
Contents
Aducanumab Still Needs to Prove Itself, Researchers Say
www.alzforum.org/news/research-news/aducanumab-still-needs-prove-itself-researchers-say
2020年11月20日
アデュカヌマブの効果はまだ実証されていないと研究者が発表
バイオジェンのアデュカヌマブは、アルツハイマー病の進行を遅らせるために承認された最初の薬になるのか?バイオジェンの生物学的製剤のライセンス申請は、11月6日に開催された米国食品医薬品局の諮問委員会で逆風にさらされた。この諮問委員会では、神経疾患の新規治療法の開発を担当するビリー・ダン氏が主導した(11月2020日付ニュース)。諮問委員会では、神経領域の新薬開発を指揮するビリー・ダン氏が中心となり、生物統計学的評価と神経学的評価の両面から、有効性に関するデータを評価した。これには、委員会だけでなく、全国から集まった多くの科学者たちも戸惑いを隠せなかった。
・専門家は、ENGAGEのネガティブなデータは無視できないと言っている。
・委員会や他の専門家は、承認には時期尚早であると判断し、確認試験を推奨している。
FDAの神経学的レビューとは異なり、生物統計学的レビューは、1日がかりの会議では発表の枠がなかった。しかし、委員会に参加した独立した専門家たちは、FDAのウェブサイトにアップロードされている背景資料の中で、このレビューを読み、説得力のある内容であると判断した。専門家たちは、デュカヌマブの有効性に関するデータが弱いことに同意した。彼らは、スポンサーとFDAのプレゼンターが「時間を忘れて話し」、彼らの質問に十分に答えなかったことを不満に思っていた。彼らは満場一致で承認に反対した。今後の決定はFDAに委ねられている。
アルツハイマー病の研究者はどう考えているのか?アルツハイマー病の研究者は、まだ多くの疑問を持っている。多くの研究者は、この抗体がアミロイドの病態の進行を遅らせると考えている。しかし、意味のある臨床効果があるのか、Aβの下流にある病理マーカーに対する効果はどうなのか、どのような患者層を対象とするのが最適なのか、などの疑問がある。これらの問題を解決するために、彼らはもう1つの試験を望んでいる。一部の人はオフレコで発言したが、他の多くの人はこの記事に名前を使わないでほしいと言った。
フロリダ大学ゲインズビル校のTodd Golde氏はAlzforumに対し、「現在のアデュカヌマブの試験では有効性を証明できない」と述べ、多数派の意見を示した。Golde氏は、「ADの初期臨床段階では、ごくわずかな有益な効果があるかもしれないが、安全性、コスト、アクセスの問題を考えると、我々が期待していたホームランにはほど遠い」と述べている(コメント全文は以下)。同様に、ワシントン大学セントルイス校のDavid Holtzman氏は、「矛盾した結果を説明するバイオジェン社の事後分析は、推測であり不確実であると述べている。アデュカヌマブのさらなる評価が必要であると考える」とホルツマン氏は書いている(以下に詳細を記載)。
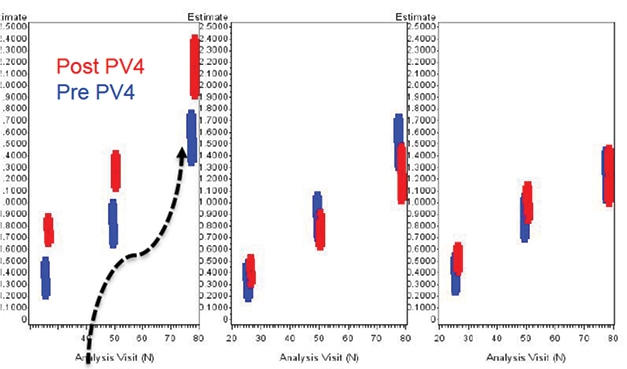
プラセボの悪化
EMERGE試験では、修正案4が発効した後、プラセボ群(左)でランダムに悪化したことで、治療効果があるように錯覚したのではないか?低用量群(中)と高用量群(右)では、主要評価項目のスコアが修正前(青棒)から修正後(赤棒)まで変化しなかった。[FDAウェブサイトより].
バイオジェン社のライセンス申請では、中止となったEMERGE試験の結果をアピールしていた。10mg/kgの持続投与により、主要評価項目であるCDR-SBの認知機能の低下が78週時点で23%抑制された。また、二次評価項目である認知機能の低下も約4分の1に抑制された。バイオジェン社は、PETによるアミロイドプラークの劇的な減少と、タウPETまたは腰椎穿刺を受けたごく一部の被験者におけるタングルの減少を示唆する結果をもって、この知見を支持した。しかし、同じENGAGE試験では陰性であった。バイオジェン社は、この結果について、有効投与量である10mg/kgの総曝露量が少なかったためとしている。APOE4キャリアに対する最大投与量は試験の途中で引き上げられ、事後解析では、最も多くのアデュカヌマブを投与されたENGAGE試験参加者のサブセットがEMERGE試験参加者と同様の反応を示した(2019年10月のニュース 2019年12月のカンファレンスニュース)。
肯定的な知見に対して提起された注意点
FDAの諮問委員会では、この分析結果に疑問が投げかけられた。パネリストたちはコメントや質問の中で、FDAの生物統計学者であるTristan Massie氏のレビューに何度も言及した(ナレーション付きスライドプレゼンテーション3,FDAウェブサイトから入手可能)。Massie氏によると、EMERGEのデータは精査に耐えられないとのことである。この試験におけるポジティブなシグナルは、一部はプラセボ群の減少が加速されたために生じたものであり、一部は薬を服用していた参加者にARIAの副作用が生じたために不用意に盲検化されたために生じたものであると考えられるという。
この2つの問題は、APOE4キャリアーの扱い方に起因する。キャリアーは参加者の約3分の2を占めていた。キャリアーはARIAを起こしやすいので、当初のプロトコルでは最大6mg/kgのアデュカヌマブまで漸増することになっていたが、プロトコル修正4でキャリアーは10mg/kgまで漸増できるように修正された。バイオジェン社の研究者は、このような曝露量の増加により、10mg/kgのEMERGE群で78週目に見られた有効性のシグナルが生じたと主張している。
一方、Massie氏の分析では、修正条項4の発効後、高用量群のCDR-SBの全体的な低下率にはほとんど変化が見られなかった。むしろ、EMERGEのプラセボ群は、修正前の平均1.51ポイントから修正後の平均1.75ポイントへと、修正後の方がより急峻に低下したことがわかった(上図参照)。このように、明らかな有効性のシグナルは、プラセボ群の変動と「絡み合っている」と彼は言う。Massie氏は、「試験結果の全体的なパターンは、用量増量補正後の302試験(EMERGE)で、プラセボがランダムに悪化したことで説明できる」と提案した。
諮問委員会はこれらのデータを問題視した。質疑応答では、バイオジェン社のCraig Mallinckrodt氏がMassie氏の結論に異議を唱え、同社の分析ではプラセボ減少の差は最小限の影響しかないとした。議論は時間切れとなったが、パネリストたちは納得しないままだった。
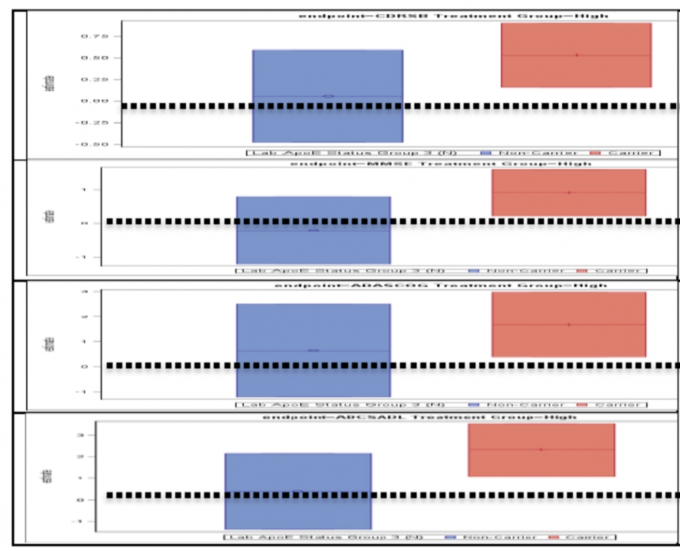
APOE4だけが恩恵を受けたのか?
