
Contents
Amyotrophic lateral sclerosis: a clinical review
オンラインで公開2020年7月7日
P. Masrori 1, 2, 3 and P. Van Dammec responsonding author 1, 2, 3
要旨
筋萎縮性側索硬化症(ALS)は、主に運動系に影響を及ぼす神経変性疾患であるが、最近では運動以外の症状も認められるようになってきている。運動皮質、脳幹核、脊髄前角に存在する上下の運動ニューロンが失われることにより、進行性の筋力低下と消耗が生じる。多くの場合、ALSは局所的に発症するが、その後、体のさまざまな部位に広がり、呼吸筋の障害により、生存期間は発症後2~5年に制限される。また、最大50%の患者さんでは、行動の変化、遂行機能障害、言語障害などの運動外症状が見られる。10%~15%の患者さんでは、これらの問題が前頭側頭型認知症(FTD)の臨床的基準を満たすほど深刻になる。
ALS患者の10%は、家族歴から常染色体優性遺伝であることが示唆されている。残りの90%は、家族に発症者がおらず、散発性ALSに分類される。
ALSの原因は不均一で、一部しか解明されていない。現在までに20以上の遺伝子がALSと関連していると言われている。最も一般的な遺伝子原因は、C9orf72遺伝子の6塩基反復拡張であり、家族性ALSの30~50%、散発性ALSの7%の原因となっている。また、C9orf72は前頭側頭型認知症の原因としてもよく知られており、ALSとFTDの分子レベルでの重複が強調されている。
現在のところ、ALSの治療法は確立されておらず、栄養・呼吸器系のサポートや症状の管理を含む集学的治療が治療の要となっている。本総説では,ALSの疫学,病因,病態,臨床的特徴,鑑別診断,検査,治療,将来の展望など,さまざまな側面からALSを論じている。
キーワード
筋萎縮性側索硬化症、散発性および家族性ALS、TDP-43病理
はじめに
筋萎縮性側索硬化症(ALS)は,1869年にJean-Martin Charcotによって純粋な運動ニューロン疾患として定義されたが,現在では臨床的,遺伝的,神経病理学的なレベルで疾患の不均一性を伴う多系統の神経変性疾患として認識されている[1, 2, 3]。
ALSの臨床症状は、成人になってから発症する局所的な筋力低下と消耗で構成されており、病気の進行とともに広がる傾向がある。筋力低下は最も一般的に四肢の筋肉から始まり、近位筋よりも遠位筋に多く見られる。約25~30%の症例では、構音障害、嚥下障害、発声障害、またはまれに咬合筋の筋力低下を伴う水疱性の発症が認められる。ALSは、発症年齢、発症部位、病状の進行速度に大きなばらつきがある。ほとんどの患者さんでは、ALSは容赦なく進行し、生存期間の中央値は発症後約3年で、死亡原因のほとんどは呼吸不全である。約50%の患者さんは、運動障害に加えて、ある程度の運動外症状を呈する。10%~15%の症例では、前頭側頭型認知症(FTD)と追加診断され [4]、35%~40%の患者では軽度の行動および/または認知機能の変化が見られる。FTDは、前頭葉および前側頭葉の変性を特徴とし、臨床的には、行動の変化、遂行機能の障害、および/または言語障害を示する[5]。現在、ALSとFTDは、両神経変性疾患の基礎となる分子メカニズムが重複していることから、スペクトルの両端に位置すると考えられている[6]。
遺伝子レベルでもALSにはかなりの異質性があり、20以上の遺伝子がALSと関連していると言われている。最も一般的な遺伝子原因は、第9染色体のオープンリーディングフレーム72(C9orf72)におけるヘキサヌクレオチド拡張と、スーパーオキシドディスムターゼ1(SOD1)TAR DNA結合タンパク質43(TARDBP)FUS(fused in sarcoma)およびTANK結合キナーゼ1(TBK1)の変異である。これらを合わせると、全患者の約15%を説明することになる[1, 2, 3]。
