, 2013)で入手可能である。その効果は、エネルギー代謝と神経保護に関連している(Chi and Sauve, 2013)。NRが細胞に入ると、少なくとも2種類の代謝経路でNAD+に変換される。1つ目は、その2つのアイソフォーム(NRK1とNRK2)のニコチンアミドリボシドキナーゼ(NRKs)(Chi and Sauve, 2013)の関与を必要とし(Bieganowski and Brenner, 2004)、2つ目は、プリンヌクレオシドホスホリラーゼ(PNP)とニコチンアミドホスホリボシル転移酵素(NAMPT)(Burgos et al, 2013)の作用(図2)である(図2)。
NRのNAD+への変換は、筋肉や脳の動物組織で観察されており(Chi and Sauve, 2013)、NRの外因性補充による治療は、NAD+の細胞内濃度を高め、その有益な効果を促進することができる(Braidyら、2008)(図2)。NRは老化および関連する疾患に対する保護を強化し、複数の動物モデルにおいて長寿に肯定的な結果をもたらし(Mouchiroudら、2013;Poljsak and Milisav、2016)、最近、NRが興奮毒性による軸索変性から保護することが報告されている(Vaurら、2017)。NRなどの前駆体の補充によるNAD+レベルの増加は、マウスのミトコンドリアと筋肉機能およびニューロン細胞の機能を改善する(Mendelsohn and Larrick, 2017)。400mg/kg/日の合成NRを栄養補給したマウスで行われた研究では、筋肉と肝臓の組織でNAD+レベルが増加した(Cantó et al.、2012)。NRはNAD+/NADH比を増加させるため、ミトコンドリア経路の酸化能の改善に関連する可能性があり、神経変性疾患などのミトコンドリア機能障害および酸化ストレスに関連する疾患において注目すべき治療戦略となり得る(Cantóら、2012年)。NAD+レベルの改善は、酸化ストレスに対するより大きなミトコンドリア抵抗性を提供し(Yang T. et al., 2007)、SIRT3の保護機能(Hafner et al., 2010)により細胞死を防ぎ、ROSの「解毒」の強化を担うSOD(Chen et al., 2011)を活性化する(図2)。
NRの補給は、マウスおよびヒトにおいて安全であり、毒性が少なく、高いバイオアベイラビリティと血液脳関門を通過する高い能力を有する(Trammellら、2016;Airhartら、2017;Martensら、2018)(表2)。
ニコチンアミドリボシド
神経変性疾患は、筋肉量と筋力の進行性低下およびそれらの弱化の増加と関連している(Gonzálezら、2017年;Van Dammeら、2017年;Dahlqvistら、2019年)。筋収縮性の機能不全は、ALSのSOD1G93Aマウスモデルにおける運動単位の連結性の喪失に先行し(Wierら、2019)、神経変性が筋質の決定的リスク要因の1つである(Di Pietroら、2017; Moonら、2018)。NRの投与は、老化したマウスの筋機能を改善し(Cantóら、2012)、NAMPTを特異的に除去し、NAD+の筋肉内含量を85%減少させたノックアウトマウスでは、筋力や骨格筋抵抗に効果がある(Frederickら、2016)。これは、NAD+のホメオスタシスの維持が、筋肉量とその収縮機能にとって重要であることを示していると考えられる(Mendelsohn and Larrick, 2014; Frederick et al.、2016)。NRは、筋肉および脳幹細胞の若返り(Ryuら、2016)、神経新生および筋肉機能の増加(Zhangら、2016)を誘導する。NAD+は筋肉の老化の基本的な調節因子であり、NRで処理した後、若いマウスと古いマウスの両方で筋肉の再生の加速があり、老化マウスのランニングタイム、最大距離、四肢の握力の改善と細胞周期遺伝子の発現が増加する(Zhang et al.、2016年)。
PGC-1αのレベルはアルツハイマー病(AD)で低下し、これはβ-アミロイド斑の形成とこの物質のより大きな沈着(ADの主な特徴)に関連している(Wuら、2006;Quinら、2009)。ADの動物モデルで行われた研究では、NAD+のレベルとアミロイド物質の毒性の減少の間に関係があることが立証された(Kimら、2007;Quinら、2009)。このため、NRの栄養補給に基づく治療戦略は、AD症例において、認知症の発症および進行を阻止し(Rodgersら、2005)、Tg2576マウス(ALSのマウスモデル)で示されたPGC-1αの機能促進による認知機能およびシナプス可塑性を改善するために興味深いものになる可能性がある(Gongら、2013)。