臨床結果指標であるCDR-SB、MMSE、ADAS-Cog13,ADCS-ADL(上から下)において、APOE4遺伝子保有者(赤)はプラセボ群(点線)よりも良好であり、非保有者(青)はプラセボ群(点線)よりも良好ではなかった。[FDAウェブサイトより].
Massie氏の分析では、盲検化を解除したことによる潜在的な影響にも注目した。高用量投与群の約3分の1がARIAを経験した。そのため、投与を一時中断し、症状が治まるまでMRIでモニタリングする必要があった。このため、被験者は自分が薬物を使用している可能性が高いことを認識し、一部の医師も同様に認識した。このことが、CDR-SBや日常生活動作(ADL)の評価に影響を与えている可能性がある。そもそもCDR-SBやADLの評価は、患者さんやケアパートナーからの情報に基づいて行われるため、やや主観的なものである。さらにMassie氏は、EMERGEの治療効果はAPOE4キャリアのみに認められ、非キャリアはプラセボよりも薬剤の方が悪い傾向にあったと述べている(上図参照)。
非キャリアーでは、最大10mg/kgの投与量が全体的に多く、効果が認められなかったことから、彼らには治療効果がないか、あるいは投与量の問題はバイオジェン社が主張するほど重要ではないことが示唆されるとMassie氏は述べている。また、APOE4サブグループの中で、薬剤に対する反応が最も大きかったグループが、最も盲検化されていたことから、これはプラセボ効果であったのではないかと考えられている。
パネリストもこの点を問題視していた。メリーランド州ベセスダにある国立老化研究所のMadhav Thambisetty氏は、この問題を「大きな懸念」と呼んだ。Mallinckrodt社は、ARIA後のデータを参加者から除外しても、試験全体の結果は変わらないと反論した。しかし、Massie氏は、このデータを除外すると治療群のバランスが崩れるため、盲検化の効果を十分に補正できないと指摘した。
諮問委員会は、EMERGEのデータについて他の懸念を持っていた。無益性解析によって試験が終了したため、参加者の約半数が78週目のデータを持っていなかったのである。2019年秋、バイオジェン社とFDAは共同で試験終了のシミュレーションを行い、欠損データをインプットした。しかし、このようなアプローチは、エラーの可能性を拡大する。「シミュレーションされた完了試験は、真に完了した試験よりも常に不確実性が高い 」とMassie氏は述べた。
認知機能の改善は、78週目の最終時点で、しかも高用量群でのみ認められたとMassie氏は指摘する。プラセボと比較して変化が認められたのは1つの時点だけであるため、この治療法が疾患の進行を遅らせることを証明することはできない、とMassie氏は述べている。さらに、解析計画では、低用量がプラセボよりも優れている場合にのみ、副次的な結果が意味を持つとされていた。このような優先順位の付け方は、Type 1エラーを抑制するのに役立つ。Massie氏によると、低用量ではプラセボを上回る結果が得られなかったため、これらの副次的アウトカムに関する知見は有意とは言えないとのことである。
Thambisetty氏をはじめとする委員会メンバーは、主張されているベネフィットの大きさは小さく、18ヵ月間のCDR-SBで0.40ポイントの差であることを強調した。パネリストたちは、治療費や副作用を考えると、この程度のスローイングに意味があるのか疑問を呈した。投票の結果、11人のパネリストのうち、EMERGEのエビデンスに説得力があるとしたのは1人だけで、8人が否定し、2人が不確かであるとした。
参考
アルツハイマー病の臨床試験の主要なアウトカム指標としてボックスの臨床認知症評価合計を使用する理由
pubmed.ncbi.nlm.nih.gov/22658286/
Clinical Dementia Rating(CDR)[14]は、ADおよびMCIの臨床試験で20年以上にわたって使用されている、十分に検証された尺度である。CDRは、認知の3つの領域(記憶、方向性、判断/問題解決)と機能の3つの領域(地域活動、家庭/趣味、身の回りの世話)を、訓練された評価者による被験者と同伴者/情報提供者との構造化された相互インタビューを用いて評価し、標準的な方法で採点する。テストされた6つの領域(範囲は0~3)のスコアは合計することができ(CDR Sum of Boxes,CDR-SB),得られた情報を総合的なスコアに統合するためのアルゴリズムが使用され,ここでは「CDR Global」スコアと呼ばれている[15].CDRには、被験者および情報提供者との構造化された話し合いが含まれる。CDRには,被験者と情報提供者との構造化された対話が含まれており,評価者が被験者のベースラインパフォーマンスの遠隔の詳細を覚えていたり,被験者のベースラインからの臨床的変化を評価したりする必要がないという点で,1年以上の試験に有利である。
CDR-SBの総得点の変化とMMSEの変化との間の相関は20.46,CDR-SBとADAS-cogとの間の相関は0.42,CDR-SBとADCS-ADLとの間の相関は0.50であり、CDR-SBの総得点の変化と認知および機能の両指標の低下との間には「中程度の」相関があることが示された。
軽度アルツハイマー病の典型的な患者におけるCDR-SB疾患の進行率は、それぞれ約0.5および1.4ポイント/年
www.ncbi.nlm.nih.gov/pmc/articles/PMC4049432/
否定的な結果を否定することはできない
委員会の最大の問題点の1つは、ENGAGE試験の否定的な結果を無視しようとしていると思われたことである。バイオジェン社の申請書とFDAの神経学者Kevin Krudys氏による臨床レビュー(FDAプレゼンテーション1)は、否定的な所見は不十分な投与量によって説明できるものであり、肯定的なEMERGE試験のデータを損なうべきではないと主張した。しかし、パネリストはこれを真っ向から否定した。
ワシントン大学(シアトル)の生物統計学者Scott Emerson氏は、「効かない薬の場合、2つの同じ研究がこのように不一致を起こし、1つの研究で明らかな利益を示す可能性は40%である」と述べている。エマーソン氏は、EMERGEのp値は、否定的な研究を考慮に入れて調整する必要があると指摘した。メリーランド州ボルチモアにあるジョンズ・ホプキンス・ブルームバーグ公衆衛生大学院の薬効専門家であるCaleb Alexander氏は、「EMERGEを単独で見ることはできない」と述べた。他のパネリストも同意見で、ENGAGEが有効性に疑問を投げかけているとしたのが10名、不明としたのが1名であった。
バイオジェン社は、ENGAGE試験の結果を無視するために2つ目の事後報告を行った。すなわち、この試験の治療群には進行の早い患者が多く含まれており、彼らの急速な減少が他の群の治療効果を覆い隠していると主張したのである。この点についても、委員会は納得しなかった。アレグザンダー氏は、数百人のグループの中に数人の進行の速い人がいるだけだと反論した。「私にはそうは思えない。証拠があるとは思えない」と述べた。Alexanderは、「ENGAGEレポートの結果に反して、異常に多くの説明がなされている」と嘆いている。Massie氏の分析によると、急激に減少した異常値を除外しても、ENGAGE高用量群の全体的な所見は変わらなかった。
バイオジェン社の申請ではさらに、フェーズ1bのPRIME試験で得られた良好な臨床データが承認の裏付けとなると主張している。前駆期または軽度のアルツハイマー病患者165人を対象としたその試験では、CDR-SBの低下が薬剤投与6カ月後に鈍化し、最高用量の10mg/kg群で最大の効果が得られた(2015年3月のカンファレンスニュース 2016年9月のニュース 2016年12月のカンファレンスニュース)。この試験は認知機能のベネフィットを検出するための検出力はなく、主要な試験担当者は以前、用量漸増試験における二重盲検法の問題点を指摘していた(2015年11月の学会ニュースのQ&A)。
Massie氏は、PRIMEで得られた知見の多くは、実際に第3相試験と不一致であると判断した。例えば、PRIMEではAPOE4キャリアーは非キャリアーよりも悪い結果となったが、EMERGEでは良い結果となった。また、PRIMEでは明確な用量依存性は認められず、3mg/kgの投与者は6mg/kgの投与者よりも良好であった。重要なのは、試験中に標準的なAD治療薬を開始した被験者がいたことで、これが認知機能のスコアに影響を与えた可能性があることである。これらの参加者を解析から除外したところ、結果はもはや有意ではなく、p値は0.04から0.1になったとMassie氏は計算している。PRIMEのデータは、しっかりとした薬効を示すものではない、とMassie氏は結論づけた。Massie氏は、「このデータが、大規模かつ良好な対照の失敗した臨床試験を覆す理由はない」と述べている。
諮問委員会は再びこの意見に同意した。ハーバード・メディカル・スクールの医薬品承認の専門家であるAaron Kesselheim氏は、PRIME試験は臨床効果を実証するためにデザインされたものではないため、そのデータを第3相試験の裏付けとして使用することはできないと指摘した。この結果、11名の委員のうち7名がこのデータを支持することに反対し、残りの4名は不明となった。
バイオジェン社は、今すぐにでもこの薬を市場に投入することを約束している。Samantha Budd HaeberleinはAlzforumに次のようなコメントを寄せている。「諮問委員会の勧告が、臨床的証拠と患者の経験を無視したことに失望している。」我々は、委員会から提起された統計的な質問に迅速に対応し、社会的に必要とされている明確な説明を行うつもりである。
誰が得をするのか?そして、そのコストは?