ALSの最も一般的な神経病理学的特徴は,TARDBPによってコードされるタンパク質であるTDP-43の細胞質内凝集であり,これはALS患者の95%以上に認められる[7]。TDP-43包接体は、TARDBPの変異を有する患者に特有のものではなく、C9orf72の拡大やTBK1の変異を有する患者や、散発性ALS(sALS)の患者にも見られる。TDP-43は基底状態では主に核に局在しているが、ALSでは細胞質に誤局在して凝集体を形成し、リン酸化される。また、SOD1やFUSなどの他の凝集性タンパク質は、それぞれSOD1やFUSの変異を持つ患者に見られる。C9orf72の6塩基反復拡大を持つ患者では、GGGGCC反復から翻訳されるジペプチド反復タンパク質の蓄積が見られるが、この反復は遺伝子の非コード領域に位置している。
ALSの診断は、臨床診断であり、他の説明が見つからない進行性の筋力低下を伴う患者において、上位運動ニューロン(UMN)と下位運動ニューロン(LMN)の両方の徴候が存在することに基づいている。ほとんどの臨床医は、改訂されたEl Escorial基準[8]やAwajiアルゴリズム[9]は感度が低く、むしろ疾患の進行を捉え、間接的にしか診断の確実性が得られないため、これらの基準には依存していない[10]。さらに、これらの基準は、臨床試験に参加する患者を選ぶための研究目的で開発されたものである。ALSおよび関連する運動ニューロン疾患のサブタイプの臨床診断基準の必要性は高く、診断の遅れを軽減することが求められているが、残念ながら発症後1年を経過することも少なくない。最近、ALSの新しい簡易診断基準が提案されており、UMNとLMNの機能障害が体の1つの部位で組み合わされているか、LMNの機能障害が少なくとも2つの部位で組み合わされていることが必要とされている[11]。これによって診断の遅れが軽減されるかどうかは、さらなる研究が必要である。
ALSの治療薬として欧州医薬品庁から承認されているのは、グルタミン酸拮抗薬であるリルゾールのみであり、ALSの生存率を高める効果は小さいながらも認められている[12]。ALSの基礎となる原因や疾患のメカニズムに関する知識は増え続けているにもかかわらず、40件以上の無作為化臨床試験が否定されている[13]。このように成功していない理由は数多く考えられるが、関係する根本的な原因や疾患メカニズムにかかわらず、ALSを1つの疾患として扱うことは、その1つであるかもしれない。
疫学
筋萎縮性側索硬化症は、ヨーロッパでは年間10万人あたり1.75~3人の発症率と10万人あたり10~12人の有病率が推定されているが、地理的に大きな違いがある[14, 15, 16]。発生率は、ALS発症のリスクが最も高い年齢層(45~75歳)では、10万人あたり年間4~8人となっている。症状が出るまでの平均年齢は様々である。sALSでは58~63歳、家族性ALS(fALS)では40~60歳である[14]。ALS発症の生涯累積リスクは、男性では1:350,女性では1:400と推定されている[17, 18]。男性は女性に比べて散発性四肢発症型ALSの発症リスクが高く、世界的な男女比は1.2-1.5である[19]。
病因 Aetiology
ALSは、他の神経変性疾患と同様に、遺伝的要因、環境的要因、加齢に伴う機能障害が組み合わさって発症すると考えられている。遺伝子レベルでは、これまでに20以上の遺伝子がALSと関連しているとされており、今後もさらに多くの遺伝的要因が発見されることが予想されている。ALSの遺伝子構造は複雑で、現在、効果の大きい単遺伝性変異が患者の約15%を説明しているが、効果の小さい、または中程度の効果を持つ一般的な遺伝子変異や希少な遺伝子変異もALSの発症リスクに寄与しているようである。ALSの全体的な遺伝率は高く、sALS患者の遺伝率は30%~60%と推定されている[18, 20]。ALS患者の一親等の親族では、ALS発症のリスクが2倍になる[20]。
