
2016年8月20日 改正2017年1月22日
受理 2017年1月28日
概要
背景
アルツハイマー病は、認知機能の漸進的な低下を特徴とする神経変性疾患であり、世界的に主要な医療課題となっている。アルツハイマー病の病態には、酸化ストレスを介したミトコンドリア機能障害が重要な役割を果たしていることが示唆されている。
したがって、増加した酸化ストレスに応答する抗酸化酵素の生理的活性化は、神経病理を予防すると考えられている。それらの内因性防御の一つは、NAD(P)Hキノンオキシドレダクターゼ1である。NQO1は細胞質のホモ二量体フラボタンパク質であり、キノンや関連分子の溶解性と排泄性を低下させることを目的とした二電子還元を触媒する。第二相ストレス応答タンパク質としての役割に沿って、NQO1の発現の変化は、アルツハイマー病を含むいくつかの病理学的状態や障害と関連している。
結論
本レビューでは、NQO1 と アルツハイマー病 病理との関連性をまとめた。この関連性を理解することで、この疾患の病態についての更なる洞察が得られるであろう。さらに重要なことは、NQO1 の発現や活性に影響を与える薬剤が最近注目されていることであり、このアプローチが アルツハイマー病 治療のための新たな治療法の選択肢につながる可能性があることを示唆している。
1. はじめに
アルツハイマー病は、認知機能の漸進的な低下を特徴とする神経変性疾患であり、世界的に医療への大きな課題となっている[1]。アルツハイマー病の病理学的特徴は、細胞外アミロイドβペプチド(アミロイドβ)斑や細胞内神経原線維抗原(NFT)の脳への沈着を含む。
アルツハイマー病は典型的に遅発性の散発性(ほとんどが65歳以上の患者に見られる)または早期発症の家族性(ほとんどが55歳以前の患者に見られる)の場合がある。アルツハイマー病症例の90~95%以上は散発性であるが、10%未満の症例は、アミロイド前駆体タンパク質(アミロイドβPP)やプレセニリン(PS1,PS2)のようなアミロイドβペプチドの産生に関与する遺伝子の遺伝子変異に関連している[2]。
散発性アルツハイマー病症例は、多くの遺伝的危険因子と関連していることがわかっている。最もよく研究されているのは、アポリポ蛋白E(ApoE)のε4対立遺伝子だ[2]。有益な証拠は、ApoEのε4対立遺伝子がアミロイドβ代謝を低下させ、その後にアミロイドβ蓄積を引き起こすことでアルツハイマー病のリスクを増加させる可能性を示唆している[3]。
これに関連して、ApoE ε4対立遺伝子は散発性アルツハイマー病患者の脳内アミロイドβ沈着リスクの増加と強く関連していることが報告されている[4]が、APOE ε4対立遺伝子の欠損はマウスにおけるアミロイドβ沈着を減少させることが示されている[5]。
全体的に、欠損したアミロイドβ処理は、アルツハイマー病の病理学的な原因となる主要な因子として同定されている[1]。アミロイド生成経路によると、神経毒性のあるアミロイドβ(1-42)ペプチドは、セクレターゼ(βおよびγ)と呼ばれるプロテアーゼを介して、血漿膜に存在するアミロイドβPPのタンパク質分解的な切断によって生成される[6]。
特に、アミロイドβの42アミノ酸型は凝集して不溶性または線維性のアミロイドβオリゴマーを形成しやすく、細胞毒性があり、神経細胞の欠損、シナプス機能の低下、認知機能の低下に寄与している[7,8]。最近では、このアミロイド生成経路はアミロイドβカスケード仮説と呼ばれている。
これによると、アルツハイマー病はアミロイドβの産生量の増加とクリアランスの減少に関連しており、その結果、このペプチドが蓄積され、アルツハイマー病のその後の病因を誘発することになる[9]。また、酸化ストレスによってアミロイドβのオリゴマー形成が亢進することも示唆されている[8]。
2. 酸化ストレスとアルツハイマー病
アルツハイマー病におけるアミロイドβ凝集およびNFT形成が活性酸素種(ROS)によって媒介されていることを示す良好な証拠がある[10-15]。活性酸素は、その濃度に依存して細胞機能に有益な効果だけでなく有害な効果も持つことが知られている[16]。
低濃度では、活性酸素は酸化還元依存性シグナル伝達や酸化還元依存性転写因子を介して細胞機能を制御することができる[16, 17]。しかし、高濃度では、活性酸素は、タンパク質、脂質、DNAなどの細胞のマクロ分子にダメージを与える影響の結果として、重要な細胞プロセスを損なう。
したがって、活性酸素の産生と除去のバランスが正常な細胞機能に不可欠である。活性酸素レベルの恒常性のバランスが崩れると、酸化ストレスとそれに続く病理学的状態を引き起こす可能性がある。実際、高齢化した集団は、内因性抗酸化システムのパフォーマンスが低下しているため、酸化ストレスに対してより脆弱である[18]。