このデータは、どのような患者がアデュカヌマブの恩恵を受けるかについて、パネリストと聴衆を困惑させた。EMERGEの効果はAPOE4キャリアに限定されているように思われるだけでなく、年齢、性別、病期、さらには国籍との相互作用も説明できなかった。Massie氏の解析では、主要な臨床的有用性は70歳以上の高齢者に認められ、若年者はプラセボ群と有意差がなかった。また、男性の方が女性よりも効果があった。また、軽度のADの人は前駆症状の人よりも効果があった。これは、早期に治療すればするほど良いという概念に反しているように思われる。アデュカヌマブでなぜ一部の患者だけが良好な結果を得られるのか、その特徴を明らかにすることは、薬剤の適応を決定する上で非常に重要である、とEmerson氏は主張した。
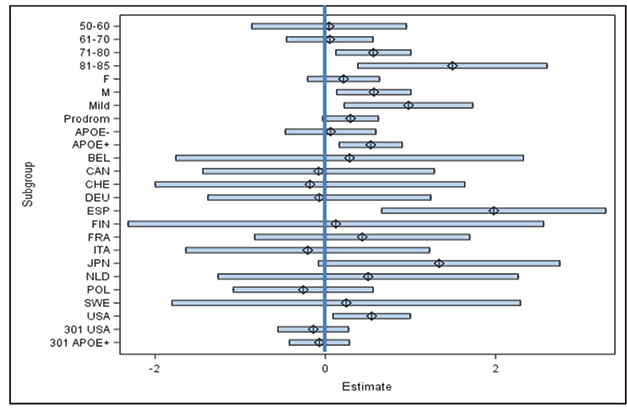
あちこちにデータがある?
EMERGEにおけるサブグループ効果のフォレストプロットでは、高齢者、男性、APOE4保有者、より進行した疾患の人にアデュカヌマブがよく効いているように見える。国別では、スペインでは効いたがイタリアでは効かなかった、オランダでは効いたがドイツでは効かなかった、米国では効いたがカナダでは効かなかった、といったことが考えられる。[FDAウェブサイトより]
EMERGEのデータでは、国による違いが顕著であった。スペインと日本の参加者には大きな効果があり、ベルギー、スウェーデン、フィンランド、フランス、オランダ、米国の参加者には中程度の効果があり、その他の国の参加者には効果がなかった(上図参照)。米国では、CDR-SBの効果を示すエラーバーの幅が狭く、フィンランドでは、エラーバーの幅がフォレストプロットの全範囲に及んでいる。ENGAGEでは、国ごとの違いも顕著であったが、台湾の施設が最も広いエラーバーを掲示していたように、国をまたいでった。
アデュカヌマブには副作用があるため、これらの問題は重要だ。Massie氏の分析では、これまであまり議論されてこなかったものとして、EMERGEの高用量投与群では転倒のリスクが33%高かったことが明らかになった。その理由は明らかではない。
一方、ARIAは治療でよく見られる。多くの場合、無症状であるが、頭痛や混乱を引き起こすことがある。カリフォルニア大学サンフランシスコ校のLawren VandeVrede氏が率いる研究者は最近、APOE4のホモ接合体で、アデュカヌマブ投与中に重度の頭痛、高血圧、てんかん様脳活動を発症したENGAGE参加者の事例を紹介した。彼は読書が困難になり、モントリオール認知機能評価の認知機能スコアが急落した。彼は、ステロイドに加え、抗てんかん薬と高血圧症治療薬で治療を受けた。6カ月後、彼の認知能力はベースラインに戻った(VandeVrede er al 2020)。
この参加者であるポートランドのオレゴン健康科学大学の退職した神経学者Daniel Gibbs氏は、どちらかというと、試練の後は認知的に鋭くなったとAlzforumに語っている。「副作用についてはあまり心配していない。私のARIAの症例は、これまでに報告された中で最も重篤なものだと思うが、私は完全に回復したし、むしろ恩恵を受けたかもしれない」と書いている。彼は、アデュカヌマブについて複雑な思いを抱いており、神経細胞が失われる前の予防的なパラダイムで最も効果を発揮するだろうと考えている。「諮問委員会が承認を推奨しなかったことには失望したが、驚きはしないであった。今後、APOE4の未発症者を対象としたアデュカヌマブ試験が検討されることを期待している」とGibbs氏は述べている(コメント全文は以下)。バイオジェン社は初期段階の試験を計画していたが 2019年に中止し、当時の現場のリーダーたちは悔しがってた。ギブスは、アルツハイマー病を発症した経験について公に書いている(Gibbs, 2019)。
そして、このような状況を現場はどう考えているのだろうか。
Alzforumが接触したアルツハイマー病研究者のほとんどは、諮問委員会に賛同し、アデュカヌマブは臨床使用の準備ができていないことに同意した。アルツフォーラムが直接聞いたところ、21人の研究者のうち18人は、アデュカヌマブが承認されるにはもっとデータが必要だと考えており、2人は確信が持てないと答えた。カリフォルニア大学アーバイン校で統計学の学科長を務めるDaniel Gillen氏は、Alzforumに次のようなコメントを寄せている(コメント全文は以下)。「私は、FDAが十分にデザインされ、十分にコントロールされた無作為化臨床試験を再度要求することを望みます」。
カリフォルニア大学デービス校のCharles DeCarli氏は、アデュカヌマブはプラークに劇的な効果があることから、二次予防試験でアミロイド仮説を検証するための重要な薬剤になると考えている。「しかし、LOADに普遍的に使用するには十分な効果が得られていない」とDeCarli氏は指摘する。
アデュカヌマブは点滴をしなければならないため、費用とロジスティックも考慮しなければならない。ワシントン大学のJohn Morris氏はAlzforumに次のように書いている。ワシントン大学のJohn Morris氏はAlzforumに次のように書いている。「臨床現場では、患者の診断と治療を可能にするための新しいリソースとトレーニングが必要である。「もし薬が効かなければ、これらのリソースは無駄になってしまう」とMorris氏は述べている(以下のコメントを参照)。
ワシントンD.C.にあるNational Center for Health ResearchのDiana Zuckerman氏は、今回アデュカヌマブが承認されたことで、他のAD試験の募集が困難になる可能性があると指摘している。なぜなら、患者さんは新しい薬を試すよりも、承認された薬を服用することを好むからである。バイオファーマ業界のニュースレターが最近行った1,000人以上を対象とした世論調査では、71%がバイオジェン社の現在の申請を却下すべきだと考えてた(Endpoint News)。
FDAのウェブサイトでは、会議の前後に提出されたパブリックコメントは、2対1で承認に反対であった。批判の多くは、データの不確かさを懸念する神経科医からのもので、アルツハイマー病患者の親族の一人は、「次に承認される薬は、間違いなく効くことが証明される必要がある」として、申請を却下することを推奨していた。賛成意見は、アルツハイマー病患者および介護者、プライマリーケア医、アドボカシーグループ、そして医療画像診断グループから寄せられたもので、アデュカヌマブが承認されれば、Centers for Medicare and Medicaid ServicesがアミロイドPET検査の償還を行うようになると指摘している。
南カリフォルニア大学ロサンゼルス校のLon Schneider氏は、FDAに提出したコメントの中で、「アデュカヌマブが実質的に有効であり、この文脈において安全であると表現することは、経済的にも人的資源の面でも無駄であることは言うまでもなく、誤解を招き、最終的には多くのアルツハイマー病患者さんを傷つけることになる 」と書いている。
同様に、ミネソタ州ロチェスターにあるメイヨークリニックのDavid Knopman氏は、FDAに次のように書いている。「私は、この病気を実質的に遅らせたり、元に戻したりする本物の治療法を切に望んでいる。しかし、アデュカヌマブがそのような治療法であり、アルツハイマー病患者に何らかの利益をもたらすという証拠は、ひどく弱いものである。」 バイオジェン社は、高用量のアデュカヌマブで3回目の臨床試験を行う必要がある。バイオジェン社は、高用量のアデュカヌマブを用いた3回目の臨床試験を行う必要がある。Knopman氏は、メイヨー大学の同僚であるDavid Jones氏、スタンフォード大学のMichael Greicius氏とともに、会議の数日前にその懸念を発表していた(Knopman er al 2020)。
また、この評価は厳しすぎると考える人もいる。サンディエゴのUSCのPaul AisenはAlzforumに次のように書いている。「会議の雰囲気とその後の議論は、データの中の重要でポジティブなシグナルを否定するものであった」。ラスベガスにあるCleveland Clinic Lou Ruvo Center for Brain HealthのMarwan Sabbagh氏とネバダ大学ラスベガス校のJeffrey Cummings氏は、Knopman氏の記事への回答の中で、アデュカヌマブを承認することで、AD治療薬への投資と開発が促進される可能性があると指摘した(Sabbagh and Cummings, 2020)。