常染色体優性遺伝によるALSの発症
1993年に最初のALS関連遺伝子が発見された。SOD1は、fALSの20%、sALSの1~2%の原因となる遺伝子である[21]。この遺伝子の変異は、SOD1の機能が失われることでALSを引き起こすのではなく、タンパク質が凝集しやすくなり、複数の重要な細胞機能が阻害されることでALSを引き起こす。
2008年と2009年には、RNA結合タンパク質であるTDP-43とFUSをコードする遺伝子、TARDBPとFUSの変異が発見された。これらの変異は、fALSの3〜5%、sALSの1%未満の原因となっている[22, 23, 24, 25]。2011年にはC9orf72が発見され、FALSの30〜50%、SALSの7〜10%の原因となっている[26, 27]。C9orf72のヘキサヌクレオチドリピートの拡大を有する患者は、バルバル発症のALSになる可能性が高く、認知障害や行動障害も有している。
TBK1の変異は、おそらく常染色体優性ALSの5番目に多い原因であり、ALS患者の約1%を占めるが[28, 29]、ALS-FTD患者の最大10%を占める[30]。
ほとんどのSOD1の変異は高い浸透率を持っているが、前述の他の遺伝子は浸透率が低いことが知られており、遺伝カウンセリングを複雑にしている。まれに、これらの遺伝子のうち2つ以上の遺伝子に変異がある患者がいるが、これはALSがオリゴゲンであることを示唆している[31]。
次世代シークエンスを用いて、追加の遺伝子におけるいくつかの希少な変異が同定されている[1, 2, 3]。これらの遺伝子の変異がALSの原因と特定されることはほとんどないが、いくつかの新しい疾患経路に集まっているようである(図1)。
図1 ALS遺伝子の病原性パスウェイへのクラスタリング
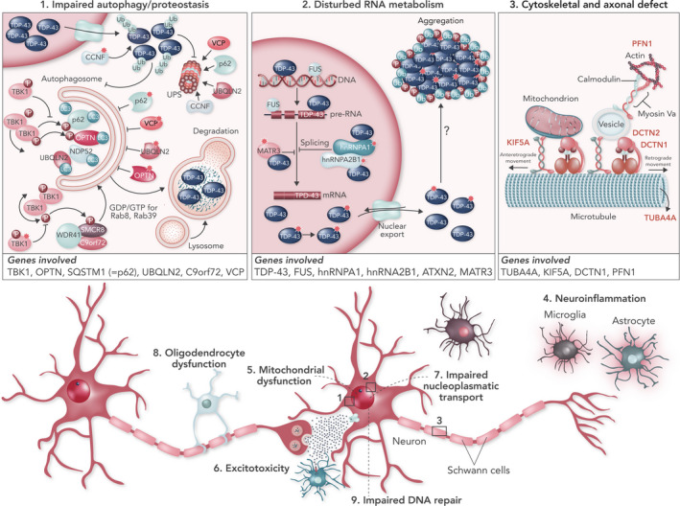
(1) TBK-1,OPTN、SQSTM1 (= p62)、UBQLN2,C9orf72,VCPの変異は、タンパク質分解経路に影響を与え、TDP-43の蓄積に寄与していると考えられる。(2) TARDBP、FUS、MATR3,TIA1,hnRNPA1,hnRNA2B1,ATXN2の変異は、いずれもRNA代謝に影響を与えている可能性がある。(3) TUBA4A、PFN1,KIF5A、DCTN1の変異は、細胞骨格のダイナミクスや軸索輸送に影響を与える。
筋萎縮性側索硬化症の危険因子
筋萎縮性側索硬化症の遺伝的危険因子はわずかにしか特定されていない。リスクのある遺伝子型はUNC13Aであり[32]、ATXN2の中間反復拡大はALSになるリスクを増加させる[33, 34]。
遺伝的要因以外では、年齢と男性の性別がALSのリスクを高めている。いくつかの研究では、喫煙、肥満度、運動、金属、農薬、β-メチルアミノ-L-アラニン、頭部外傷、ウイルス感染など、ALSの環境的な危険因子が示唆されている[35, 36, 37]。しかし、これらの要因とALSとの因果関係はまだ確立されていない。
発症機序 Pathogenesis
ALSの神経病理学的特徴は、神経筋接合部の消失、軸索の後退、それに続くUMNおよびLMNの細胞死であり、これらはアストログリア症とミクログリア症に囲まれており、生き残った神経細胞にはユビキチン陽性の封入体が観察される。ALS患者の95%以上において、TDP-43がこれらの封入体の主成分であることがわかっている[7]。TDP-43は、RNAおよびDNAに結合するタンパク質であり、転写、スプライシング、マイクロRNAの成熟、RNAの輸送、ストレスグラニュールの形成など、複数のプロセスに関与している。TDP-43は、核と細胞質の間を行き来することができるが、その局在は主に核である。細胞質への誤局在は、細胞質のタンパク質凝集とともにTDP-43の核での枯渇につながり、ALSの特徴となっている[38]。
