
Contents
Ammonia as a Potential Neurotoxic Factor in Alzheimer’s Disease
www.ncbi.nlm.nih.gov/pmc/articles/PMC4976099/
要旨
アンモニアは、中枢神経系に深刻な悪影響を及ぼす強力な神経毒であることが知られている。アルツハイマー病のような神経障害を持つ患者の脳内では、過剰なアンモニアレベルが検出されている。そのため、アンモニアはアルツハイマー病の進行に寄与する因子である可能性がある。
本総説では、アンモニアの毒性と考えられるアンモニア輸送タンパク質について紹介する。また、アンモニアがどのようにアルツハイマー病と関連しているかについても仮説を立てる。さらに、アンモニアがアルツハイマー病の進行に寄与する重要な因子であるという仮説を支持する証拠を論じる。
最後に、エネルギー代謝、ミトコンドリア機能、炎症反応、興奮性グルタミン酸、GABA作動性神経伝達、記憶に焦点を当てた新旧の実験的証拠をまとめ、アンモニアがアルツハイマー病に関連しているという仮説を支持する。
キーワード
アンモニア、アンモニア輸送体、毒性、アルツハイマー病、エネルギー代謝、ミトコンドリア機能不全、グルタミン酸、GABA作動性
序論
すべての生物は、細胞の代謝の副産物としてアンモニアを生成する。高濃度では、アンモニアは有毒であり、細胞に有害な影響を及ぼす(Cooper and Plum, 1987)。影響には、細胞エネルギー代謝の混乱、ミトコンドリア機能不全、炎症反応の調節、およびニューロンにおける神経伝達が含まれる。
既存の証拠は、脳内のアンモニアの蓄積が神経細胞の機能に影響を与え、いくつかの神経学的異常を引き起こす可能性があることを示唆している。したがって、アンモニアはアルツハイマー病の原因因子である可能性があり、病気の進行に関与している可能性がある。
1993年、Seilerはアンモニアとアルツハイマー病の関連性についての仮説を初めて発表した(Seiler, 1993)。しかし、それ以来、アルツハイマー病脳内でのアンモニアの病態生理学的役割を直接示した研究はほとんどなかった。
このレビューでは、様々な形態のアンモニアの毒性と輸送について簡単に記述されている。アルツハイマー病に関連する因子も強調され、それに基づいてアルツハイマー病へのアンモニアの寄与を論じている。
脳内アンモニアの発生源
このレビューでは、「アンモニア」という用語は、2つの化学種(NH+4とNH3)を指し、特定の分子形態を指す場合は、「NH+4」または「NH3」を使用する。哺乳類の脳では、アンモニアは主に神経伝達物質であるグルタミン酸、アスパラギン酸、モノアミンの代謝に由来する。脳内では、アンモニアは、内因性および外因性の2つの主要な経路;図(図1;1;Seiller,1993,2002;O’Donnnell,1997)に由来する。
脳アンモニアの内因性源には、タンパク質の加水分解、アミノ酸(例えば、グルタミン、アスパラギン、グリシン)の分解およびヘキサミンの分解、アミノプリン、アミノピリミジンの脱アミノ化、および一級アミンの酸化的脱アミノ化が含まれる。1つの内因性の原因は、大脳皮質内の過剰なアンモニア濃度をもたらすグルコース代謝異常に由来する(Hoyer et al 1988)。
肝機能障害とは別に、アンモニアはまた、グルタミン合成の活性の大幅な低下に起因する脳代謝または解毒プロセスの欠乏から発生する可能性がある(Suarez et al 2002)。脳アンモニアの別の発生源は、プリンヌクレオチドを調節し、AMPをイノシン一リン酸およびアンモニアに変換するアデノシン-3-リン酸(AMP)デアミナーゼである。
1998年、シムズらは、アデノシン-3-リン酸(AMP)デアミナーゼの活性は、コントロール個体と比較して、アルツハイマー病脳で約2倍大きいことを発見した(シムズ et al 1998)。これらの結果は、AMPデアミナーゼの過剰活性が、アルツハイマー病におけるグルコース代謝不足時のアンモニアレベル上昇の原因である可能性があるという仮定につながった(Sims et al 1998)。さらに、モノアミン酸化酵素(MAO)は、神経伝達物質や非伝達物質モノアミンの分解に起因するアンモニア生成のプロセスに、より低い程度に関与している可能性がある。
図1 脳内のアンモニアの発生源、輸送、代謝を図式化したもの
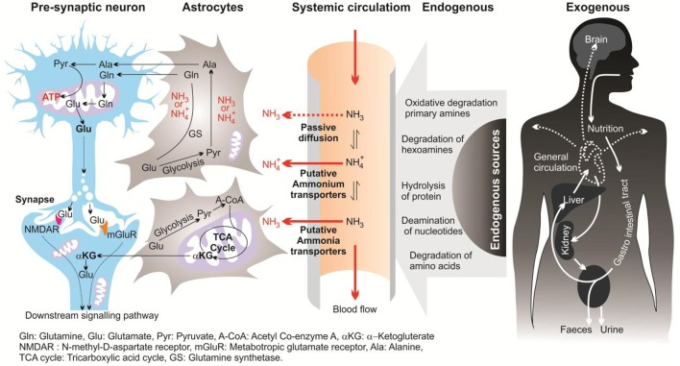
外因性の発生源は、細菌による尿素の分解とアミノ酸の脱アミノ化の結果、消化管内で大量のアンモニアを生成する(Marcaggi and Coles, 2001)。尿素サイクルの障害と肝尿素形成不全、先天的な代謝のエラー、腸内細菌感染は、脳へのアンモニアの蓄積の主な原因である(図.2)。