妥協点を探る人もいた。フェニックスにあるBanner Alzheimer InstituteのPierre Tariot氏は、おそらくFDAは数年にわたる試験ではなく、迅速な確認試験を要求できるのではないかと述べた。諮問委員会では、ワシントン大学の生物統計学者であるEmerson氏が、治験参加者やその介護者が「薬を飲んでいる間は認知機能が維持されていたが、薬をやめると落ち込んだ」と証言していることを取り上げた。エマーソンは、無作為化投与中止試験を行えば、より明確な効果の証拠が得られるのではないかと提案した。この研究デザインでは、すべての参加者が薬物投与を開始し、効果が得られない人は試験から除外される。その後、一部の参加者はプラセボに切り替えられ、症状が悪化すれば薬物の効果があったことになる。このデザインでは、反応した人の数が多いため、異種集団での治療効果を確認しやすく、また、プラセボ投与の時間を最小限に抑えられるため、リクルートにも有利である。
「個人的には、この治療法が成功することを願っている」と、エマーソンは、自身がアルツハイマー病の家族を介護している経験を踏まえて付け加えた。
コメント
Samantha Budd Haeberlein
バイオジェン
投稿日: 2020年11月20日
諮問委員会の勧告が、臨床的証拠と患者の経験を無視したことに失望しています。私たちは、委員会で提起された統計学的な質問に迅速に対応し、コミュニティにとって必要な明確化をもたらします。
私たちは、臨床データと統計手法の完全性を強く支持します。302試験(EMERGE)で実証され、103試験(PRIME)で支持されたように、データはaducanumabの臨床効果の実質的な証拠を提供しています。また、301試験(ENGAGE試験)の結果が一部不一致であっても、302試験の説得力を大きく損なうものではないというFDAの評価を支持します。当グループは、FDAがaducanumabのレビューを完了するまで、引き続き協力していきます。
Daniel Gibbs
投稿日: 2020年11月20日
私はアルツハイマー病の退職した神経科医です。私はAPOE-4ホモ接合体で、ENGAGE試験に参加しました。18ヶ月間の二重盲検法ではプラセボ群でしたが、その後、非盲検法での継続投与に入りました。毎月3回目の投与を受けてから約2週間後に頭痛がひどくなり、軽度の読書障害が出始めるまでは、軽い頭痛を除いて目立った副作用はありませんでした。4回目の投与から数日後、突然、激しい頭痛と脳症が発症しました。地元の病院のICUに入院して血圧をコントロールし、脳卒中とPRESの診断を受けましたが、朝のMRIでは、アミロイド関連画像異常(ARIA)に一致する局所的な浮腫と微小な出血が広範囲に見られました。
血圧はコントロールされましたが、MRIの画像だけでなく、脳症や読解力の問題も1ヶ月間に渡って悪化し、大量のステロイド剤の点滴による治療が必要となりました。私の症状は急速に回復し、MRIは5~6ヶ月で(微小出血によるヘモジデリンの残存を除いて)ベースラインに戻りました。
認知機能はベースラインよりも向上していると感じましたが、Nが1であっても意味がないことを理解しています。その後のアミロイドPETでは、ARIAが悪化した部分のアミロイドが減少していましたが、タウPETでは3年前の前回のPETと比べて改善は見られませんでした。それどころか、タウは進行していました。
アデュカヌマブについては複雑な心境です。諮問委員会が承認を推奨しなかったことには失望しましたが、驚きはしませんでした。アデュカヌマブは、特にAPOE-4キャリアの方のβアミロイドの負担を軽減するという点では、これまでで最も効果的な抗アミロイド治療薬ですが、認知機能の低下を遅らせる効果は、ほぼ期待できません。
このことは、アミロイド仮説が正しくないことを示すものに他ならないと主張する人もいる。確かにそうかもしれませんが、私は、抗アミロイド薬は、認知機能障害が始まる前、つまり神経細胞が失われる前に使用しなければ効果が得られない可能性が高いと考えています。幸いなことに、この仮説を検証するために、発症前の被験者を使った研究が少なくとも2つ進行中です。将来的には、APOE-4を用いたアデュカヌマブの研究でも、発症前の被験者が検討されることを期待しています。
副作用についてはあまり心配していません。私のARIAはこれまで報告された中で最も重篤な症例だと思いますが、私は完全に回復しましたし、恩恵を受けたかもしれません。私の知る限り、ARIAで亡くなった人はいません。ARIAは、血液脳関門を開き、MABがアミロイドに関与するために実際に必要なのではないかと思います。
UCSF
投稿日: 2020年11月20日
難しい状況で、どちらの立場もわかります。パブリックコメントの期間中に患者さんが見事に表現していたように、治療法は緊急に必要であり、有望と思われる治療法へのアクセスを遅らせることは信じられないほど残念なことです。一方で、臨床効果を裏付けるデータの強さには大きな懸念があり、特にEMERGEとENGAGEの間の不一致は、高用量レジメンに基づく第3相試験でなければ明確に解決できない問題です。
現在のデータに基づいてFDAが承認されると、臨床医や患者さんが治療法を決定する際に混乱や不確実性が生じるのではないかと心配しています。最終的には、私たちは疾患修飾治療の頂点にいるという希望を持っていますが、その日が来るのは私たちが望むよりも少し遅いように思えます。
John Morris
ワシントン・ユニバーシティ・スクール・オブ・メディスン
投稿日: 2020年11月20日
37年間、アルツハイマー病の認知症患者さんを介護し、真に効果的な治療法の開発を進めるために臨床研究やトランスレーショナルリサーチを行ってきた私にとって、メカニズムベースの候補薬であるアデュカヌマブがついにスポンサーからFDAに提出され、登録を検討することになったことは非常に喜ばしいことです。もし、本当に有効な抗アルツハイマー病治療薬ができたとしたら、それはこの分野にとって本当に驚くべき進歩であり、患者さんとそのご家族に大きな希望を与えることになるでしょう。
しかし、バイオジェン社がスポンサーとなって実施したアデュカヌマブのENGAGE試験とEMERGE試験のデータを確認していないことをお断りしておきますが、1つの試験で明らかに良好な結果が得られたにもかかわらず、同じように実施された2つ目の試験で同じ結果が得られなかったからといって、この薬が承認されるとは、私には納得がいきません。次のステップとしては、少なくとも1つの厳格な試験を行い、どちらの結果が実際に支持されるかを知ることが適切と思われます。
効果的な治療法が切実に求められていることから、現在のように相反するデータがあっても、アデュカヌマブを承認しようとする気持ちがあるのは理解できます。しかし、万が一、承認されても効果がないことが判明した場合には、様々な弊害が生じる可能性があります。例えば、本当に効果のある治療法を臨床に取り入れるための準備が今のところできていないため、承認されたプロトコルに従って患者を適切に診断・治療する能力を大幅に向上させるために、莫大な資源が必要となります。もし薬が効かなければ、その資源は無駄になってしまいます。
Paul Aisen
USC アルツハイマー病治療研究機関
投稿日: 2020年11月20日
FDAの諮問委員会の会議とその余韻に落胆しています。提出されたデータは難易度が高く、中断された2つの第3相試験では不一致の結果が出ています。しかし、この会議とその後の議論の基調は、データに含まれる重要でポジティブなシグナルを概ね否定するものでした。EMERGEは、ADの疾患修飾を支持する、これまでで最も有望なデータを提供しています。一次解析では、試験が中断されたにもかかわらず、高用量投与による臨床進行に対する緩やかな有益性が示されており、すべての主要な副次評価項目の結果、および下流の病理のバイオマーカーによって裏付けられています。ENGAGE試験では陰性となりましたが、この不一致は、大規模な修正に対する試験のタイミングの違いにより、薬剤への曝露が減少したことで一部説明できます。