脳内の神経細胞は、特に7つの理由でこのイベントに影響を受けやすく、したがって、高齢者におけるアルツハイマー病のような神経疾患の高い罹患率を少なくとも部分的に説明している[19]。まず第一に、定義上、マイトシス後の細胞は再生することができない [20]。
さらに、彼らの高い代謝率と多量の多価不飽和脂肪酸の存在は、脂質過酸化の活性酸素誘発自己増殖連鎖反応にそれらを作りやすい[20]。最後に、ニューロンは他のガンに比べて限られた抗酸化防御システムを有している [20]。したがって、驚くことではないが、酸化ストレスは、アルツハイマー病 [8,20]の病因の中心であると考えられている。
ヒト神経芽腫SH-SY5Y細胞では、100-250μMの過酸化水素による酸化ストレスが細胞内のアミロイドβレベルの有意な増加を引き起こした[11]。アミロイドβの沈着に加えて、酸化ストレスはNFTの蓄積も促進する[21]。
NFTは、微小管関連タンパク質であるタウを主要な構成要素とするペアらせん状フィラメント(PHF)の束で構成されている[21]。タウのリン酸化は酸化ストレスの結果として起こり、アルツハイマー病の影響を受けたニューロンにおける異常な凝集とこのタンパク質の機能障害に重要であると考えられている[21]。
慢性的な軽度の酸化ストレスの試験管内試験モデルでは、活性酸素は時間依存的にPHF-1エピトープ(セリン399/404)でのタウリン酸化レベルを増加させた[21]。さらに、アルツハイマー病の動物モデルでは、酸化ストレスのレベルの上昇がアミロイドβ沈着、タウの高リン酸化、認知機能の障害に先行することが繰り返し実証されている[8, 12, 22-24]。
確かに、酸化的損傷のバイオマーカーのレベルの増加は、アルツハイマー病患者や高齢者の脳や組織で示されている[25-27]。これらのバイオマーカーは、脂質過酸化(TBARS:チオバルビツール酸反応性物質、マロンジアルデヒド)タンパク質酸化(タンパク質カルボニルと硝酸化)DNA酸化(8HdG:8-ヒドロキシ-2-デオキシグアノシン)GSH系の欠陥、およびスーパーオキシドジスムターゼ活性の障害の存在を示している[25]。
興味深いことに、酸化ストレスのこれらのバイオマーカーは、アルツハイマー病によって影響を受ける脳領域に対応しており、これらのバイオマーカーの量は、疾患の重症度と相関している[28, 29]。それは、アルツハイマー病では、活性酸素誘発性の高分子損傷は、タンパク質のミスフォールディングとアミロイドβとタウを含むミスフォールディングされたプロテインのその後の凝集につながると考えられている[29]。
* *
アルツハイマー病病理学の不可欠な部分としての活性酸素は、トランスジェニックアルツハイマー病動物モデルの血漿、尿および脳組織中の酸化ストレスバイオマーカーの存在によってサポートされている[24, 30]。これらのADマウスモデルにおける抗酸化防御への追加的な障害は、酸化ストレスレベルを上昇させるだけでなく、正常なADマウスと比較してアミロイドβの沈着も亢進させた[31]。
対照的に、トランスジェニックアルツハイマー病動物における内因性抗酸化系(Mn-SOD/Cu-Zn SOD)の過剰発現は、酸化ストレスを減少させ、脳プラーク負担を減少させ、記憶障害を回復させた[22, 32]。アルツハイマー病 脳における酸化マーカーの上昇に加えて、Vit E, C, A などの非酵素性抗酸化物質のレベルが有意に低下し、SOD、グルタチオンペルオキシダーゼ、還元酵素などの抗酸化酵素の活性が低下したことが報告されている [33]。これに関連して、これらの抗酸化物質のレベルが低いと、神経症状、認知機能障害、およびアルツハイマー病発症のリスクの増加を引き起こす。
同様に、別の抗酸化物質CoQ10(ユビキノン)もまた、酸化ストレスとアミロイドプラーク負担を減少させ、トランスジェニックADマウスにおける認知行動を改善することが示されている[34-36]。げっ歯類の主な生理的キノンはCoQ10ではなく、むしろCoQ9であるとして、しかし、ヒトの状況にこれらの知見の適用可能性は不明のままである。
実際、CoQ10の吸収に固有の限界があり、血液脳関門輸送や標的部位へのアクセスが不確実であること、適切にコントロールされた臨床試験が行われていないことから、現時点ではこのアプローチは疑問視されている[37, 38]。
CoQ10と構造的に類似しているもう一つのキノンはイデベノンであり、CoQ10と比較して高い水溶性と優れた経口バイオアベイラビリティーを有する[39]。この薬剤は、アルツハイマー病患者を対象とした複数の臨床試験で試験された。