ALSの発症には、
- プロテオスタシスの破綻
- 興奮毒性
- 神経炎症
- ミトコンドリア機能不全と酸化ストレス
- オリゴデンドロサイト機能不全
- 細胞骨格の障害と軸索輸送障害
- RNA代謝障害
- 核細胞質輸送障害
- DNA修復障害
など、複数の分子経路が関与している[2, 39]。興味深いことに,ALSに関連する遺伝子の多くは,タンパク質の品質管理と分解,RNAの代謝,細胞骨格と軸索の輸送という重要な経路に集まっているようである(図1)。
プロテオスタシスの破綻
タンパク質の凝集体、あるいはその前駆体であるオリゴマー複合体は、正常なタンパク質のホメオスタシスを乱し、細胞のストレスを引き起こす。分子シャペロンは、ミスフォールドしたタンパク質のリフォールディングを助けるが、細胞内にミスフォールドしたタンパク質が過剰に蓄積されると、ユビキチン-プロテアソーム系を介してユビキチン化された後、分解の対象となるだろう。また、タンパク質の凝集体は、p62(sequestosome 1)に結合した後、オートファジー経路によるリソソーム分解を受けることもある。
複数のALS関連遺伝子から、タンパク質の凝集と分解の障害がALS発症の鍵となることが示唆されている。実際、ユビキリン-2(UBQLN2)は、ユビキチン化されたタンパク質のプロテアソームへの送達に役割を持っている[40]。他にも、ユビキチン化されたカーゴやファゴフォア膜と相互作用するタンパク質をコードしていることから、オートファジー経路のカーゴ認識に関与する遺伝子にいくつかの変異が見られる。例えば、
- SQSTM1(ユビキチン化されたタンパク質をファゴフォアに標的化するタンパク質p62をコードしている)[41]
- optineurin(OPTN、オートファジーの受容体として機能する)[42]
- TBK1(リン酸化によりOPTNを活性化する)[29]
- valosin-containing protein(VCP)[43]
- C9orf72タンパク質[44]
などが挙げられる。
RNA代謝の乱れ
ALSの発症には、非常に多くのRNA結合タンパク質が関与している。2つの関連するRNA結合タンパク質であるTDP-43とFUSの遺伝子に変異があることが判明したことで、RNA代謝の調節障害というメカニズムがALSに導入された[45]。さらに、アンジオジェニン(ANG)セナタキシン(STX)マトリン-3(MATR3)異種核リボ核タンパク質A1(hnRNPA1)およびA2B1(hnRNPA2B1)アタキシン-2(ATXN2)などの他のRNA結合タンパク質にも変異があることから、RNA代謝の乱れがALSに重要な役割を果たしている可能性があると考えられている[46]。正常な状態では、これらのタンパク質は主に核内に存在し、転写、スプライシング、ノンコーディングRNAの代謝、マイクロRNAの生合成などに重要な機能を果たしている。そのため、核を破壊すると、有害なトランスクリプトームの異常を引き起こす可能性がある。また、細胞質への誤局在化や凝集も同様に毒性を引き起こす可能性がある。
細胞骨格の乱れと軸索の輸送障害
ALSにおけるいくつかの遺伝的要因は、細胞骨格の完全性と軸索輸送の重要性を示唆している[47]。プロフィリン-1(PFN1)とチューブリンα-4A(TUBA4A)の変異は、ALSを引き起こすことは稀であるが、チューブリンネットワークを不安定にし、軸索輸送の欠損を引き起こすことがわかった。ダイナクチン複合体は、ダイニンモーターの重要な活性化因子であり、カーゴの結合を安定化させ、モーターの機能を調節する。ダイナクチン複合体のサブユニットであるダイナクチン1(DCTN1)をコードする遺伝子の点変異は,ALSやFTDの原因となることがある[48, 49].キネシン重鎖アイソフォーム5A(KIF5A)にコードされるキネシン-1のC末端の変異は,微小管に沿ったカーゴの前向輸送を損なう可能性がある[50, 51].