これまでの証拠は、アンモニアが肝性脳症(HE)の主要な病原因子であり、肝不全の主要な神経栄養因子であることを示している(Häussinger and Schliess, 2008; Lemberg and Fernandez, 2009)。さらに、いくつかの研究では、哺乳類の過剰なアンモニアレベルは、細胞の腫れと最終的に細胞死(バターワース 2002)につながるものは、アストロサイトでグルタミンの毒性蓄積に起因するアルツハイマー病に関連していることを示唆している。しかし、アルツハイマー病 の病理学のアンモニアの役割のための証拠はまだ具体的ではない。
図2
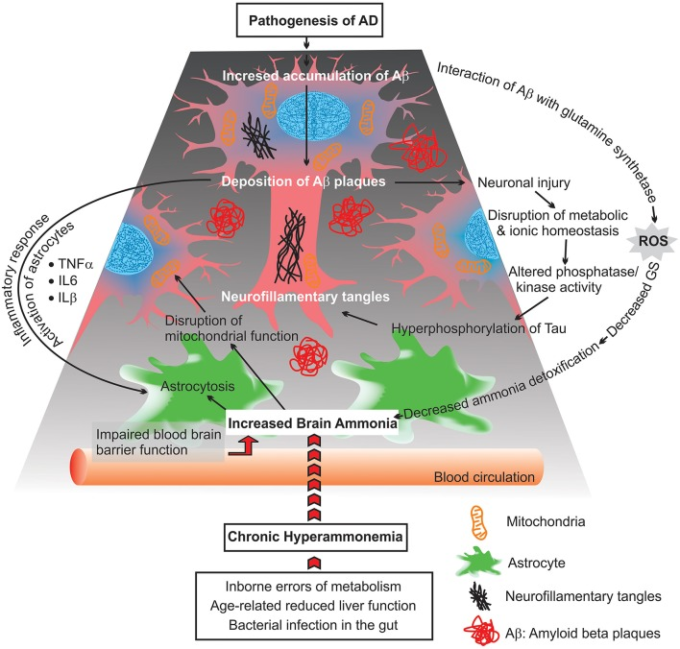
慢性高アンモニア血症は、アストロサイトや神経細胞の障害が進行し、ミトコンドリアの機能不全を引き起こすと考えられている。
アンモニアの毒性
アンモニアは、細胞アミノ酸代謝の主要な最終生成物である(Wright, 1995)。アンモニアは、サブミリモル濃度であっても動物において非常に毒性の高い物質である(Marcaida et al 1992; Britto and Kronzucker 2002)。アンモニアは、媒体の温度および塩分濃度に依存して、pKが9.2〜9.8の弱塩基である(Cameron and Heisler, 1983)。生理的なpH(~7.4)の体液中では、アンモニアの主要な割合(約99%)はNH+4として現れ、残りはNH3として現れる(図(図1).1)。
アンモニアの両方の形態、NH3とNH+4は、潜在的に細胞質と体液のpHバランスを乱すことによって有毒な効果を持っている(エリクソン、1985)。
NH3 は小さいサイズと荷電していない状態のため、脂質二重膜を横切ってリソソームのような酸性小胞に分圧勾配 (ΔPNH3) で拡散し、ゴルジ小胞とリソソームプロテアーゼの適切な機能を損なう可能性がある。これは、NH3がオルガネラ内のpHを正常な動作に必要な最適なpHから遠ざけて変化させるためである (Seglen, 1983)。
アンモニアはグルタミン酸の脱アミノ化により生成され、その毒性はミトコンドリアの内側の膜を横切るH+勾配の破壊に起因する。相対的なアルカリ性のため、ミトコンドリアのpHは細胞質のpHと比較して、マトリックスからミトコンドリア間の空間にΔPNH3が外向きに誘導される。したがって、NH3はこの勾配に沿ってミトコンドリアマトリックスを出て、膜間空間でH+と結合し、それによってATP合成に必要なH+勾配を排除する (Cooper and Plum, 1987)。したがって、pHの低下は、アンモニアがH+勾配-アンカプラーとして作用する酸化的リン酸化を駆動する(O’Donnell, 1997)。さらに、水和されたNH+4イオンとK+イオンは同じイオン半径1.45Åを有する(Knepper et al 1989; Weiner and Hamm 2007)ことから、K+チャネルのK+結合部位での競争が生じる可能性がある。この競争は、哺乳類ニューロンにおけるニューロンの興奮性および膜電位に影響を与える(Cooper and Plum, 1987)。
また、高アンモニア濃度が海馬ニューロンを脱分極させることも実証されている(Bosoi and Rose, 2009)。また、アンモニア濃度の上昇は、血液脳関門(BBB)形態の変化(Laursen and Diemer, 1979)アストロサイトおよびニューロン形態の変化(Gregorios et al 1985)肝性脳症(Butterworth, 2002)など、中枢神経系に大きなダメージを与える。
さらに、哺乳類におけるアンモニアレベルの上昇は、アストロサイトにおけるグルタミンの毒性蓄積のためにアルツハイマー病に関連しており、それは細胞の腫れ、最終的には細胞死につながる(Butterworth 2002)。ミクログリアとアストログリオーマの細胞株では、アンモニアは貪食やエンドサイトーシスなどの主要な機能活性に影響を与える。さらに、アンモニアはサイトカインの分泌を修飾し、リソソソームヒドロラーゼの活性を上昇させる(Atanassov et al 1994, 1995)。さらに、アンモニウムイオンは、α-ケトグルタル酸デヒドロゲナーゼやイソクエン酸デヒドロゲナーゼなどのタンパク質代謝に関与する重要な酵素を阻害し、最終的にフリーラジカルの発生を引き起こす(Cooper and Plum, 1987)。さらに、アンモニア濃度の上昇は、抗酸化酵素の活性を低下させ、ミトコンドリア電子輸送鎖(ETC)を阻害する結果となる(Murthy er al)。