3xTgAD/Polß±マウスモデル(ヒトのADの主な特徴を悪化させるDNA修復欠損マウス)における生理病理および細胞メカニズムに対するNRの効果を分析した後、NRを補充することにより、NAD+/NADH比が有意に増加し、神経炎症、アポトーシスおよびDNAへの損傷が減少し、神経新生が増加し、シナプス可塑性が回復し、学習能力が改善し、記憶障害が逆転されたことが観察された(Hou et al.、2017a)。NRによる治療は、タウタンパク質(pTau)(AD患者の脳に蓄積し、この疾患の特徴である形態)のリン酸化を低減することによって、ADのトランスジェニックマウスの認知力を改善する(Greenら、2008;Rübら、2017;Nizynskiら、2017)。PDの場合、NRを用いたNAD+の補充は、PDのショウジョウバエモデルにおけるドーパミン作動性ニューロンの損失および運動機能の低下に関連する老化を防ぐ(Sidranskyら、2009年)。NRは、PDニューロンのミトコンドリア機能を改善し、ミトコンドリア生合成のマーカーを増加させ、mtROS(ミトコンドリアROS)の産生を著しく減少させ、ミトコンドリア膜電位を低下させ、NAD+およびNADHのレベルを増加させる(Schöndorfら、2018年)。
ほとんどの神経変性疾患(AD、PD、ALS)では、軸索変性が起こる(Raffら、2002年、Coleman、2005年)。NRなどの前駆体の使用によるNAD+合成の増加は、軸索保護を促進する(Wangら、2005年;Sasakiら、2006年)。NAD+回復経路の改善は、ALSに関連するSOD1変異を発現する初代アストロサイトの毒性を逆転させ、ROSのミトコンドリア生成を減少させ、神経毒性を逆転させる(Harlanら、2016年)。興奮毒性は、ALSのようなほとんどの神経変性疾患で起こるプロセスであり、神経細胞における強いNAD+の枯渇に関連している。NRは興奮毒性に起因する軸索の生成から保護する(Vaurら、2017年)。
デュシェンヌ型筋ジストロフィー病では、NRの補充は、ノックアウトマウスモデルにおける骨格筋NAMPTの進行性摩耗を逆転し、抵抗性を改善する(Ryu et al.)
プテロスチルベンと神経変性疾患
ポリフェノールは、植物に存在する有機代謝物であり(Cory et al., 2018)、様々な食事源に広く分布している(Aherne and O’Brien, 2002; Kim et al, 2009):果物、野菜、豆類、全粒粉、種子、ナッツ、エキストラバージンオリーブオイル、赤ワイン、コーヒー、紅茶、チョコレート、ハーブ、スパイス(Rusconi and Conti, 2010; Fu et al, 2011; Regueiro et al, 2014; Vallverdú-Queralt et al, 2014, 2015; Talhaoui et al, 2016)。エネルギー寄与がゼロであるにもかかわらず(Ruizら、2009)、代謝、慢性疾患および細胞増殖に対する調節作用(Tressera-Rimbauら、2017)により、複数の好ましい健康効果(GanesanおよびXu、2017;Szwajgierら、2017;RenaudおよびMartinoli、2019)と関連する生物活性食事成分として作用する。ポリフェノールは、o-ジフェノール基、4-オキソ関数と共役の2-3二重結合、3位と5位に水酸基(OH)という、最大の抗酸化作用を持つ化合物となる特性を持つ化学構造を持っている(Ruizら、2009年)。それらは抗酸化作用(Hua and Rong, 2016; Tarique et al., 2016)、抗炎症作用(Oliviero et al., 2018)、抗発癌性(Niedzwiecki et al., 2016)、抗アレルギー作用(Juríková et al., 2015)、抗生物質(Xie et al., 2017)および免疫調節(Nour et al., 2018)性質を有している。
ポリフェノールは、神経変性疾患(Bhullar and Vasantha, 2013; Raskin et al., 2015; Reinisalo et al., 2015; Carvalho et al., 2018)、がんおよび心血管疾患(Crozier et al., 2009; Bulotta et al., 2014; Lamuela-Raventós et al., 2014)などの酸化ストレスを提示する病態における有用な治療戦略たり得る。栄養が認知またはCNS疾患の進行などのプロセスを調節する因子であることを考慮すると(Gustafsonら、2015;Huhnら、2015)、ポリフェノールは最近、以下のことに関連している:予防、酸化的損傷の修復(Lange and Li、2018)、シナプス可塑性、ニューロンシグナル伝達およびオートファジー(Miller and Shukitt-Hale, 2012;Poulose et al.、2014)。