臨床的特徴
臨床症状
ALSの特徴は、進行性の筋力低下であり、筋萎縮、筋束、筋痙攣、筋硬直を伴う動作の緩慢さを伴う。ALSの筋力低下の発症は通常、局所的であり、典型的には隣接する体の部位に広がっていく。このパターンは、脊髄セグメントおよび運動皮質内での神経解剖学的な伝播を伴う、運動系内での疾患病理の広がりと一致する[52]。
本疾患は、通常、片側の遠位筋の筋力低下および上肢または下肢の筋肉の萎縮を呈する(脊髄性ALS、約3分の2の患者)または大腿筋の筋力低下を呈する(大腿筋性ALS、約3分の1の患者)。上肢の発症は利き手が最も多く[53]、足底筋は下腿筋よりも影響を受けやすく(これはスプリットハンド症候群と呼ばれている)[54]、第一骨間筋が早期に侵され、指の伸筋は指の屈筋よりも影響を受けやすい[55]。下肢では、前脛骨筋は腓腹筋よりも早期に、ハムストリングスは大腿四頭筋よりも早期に侵されるのが一般的である[56]。
Bulbar onset ALSは、最も一般的には構音障害または嚥下障害を呈し、あまり一般的ではないが、発声障害、または閉口や咀嚼障害の減少を伴う。頭部が下がるような軸索の筋力低下や姿勢の問題は、病気の後期にはよく見られるが、それが症状として現れることはまれである。約3分の1の患者では、コントロールできない笑いや泣きの発作(仮性球麻痺と呼ばれる)が起こることがある [57]。
一部の患者では、筋力低下に先立って、筋緊張、筋痙攣、または軽度の体重減少が認められている。
神経学的検査では、典型的なALS患者では、UMNとLMNの病変の兆候が組み合わさって認められる。LMN病変の徴候には、筋力低下、萎縮、筋痙攣、筋緊張の低下などがある。UMNの病変としては、反射亢進(または萎縮した筋肉に反射が残る)筋緊張の増大(特に上肢屈筋と下肢伸筋)動作の緩慢さ(例:舌の動き)などが挙げられる。
大多数の患者さんは、脊髄や水疱から発症する古典的なALSの表現型を持っているが、臨床的には、ALSは運動機能や運動外の症状が異なる異質な症候群であることがますます認識されている(図2)。疾患自体の運動症状にもかなりの不均一性があり、運動症状には様々な程度の前頭側頭骨の病変が伴うことがある。そのため、この病気の表現型は様々で、病状の経過も異なる。異なるALSの表現型に対する広く受け入れられた臨床基準は存在しないが、ALSの疾患の異質性を説明するために、普遍的に受け入れられる用語を用いた新しい分類システムの必要性が高まっている[58]。
図2 ALSの表現型の提示
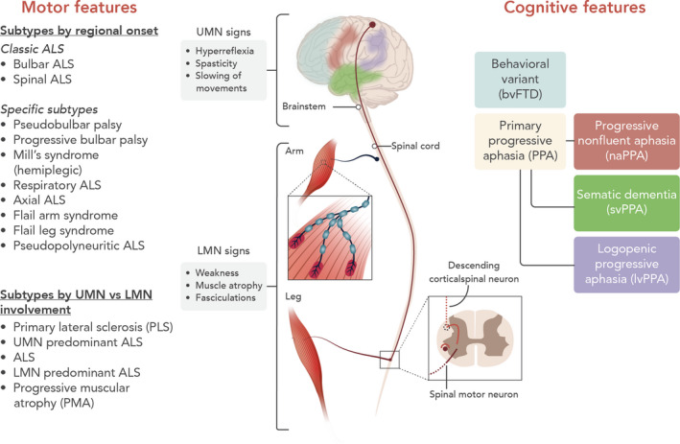
ALSの運動機能は、局所的な分布とUMNとLMNの相対的な関与が異なる。認知・行動面での特徴は、最大で50%の患者に認められる。
筋萎縮性側索硬化症の表現型
筋萎縮性側索硬化症の表現型には様々なものがあり、それらは主にUMNとLMNの相対的な病変と病変の部位分布に基づいて分類される(図2)[3, 58]。ALSのサブタイプによって生命予後が大きく異なるため、異なる運動表現型を認識することは重要である[59]。さらに、様々な程度の認知障害や行動障害が存在する可能性がある。
UMNとLMNの相対的関与に基づくALSのサブタイプ
古典的なALSでは、UMNとLMNの複合的な消失の兆候が1つ以上の身体領域に見られ、運動ニューロン疾患を呈するほとんどの患者が古典的なALSと分類される。
原発性側索硬化症(PLS)は、臨床検査で孤立したUMN徴候を伴う進行性の痙攣と動作の緩慢さが特徴である。症状発現から4年後には、筋萎縮や目に見える筋収縮、筋電図(EMG)での除神経の兆候はないはずである[60]。最も一般的には、症状は下肢に対称的に始まるが、水疱部にも始まることがある。PLSは、全運動ニューロン疾患の3~5%を占めている。PLSは、通常、発症後3~4年以内にALSに移行する可能性がある。PLS患者の生存期間の中央値は20年以上である。UMN優位のALS患者は、LMN病変の特徴をいくつか示するが、UMN病変に比べてはるかに少ない。PLSに比べて生存期間は短いであるが、古典的なALSに比べて病状の進行は緩やかである。
下部運動ニューロン優位のALS患者は、UMNの徴候が非常に限られており、病状の進行速度が異なることがある。進行性筋萎縮症は、UMN機能障害の臨床的証拠を伴わない孤立したLMN徴候が進行することを特徴とするが、進行性筋萎縮症患者の最大30%が追跡調査中にUMN徴候を発症する。