ラット脳では、高アンモニア濃度がミトコンドリアと相互作用し、ETCの複合体I-IVを阻害することが示されている(Veauvy et al 2002)。Marcaidaらは、アンモニアの毒性が脳内のN-メチル-D-アスパラギン酸(NMDA)型グルタミン酸受容体の過剰な活性化によって媒介されるという証拠を発見した。その結果、脳ATPは枯渇し、細胞内Ca2+は細胞外K+のその後の増加に伴って増加し、細胞死につながる(Marcaida et al 1992)。
さらに、神経毒性は、ニューロンシナプスからのグルタミン酸の除去を担うアストロサイトEAAT-1(GLAST)およびEAAT-2(GLT-1)トランスポーターに対するアンモニアの直接的な抑制効果によって媒介される(Knecht et al 1997; Norenberg et al 1997; Chan et al 2000)。哺乳類を含むほとんどの種では、体液中のアンモニア濃度は通常低濃度(約50〜250μM)である(Cooper and Plum, 1987)。1 mMを超える濃度は、通常、哺乳類の細胞に対して毒性がある(Hrnjez et al 1999)。その毒性のため、細胞および体液中のアンモニア濃度を許容範囲内に維持し、正常な全身機能を確保するためには、効果的なアンモニア解毒または排泄システムが極めて重要である。
アンモニア輸送体
rhesus たんぱく質(Rh)
アンモニア誘発ストレスから脳を保護するためには、アンモニア輸送系に仮説的に関与しているアンモニア輸送体の具体的な役割を理解することが重要である。ヒトにおけるアンモニア輸送タンパク質は、rhesus (Rh)タンパク質である。RhAG、RhBG、RhCGである。赤血球中の総アンモニア濃度が血漿アンモニア濃度の3倍以上であることが示されている(Huizenga et al 1994)。RhAG(赤血球-Rh)複合体は、赤血球(RBC)内でアンモニアを輸送することにより、総血中アンモニア濃度を低く保つ役割を果たしていると考えられる(Huanu et al 2004)。哺乳類では、RhAGは赤血球や赤血球組織に存在する(中田 et al 2007)。RhBGおよびRhCG(非erythroid Rh)タンパク質は、脳、腎臓、肝臓、皮膚などの様々な臓器、より具体的にはアンモニアの産生および排泄が重要な場所に分布している(Liu et al 2000; Weiner and Verlander, 2003)。ニジマス(Oncorhynchus mykiss)の脳の Rh タンパク質の遺伝子発現は、アンモニア誘発ストレスを受けると有意に増加した(Nawata and Wood, 2009)。このことは、脳内 Rh タンパク質がアンモニア排泄過程の少なくとも一部に関与していることを示唆している。
脊椎動物のRhタンパク質の機能発現研究では、輸送される分子種(NH3またはNH+4)が異なるため、正確には明らかにされていない。HeLa細胞で発現した場合、ヒト赤血球RhAGは、両方のタイプの分子種を輸送するように見える(Benjelloun et al 2005)。トレーサー研究では、NH+4はRhタンパク質を介してBBBを越えて血漿から脳に輸送されることが提案されている(Ott and Larsen, 2004)。ヒトRhタンパク質の生理的役割は、内因性アンモニア輸送体を欠失した酵母株(triple-MepΔ;3つのアンモニウム輸送体すべてを欠失)でRhCGを発現させることによって明らかにされた。アンモニアが窒素の唯一の供給源である培地上でヒトRhを発現するtriple-MepΔ細胞の成長は、RhCGが酵母細胞内でアンモニウムを輸送する能力があることを示した(Marini et al 2000)。しかし、Rhファミリーのメンバーの輸送特異性に関する議論は現在進行中である。最近、RhCGのX線結晶構造解析により、Rhタンパク質のモノマーには疎水性の細孔要素が含まれており、3量体複合体のタンパク質形態はガス状のアンモニア(NH3)の通過を促進することが明らかになった(Gruswitz et al 2010)。さらに、トポロジー解析の結果、12個の膜貫通ドメインの構造はすべてのRhタンパク質で保存されていることが明らかになった(Huang and Peng, 2005)。哺乳類、魚類、甲殻類、線虫類、昆虫類の間でのRhタンパク質の配列アライメント解析から、Rhタンパク質は系統的に関連しており、保存されたアンモニア輸送機能を共有している可能性が高いことが示唆された(Weihrauch et al 2004; Huang and Peng, 2005; Zidi-Yahiaoui et al 2009; Adlimoghaddam et al 2016)。
アクアポリン(AQP)