FDAがポジションステートメントで述べているように、これまでの抗アミロイド療法の多くの失敗は、抗アミロイド療法を否定する理由にはなりません。これらの失敗は、成功の可能性を大幅に高めるための標的の設定、投与量、試験対象者に関する重要な教訓となっています。aducanumabのデータは、委員会の投票や最終的なFDAの決定にかかわらず、大きな前進を意味します。
ADの原因となる様々な病理学的プロセスを標的とした他の有効な治療法とともに、有益な抗アミロイド療法が臨床の場に登場するでしょう。私たちは正しい道を歩んでいるのです。
David Holtzman
ワシントン大学
投稿日: 2020年11月20日
アルツハイマー病に取り組む多くの医師、医師科学者、科学者と同様に、この分野の私の大きな目標は、アルツハイマー病で起こる認知機能の低下を防ぐ、発症を遅らせる、または遅らせる治療法を開発することです。アデュカヌマブのEMERGE試験やENGAGE試験、そしてこれらの試験に至るまでの研究では、アデュカヌマブを高用量で投与すると、ヒトの脳からかなりの量のアミロイドが除去されることが明らかになっています。私は、アミロイド除去を目的とした抗体やその他の治療法は、アミロイドーシスを発症しても症状が出る前に認知機能の低下を遅らせることができると考えています。EMERGE試験やENGAGE試験で試みられたのは、基礎的なADの病理に関連した認知機能低下がすでにある人を治療することでした。
EMERGEとENGAGEでは、脳にアミロイドが沈着している超軽度/軽度の認知症/軽度の認知機能障害者を無作為化対照臨床試験に登録し、78週間の試験での臨床的認知症評価(CDR)のsum of box(SB)を主要評価項目とした。EMERGE試験の主要評価項目では、最高用量(10mg/kg)でCDR-SBの低下を統計学的に有意に遅らせる(22%)ことが示されたが、ENGAGE試験では主要評価項目に明確な効果は見られなかった。ENGAGE試験の事後解析では、今回の陰性結果は、10mg/kg群の曝露量が少なかったことや、その他の要因による可能性が示唆されています。
ADのような非常に一般的で、非常に重要かつ影響力のある疾患では、RCTが行われ、主要な結果が患者さんとそのご家族にとって明確で影響力のある結果を示すことが重要です。ENGAGE試験の主要評価項目がマイナスであったことは、抗体への曝露量やその他の要因によって説明できるのではないかという議論がなされていますが、私にはこれは非常に不確実で推測に過ぎません。
要するに、EMERGE試験では、重要な臨床結果(CDR-SB)について、最高用量で非常に小さいながらも統計学的に有意な効果が認められたということです。ENGAGE試験では、主要評価項目で陰性となっています。
私は、文字通り何百万人もの人々が対象となり、米国だけでも年間200億ドル以上の費用がかかると推定される治療法を、有益である可能性が高いかどうかが明確でないにもかかわらず承認するのは大きな間違いだと思います。アデュカヌマブがADによる超軽度認知症・軽度認知機能障害の人々に有益であるか否かをどちらかに確信するためには、さらなる評価が必要だと思います。
このような人々を対象とした新しい治療法にも、同様の基準が適用されるべきだと思います。ADの患者さんを診察し、その影響を知っている医師や医療従事者と同様に、私も効果のある治療法を望んでいます。そのような新しい治療法が意味のある効果を持つことを確認する必要があります。aducanumabのこれまでの結果は、明らかにそうではありませんでした。
Todd E. Golde
フロリダ大学医学部医学科
投稿日: 2020年11月20日
Knopman氏やSchneider氏らが発表した、現在のアデュカヌマブの臨床試験では有効性を証明できないという研究結果に同意します。善意の多くの関係者がこれを推し進めようとしているのが心配ですが、これは危険な道です。データに頼る必要があるのです。全体的に見て、私の「ゲシュタルト」は、ADのこの初期臨床段階では、非常にささやかな有益な効果があるかもしれないが、安全性、コスト、そして可能性のあるアクセス問題を考慮すると、これは我々が期待していたホームランにはほど遠いということです。もしかしたら、バントシングルを試みて、一塁で非常に惜しい判定がなされたようなものでしょうか(しかし、ほとんどの人が打者はアウトだと言っています)。
私が見た中では、まだはっきりとした答えが出ていない別の懸念があります。まず、アミロイドリガンド結合の減少と、脳内の真の病的な生化学的Aβとの間には、どのような関係があるのでしょうか。もしそれが強いものであれば、この薬剤は厳密な二次予防試験で評価されるべきだと思います。
第二に、高用量での副作用を考えると、評価者側と参加者側の両方からの盲検化の完全性を心配しています。これについては、正式に対処されたものを見たことがありません。
3つ目は、第1b相試験でPETによるアミロイドリガンド結合の強い減少が見られた人たちの長期的な結果です。もし、本当に大きな効果があったのであれば、これらの人たちは、病気の臨床経過がかなり大きく変わっているはずです。しかし、E4キャリアーが依然として良好な状態にあるのであれば、私はそれを知りたいと思います。承認を得るための18ヵ月後の疾患修飾に対する認知機能とADLのベネフィットの目標は、かなり控えめです。これらの目標を達成しても、持続的な効果が得られない薬は、公衆衛生上あまり有益ではありません。繰り返しになりますが、小規模で長期間の試験を行えば、実際にはもっと多くのことがわかるかもしれませんし、登録が容易なために基本的には同じ時間で済むかもしれません。
これは残念なことですが、予測できる結果です。私や他の研究者が複数の論文で述べているように、このデータはアミロイド仮説を弱めるものではありません。ただ、病気の経過を変えようとする人については、もっとよく考える必要があるということです。私は何年も前から、”早期AD “という言葉を使うのはやめるべきだと考えています。確かに、症状的には早期ですが、病理学的には後期なのです。症状が出るということは、脳が臓器不全に陥り始めているということです。ADの症状のある段階を臓器不全と考えると、症状のある段階での治療は、今までとは根本的に異なるアプローチになります。
Russell Swerdlow
カンザス大学
投稿日: 2020年11月20日
ここではいくつかのレイヤーを考慮する必要があります。対照的な第3相試験の結果を考えると、積極的な介入が低下速度を減衰させたと確信するのは難しい。否定的な研究結果ではなかったとしても、何が原因で減少したのかを考えることは重要です。衰退率のわずかな減少は、積極的な介入を受けた人たちの真の利益を反映している可能性があるが、最も急速に進行した人たちをグループから排除することによって生じる可能性もある。
FDAの説明文書では、一般的にADのAPOE4キャリアはAPOE4非キャリアよりも急速には衰えないとする2つの論文が引用されているが、その2つの論文のうち1つが、APOE4キャリアのMCIは実際に急速に衰えると報告していることは示されていない(Petersen et al 2005年の図1Cでも示唆されている)。
APOE混合物のシフトがグループ内での衰えの減衰というイメージを生み出すかどうかについてさらに詳しく説明すると、この問題はbapineuzumabで以前にも取り上げられたが、スポンサーはAPOE4キャリアのみ、あるいはAPOE3キャリアのみを対象とした別の第3相試験を実施することでこの問題を解決した。ご存知のように、これらの試験では否定的な結論が出ている。この問題は、最近のBAN2401でも取り上げられたが、APOE4キャリアに重大な有害事象を戦略的に引き起こし、APOE4キャリアの盲検化にもつながる薬剤を扱っている以上、再び取り上げられることは間違いない。
参考文献
Barnes DE, Yaffe K. Vitamin E and Donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005 Sep 1;353(9):951-2; author reply 951-2. PubMed.