重要な医薬活性を有するポリフェノールのうち、スチルベン(Dvorakova and Landa, 2017)は、1,2-ジフェニルエチレンコアを有するポリフェノール構造の非フラボノイド植物化学物質群で、植物においてフェニルプロパノイド経路(Sirerol et al, 2016)経由で自然に生成されて、真菌感染や毒素から植物を保護する(Akinwumi et al, 2018)。様々な位置に様々な置換基を付加できる共通の構造を持ち、酸性で両親媒性の性質を持つため、様々な形態が存在する(Neves et al.) トランス異性体が最も一般的な提示形態であるが(最も安定であるため)、シス異性体も見出される(Rivière et al.) それらに起因する特性は、抗酸化、抗炎症、神経保護、心臓保護、抗発癌性(Lange and Li, 2018)、高脂血症および抗糖尿病(Voloshyna et al, 2013; Szkudelski and Szkudelska, 2015)である。それらは、神経変性疾患の分野で大きな実用性の可能性を持っている。それらは、脳内のアミロイド斑の形成を減少させ、活性酸素の産生を減少させ、以下のような他の状況において関心を持たれる可能性がある:虚血再灌流傷害、アテローム性動脈硬化、糖尿病、癌、肥満、血小板凝集、血圧、色素脱失および心筋細胞と心肥大(Thandapilly et al, 2011; Gomez-Zorita et al., 2013; Zordoky et al., 2015; Guo et al., 2016; Akinwumi et al., 2018)がある。
最も研究されているスチルベンの1つは、心血管の健康におけるその利点により、レスベラトロール(RES)(3,5,4′-トリヒドロキシ-トランス-スチルベン)であった(Zordokyら、2015; Bonnefont-Rousselot、2016; Dyckら、2019). しかし、最近の研究では、1つのヒドロキシル基と2つのメトキシ基を有するジメチル化天然スチルベン(Albassam and Frye, 2019)のプテロスチルベン(PTER)(トランス-3,5-ジメトキシ-4ヒドロキシスチルベン)に興味が集中している(Cichocki et al, 2008; Estrela et al, 2013; Traversi et al., 2016)、RES(3水酸基)に関して、より高い経口バイオアベイラビリティ、半減期、親油性および標的組織への高い透過性(Kapetanovic et al., 2011; Yeo et al., 2013)を提供する。さらに、PTERは、第2相肝代謝の影響を受けにくい(Zhou et al.、2016)。これらの特定の特性は、その生物学的可能性を改善し(Yeoら、2013)、in vivoにおけるその高いバイオアベイラビリティは、RESに対する利点である(Chiouら、2011;Careyら、2013;Linら、2016)。PTERは、RESの20%と比較して80%のバイオアベイラビリティを示し、PTER及びPTER硫酸塩の血漿レベルは、マウスモデルにおいてRES及びRES硫酸塩の血漿レベルよりも有意に高かった(Kapetanovicら、2011年)。PTERとRESを同用量で雌雄マウスに8週間投与したところ、PTERはRESよりも血清中及び脳内で高濃度に達した(Changら、2012年)。
PTERの高い親油性により、血液脳関門を容易に通過し(Temsamaniら、2015)、その結果、RESよりも優れた神経保護作用が得られる(Chooら、2014)。ADにおいて、PTERはRESよりも優れた神経保護作用を示し(Changら、2012)、5-リポキシゲナーゼ(5-LOX)に対する最も高い阻害能を有するスチルベンであり(Kutilら、2015)、脂質およびタンパク質酸化のレベルを低下させる(Czpaskiら、2012)。RESよりも強力な抗がん剤および抗炎症剤であり(Chiouら、2011)、有望な(Chooら、2014)かつ安全な(Richeら、2013)治療戦略と思われるものについて、主要標的器官(脳、肝臓、腎臓、心臓および肺)間で広く分布している。オスとメスのスイスマウスを用いた研究では、PTERの亜慢性毒性と考えられる副作用を調べ、最高量を投与しても、PTERは毒性がないと結論付けている(Ruiz et al.)