アクアポリン(AQP)は、水の輸送のためのチャネルとして動作する膜タンパク質である。AQPファミリーの一部のメンバーは、グリセロール、NH3,尿素、NO、O2,CO2,H2O2,As(OH)3などの他の分子にも透過性がある。アンモニア輸送能力は、哺乳類アクアポリンファミリーの4つのメンバー、AQP3,AQP7,AQP8,およびAQP9について、Xenopus卵母細胞で発現させた場合に確認された(Saparov et al 2007;Litman et al 2009)。遺伝子発現解析の結果、Spf/GFAP-EGEPマウスのアストロサイトにおいて、AQP-4の発現がダウンレギュレートされていることが示された。しかし、ラットの急性肝不全モデルでは、AQP-4のタンパク質発現レベルが有意に上昇し、これはアストロサイトの腫脹の発症に先行するように思われた。
したがって、アストロサイトは、AQP-4の発現レベルの変化によって血中アンモニア濃度の上昇に反応する可能性があると考えられる(Rao et al 2010)。さらに、培養アストロサイトにおけるAQP-4遺伝子をノックアウトすると、アンモニア誘発細胞の腫れを防止できることが示された(Rama Rao et al 2014)。
V型H+-ATPアーゼ(V-ATPアーゼ)
アンモニアを輸送する別の方法は、液胞型H+-ATPase (V-ATPase)を介して起こる (Weihrauch et al 2002)。このトランスポーター自体はアンモニア輸送には直接関与していないが、V-ATPaseを介して上皮の外側にプロトンをポンピングすると(pHを下げると)外向きのΔPNH3が生成され、このΔPNH3は、受動的な膜拡散を介して、あるいは潜在的にRhesusタンパク質のようなNH3透過性チャネルを介して、膜を横切ってNH3の排泄を促進する (Nawata et al 2007; Musa-Aziz et al 2009; Gruswitz et al 2010). このトランスポーターは脳組織で高い発現レベルで局在しており、これは、そのハウスキーピングの役割を超えて神経組織での特定の役割を明らかにしている可能性がある。例えば、試験管内試験でのアンモニア処理は、ラット脳のシナプス小胞におけるH+-ATPaseの活性を刺激する(Albrecht er al)。
Na+/H+交換体(N肝性脳症)
Na+/H+交換体(N肝性脳症)アイソフォームは哺乳類の中枢神経系に広く分布している。これは、H+と交換するために、細胞膜に局在化したN肝性脳症を介して細胞質へのNa+の濃度勾配を下降させる動きにつながっている。すべての細胞は積極的に細胞内pHを調節しており、N肝性脳症は潜在的に酸塩基調節に関与している。例えば、N肝性脳症-1は、ニューロンやアストロサイトで高発現しており、細胞内pH調節や細胞容積の調節に寄与している(Pizzonia er al)。 細胞のpH調節に加えて、N肝性脳症sは脂質二重膜の酸性化を促進し、それによって近位尿細管で示唆されているようにアンモニアの捕捉を助けると考えられる(Hamm and Simon, 1990)。しかし、プロトン輸送が哺乳類アストロサイトのアンモニア捕捉を助けるかどうかは完全には理解されていない。
NH+4の輸送
アンモニアのイオン形態(NH+4)は、生体膜に沿って拡散することはできないが、タイトジャンクションのイオン透過性に基づく傍細胞経路を介して電気化学的勾配を越えて上皮に浸透することができる。さらに、水和されたNH+4とK+は、サイズ及びイオン半径が類似しているので(Knepper et al 1989; Weiner and Hamm, 1989; Weiner er al)。 1989; Weiner and Hamm, 2007)NH+4は、ある程度までは、Na+/K+-ATPase(NKA)K+チャネル、およびNa+/K+/2Cl-共輸送体(NKCC)のようなK+輸送タンパク質において、K+と競合し、基質としてK+に置き換わることができる(Marcaggi and Coles, 2001; Weiner and Hamm, 2007; Larsen et al 2014; Adlimoghaddam et al 2015; Hertz et al 2015)。
Na+/K+-ATPアーゼ(NKA