6つの臨床試験のうち、5つの試験は、1つの研究はアルツハイマー病患者で保護効果を示すことができなかったが、アルツハイマー病におけるイデベノンの有益な効果を示した。成功した試験にもかかわらず、規制当局は、アルツハイマー病におけるイデベノンの治療的使用を支持するには不十分なデータであると判断した[40-44]。イデベノンが単にCoQ10のような抗酸化物質なのか、それとも全く異なる機能を持つ医薬品なのか、文献ではまだいくつかの論争があることに注意しなければならない[39]。
* *
ミトコンドリア電子輸送鎖(ETC)が細胞内の活性酸素の大部分(80-90%)を発生させていることに注意することが重要である。
同時に、これらの小器官の機能は酸化ストレスの影響を非常に受けやすい[45]。ミトコンドリアの機能は加齢とともに低下するという良い証拠がある[46]。したがって、ミトコンドリア機能不全は、驚くことではないが、アルツハイマー病を含む多くの加齢に関連した神経再生疾患に関与している[8, 47]。
脳グルコース代謝の変化は、アルツハイマー病患者の海馬および皮質ニューロンで検出されている[48-50]。脳内グルコース代謝の変化の2つの側面は、脳のインスリン応答性の障害の結果としての異常なグルコース輸送と、ミトコンドリアの機能不全の結果としての細胞内グルコース代謝の変化に関連していると提案されている[49, 50]。
この考えと一致して、2型糖尿病患者は健康な人と比較してアルツハイマー病発症のリスクが高いことを示している[51]。実際、インスリンシグナル伝達は、思考や記憶を含む様々な脳機能に影響を与えることがますます認識されている;アルツハイマー病でも障害されている機能[51]。
アルツハイマー病におけるミトコンドリア機能不全は、アルツハイマー病患者の脳内のチトクロームCオキシダーゼなどのミトコンドリア酸化的ホスホリゼーション(OXPHOS)の主要な酵素の活性低下によって明らかになっている[52]。さらに、ピルビン酸デヒドロゲナーゼ、イソクエン酸デヒドロゲナーゼ、α-ケトグルタル酸デヒドロゲナーゼを含むクレブのサイクル酵素の障害がアルツハイマー病の脳組織で報告されている[53]。
同様に、トランスジェニックADマウスでは、過剰な活性酸素産生、ミトコンドリア抗酸化活性の低下、ミトコンドリア膜電位の低下、アポトーシスが報告されており、神経変性に寄与していると考えられている[54, 55]。
さらに、アルツハイマー病におけるミトコンドリア機能不全の最も直接的な証拠は、アルツハイマー病脳におけるミトコンドリアDNA(mtDNA)変異の存在である[33]。健常者と比較してアルツハイマー病患者の脳組織は、mtDNAのコントロール領域(CR)内での突然変異の割合の顕著な増加を示している[56]。
アルツハイマー病患者の脳の65%がCR変異T414Gに陽性であったのに対し、この変異は健常者の脳には見られなかった。実際、T414C、T4774,およびT477C、T146C、T195Cを含むアルツハイマー病で報告されている他のCR変異を含むいくつかのCR変異はアルツハイマー病に特異的である[56]。これらの変異は、mtDNAのL鎖転写(ND6)および/またはH鎖複製領域に位置している。したがって、これらの変異は、再誘導されたmtDNAコピー数に直結している[56]。
しかしながら、アルツハイマー病患者で観察されるこれらのヘテロプラスミック変異のいくつかがアルツハイマー病病理学的に選択的であるか、あるいは一般集団においてもホモプラスミックな状態で発生するかどうかについては、いくつかの論争がある[57]。
他のmtDNA変異には、アルツハイマー病患者の5.2%に見られるヌクレオチド4336に位置するtRNAGln遺伝子変異や、遅発性アルツハイマー病患者の2%に見られるヌクレオチド3397に位置するND1遺伝子変異が含まれている[58, 59]。共通の4977 mtDNA欠失の定量化は、アルツハイマー病患者の大脳皮質におけるこの突然変異のレベルが対照脳と比較して平均15倍高いことを示した[58]。
興味深いことに、アミロイドβ自体がミトコンドリアの機能を変化させることが実証されている[20]。アミロイドβは、アルツハイマー病患者、トランスジェニックADマウス、変異ヒトアミロイドβPPを発現する神経芽腫細胞の脳ミトコンドリアで検出された[60, 61]。
このミトコンドリアアミロイドβは、ミトコンドリア代謝の障害、ミトコンドリアレスピラトリー鎖の酵素活性の低下、ミトコンドリアダイナミクスの異常、mtDNA変異の存在と関連していた[61-63]。この直接的な効果は、アルツハイマー病のトランスジェニックマウスモデルにおいて、アミロイドβのレベルが過酸化水素のレベルと直接相関しているという観察によって明らかにされている[60]。