運動ニューロン疾患の病変部位の分布に基づくサブタイプ
Bulbar ALSは、ALSの壊滅的な亜種であり、急速な衰退と発症からの生存期間中央値が2年であることが特徴である。Bulbar UMNの機能障害は、遅々として進まない、歪んだ音声を特徴とする痙性構音障害をもたらす。一方、LMNの機能障害は、舌の衰えと筋収縮を特徴とし、弛緩性構音障害と嚥下障害を伴う。ALS患者の約30%が口唇部の症状を呈するが、大多数のALS患者は最終的に発話および嚥下障害を患います。
仮性球麻痺は、顔の表情がない(無表情顔)痙性の構音障害、咀嚼困難、嚥下困難、痙性による舌の突出が特徴であるが、舌の筋収縮や消耗はない[61]。これはUMNの病変に関するものであるため、顎の痙攣は誇張されているか、間欠性である。この疾患は、LMNが侵される進行性水疱性麻痺と区別する必要があるが、この症候群に関する文献のコンセンサスは得られていない。
ミル症候群(片麻痺変型)は、片麻痺性または非対称性の病変パターンを示す。症状は徐々に進行し、進行は下降よりも上昇が多く、麻痺は顔面筋にも及ぶことがある。錐体外路症状は、通常、片麻痺の側で優勢である。
約3%の患者様は、横隔膜の脱力感(例:労作時の呼吸困難、安静時の呼吸困難、起立性呼吸困難)を初期症状として呈する(呼吸器ALS)。呼吸器系で発症した患者さんは予後が悪い。軸索型ALSでは、傍脊椎筋から発症し、前かがみの姿勢が症状として現れる。
フレイルアームALS(上腕筋萎縮性片麻痺、man-in-the-barrel症候群、Vulpian-Bernhardt症候群)は、進行性の主にLMNパターンの上肢の筋力低下であり、ほとんど対称的な筋力低下のパターンで、典型的には近位筋から始まり、遠位筋へと進行する。最大で77%の方に烏野症状が現れる。男性の割合が高い(男女比3:1)[62]。Flail leg ALSは、進行性、非対称性、主にLMNパターンの筋力低下であり、遠位に発症した下肢の筋力低下および消耗を伴う。発症後12ヶ月以内には上肢や水疱部に顕著な脱力感や消耗はなく、進行は古典的なALSに比べてやや遅い。Pseudopolyneuritic ALSは、下肢の遠位部の筋力低下とアキレス腱反射の欠如が特徴であり、末梢神経障害とは区別する必要がある。
前頭側頭骨の病変に基づくALSのサブタイプ
FTDは、アルツハイマー型認知症に次いで、65歳未満の患者様に最も多く見られる認知症の原因である。ALS患者の約50%では、変性プロセスが前頭葉や前側頭葉にまで及ぶことがあり、様々な程度の遂行機能障害、言語障害、行動変化を引き起こす(図2)。特に求めなければ、これらの変化に気づかないこともある。Edinburgh cognitive and behavioural ALS screenは、前頭側頭葉の機能障害を特定するための有用なスクリーニング検査である[63]。約50%の患者は認知機能が正常であるが、約10~15%の患者では、行動変容型FTDの基準または原発性進行性失語症の基準を満たす場合、ALS-FTDの診断が可能である(表1)。ALS-行動障害は、行動変容型FTDの6つの基準のうち2つを満たすだけでよい。認知障害や行動障害を伴わないALSは、非遂行的な2つの領域(記憶または視空間機能)の機能障害と関連しており、一方、ALS-認知障害は、遂行機能に関する2つのテストの機能障害と関連している[64]。
表1 FTD の基準
| 障害 | 変異体 | 臨床診断 | イメージング(脳の18 F FDG PET / CT) |
|---|---|---|---|
| 原発性進行性失語症(PPA) | 非流暢な失文法異型原発性進行性失語症(naPPA) |
少なくとも一つの:
|
左前頭葉の下、弁蓋および島の部分を含む前部シルビウス周囲萎縮の萎縮 |
| 原発性進行性失語症(svPPA)のセマンティック変異体 |
|
外側および腹側表面ならびに前海馬および扁桃体に影響を与える左前側頭葉萎縮の萎縮 | |
| Logopenic変異体原発性進行性失語症(lv-PPA) |
|
左後部シルビウス周囲または頭頂葉の萎縮 | |
| 行動変種前頭側頭型認知症(bvFTD) |
少なくとも3つ:
|
特に右半球における前頭前野または前頭葉皮質の喪失 |