基底側に局在するNa+/K+-ATPase (NKA)は、ATPを加水分解して細胞質から3つのNa+イオンを細胞外に送り出すと同時に、2つのK+を細胞内に送り込む(Skou, 1957)。NKAは、Na+の電気化学的勾配を生じさせ、また、多くの経上皮輸送プロセスにとって重要な負の膜電位を発生させる。これらのプロセスは、細胞の浸透圧を維持し、NKAのような様々なナトリウム依存性トランスポーターにエネルギーを与えることに有利である(HuおよびKaplan,2000;Kaplan,2002)。アンモニア輸送プロセスにおけるNKAの関与は、哺乳類アストロサイトを含む多くの種および様々な組織において示されてきた。さらに、ラットアストロサイト培養物からの酵素活性測定により、NKAもまた、K+に代わる基質としてNH+4を受け入れ、それによってNH+4の活性輸送(すなわち、体液から細胞質への輸送;Chan et al 2013;Rangroo Thrane et al 2013)に直接関与していることが明らかにされた。アンモニア誘発アストロサイト培養物からのタンパク質およびmRNA発現解析は、高アンモニア濃度に応答してNKAがアップレギュレートされたことを示し、アンモニア輸送機構におけるNKAの重要な役割を示唆している(Xue et al 2010)。さらに、オウアバイン阻害剤を用いてNKAを阻害すると、アンモニア誘発性アストロサイトの膨潤が減少し、NKAがアンモニアの恒常性と細胞の膨潤に関与していることが示唆された(Dai et al 2013; Song and Du, 2014)。
K+チャネル
K+チャネルは、そのユビキタスな細胞内での存在により、膜貫通型のNH+4輸送を媒介する重要な候補の一つであると考えられている。前述のK+とNH+4の競合に基づき、NH+4はバリウム阻害性のK+チャネルを介してBBBを透過することが示唆されている (Ott and Larsen, 2004)。さらに、培養アストロサイトでは、高アンモニア血症の条件下で内向きK+チャネル遺伝子(Kir4.1とKir5.1)が有意にダウンレギュレーションすることが示されている(Lichter-Konecki er al)。 これらの結果から、K+チャネルの変化は、血中NH+4濃度の上昇に対するアストロサイトの保護反応を示すか、または脳外K+および血漿中K+濃度の上昇に反応することが示唆された。このように、脳内K+濃度の変化は、高アンモニア血症時および高アンモニア血症後の神経細胞活動およびネットワーク活動に重要な影響を与えている可能性がある。今後、脳内K+チャネルを介したアンモニアの輸送に関わるメカニズムの詳細を調べるためには、さらなる研究が必要であると考えられる。
Na+/K+/2Cl-共輸送体(NKCC
基底側または先端側に局在するNa+/K+/2Cl-共輸送体(NKCC)は、Na+, K+, 2Cl-をエレクトロンニュートラルな方法で輸送する。NKCCの2つのアイソフォーム(1と2)がいくつかの細胞や組織で同定されている。哺乳類では、NKCC1はアストロサイトや神経細胞など多くの細胞に存在し、NKCC2は主に腎臓に存在している。
最近の研究では、哺乳類の脳組織や脳細胞培養物中のNKCC1がNH+4を基質として受容し、K+を副題としていることが明らかになり、アストロサイトにおけるアンモニア輸送システムにおけるNKCCの重要性が示された。NKCC1は、単離されたアストロサイトにおいてNH+4を輸送することが示されている(Jayakumar et al 2008)。最近の研究では、ラットの培養アストロサイトにおいても、NH+4曝露に応答してNKCC1が活性化されることが示されている。したがって、NKCCの活性化の増加はアストロサイトの膨潤と関連しており、このプロセスはNKCC阻害剤によってブロックされた(Jayakumar et al 2008; Rangroo Thrane et al 2013)。これらの研究は、アンモニアの恒常性とアストロサイトの膨潤におけるNKCCの役割を強調している。
比較研究と窒素輸送の必要性
全体として、アンモニア毒性による窒素輸送系の制御異常やアンモニアトランスポーターの発現・機能の変化は、脳のアンモニア恒常性と機能に影響を与え、アルツハイマー病脳の重篤な神経細胞障害につながる可能性がある。以上のように、アンモニアトランスポーターの発現の変化は、アンモニアの恒常性と細胞の膨潤に重要な役割を果たしている可能性が高いと考えられるが、アンモニアトランスポーター機能の変化とアルツハイマー病との関連性については、まだ明らかにされていない。したがって、アルツハイマー病と正常脳における有害アンモニアの輸送に関与するメカニズムの詳細を調べるためには、さらなる研究が必要であると考えられる。アンモニア輸送系に関連する臨床病理学的メカニズムの解明は、アルツハイマー病の病因との共通のリンクを提供する可能性がある。これらの知見は、全身のアンモニア濃度を上昇させ、最終的にはアルツハイマー病における致死的な脳障害を引き起こす危険なアンモニアの流入を修正するための治療薬の開発に極めて重要である。
アルツハイマー病