Bruno Pietro Imbimbo
Chiesi Farmaceutici S.p.A.
投稿日: 2020年11月20日
私は、Tristan Massie氏がFDA/Biogen社の文書の統計セクションで提起した問題について、また、David Knopman氏らがAlzheimers & Dementia誌に掲載された最近の論文で提起した方法論的および臨床的な問題について、それぞれ同意します。
私は、両試験には以下のような大きな問題があると考えています。
1. 有効性と高用量での有害事象の両方について、多くの患者が試験を完了していないこと。
2. 特に高用量での盲検化は、ほとんどがARIAによるものである。
3. 効果量が限られており、認知・臨床尺度の主観性と逆相関している。基本的に、EMERGEの平均効果量は、評価者による主観的な評価項目が多い尺度ほど大きく、客観的な尺度であるMMSEでは効果量が小さい….MORE。
4. 10mg/kg群に速い減退が見られたことは、BACE1阻害剤やγセクレターゼ阻害剤で見られたのと同様に、一部の患者に高用量で有害な作用があることを示していると解釈される。我々は、ADにおける他のいくつかのモノクローナル抗体(オリゴマーAβ種を標的とすると主張しているもの(crenezumab)を含む)との関連で、aducanumabの話を考えるべきである。モノマー、オリゴマー、プロトフィブリル、フィブリルは平衡状態にあり、主に特定のAβ種に作用する薬剤は、他の種にも影響を及ぼす。この点を考慮すると、Aβの種に対する親和性プロファイルが異なるモノクローナル抗体で臨床失敗が見られた理由も説明できる。 今回、FDAがアデュカヌマブを承認した場合、次のようなことになる。1)FDAが、無益なために中断された2つの重要な第3相試験に基づいて薬剤を承認したのは初めてのことである。
Douglas Galasko
カリフォルニア大学サンディエゴ校
投稿日: 2020年11月20日
私はバイオジェン社の有料コンサルタントなので、FDAのアデュカヌマブの審議について直接コメントすることはありません。ここでは、アデュカヌマブの臨床試験やその他のデータをもとに、今後のAD治療薬に影響を与える可能性のある一般的な疑問を提起します。
MCIや軽度ADにおいて、CDR-SBを主要評価項目とした場合、どの程度の傾きの変化が意味を持つのか?認知能力や機能的能力をより長く維持することは、(たとえ記憶障害が残ったとしても)患者さんやそのご家族にとって価値のあることだと思われますが、どの程度の臨床的変化が意味のあることなのか、コンセンサスは得られていません。しかし、どの程度の臨床的変化が有意義であるかについては、意見が一致していません。
より大きな効果の大きさを明らかにする可能性があるために、この集団を対象とした試験は18ヵ月よりも長く行うべきでしょうか?人気のある18ヵ月デザインは、自然史研究というよりは習慣から生まれたような恣意的な期間である。そして ADの初期段階において、ADAS-cogに代わる新しい認知機能検査が必要な時期なのでしょうか?
ARIAは、CDRとADL評価を提供する人々の盲点をついています。FDAの審査官の中には、これが大きな問題であると考える人もいましたが、アデュカヌマブ試験のブリーフィング・ドキュメントによると、ARIA後の評価をすべて省略しても、結果は変わりませんでした。ARIAは、アミロイドを除去する抗体にとって、盲検化できない可能性があるため、さらなる議論が必要である。
アミロイド負荷はADの認知機能の測定値とは相関しないため、除去の程度が臨床結果と直接相関するとは考えられません。沈着したアミロイドを除去した後、脳内で何が起こっているのか、また、除去した後、どのくらいの期間追跡調査をすれば、臨床的に効果があるかどうかを評価することができるのか、理解できていません。アミロイド除去が神経変性に影響を与えるかどうかを評価するために使用できるバイオマーカーがあり、DIAN-TU gantenerumab試験の脳脊髄液データはこの主張を支持している。アデュカヌマブの臨床試験において、血漿中のP-tauやNfLのバイオマーカーがどのようになるかは非常に興味深いことであり、血漿中のバイオマーカーの出現は、今後の臨床試験に劇的な影響を与えるでしょう。
諮問委員会は代用マーカーについて何度か言及していますが、アミロイドPETだけでは臨床試験での代用マーカーとしての役割は果たせないでしょう。タウPET、脳脊髄液や血漿中のP-tauバイオマーカー、シナプスバイオマーカーなどを提案したいところですが、これらはサロゲートというよりも、抗アミロイド試験における標的の関与による下流効果のマーカーと考えた方が保守的です。そのため、ADスペクトラムに沿った症状のある患者を対象とした試験で、臨床的有用性を示す必要性が残っている。
Daniel Gillen
カリフォルニア大学アーバイン校
投稿日: 2020年11月20日
この臨床プログラムには複数の問題があり、アデュカヌマブの有効性を評価することは困難です。これらの多くは、統計学的および臨床的観点から非常に技術的なものです。そのすべてを説明することはしませんが、諮問委員会の会議でも取り上げられた2つの大きなポイントに焦点を当てます。
まず明らかな問題は、301と302の2つの並行したフェーズ3試験の結果が食い違っていたことです。一般的に、FDAの承認には2つの独立した確認試験が必要です。これは通常、1つは統計的に有意であり、もう1つは有効性の裏付けとなる証拠を提供し、治療効果の点推定値を有意な試験のものと一致させることを意味します。アンメットニーズがある場合(今回はこれに該当する)治療効果の推定値が臨床的に意味があり、その結果が非常に有意であれば(例えば、p値が0.001以下)FDAは1つの確認試験に基づいて迅速な承認を与えることができる。
aducanumabの問題点は、302試験が無益であるとして早期に中止され、その後、有益な効果を示したという事実に加えて、2つの試験の結果が一致せず、全体として有効性を示す説得力のある証拠が得られなかったことにあります。この会議でFDAは、最初の論点で委員に302試験のみを検討するよう求めました。これは、多くの委員から不適切な姿勢であるとの意見が出されましたが、私も同感です。
302試験で得られた有効性の科学的根拠を評価する際に、301試験の結果を簡単になかったことにすることはできません。これらの研究は非常に類似しており、私の意見では、優れた科学者であれば、301の結果を考慮せずに302の結果を「選択」するべきではありません。FDAの統計学者の報告書は、302の結果を301の結果と照らし合わせて解釈すべきだと正しく指摘しています。そうすると、この2つの試験を全体として見た場合、上述の2つの状況のいずれにおいても、承認に必要な科学的・統計的証拠のレベルを満たしていないと私は考えます。
スポンサーは、「有効性の裏付けとなる独立した試験」を提供するために、103試験の結果を提示した。この議論の問題点は、103試験は301および302試験に先行する試験であり、それらの設計に影響を与えたことである。したがって、301および302試験の実施は、ほぼ確実に103試験の結果を条件としていた。したがって、私の考えでは、103試験は、承認を正当化するための裏付けとなる証拠を提供する独立した確認試験とは、どう考えても考えられません。