逆に、ミトコンドリア媒介の活性酸素はまた、試験管内試験および生体内試験でアミロイドβの産生を増加させる可能性がある[64]。ミトコンドリア複合体IおよびIIIのインヒビターは、ミトコンドリア機能不全および活性酸素産生を誘導する一方で、アミロイドβ産生を増強する。
同時に、抗酸化物質はミトコンドリア機能不全を改善し、ROS産生はアミロイドβ産生を減少させることができる[64]。さらに、ミトコンドリア複合体I欠損マウスまたは複合体I阻害剤で処置されたADマウスは、アミロイドβ産生の増加を示し、これは、ミトコンドリア由来の活性酸素がアミロイドβ産生を誘発し、その後のアルツハイマー病のパトコジェネシスを誘発することができるという見解を支持するものである[64]。
この一連の事象と一致するように、ミトコンドリアの呼吸障害と酸化的損傷の増加がアミロイドβプラークの出現前に起こることが報告されており、これはミトコンドリアの障害が病気の進行の初期に起こり、アルツハイマー病の重要な病理学的因子である可能性が高いことを強く示唆している[48, 60]。
* *
ミトコンドリア機能不全とインスリンシグナルの不完全性の結果として、サイトカインの放出、ミクログリアとアストロサイトの活性化、血管収縮の増加、アミロイドβの生成、血液脳関門の障害を含む炎症性および酸化ストレス反応が引き金となると考えられている。これらのイベントは、その後、脳-ブラール血流、シナプス機能、灰白質と白質の両方のニューロンの完全性を減少させ、その後、認知機能障害とアルツハイマー病につながる[65, 66]。
* *
これらの知見をまとめると、ミトコンドリア機能不全が発生させた酸化ストレスがアルツハイマー病の主要な危険因子であることが示唆される。したがって、増加した酸化ストレスに対抗する抗酸化酵素の生理的または薬理学的活性化は、神経病理学の予防に不可欠である。急性の生理的抗酸化反応の一つは、NADPHキノンオキシドレダクターゼ1(NQO1)のアップレギュレーションである。
3. NQO1
NQO1は、NAD(P)Hを電子供与体として使用することにより、キノン、クワイノニイミン、ニトロ芳香族、グルタチオン置換ナフトキノン、ジクロロフェノールリンドフェノール(DCPIP)およびアゾ色素の二電子還元を触媒する細胞質のFAD依存性フラボタンパク質である。
この2電子再還元により、例えば、酸化ストレスの原因となる反応性セミキノンの形成を防ぐことができる[67]。この触媒的役割に加えて、NQO1は心筋などのSODレベルが比較的低い組織でスーパーオキシドラジカルを直接消去することが報告されている[68]。
NQO1はまた、20Sプロテアソーム分解を防ぐことにより、腫瘍抑制タンパク質p53の安定化にも不可欠であり、これは癌の発生からの保護に不可欠なメカニズムである[68]。
* *
NQO1は、心臓、肝臓、肺、乳房、大腸、血管内皮、脂肪、角膜、水晶体上皮、網膜色素上皮(RPE)視神経、神経線維組織で高発現している[69]。一部の組織では基底レベルが上昇しているにもかかわらず、NOQ1タンパク質レベルは一般的に非常に誘導性が高く、NQO1の発現は、核内因子エリスロイド2関連因子2(Nrf2)とケルチ様ECH関連タンパク質1(Keap1)からなるタンパク質複合体によって制御されている(図1)[67]。
通常の生理的条件下では、細胞質のNrf2はKeap1を介してユビキチンリガーゼ系(カリン3-塩基-E3リガーゼ)に結合し、Nrf2の連続的なユビキチン化とプロテアソーム分解を促進する[67]。酸化ストレス下では、Keap1の257,273,288,297の位置にある反応性の高いシステインはラジカルに攻撃されやすく、Nrf2との複合体を不安定化させる[68, 70, 71]。
その結果、Nrf2はユビキチン化されるのではなく、核内に蓄積され、標的遺伝子のプロモーター領域にある抗酸化応答エレメント(ARE)と結合する。この結合は、ヘムオキシゲナーゼ(HO-1)γ-グルタミル-システイン-リガーゼ(GCL)グルタチオンS-トランスフェラーゼ(GST)グルタチオンペルオキシダーゼ(GPX)およびNAD(P)H:キノンオキシドレダクターゼ1(NQO1)などの広範な細胞保護遺伝子の転写の増加を促進する [70, 72]。
NQO1の誘導またはノックダウンは、酸化ストレスのレベルの低下または増加に直接関連しているため、プロオキシダント状態に対するこの細胞応答の重要な部分は、NQO1に起因している可能性がある[68]。ストレス応答タンパク質としての役割に沿って、NQO1の発現は、アルツハイマー病を含むいくつかの病理学的状態や障害と関連している。
また、いくつかの研究では、NQO1と特異的に共発現しているHO-1の神経保護効果も報告されている。また、HO-1のアップレギュレーションはアルツハイマー病でも報告されているが([73, 74]でレビュー)他の研究では、鉄を介した酸化ストレスの増加に起因するグリア細胞に対するHO-1のアップレギュレーションの有害な効果が報告されている[75-78]。