現在、アルツハイマー病は世界で最も一般的な進行性の神経変性疾患である(Sperling et al 2011)。臨床的には、シナプス可塑性、学習、記憶、および他のいくつかの認知機能の障害によって特徴づけられる(Albert, 1996)。神経組織学的には、アルツハイマー病は、高リン酸化タウタンパク質の組成とアミロイドβ(アミロイドβ)の沈着から凝集する細胞外老人斑(SP)の蓄積から形成された細胞内神経原線維絡み(NFT)の発達によって特徴づけられる(Price and Morris, 1999; Sperling et al 2011)。この疾患のもう一つの組織学的特徴は、SPにおけるアミロイドβ前駆体タンパク質(アミロイドβPP)の好ましくない代謝、およびその後の損傷した軸索におけるアミロイドβPPの蓄積である。アミロイドβPPの過剰発現は、シナプス障害を引き起こす高リン酸化タウを含むイベントのカスケードを生成する(Ward et al 2012)。
タウの高リン酸化(Grundke-Iqbal et al 1986)およびアミロイドβ沈着(HardyおよびSelkoe 2002)の他に、他の病理学的異常としては、転写異常(PastorcicおよびDas 2007)および神経炎症性プロセスの改変(Granic et al 2009)およびアストログリア症(Akude et al 2011)が挙げられる。アルツハイマー病の病因と神経病因は複雑であり、様々なタンパク質が関与する多因子性の神経変性疾患と考えた方が良いことを示唆している(Carreiras et al 2013)。アルツハイマー病の原因因子については、エネルギー代謝障害、ミトコンドリア機能不全(Hoyer, 1998)神経伝達物質受容体系の変化(GABA、グルタミン酸、MAOなど;Sims er al)。 1998; Jones, 2002)マイクロRNA欠損(Nixon, 2013)細胞周期再突入(Bonda et al 2010)コリン作動性欠損(Pinto et al 2011)神経免疫調節(Akude et al 2011)およびカルシウムホメオスタシスの欠損(Berridge, 2010)などが挙げられる。
アルツハイマー病の病態に関連して研究されてきた神経毒性物質の中で、強力な神経毒としてのアンモニアの影響は、それに値するほど注目されなかった。本レビューでは、アルツハイマー病におけるアンモニアの病因について、糖代謝の欠乏、ミトコンドリア機能不全、グルタミン酸作動性神経伝達とGABA作動性神経伝達の障害、炎症反応の調節障害、記憶機能障害など、いくつかの仮説が挙げられている。
アルツハイマー病と高アンモニア血症の状態でのエネルギー代謝とミトコンドリアの障害
グルコースは脳内の主なエネルギー源であり、グルコース代謝の機能不全は重要な病態生理学的結果をもたらす。いくつかの研究では、認知症の脳における解糖過程の有意な減少が示されている(Meier-Ruge et al 1994; Simpson et al 1994; Hoyer 2000,2004)。グルコース代謝の調節障害は、グルコーストランスポーター(SimpsonおよびDavies、1994)ヘキソキナーゼ(MarcusおよびFreedman、1997)ピルビン酸脱水素酵素(PDH)(Bubber et al 2005)およびアルツハイマー病対対照個体におけるトリカルボン酸(TCA)サイクルの酵素の酵素活性を比較することによって実証されている(Kosenko et al 2014)。
高アンモニア濃度は、マレイン酸塩シャトル(MAS)の活性の低下を引き起こすグルタミン酸濃度の低下とアストロサイトグルタミンの上昇コンテンツにつながる。障害されたMASの結果として、アストロサイトのピルビン酸/乳酸比が減少する。MAS活性とは無関係に、アストロサイトとニューロンの両方で高アンモニア濃度は、PDHの阻害につながるTCAサイクルにおけるα-ケトグルタル酸の脱炭酸を阻害することができる(ヘルツとカラ 2007)。
グルコース代謝の不足の横に、ミトコンドリアの機能性は、アルツハイマー病の脳に影響を受ける。これは含まれている:活性酸素種(ROS)産生の増加、ミトコンドリアの分裂と融合の間のバランスの崩壊、ミトコンドリアの形態の変化、ミトコンドリア酵素の障害、およびミトコンドリア軸輸送の減少率(図(図2;2;朱 et al 2013;カドニック et al 2015)。