PTERの分子機構
PTERの生理活性としては、抗酸化・抗炎症活性、細胞内カルシウムの回復能力、認知機能(Li Y. R. et al., 2018)などが知られている(図3)。
図3 プテロスチルベンの性質と作用機序
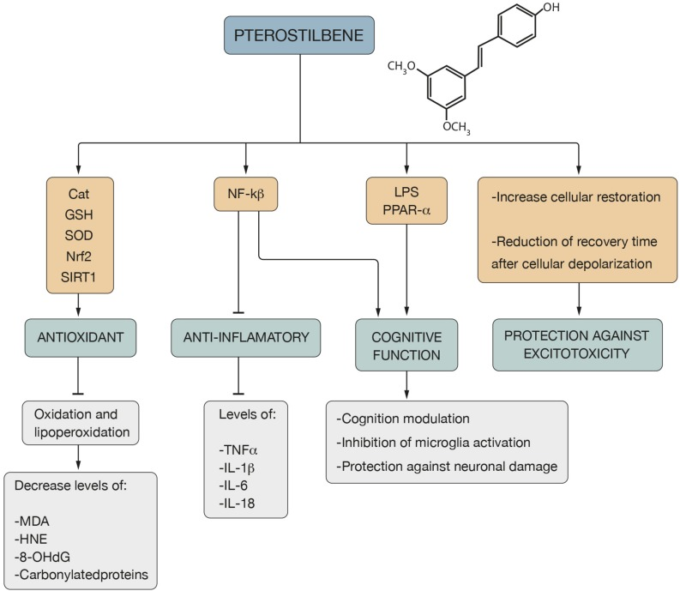
Pterの抗酸化機構は、Cat、GSH、SOD、Nrf2、SIRT1経路と関連しており、酸化および脂質過酸化過程の抑制をもたらし、MDA、HNE、8-OHdGおよび炭化タンパク質のレベルを低下させる。NF-kßに対する作用は、TNF-αおよびIL-1β、IL-6およびIL-18などのインターロイキンのレベルの減少による抗炎症効果を伴う。NF-kß、LPS、PPAR-αに対する役割は、認知機能の調節、ミクログリアの活性化の抑制、神経細胞の損傷に対する保護を通して表されるPterの認知機能を媒介するものである。さらに、Pterは、細胞の復元力の増加および細胞の脱分極後の回復時間の短縮により、興奮毒性に対する保護を提示す
抗酸化作用 PTERは、多くの種類のフリーラジカルに対する抗酸化作用が際立っている。HO、H2O2 (Castellani et al., 2008; Mikstacka et al., 2010; Acharya and Ghaskadbi, 2013) and NO∙ (Pan et al., 2008; Zhang et al., 2012)である。また、金属誘導ラジカルを中和し (Rossi et al., 2013; Saw et al., 2014) 、酸化および脂質過酸化プロセスを抑制することができ (Rimando et al., 2002) 、カルボニル化タンパク質および酸化副産物の減少を引き起こす。MDA、HNEおよび8-OHdG(Li Y. R. et al.、2018)(図3)。PTERの投与は、CAT、GSHのレベルおよびSODの活性を上げることにより、脳の抗酸化防御を改善する(Naikら、2017年)(図3)。また、PTERを添加した食事は、SOD2の発現を増加させる(Ding et al.) PTERの前処理は、P7ラットモデルの低酸素虚血性脳損傷においてSOD活性を増加させる強力な抗酸化機能を有する(Li D. et al.、2016)。PTERの投与は、神経細胞におけるNrf2シグナル伝達経路を活性化することにより、ROSを低減し、SODおよびGSHの機能を増加させることにより、グルタミン酸による酸化ストレス損傷を軽減する(Wang et al., 2016; Bhakkiyalakshmi et al., 2018; Li Y. R. et al, 2018) (図3).