この有害な役割と一致して、グリアHO-1過剰活動の抑制は、アルツハイマー病の試験管内試験と生体内試験の両方のモデルでアルツハイマー病関連の行動障害や神経病理学的変化を減衰させた[79,80]。さらに、NQO1とは異なり、ヒトAD患者におけるいくつかの報告された関連研究は、増加したアルツハイマー病リスク[81,82]とHO-1多型をリンクしていない。したがって、本レビューでは、NQO1とアルツハイマー病の間の関連性に焦点を当てている。
図1
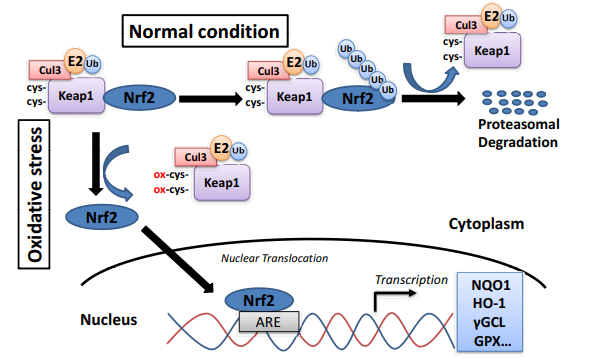
図(1)は、酸化ストレスに対するNrf2依存性のサイト保護遺伝子の誘導を示している。酸化ストレスに応答したサイト保護遺伝子のNrf2依存的誘導の模式図。酸化ストレスはKeap1内のシステイン残基を酸化させ、Nrf2-Keap1複合体を崩壊させる。
これはNrf2の安定化を引き起こし、その結果、NQO1などのARE要素制御細胞保護遺伝子の転写活性化を誘導する。Cul 3:カリン3;E2:ユビキチン結合酵素;keap1:ケルヒ様ECH関連蛋白質1;Nrf2:核内因子-エリスロイド2関連因子2;Cys:システイン;ox:酸化剤;ARE.抗酸化反応エレメント;NQO1:NADPHキノンオキシドレダクターゼ1;HO1:ヘメオキシゲナーゼ1;γGCL:γ-グルタミル-システイン-リガーゼ;GPX:グルタチオンペルオキシダーゼ。
4. NQO1とアルツハイマー病
数多くの研究が、アルツハイマー病 における NQO1 発現のレベルと部位を調べることにより、アルツハイマー病 の病理学における NQO1 の可能性のある役割を調査していた。全体的に、NQO1発現の増加レベルがアルツハイマー病患者の脳で報告されている [83-85]。
このNQO1のアップレギュレーションは、一般的にアルツハイマー病に関連する酸化ストレスへの神経保護反応として再評価されている。アルツハイマー病 患者の脳を年齢をマッチさせた若い対照群と比較したとき、上昇した NQO1 発現は主に海馬ニューロンの細胞質のような アルツハイマー病 病理に影響された領域で観察された(47)。同時に、健康な年齢にマッチした若年者では、同じ神経細胞集団においてNQO1発現は有意に低かった[83]。
同様に、アルツハイマー病患者の海馬切片の組織化学染色では、タウ免疫染色を示した錐体ニューロン集団において、NQO1タンパク質レベルと酵素活性の両方のレベルが上昇していることが示された[84]。NQO1 酵素活性が アルツハイマー病 病理の領域と密接に共局在するという更なる証拠は、前頭皮質のような アルツハイマー病 に一般的に影響を受ける領域で NQO1 酵素活性と免疫組織化学染色を測定し、後頭皮質、小脳、黒質などの アルツハイマー病 に影響を受けていない領域と比較した研究から来ている[85]。
この研究では、NQO1 の小脳酵素活性に対する前頭前野の比率がコン トロールと比較して アルツハイマー病 患者で有意に増加していることが明らかになった。さらに、アルツハイマー病患者の前頭前野に存在していたNQO1染色は、対照個体には存在しなかった[85]。
実際、3xTg-ADマウスにおけるNQO1発現の上昇は、脳内アミロイドβ免疫反応や他の抗酸化酵素の発現増加に先行していた。このことは、少なくとも3xTg-ADマウスでは、NQO1のアップレギュレーションが疾患病理の最初の指標の一つであることを示唆している。
アルツハイマー病におけるNQO1レベルの増加とは対照的に、いくつかの研究では、アルツハイマー病患者におけるNrf2およびNQO1などのNrf2標的遺伝子の発現低下も報告されている[86, 87]。Nrf2の発現を解析したところ、対照群の海馬ニューロンでは主に核内局在と一部の細胞質局在が認められた。
対照的に、アルツハイマー病患者ではNrf2の細胞質局在のみが観察された [88]。Nrf2を標的とした遺伝子の活性化は、アルツハイマー病患者では病期に応じて異なる制御を受けている [29, 88]。
初期段階では、Nrf2依存性遺伝子の発現は、活性酸素に対する初期の防御的な細胞メカニズムの結果として増加する。しかし、疾患が進行するにつれて、酸化ストレスは増加し続け、Nrf2依存性遺伝子の発現は減少するか、停滞したままである[89]。
3xTg-ADマウスでは、病気の初期段階では海馬と大脳皮質でNQO1タンパク質が有意に増加しており、2ヵ月後にはNrf2依存性遺伝子の発現が低下または停滞したままであった[86]。しかし、生後6ヶ月までには、海馬・大脳皮質のNQO1レベルは有意に低下していた[86]。