ミトコンドリアの調節は一般的に遺伝的に固有であるという仮説が立てられているが、ミトコンドリアの活性は、アンモニアなどの他の神経毒性因子の影響を受けている可能性がある。いくつかの研究は、アンモニアが細胞の生体エネルギー機械の様々な部分を損なうことを示している。例えば、いくつかのETC酵素、ミトコンドリアのチトクロームcオキシダーゼ、グルタチオンペルオキシダーゼ、およびスーパーオキシダーゼジスムターゼの活性は、アンモニア処理した脳では有意に低下する(Kosenko et al 1997,1999,2004,2007;Qureshi et al 1998;Esteves et al 2009)。また、スーパーオキシダーゼ、活性酸素、ポリ(ADP-リボース)ポリメラーゼ(PARP)の活性は、アンモニア誘発ストレス条件で脳のミトコンドリアで増加した(Kosenko et al 2003, 2004; Moreira et al 2008)。既存のエビデンスは、アルツハイマー病ではエネルギー代謝が低下しており、アンモニアがアルツハイマー病におけるエネルギー代謝の混乱(すなわち、ミトコンドリアの機能不全)に関与していることを示している。しかし、ミトコンドリアが高アンモニア濃度によってどのように影響を受けるか、アルツハイマー病 vs. 正常な哺乳類の脳細胞がどのようにエネルギー不足を処理するか、そしてこれらの小器官が脳への有毒アンモニアの大量流入からどのように自分自身を保護するかについてのより良い理解を得るためには、より多くの研究が必要である。
興奮性グルタミン酸作動性およびGABA作動性神経伝達に対するアンモニアの影響
アストロサイトの重要な役割の一つは、過剰なアンモニア(NH3)とグルタミン酸(Glu)を取り込み、アデノシン三リン酸依存性グルタミン合成酵素(GS)を介してグルタミン(Gln)に変換することで、興奮毒性からニューロンを保護することである。肝臓および神経細胞内では、Glnはリン酸依存性グルタミナーゼを介して加水分解され、Gluおよびアンモニア(NH3)に変換される(Zielke et al 1989; Smith, 1990)。
肝性脳症を有する個体では、興奮性神経伝達と抑制性神経伝達の間のバランスが欠如していることが示されている。主な阻害は、グルタミン酸受容体の発現低下によるものであり、これはグルタミン酸緊張の低下をもたらす。さらに、肝性脳症患者におけるグルタミン酸トランスポーター(Glt-1)の阻害は、Gluの過剰なシナプス外蓄積に続くアストロサイトへのGlu再取り込みの減少をもたらす(Albrecht and Jones, 1999)。
さらに、アルツハイマー病脳におけるアンモニア代謝異常は、アストロサイトのグルタミン合成酵素活性の低下と相関していた(Suarez et al 2002)。アンモニア誘発ストレス状態におけるアストロサイトーシスの発現レベルの変化は、アストログリアの形態(アストロサイトーシス)を変化させ、神経細胞の機能に影響を及ぼす可能性がある(図2)2)。グルタミン合成酵素の制御の変化は、Glu-Glnサイクルがアルツハイマー病において異なる障害を受けている可能性を示唆している。さらに、グルタミン合成酵素の活性の低下は、アルツハイマー病脳の皮質におけるアミロイドβおよびSPの細胞外堆積物の密度と関連している(Le Prince et al 1995)。アミロイドβはグルタミン合成酵素と相互作用し、この酵素の酸化的不活性化を誘導し、アミロイドβの神経毒性を高めることが実証された。これらの知見と一致して、アンモニア解毒障害(グルタミン合成酵素活性の変化による)とアルツハイマー病脳におけるアミロイドプラーク形成との間には関連性があることが示唆されている(Aksenov et al 1997年;Robinson 2000)。
アンモニアのグルタミン酸緊張に対する効果に加えて、アンモニアはまた、脳内のガンマ-アミノ酪酸(GABA)システムを変化させる可能性がある。GABAは抑制性神経伝達を媒介する因子の一つである。たとえば、アンモニアの過剰なレベルは、GABA の放出を増加させ、これは、アルツハイマー病 の GABA 神経系の強化につながる。このように、アンモニアによって引き起こされる神経伝達のアンバランスが、アルツハイマー病における認知障害の原因となっている可能性がある(Seiler, 2002; Rama Rao et al 2010)。しかし、アンモニアがどのようにしてアルツハイマー病の症状に寄与するのか、そのメカニズムはまだ十分に定義されていない。
アルツハイマー病におけるアンモニアが引き金となる炎症性反応
脳内アンモニアの上昇は、ミクログリア、アストログリオーマ、アストロサイト、ニューロンによるサイトカインや炎症性タンパク質の放出の変化を引き起こす重要な炎症プロセスに影響を与えることができる(図(図2).2)。さらに、アンモニアレベルの増加は、核内因子κB(NF-κB)のような異なるシグナル伝達分子を介して、神経細胞の変性に関連するアポトーシスを誘導する(Buzanska et al 2000)。NF-κBは、炎症性応答、自然免疫、アポトーシス、およびミトコンドリア機能不全の調節において重要な役割を有する転写因子である(BarnesおよびAdcock、1997;Henderson et al 2002;Sinke et al 2008;Shi et al 2014)。