PTERの強力な抗酸化機構は、SIRT1シグナル活性化(Li et al., 2015; Guo et al., 2016; Liu et al., 2017)と関連しており、雄スプラグドーリーラットの虚血再灌流障害によって引き起こされる骨格筋酸化ストレス損傷およびミトコンドリア機能障害の減弱につながる(Cheng et al.、2016)。PTERはNrf2に作用し(Sawら、2014;Zhangら、2014;Xueら、2017)、Nrf2シグナルの活性化を通じてin vitroで高グルコース誘発中枢神経系損傷を減弱し、ミトコンドリア機能障害由来の酸化ストレスに対する保護作用を示す(Yangら、2017)。
抗炎症活性 PTERは、ROS産生の抑制により神経炎から神経細胞を保護することができる抗炎症剤(Remsbergら、2008;Pouloseら、2015;Kosuruら、2016)として認識されている(Shinら、2010)。NF-kBに作用し、様々な炎症性サイトカインの作用を抑制する。TNF-α、IL-1β、IL-6、IL-18(Cichocki et al., 2008; Paul et al., 2009; Hou et al., 2014; El-Sayed et al., 2015)(図3)。PTERは、NLRP3/カスパーゼ-1インフラマソーム経路を不活性化することでミクログリアにおけるアミロイドβ誘発性の神経炎症を抑制し、IL-6、IL-1βおよびTNF-αの分泌レベルを低下させて、神経炎症反応を減衰させることから、神経変性疾患における有用な治療戦略となる可能性があると考えられる (Li Q. et al., 2018; Seo et al., 2018)。
細胞内カルシウムの復元能力 PTERは、細胞脱分極後の回復時間を短縮することにより、細胞内カルシウムの復元能力を高めることができる(Josephら、2008年)。細胞内の活性酸素レベルの増加は、細胞膜の損傷、カルシウムのホメオスタシス障害(Thibaultら、2007;Josephら、2011)および興奮毒性の増加(Rzheshevsky、2014)に関連していることが観察されているので、それは興味深い側面である興奮毒性から保護する(図3)。Sprague-Dawley ラットモデルでは、PTERは、アセチルコリンエステラーゼの活性の低下によりコリン作動性伝達を改善し、細胞膜電位の維持を示すATPase(Na+、K+、Ca2+、Mg2+)の作用が増加する(Naik et al.、2017年)。
認知機能 老いたフィッシャーラットの老化の影響を逆転させるのにPTERが有効かどうかを判断した結果、記憶機能は海馬のPTERレベルに関連しており、この抗酸化物質を補充した食事は認知行動の障害を逆転させるのに有効であると結論付けられた(Joseph et al.) PTERはリポポリサッカライド(LPS)誘発の学習・記憶障害を減弱させ、これはミクログリアの活性化の抑制と関連しており、したがって神経細胞の損傷に対する保護効果がある(Houら、2014年)(図3)。Sprague-Dawley ラットの脳室内投与ストレプトゾトシン誘発記憶力低下における記憶喪失を軽減し(Naik et al., 2017)、ヒト神経細胞SH-SY5Y細胞においてエストロゲン受容体シグナル伝達経路を介した酸化的毒性に対する神経保護を仲介する(Song et al., 2015)。
ADでは、PTERを補充することでpTauのリン酸化が減少する(Porquetら、2013)。PTERの誘導体は、抗酸化作用とβアミロイドプラークの凝集に対する阻害作用、コリンエステラーゼ阻害作用を示し、AD患者(Li Y. et al., 2016)や血管性認知症(Lange and Li, 2018)の治療に有用であると考えられる。