このNQO1発現パターンは、3ヵ月後には核内Nrf2レベルが有意に増加し、15ヵ月後には3xTg-ADマウスモデルの大脳皮質でレベルが減少していることによって裏付けられている[87]。Nrf2依存性遺伝子発現の保護的役割と一致するように、Nrf2 -/-マウスと変異APP/PS1マウスを交配した場合、変異アミロイドβPP/PS1マウスと比較して、アミロイドβおよびアミロイドβPPの細胞内レベルが有意に増加しており、Nrf2の不活性化がアルツハイマー病の病態を引き起こすことを示唆している[90]。
疾患の後期段階でのNQO1の減少のさらなる証拠は、3xTg-ADモデルで血漿膜酸化還元系(PMRS)活性を評価した研究から得られている[91]。PMRSは、NQO1,チトクロームb5リダクトターゼ、コエンザイムQ10を含む多くの重要な構成要素で構成され、細胞全体の酸化還元状態を維持している[92]。
このマウスモデルでは、酸化ストレスの増加は、酸化ストレスを減少させることでアミロイドβの毒性から神経細胞を保護するPMRSの機能を低下させていた[91]。3xTg-ADマウスの海馬と大脳皮質では、PMRSの重要な酵素の一つであるNQO1がダウンレギュレーションされているのに対し、PMRSの構成要素を過剰発現している神経細胞では、アミロイドβ毒性に対する抵抗性が向上していることが示されている[91]。
NQO1酵素活性とタンパク質発現を調べる以外にも、アルツハイマー病患者ではいくつかの遺伝子関連研究が行われてきた。ヒトでは、NQO1遺伝子は多型である[93]が、NQO1*2と呼ばれる最も頻度が高く、最もよく研究されている多型である。
NQO1*2(C609TまたはPro187Ser)は、1つのヌクレオチド多型であり、NQO1 cDNAの609位のCからTへの変化であり、プロテインのアミノ酸187でプロリンからセリンへの置換をもたらす。この多型はタンパク質を不安定化させ、それにより細胞全体の活性を低下させる[94]。
ヘテロ接合体キャリア(C/T)は、C/C遺伝子型のキャリアと比較して約50%のNQO1活性しか示さず、ホモ接合体キャリア(T/T)は、非常に低い~検出不可能な残留活性しか保有していない[95, 96]。
NQO1*2多型は、いくつかの病態や障害と関連している[86, 97-100]。NQO1*2の有病率における民族間の差異が広く研究されている[93,101]。中国では、人口の50%近くがヘテロ接合体で最大22%がホモ接合体であるのに対し、白人ではヘテロ接合体で最大33%、ホモ接合体で最大5%がNQO1*2であることがわかっている(表1)。
つまり、白人に比べて中国人では、NQO1活性を持たないホモ接合体キャリアが400%近くも多いことになる。NQO1※2とアルツハイマー病との関連性については、主に中国人を対象に研究が進められていた。大半の研究では、NQO1*2多型とアルツハイマー病病理との関連が示されている[102-104]。
中国人集団の散発性アルツハイマー病症例92例と正常対照者108例を対象にNQO1*2の存在を分析したところ、C/T遺伝子型とT/T遺伝子型の両方でT対立遺伝子の頻度が健常者に比べて散発性アルツハイマー病症例の方が有意に高かった[102]。
同様の結果は、65人の散発性アルツハイマー病患者、21人の認知機能障害患者、110人の認知機能障害のない対照者を対象とした研究でも観察されており、NQO1*2多型が実際に中国人集団における認知機能障害と散発性アルツハイマー病の両方の危険因子である可能性を示唆している。
[103]。 これらの結果と一致するように、APOE ε4非キャリアおよび65歳以上のアルツハイマー病症例では、対照と比較してTアレル頻度が高く、C/C遺伝子型頻度が有意に低いことが明らかになった[104]。しかし、中国の後期発症アルツハイマー病患者(アルツハイマー病患者104名、対照128名)では、アルツハイマー病患者と対照との間に対立遺伝子頻度の差は報告されていない[105]。
この結果は、遅発性アルツハイマー病の重要な危険因子と考えられているAPOE遺伝子型(ε4対立遺伝子)で層別化しても変化しなかった[105]。全体として、これらの結果は、C/C遺伝子型がアルツハイマー病の進行を防ぐ可能性がある一方で、T対立遺伝子は遅発型アルツハイマー病の危険因子である可能性があることを示唆している[104]。
以前に実証されたように[102, 104]、NQO1*2多型の存在はNQO1タンパク質レベルを低下させ、したがってアルツハイマー病患者におけるNQO1の全体的な活性を低下させる。アルツハイマー病患者から採取した海馬の50%では、NQO1レベルは検出不可能であり、これはNQO1*2多型の存在と関連していた[98]。