これらには、誘導性一酸化窒素合成酵素(iNOS)一酸化窒素(NO)NADPHオキシダーゼ(NOX)スーパーオキシドおよびペルオキシナイトライト、ホスホリパーゼA2(PLA2)およびサイコオキシゲナーゼ2(COX-2)が含まれ、これらはアストロサイトの膨潤を誘導することが可能であることが証明されている。また、最近の研究では、アンモニアがiNOS、NOX、PLA2,COX-2を活性化し、後述の酵素を阻害することで、アンモニアによって誘導されるアストロサイトの腫脹が有意に減少することが示されている。さらに、免疫組織化学的解析により、アンモニア処理したアストロサイト培養物はNF-κB活性を増加させ、アストロサイトの膨潤を増加させることが示されている。BAY 11-7082によるNF-κB活性の遮断は、培養アストロサイトのアンモニア誘発性膨潤を減少させる結果となった(Sinke et al 2008; Rama Rao et al 2010; Rao et al 2010)。これらの知見は、NF-κB の活性化、ひいてはアストロサイトの膨潤にアンモニアが重要な役割を果たしていることを示唆している。しかし、NF-κB シグナル伝達経路がアストロサイトスウェリングにどのように寄与しているのか、そのメカニズムは完全には解明されていない。このように、NF-κB シグナル伝達経路がどのようにしてアストロサイトの腫脹に寄与しているのかは完全には解明されていない。
炎症性反応の活性化が アルツハイマー病 の病理学的特徴であることが示されている。アルツハイマー病における神経炎症は、IL-6,IL-1β、およびTNF-αなどの炎症性サイトカインの活性の増加によって特徴づけられる。神経炎症は、アルツハイマー病と高アンモニア血症の両方の重要な因子の一つであるため、NF-κBなどの神経炎症性メディエーターを標的とすることは、アンモニアレベルの上昇に伴う神経学的異常の治療に有効な戦略を提供する可能性があるという仮説が立てられている。
アンモニアと記憶
記憶障害は、アルツハイマー病の主要な神経病理学的特徴の一つである(アルバート、1996)。アンモニア濃度の上昇が進行的に障害された精神状態(認知、空間学習、および記憶機能障害)につながる。2000年には、Aguilarらは、高いアンモニア濃度にラット海馬スライスの露出は、その後、動物の記憶や条件付き学習を損なうNMDA受容体を妥協したことを示した(Aguilar et al 2000)。アンモニアの高レベルによって生成される学習障害の他の可能性のあるメカニズムは、ほとんどの場合、ニューロンのグルタミン酸一酸化窒素(NO) ・環状GMP経路の減少を伴う。興味深いことに、NO形成の減少はアルツハイマー病認知症患者の知的機能障害と相関していたが、血管性認知症患者では相関していなかった(Kosenko et al 2014)。
さらに、慢性的なアンモニア暴露は、ニューロステロイド代謝を介して認知機能に影響を与えることが示唆されている。アンモニアはニューロステロイドの合成を阻害し、記憶障害に関与していると考えられている。また、記憶形成に決定的に関与すると考えられている海馬の長期増強(LTP)を阻害することも示されている。最終的には、アンモニアはNMDA受容体の過剰な活性化を介してニューロステロイド代謝を調節し、GABA受容体を介してLTPを阻害することが考えられる(泉 et al 2013)。
おわりに
アンモニアとアルツハイマー病の間に関連があるかもしれないと示唆する最初の仮説が発表されてからちょうど20年以上が経過した(Seiler, 1993)。それ以来、アルツハイマー病脳におけるアンモニアの病態生理学的役割を支持する直接的な研究はほとんどなかった。しかし、アルツハイマー病では高濃度の脳アンモニアが検出されていることから、アルツハイマー病 vs. コントロールの脳細胞におけるアンモニア輸送系の生理学的・分子的メカニズムを明らかにすることで、アルツハイマー病でアンモニア輸送が変化しているかどうかをよりよく理解することができるようになるであろう。アルツハイマー病における窒素輸送機構とその制御に関する知識を得ることは、医療分野に直接関連する。
アンモニアは、おそらくアルツハイマー病の主な原因ではないが、強力な神経毒として作用し、エネルギー代謝の障害、ミトコンドリアの機能不全、炎症反応の調節障害、記憶機能障害などの様々な生物学的経路に影響を与える。興味深いことに、これらの経路は、アルツハイマー病の発生や進行にも関与している。また、これらの要因はアルツハイマー病においても観察されている。このように、アンモニアの毒性とアルツハイマー病の関連性を調べるためには、さらなる研究が必要とされている。このような関連性を調べることは、アルツハイマー病脳内の危険なアンモニアの流入を改善する治療薬の開発や、アルツハイマー病脳内の致死的な脳細胞障害の予防に大きく貢献すると考えられる。