以上の2つの主な理由に加え、他の多くの理由から、現時点ではaducanumabの承認は正当化されないと考えます。私は、この恐ろしい病気を治療するためのアンメットニーズを非常によく理解しており、ADの予防および治療のための安全で効果的な治療法を発見するために、自分の専門的な時間の多くを捧げてきました。しかし、私は、どのような治療法であっても、承認前に安全性と有効性を示す説得力のある証拠が必要だと考えています。そうしないと、結果があまりにも大きくなってしまうからです。
私は、FDAが、302試験の結果を裏付ける証拠が得られるような、十分にデザインされ、十分にコントロールされた無作為化臨床試験を再度要求することを望みます。スポンサーがそのような試験を実施する意思があり、その結論として支持できるデータを提供できるのであれば、承認は正当化されると思いますが、それまでは必要ありません。
Eric Siemers
Siemers Integration LLC
投稿日: 2020年11月23日
2020年11月6日に開催されたFDA諮問委員会では、提出されたアデュカヌマブに対して委員が全体的に否定的な見解を示しました。しかし、この会議では、プロセス上、最も重要な問題を十分に議論することができませんでした。スポンサーは、当然のことながら、提出書類に対して肯定的な見解を示しました。驚くべきことに、FDAの臨床審査は、スポンサーの熱意を多くの点で反映し、またそれを上回るものでした。FDAからの統計的レビューは、承認を与えるべきではなく、別の試験が必要であると結論づけられたが、会議中には発表されなかった。このため、諮問委員会のメンバーは、バーチャルな会議の中で、比較的短い時間でFDAの臨床レビューと統計的レビューのバランスをとるという厄介な立場に置かれた。このように、諮問委員会のメンバーが熟考して議論したにもかかわらず、プロセス自体が最も関連性の高い質問に十分に対応できなかった可能性がある。
スポンサーとFDAの臨床審査官は、否定的な301試験の結果を考慮することなく、肯定的な302試験を考慮すべきであると強く主張しました。また、第1b相試験である103試験は、裏付けとなる証拠とみなすべきであるとした。FDAの統計審査官は、1つの陽性と1つの陰性のフェーズ3試験がある場合、3つ目の試験が必要であり、103試験の結果は無視されるべきであるという伝統的な頻出論者であった。私の考えでは、これらの議論はどちらも正しくありません。確かに、諮問委員会は、301試験を「無視」して302試験のみを検討することを期待すべきではありません。同様に、103試験は有効性試験としてデザインされたものではありませんが、これらの結果を無視すべきではありません。
スポンサー、FDAの臨床審査官、FDAの統計審査官は、それぞれの立場を支持するために様々な事後分析を行いました。これらの事後分析の議論は、残念ながら諮問委員会の会議で多くの時間を必要とした。これらの分析には興味があるかもしれませんが、事後分析は仮説検証ではなく仮説生成であるという事実は、議論の間ほとんど無視されていました。このような議論の例として、国別に評価した効果の大きさがあります。スペインでは薬が効くが、イタリアでは効かないという考えは、生物学的にはほとんど妥当ではありません。これは、火曜日生まれの人は治療の恩恵を受け、水曜日生まれの人は受けないという事後分析と同じです。したがって、プラセボ群の減少率の変化、ApoEの違い、より一貫した投与を行ったサブグループ、急激な減少が見られなかったサブグループなどの議論は、すべて大きな塩梅で受け止めるべきです。これらの解析は仮説を立てるものであり、その結果は将来の臨床試験のデザインの指針となるかもしれませんが、aducanumabの申請に関して諮問委員会が最も重要な質問に答える上での有用性は限られています。
aducanumabの承認により、将来の疾患修飾薬の開発が困難または不可能になるという議論がなされています。これはありそうにありません。今後の試験の開始時期を遅らせることについての議論もありましたが、1993年にDATATOP試験が発表されて以来、開始時期を遅らせることの限界は認識されていました(Parkinson Study Group, 1993)。遅延開始デザインには明確な利点があります。一つは、試験参加者が、最初の試験期間は薬物とプラセボに無作為に割り付けられるが、その後はすべての参加者に有効な治療が行われることを理解できることである。潜在的な試験参加者の多くは、18カ月または24カ月間のプラセボ治療の可能性を受け入れ、その後、aducanumabよりも有効性と安全性が高い可能性のある薬剤で積極的な治療を受けることを約束するでしょう。したがって、アデュカヌマブの承認が、より有望な薬剤を用いて慎重にデザインされた後続の試験への登録に大きな影響を与えることはないと思われます。
委員会にとって最も有益であったと思われる議論は、2つのピボタル試験のみに適用される伝統的な頻度論的アプローチではなく、よりベイズ的なアプローチの価値です。現在では、すべてのAβ/アミロイド標的治療薬を一括りにすべきではない、特にセクレターゼ阻害剤とモノクローナル抗体を検討する際には、幅広いコンセンサスが得られているように思われる。モノクローナル抗体ソラネズマブは、EXPEDITION試験、EXPEDITION2試験、EXPEDITION3試験において、現在では低用量と考えられている用量で、小さいながらも一貫した数値の変化を示したとされる。BAN2401の第2相試験の結果は、第2相試験のあらゆる注意点を考慮すると、有望であると思われます。他のモノクローナル抗体に関するこれらの知見であっても、aducanumab試験に割り当てられた事前確率に影響を与えるはずです。aducanumabに割り当てられた事前確率がどのようなものであっても、103,301,302試験の結果を総合すると、aducanumabの有効性がゼロであるという事後確率はどのようなものになるのかという疑問が残ります。有効性がゼロである確率の議論は、薬事承認の議論に影響を与えるはずです。
注目すべきは、FDA統計レビューの以下の段落が委員会で十分に議論されなかったことです。”2019年3月21日以降の打ち切りを伴う全ITTデータに基づく条件付検出力は、無益時までの非OTC患者が何らかの形で完了できた場合にメタ分析が陽性となることから、以下のように0.61と推定される。このようなプール解析が、条件付きパワー計算のベースとなった中間治療推定値の根拠となりました。おそらく、この計算では試験間の異質性を十分に考慮していないと思われますが、利用可能なすべての第3相エビデンスを要約しており、中間期にプールされた治療効果推定値を使用するというスポンサーの当初の計画の精神に従っています。301試験と302試験のベイズメタ分析によると、対立仮説の事後確率は0.62で、それに対応する帰無仮説の確率は0.38です。” その分析結果と有効性がゼロである確率についての追加的な議論があれば、諮問委員会での議論の深さは大幅に改善されたことでしょう。
参考文献
初期のパーキンソン病における障害の進行に対するトコフェロールとデプレニルの効果。The Parkinson Study Group. J Biol Chem. 1993 Jan 5;268(1):608-12. PubMed.