PTERは、ペルオキシソーム増殖剤活性化受容体α(PPAR-α)の発現が増加することにより、認知や酸化ストレスの調節因子となる(Changら、2012年)(図3)。SAMP8マウスモデル(散発性および加齢性ADのモデルとして検証されつつある老化促進モデル)におけるADに特徴的な機能障害の改善におけるRESおよびPTERの食事補充効果を比較した結果、RESではなくPTERが細胞ストレスおよび炎症のマーカーを調節し、PPAR-αの発現を上昇させることができると結論付けられた(Chang et al, 2012)、および認知機能の損失に対する保存的因子であるNF-kBの活性化の抑制(Inoueら、2003)(Jeongら、2012;Houら、2014)(図3)を引き起こした(図3)。
酸化ストレス保護、炎症、興奮毒性の調節、神経細胞機能の保存に関連する代謝経路の活性化により、PTERは神経変性疾患に対する保護因子である(Poulose et al.、2015)。
結論
酸化ストレスは神経炎症、ALSの発症と進行に関与している。すべての因子の複雑な相互作用により、酸化ストレスが一次的な原因であるのか、それとも神経細胞における損傷の伝播に対する二次的な効果であるのかを確定的に判断することはできない。しかし、酸化ストレスとミトコンドリア機能障害および神経炎症の間には、ATP生合成能力の低下と活性酸素レベルの上昇を引き起こす「悪循環」を促進する側面があり、明確な関係が確立されている。
現在、ALSの治療薬はないが、さまざまな抗酸化物質の使用が治療戦略として提案されている。その目的は、身体の抗酸化防御力を高め、レドックスバランスを維持することだ。しかし、これらの抗酸化物質の使用にはデメリットがあり、また、結果がほとんど得られていない、あるいは矛盾している、結論が出ていない、統計的なレベルでほとんど有意でないため、より多くの研究が必要な場合もある。ALSの発症メカニズムを考慮すると、新しい治療戦略は、ミトコンドリア、エネルギー産生、抗酸化防御レベルに関連する代謝経路を活性化することを主な目標としている。
NAD+は、エネルギー代謝、ミトコンドリア機能、脳代謝に関与しており、ALSの病態に深くかかわっている。NAD+ 前駆体の投与により NAD+ レベルを回復させることは、ミトコンドリアが酸化還元バランス の障害に対してより抵抗力を持つようになるため、ミトコンドリア機能障害および 酸化ストレス を伴う疾患において 重要な役割を果たすことが期待される。
PTERは、酸化ストレス、ミトコンドリア機能障害、炎症、細胞内カルシウムの回復、認知機能に対する保護に関連する代謝経路を活性化する能力を有するため、ALSの発症メカニズムに対する神経保護機能をもたらすという、生物学的に大きな可能性を持っている。
ヒトを対象とした無作為化二重盲検プラセボ対照試験において、NRとPTERの併用療法を繰り返し投与することで、安全かつ効果的にNAD+レベルが上昇することが明らかになった(Dellinger et al.、2017)。NRとPTERによる治療は有効かつ安全であるため、ALSの病態に作用することから、ALSの治療戦略として期待される。
利益相反
著者らは、本研究が利益相反の可能性があると解釈される商業的または金銭的関係がない状態で実施されたことを宣言する。
略語
- 3-NT 3-ニトロチロシン
- 5-LOX 5-リポキシゲナーゼ
- 7,8-DHF(7,8-ジヒドロキシフラボン) 7,8-dihydroxyflavone
- 8-OHdG 8-オキソデオキシグアノシン
- AD アルツハイマー病
- ALS 筋萎縮性側索硬化症
- CATカタラーゼ
- CNS 中枢神経系
- CoQ10 コエンザイムQ10
- COX シクロオキシゲナーゼ
- CSF脳脊髄液
- DMC ジメトキシクルクミン
- EGCG エピガロカテキンガレート