FTD(前頭側頭型認知症)18F FDG PET/CT(18F-フルオロデオキシグルコース・ポジトロン・エミッション・トモグラフィー/コンピュータ・トモグラフィー)。
予後の予測
ALSの生命予後は極めて多様である。初発時にすでに存在する様々な臨床的特徴が、生存期間の短縮と関連していることが知られている。それらには、水疱性発症、診断までの時間の短さ、急速な機能低下(例:改訂版ALS機能評価尺度(ALSFRS-R)による低下)顕著な体重(または肥満度)の減少、FTDの存在、発症時の年齢の高さ、低い強制肺活量などが含まれる。さらに、遺伝的要因も生存率に影響する。生存期間の短縮に関連する単遺伝性の原因もあるが(SOD1のAla5Val変異、C9orf72反復拡大、FUSのP525L変異)生存期間に影響を及ぼす一般的な変異や稀な変異も報告されている。例えば、UNC13aのrs12608932のCアリルのホモ接合性は、生存期間の短縮と関連している[65]。
臨床パラメータの組み合わせに基づいて、個々の患者の生存結果を推定することができる、初めての個別化予測モデルが開発された[66]。このようなツールは、臨床試験における患者の選択や層別化に有用であり、個別化されたリスクの推定や治療計画にも重要となる可能性がある。
重要な鑑別診断
ALSの診断は、典型的な病状を呈する患者においては比較的容易であり、UMNおよびLMNの変性の徴候を認識することに基づいており、症状や徴候がある領域内または他の領域へと徐々に悪化していくことが必要である(図2)。しかし、非常に早い時期に発症した場合や、病気の進行が遅い場合、あるいは中枢神経系や末梢神経系の障害を併発している場合には、診断が困難になることがある。いわゆる「ALS模倣症候群」と呼ばれる誤診の確率は、約7~8%である[67]。治療の遅れが転帰に好ましくない影響を及ぼす可能性があるため、ALS模倣症候群は除外されるべきである。
UMNまたはLMNの病変が優勢な患者では、鑑別診断の範囲が広くなる。UMN が優勢な患者では、頸部根治性脊髄症、遺伝性痙性麻痺、副腎皮質神経症、および脳腱性黄色腫症を考慮する必要がある。純粋なLMNの特徴がある場合には、神経叢症、末梢神経障害(例:伝導ブロックを伴う多巣性運動神経障害、慢性炎症性脱髄性多発神経炎、感染性神経障害)ミオパチー(例:封入体筋炎)の診断を除外すべきである。フレイルアーム型ALSは、脊髄性筋萎縮症、ケネディ病、多巣性運動ニューロパシー、単孔性筋萎縮症などの擬態と区別する必要がある。局所的に発症した頸部伸展筋力低下の場合には、重症筋無力症や局所性ミオパチーを考慮する必要がある。筋特異的チロシンキナーゼ(MuSK)筋無力症は、舌の脱力と萎縮を伴い、水疱性ALSと間違われることがある[68]。
調査
ALSの診断は、病歴、身体検査、電気診断(針による筋電図)および神経画像診断に依存する。筋電図は、臨床的に罹患している筋肉と罹患していない筋肉におけるLMNの病変を確認するための非常に有用な診断ツールである(細動電位、鋭い波、弛緩した筋肉における筋収縮電位、収縮時の慢性的な神経原性変化を伴う)[8, 69]。
バイオマーカーは、診断、予後、予測の研究において重要な役割を果たす。また,患者の層別化や臨床試験における治療効果のモニタリングにも重要な役割を果たす可能性がある。標準的な臨床診療にはまだ組み込まれていないが、脳脊髄液のニューロフィラメントレベル(特にリン酸化ニューロフィラメントヘビーサブユニット)などのいくつかのバイオマーカーは、特にごく最近に筋力低下が発症し、UMN病変の明確な徴候がない患者や、神経障害/プレキソパシー/頸髄症を併発している患者において、診断の裏付けに有用である[70, 71, 72]。
運動系に影響を及ぼす構造的病変を除外するために、脳および脊髄の磁気共鳴画像がしばしば実施される[73]。さらに、18F-FDG(フルオロデオキシグルコース)ポジトロン・エミッション・トモグラフィーがすぐに利用できる場合は、ローランド脳領域と前頭側頭の病変における代謝低下の典型的なパターンを明らかにすることができる [74, 75]。