これらの結果は、不活性化NQO1*2多型が、アルツハイマー病の進行だけでなく、タンパク質レベル、したがってNQO1の活性の主要な危険因子であることを示している。しかし、NQO1発現が正常な個体では、NQO1活性は薬物や毒素曝露などの他のいくつかの要因によって影響を受ける可能性があることに注意しなければならない。
5. NQO1の発現や活性に影響を与える因子
NQO1の構造と機能の研究により、NQO1は「ピンポン」機構を介して働く2つの活性結合部位を含むことが示された [106]。1つのサイトは電子供与体であるNAD(P)Hと結合し、もう1つのサイトはキノンなどの基質分子と結合する。
NAD(P)HがNQO1に結合すると、NAD(P)+が活性部位から放出される前に、強固に結合した補酵素フラビンアデニンジヌクレオチド(FAD)をFADH2に還元する。これにより、第二の基質が結合し、その後FADH2によって再誘導される。
主に全体的なNQO1発現(すなわちC690T NQO1多型)に影響を与える遺伝的因子および年齢に加えて、いくつかの薬物および環境因子(すなわち慢性タバコ喫煙)がNQO1活性を直接阻害することが報告されている。
最もよく研究されている、NQO1の競合阻害剤の一つは、ジクウマロール[3,3′-メチレンビス(4-ヒドロキシクマリン)]である(表2)。ジクマロールはNQO1との結合のためにNAD(P)Hと競合し、したがってFADのFADH2への還元を阻害する[106, 107]。
しかし、ジクウマロールは特異性に欠け、ミトコンドリアのアンカップリングや細胞内活性酸素の発生などのオフターゲット効果と関連している[108]。これを克服するために、多くのジクマロール様化合物が合成され、より強力でオフターゲット効果がないと主張されている[108]。
ワルファリンは、ジクマロールに構造的に関連した別のクマリン脱リバティブであり、ジクマロールに比べて効力は低いものの、NQO1を阻害する。
ワルファリンの阻害様式および相互作用部位はジクマロールと非常に類似していることが示唆されている[109]。最近、作用機序の異なる医薬品の代替品が出現しているにもかかわらず、ワルファリンは現在、経口抗凝固療法の主力であることに注意することが重要である。
世界中で何百万人もの人々がワルファリン療法を受けている[110]。ワルファリンの広範な使用と NQO1 の阻害作用、および血液脳関門を通過する能力を考えると、ワルファリンの使用は NQO1 遺伝子型が正常な患者において アルツハイマー病 発症のリスクに寄与する可能性がある。
しかし、現在のところ、この可能性のある関連性を取り上げた研究はない。ジクウマロールの可逆的な阻害とは対照的に、非可逆的なNQO1阻害剤として作用する別の非常に強力な分子はES936であるが、細胞のDNA損傷を誘発することに関連している[111]。
老化の酸化ストレス仮説では、過剰な活性酸素の産生と抗酸化力の低下が老化プロセスを促進すると仮定している。その結果、高分子の酸化損傷が増加し、そのような損傷の蓄積が徐々に細胞の機能不全を引き起こす。老化がアルツハイマー病の主要な危険因子の一つであることはよく知られている。
ストレス因子の存在下または非存在下での老化中の基底および誘導性NQO1の両方のレベルは、異なる動物モデルで研究されている[86,112-120]。しかしながら、動物におけるNQO1の基底発現の加齢に伴う変化の報告は相反するものであり、調査した細胞タイプおよび組織に応じて、発現の増加または減少の証拠がある(表3)。
それにもかかわらず、大多数の研究は、老化した生物の様々な組織におけるNQO1誘導の加齢に関連した減少を示している[113,114,117,121](表3)。
高齢マウスの肝臓、肺、小脳、網膜色素上皮、脾臓Tリンパ球の基底NQO1発現は若いマウスと比較して高かった[112-116]。一方、高齢(18~24ヶ月)ラットの大動脈および肝臓におけるNQO1発現量およびNQO1活性は、若い動物(2~12ヶ月)に比べて低かった [117, 118]。
同様に、高齢マウスの海馬、肺、アストロサイトのNQO1レベルは若いマウスと比較して低かった [86, 119, 120]。基本的なNQO1発現に加えて、加齢はNrf2の調節も障害している [122-124]。
結果として、加齢による酸化ストレスはNrf2の活性化に失敗し、活性酸素の無害化に必要な抗酸化標的遺伝子の誘導も障害する。実際、Nrf2のダウンレギュレーションは、老化したマウスの脳で報告されている[122]。Nrf2標的遺伝子発現の加齢に伴う減少による活性酸素産生の増加、抗酸化物質の枯渇、および高分子に対する酸化的損傷の増加は、ヒトおよびげっ歯類の両方で実証されている[123, 124]。