- ETC 電子伝達鎖
- F2-IsoPS F2-isoprostane
- fALS 家族性筋萎縮性側索硬化症
- GPx グルタチオンペルオキシダーゼ
- GR グルタチオンレダクターゼ
- GSH 還元型グルタチオン
- GSK-3ß グリコーゲン合成酵素キナーゼ3β
- GSSG グルタチオンジスルフィド
- H2O2 過酸化水素
- HD ハンチントン病
- HNE 4-ヒドロキシ-2-ノネナール
- IDH2 イソクエン酸デヒドロゲナーゼ
- IL-18 インターロイキン18
- IL-1ß インターロイキン1β
- IL-6 インターロイキン6
- IL-8インターロイキン-8
- iNOS 一酸化窒素合成酵素
- LDL 低密度リポ蛋白質
- LMN 下部運動ニューロン
- LOX リポキシゲナーゼ
- LPS リポポリサッカライド
- MDA マロンジアルデヒド
- MPO ミエロペルオキシダーゼ
- MS 多発性硬化症
- mtDNA ミトコンドリアDNA
- mtROS ミトコンドリア活性酸素種
- NA ニコチン酸
- NAD+ ニコチンアミド・アデニン・ジヌクレオチド
- NADPH ニコチンアミドアデニンジヌクレオチドリン酸
- NAM ニコチンアミド
- NAMPT ニコチンアミドホスホリボシルトランスフェラーゼ
- NF-kB 活性化B細胞の核因子κ-軽鎖エンハンサー
- NMN ニコチンアミドモノヌクレオチド
- NO 一酸化窒素
- NO- ニトロキシルアニオン
- NO+ ニトロソニウムカチオン
- NO ∙ 一酸化窒素ラジカル
- NOS 一酸化窒素合成酵素
- NR ニコチンアミドリボシド
- Nrf2核因子赤血球2関連因子(Nuclear Factor Erythroid 2-Related Factor
- NRK1 ニコチンアミドリボシドキナーゼ1
- NRK2 ニコチンアミドリボシドキナーゼ2
- NRKs ニコチンアミドリボシドキナーゼ
- O2 一重項酸素
- O・2スーパーオキサイドアニオンラジカル
- OH ヒドロキシル基
- OH ∙ ヒドロキシルラジカル
- ONOO・・・ペルオキシナイトライトアニオン
- 酸化ストレス 酸化ストレス
- PARPs ポリ(ADP-リボース)ポリメラーゼ
- PD パーキンソン病
- PGC-1 α ペルオキシソーム増殖剤活性化受容体γコアクチベーター1-α
- PNP プリンヌクレオシドホスホリラーゼ
- PPAR- α ペルオキシソーム増殖剤活性化受容体α
- pTau タウタンパク質
- PTER プテロスチルベン(トランス-3,5-ジメトキシ-4ヒドロキシスチルベン) Pterostilbene (trans-3,5-dimethoxy-4 hydroxystilbene)
- PUFAs 多価不飽和脂肪酸
- Q3BDG クエルセチン3-β-d-グルコシド
- RCS 銅の活性種
- RES Resveratrol (3,5,4 ′ -trihydroxy-trans-stilbene) レスベラトロール (3,5,4 ′ -trihydroxy-trans-stilbene)
- RIS 活性鉄種
- RNA リボ核酸
- RNS 活性窒素種
- RONS 活性酸素・窒素種
- ROS 活性酸素種
- SA脊髄小脳変性症
- sALS 散発性筋萎縮性側索硬化症
- SIRTサーチュイン
- SIRT1 サーチュイン1
- SIRT3 サーチュイン3
- SODスーパーオキシドジスムターゼ
- SOD1 スーパーオキシドジスムターゼ1
- SOD2 スーパーオキシドジスムターゼ2
- TAS 総合抗酸化状態
- TBARS チオバルビツール酸活性種
- TNF- α 腫瘍壊死因子(TNF)α
- UMN 上位運動ニューロン
- XO キサンチンオキシダーゼ
- 参考文献