ALSで変異が認められる最も一般的な5つの遺伝子の遺伝子検査は、家族歴が陽性の患者にルーチンで提供されている(C9orf72,SOD1,TDP-43,FUS、TBK-1)。sALS患者に対する遺伝子検査についてのコンセンサスは得られていないが、すべての患者に遺伝子検査を提供する傾向がある[76, 77]。しかし、遺伝子検査は、病原性遺伝子の変異が確認された場合に、遺伝カウンセリングが可能な場合にのみ実施すべきである。希少なALS関連遺伝子を含む遺伝子パネルも登場しているが、最も多く変異している5つの遺伝子に加えて診断の歩留まりはまだ低いままである。
治療・管理
過去数十年にわたり、ALS患者を対象とした40以上の無作為化比較試験が行われたが、病気の進行や生存に対する有益な効果を示すことができず、この病気の複雑さを物語っている[13]。欧州のほとんどの国では、リルゾールが唯一承認された疾患修飾薬となっている。リルゾール50mgを1日2回投与することで、抗グルタミン酸作用を発揮し、患者の平均生存期間を3~6カ月延長することができる[12, 78, 79]。最も一般的な副作用は、吐き気、下痢、疲労、めまい、肝機能障害などである。
最近では、フリーラジカルスカベンジャーであるエダラボンがALSで研究されている。選抜されたALS患者を対象に、エダラボン60mg/日を月に2週間ずつ静脈内投与する第3相無作為化二重盲検試験では、6ヶ月間の治療後、ALSFRS-Rのスコアの低下が有意に小さくなったことが示された[80]。この研究は、研究規模が小さいこと、研究期間が短いこと、患者の選択、生存率に関するデータがないことなどから批判されている[81]。現在までに、エダラボンは、米国、カナダ、日本、韓国、スイスでALSの治療薬として承認されているが、欧州連合では承認されていない。
もう1つの治療法として、経口チロシンキナーゼ阻害剤であるマシチニブが検討されている。4.5mg/kg/dayのマシチニブをリルゾールの追加療法として用いた無作為化比較試験では、少なくとも典型的な疾患進行を示す患者において、ALSFRS-Rの低下にプラスの効果があることが示唆されており[82]、この効果は確認試験でさらに検討される予定である。
ALS患者の疾患管理の基礎は、患者の満足度と転帰にプラスの効果をもたらす集学的治療である[83]。ALSの不快な症状のいくつかは、薬理学的および非薬理学的介入を含む対症療法の選択肢によって管理することができる[83]。
例えば、痙攣はバクロフェン、チザニジン、カンナビノイド、筋肉のストレッチで治療でき、唾液漏は抗コリン薬(アミトリプチリン、臭化グリコピロニウム、オキシブチニン)や唾液腺へのボツリヌス毒素の注射で治療できる。
筋痙攣は、マグネシウムサプリメント、硫酸キニーネ、ガバペンチン、カルバマゼピンなどに反応することがある。
情緒不安定の場合には、選択的セロトニン再取り込み阻害剤であるアミトリプチリン、ベンゾジアゼピン、デキストロメトルファン臭化水素酸塩/硫酸キニジンなどが使用できる。
食事の変更は栄養状態の改善に役立ち、カロリー摂取量が不十分な場合や嚥下が困難な場合には、胃瘻チューブを使用することができる。
言語療法は頻繁に必要であり、コミュニケーション支援(カスタマイズされたソフトウェア)も使用することができる。
呼吸不全に対しては、非侵襲的な人工呼吸が望ましい延命治療となる。すべての病期において、患者さんの個別の希望を考慮し、事前のケアプランを早期に開始する必要がある。
将来の展望
ここ数年で、ALSの精密医療の方向に向けた最初の一歩が踏み出された。ALSのいくつかの遺伝子サブタイプでは、上流の遺伝子原因を標的とした治療法が開発されている。これらの治療法の1つにアンチセンスオリゴヌクレオチド(ASO)がある。ASOは短い一本鎖のヌクレオチドで、プレmRNAやmRNAに結合して遺伝子の発現を調節したり、スプライシングを変化させたりする。ASOは、SOD1遺伝子変異やC9orf72リピート拡張を原因とするALSの前臨床モデルで成功している[84, 85]。現在、SOD1およびC9orf72を標的としたASOの髄腔内投与による臨床研究が進行中であり、その結果が待ち望まれている。顆粒球コロニー刺激因子による末梢血幹細胞、骨髄間葉系幹細胞、非神経前駆細胞などの幹細胞治療は、安全性と忍容性が高いことが証明されているが、疾患の進行に対する効果はまだわかっていない。いくつかの第2,3相臨床試験が進行中である[86, 87, 88]。全体として、発症メカニズムに基づいて症例をよりよく分類することで、特定のALSサブグループに有益な効果をもたらす標的療法が可能になり、将来、ALSが治療可能な疾患になることが期待されている。