さらに、過酸化水素とグルコースに対するNQO1誘導は、老齢ラット(24ヶ月)の培養大動脈では、若齢ラット(3ヶ月)と比較して減少していた[117]。この年齢依存性は、非ヒト霊長類モデルでも再現されており、頸動脈および血管平滑筋細胞におけるNQO1の誘導は、若い動物と比較して過酸化水素および高グルコースに反応して鈍化していた[121]。
酸化ストレス誘発剤への曝露は、若年動物(6ヶ月)と比較して、高齢マウス(21ヶ月)の肝臓、肺、小脳のNQO1レベルを上昇させることができなかった[113]。また、若年者に比べてヨウ素酸二ウムに曝露された老齢マウスの網膜色素上皮では、NQO1の誘導障害が観察された[114]。これらの結果は、基底または誘導性のNQO1レベルが加齢に伴って変化することを示している。
たばこの煙(CS)はNQO1発現を変化させるもう一つの因子である。CSは約4700の化学物質を含み、その中でフリーラジカルが最も豊富である[125]。タバコの各パフは、O2-とNO [125]を含む105のフリーラジカルを含むと報告されている。気相とタールまたは微粒子相:CSは2つの相が含まれている。
気相は、そのような一酸化窒素の高濃度など、はるかに多くの反応性である酸素と炭素中心のラジカルが含まれているが、タール相は、qui-none/ヒドロキノンペア[126]を含むいくつかの安定したラジカルが含まれている。特にキノンは、分子状酸素を還元してスーパーオキシドを生成することが知られており、これは二次事象として過酸化水素およびヒドロキシルラジカルの形成に寄与する[126]。
タバコのタール相に含まれる多環芳香族炭化水素(ベンゾ(α)-ピレン-3,6-キノン)複素環芳香族アミンおよびN-ニトロソコム化合物は、ほとんどがNQO1のような第2相酵素によって代謝される[127]。
多環芳香族炭化水素もまた、NQO1の誘導因子として知られている。CSに5時間/日暴露したマウスは、暴露1日後にNQO1誘導を示し、21日後にはより強い誘導を示した[125]。CSの微粒子相を1日2時間/日、28日間曝露すると、スプラague-Dawleyラットの肺の酸化性恒常性が変化し、その結果、NQO1/アクチン比とNQO1酵素活性が増加した[128]。
同様に、18時間のためのタバコの煙の凝縮物に一次正常ヒト気管支上皮細胞の暴露は強く非処理のコントロール細胞と比較してNQO1の発現を増加させた[129]。しかし、タバコの煙への慢性的な暴露では、HO-1とNQO1を含むNrf2ターゲット遺伝子のNrf2活性化とその後の減少の証拠がある[130, 131]。ヒト単細胞/マクロファージ細胞株(THP-1)では、24時間までのCS暴露は、核内Nrf2転座の増加とNrf2標的遺伝子HO-1の誘導を引き起こした[130]。
しかし、長期暴露(72時間)では、HO-1の発現が減少し、Keap 1とNrf2の細胞質蓄積に関連していた [130]。Nrf2 の核内転座の失敗とそれに伴う抗酸化レベルの低下は、CS 曝露 24 時間後のヒト肺上皮細胞でも実証されている [131]。
Nrf2依存性遺伝子発現の阻害と矛盾しないように、慢性的なタバコの喫煙もまた、前臨床試験および臨床試験でアルツハイマー病の病理学を誘導するための重要な危険因子として報告されている[132, 133]。この観察は、喫煙に関連した脳酸化ストレスが潜在的にアルツハイマー病病理学[132, 134]に寄与していることを示唆する報告によってサポートされている。
図2
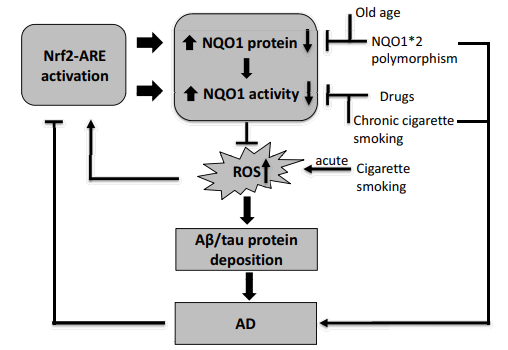
図(2)に示すように、NQO1の活性を阻害する可能性のある素因を示した。NQO1活性を阻害する可能性のある素因。AD病態の素因は、NQO1※2多型、薬物、喫煙、高齢化によるNQO1発現または活性の阻害と関連している可能性がある。
結論
ミトコンドリア機能障害を介した酸化ストレスは、アルツハイマー病の発症に関与している。アルツハイマー病における酸化ストレスの因果的役割に沿って、生理的解毒酵素NQO1の不活性化もまた、アルツハイマー病の進行にリンクされている。
NQO1活性を変化させる要因としては、頻発するC690T NQO1多型などの遺伝的素因、高齢化、タバコの煙、いくつかの薬物などが挙げられる(図2)。NQO1の発現および活性に対するそれらの影響に沿って、これらの要因はまた、アルツハイマー病の発症に役割を果たしている可能性がある。
これまでのところ、NQO1の活性とアルツハイマー病との関連を調べた研究は限られた数にとどまっているが、一貫した結果は時間依存的な関連性を示している。この関連性を理解することは、本疾患の病態をより深く理解することに貢献し、また、アルツハイマー病治療のための新たな治療法の可能性につながる可能性がある。