Stephen Salloway
ブラウン大学
投稿日: 2020年11月24日
本試験では、301および302試験の早期終了と、試験途中での投与量の増加により、データセットが不完全となり、結果の解釈が明らかに複雑になっている。しかし、これらのデータを総合すると、3つの臨床試験すべてでアミロイド斑が用量に応じて減少し、3つの臨床試験のうち2つの試験でCDR-SBなどの臨床的に意味のある効果が得られたことがわかります。
私たちはAD治療の重要な岐路に立っており、FDAがアデュカヌマブに関して難しい決断を迫られていることを理解しています。クラス最高の薬を手に入れるためには、クラス初の薬を手に入れなければなりません。アデュカヌマブが承認されれば、治療の足がかりを得ることができます。これは、AZTが承認され、その限界にもかかわらず、HIVの研究に活気を与え、強力な新しい治療法をもたらしたことと同様です。アデュカヌマブが承認されれば、アルツハイマー病にとって、17年ぶりに承認された薬剤、ADによる軽度認知障害で初めて承認された薬剤、アルツハイマー病の中核的な病態を対象とした初めての治療薬など、多くの初の試みがなされることになります。
私は、アデュカヌマブの承認にあたっては、第3相試験で用いられた臨床およびバイオマーカーの基準と密接に連携し、認知症の専門家が管理し、慎重な第4相市販後調査プログラムを必要とすることを推奨します。患者様とそのご家族は、時間と生活の質を維持し、アルツハイマー病による障害を遅らせることを緊急に必要としています。彼らにとって、潜在的な利益は、本薬が承認されない場合の確実な衰えのリスクを明らかに上回っています。患者さんが医師と一緒にアデュカヌマブが自分に適しているかどうかを判断できるように、この薬へのアクセスが提供されるべきです」と述べています。
Philip Scheltens
アルツハイマーセンター・アムステルダム、LSP認知症基金マネージング・パートナー、キングス・カレッジ・ロンドン名誉教授。
Everard (Jort) Vijverberg
アムステルダムUMC神経科/アルツハイマーセンター・脳研究センター・アムステルダム
投稿日: 2020年12月01日
aducanumabについては、すでに多くのことが語られており、あらゆる科学的議論と同様に、意見は大きく異なっています。残念ながら、FDAの公聴会で明らかになったように、議論はまるで政治のように偏ってしまっています。これは、どのような議論においても有益なことではありません。
このような科学的議論は、データやアデュカヌマブの開発がこれまで歩んできた道のりが曖昧であることを反映しています。チューリッヒの同僚からの素晴らしい発見に基づき、非常に有望なフェーズ1データでは、アミロイドPET画像上で明確な用量依存性のアミロイドプラーク除去が示され、1年間の治療で臨床効果のヒントが得られましたが、この試験ではそのような効果を見出すための検出力はありませんでした(Sevigny et al 2017)。
最も合理的で標準的な医薬品開発のステップは、最適な投与量を検討する適切な第2相試験をデザインして実施し、臨床効果を確立するための検出力を確保し、それに基づいて大規模な第3相試験を実施することであっただろう。しかし、バイオジェン社は第2相試験をスキップして、すぐに2つの大規模な第3相試験に飛び込んだが、残念ながら無駄に終わってしまった。患者が試験に参加する10~15年前にすでに発症している病気を扱う場合、早期に試験を中止してはいけないという教訓がここにもある。
バイオジェン社は、2つの「未完成」な試験を行うことになったが、そのうちの1つはポジティブで、1つはネガティブであると思われる。両試験が同じ結果を示さなかった理由について、多くの賢明で合理的な事後分析が行われましたが、301試験が主要評価項目を満たさなかったことは事実です。Eric Siemers氏がコメントで正しく指摘したように、このような事後解析は仮説を立てるためのものであり、検証のためのものではありません。
Alzforumの多くのコメントから判断できるように、「aducanumabのグラス」は半分空なのか、半分埋まっているのかという見方ができます。肯定的に考えれば、AD治療薬開発の歴史上初めて、3つの臨床試験すべてにおいて、用量に応じたアミロイド・プラークの減少を実証したという事実に異論はないと思います。このこと自体が画期的なことです。
両フェーズ3試験において、この真の疾患修飾効果を臨床的に意味のある結果に結びつけることができなかったのは、時間の問題(短すぎた)あるいは結果指標の感度の問題と考えられます。
神経学の他の分野では、このような問題があってもFDAは止めません。他の神経学分野では、バイオマーカーの効果は明らかであるが、ニーズが非常に大きく、バイオマーカーの効果が非常に説得力があるという理由で、粗い尺度では曖昧な、あるいは臨床的に意味のある結果が得られない治療法を承認してきました。例えば、多発性硬化症。1993年にベタフェロンが初めて承認されましたが、その理由は「基礎疾患に影響を与える」というものでした。あとは歴史のとおりです。その後、多発性硬化症の治療法の改善と開発に成功したことを見ると、アルツハイマー病とどう違うのかと思うかもしれません。
アミロイド低下だけでは十分ではなく、他のアプローチとの組み合わせを開発する必要があることを理解した上で、膨大なアンメットニーズ、ADの治療法を求める現場の明確かつ現在の要望と組み合わせれば、我々はaducanumabの「条件付き承認」を主張することになるでしょう。これは、EMAが過去に使用した手段でもあります。
この分野の専門家は、エキスパート・センターに所属し、NIA-AAバイオマーカーでADが証明された特定の患者(おそらくApoE4キャリアのみ)に対して、厳格なARIAと臨床モニタリングを行いながら、この薬剤を実際に使用する責任を負うことができます。最高用量を長期間使用し、定期的にバイオマーカーと有効性のモニタリングを行い、継続的にデータを収集して当局と共有することができます。これは、COVID-19ワクチンの開発で行われている「ローリングレビュー」のようなものです(ロイター通信参照)。
私たちは、これがこの分野の解決策になると考えています。患者さんにとって非常に貴重で、失ってはならない時間を何年も待つことなく、治療法を承認することができるのです。
参考文献
Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM, Sandrock A. 追記:抗体アデュカヌマブはアルツハイマー病のAβプラークを減少させます。Nature. 2017 Jun 21;546(7659):564. PubMed.
George Vradenburg
アルツハイマー病への取り組み
投稿日: 2020年12月10日
現在、単一の疾患修飾療法がないため、アルツハイマー病を経験している何百万人もの人々は、アデュカヌマブの安全性と有効性に強い関心を持っています。
諮問委員会の意見は、評価プロセスにおいて重要です。しかし、上記の記事にあるように、時間的制約から多くの質問に答えられませんでした。FDAは、アルツハイマー病治療薬に関する長年の取り組みにより、1回の諮問委員会では説明できないほど多くの情報と証拠を入手していることを認識し、継続的な評価の中でこれらの疑問に対処することを奨励します。FDAは、規制に関する大きな裁量権を持っており、その裁量権を行使することで、HIV、癌、MSなどの患者さんへのケアに大きな進歩をもたらしてきました。アルツハイマー病で自分を見失っている人々には、それ以上の価値はありません。
諮問委員会のメンバーの多くは、臨床的レビューと統計的レビューの不一致を指摘した。彼らは、最適な解釈を求めてその違いを探るのではなく、統計的レビューの長所を褒め称え、臨床的レビューを否定した。私たちは、人はp値以上のものであることに注目しています。
もし分析結果が僅差であれば、FDAは、患者さんとその医師に、現在は何もない治療法を選択する余地を与えることに賛成の判断を下すべきだと考えます。承認後に実世界でのエビデンスを収集することで、aducanumabの有効性についての理解が深まります。私たちは、その作業のパートナーとなる準備ができています。
この病気と闘う家族にとっては、一日一日が大切です。私たちのコミュニティの何千人もの人々が、自分たちだけでなく将来の世代を助けるために、18ヶ月以上の期間を臨床試験に費やしました。その結果、本薬が脳内のβアミロイドを減少させることが確認されました。私たちは、アデュカヌマブのより良いデータを何年も待つことはできませんし、現在のデータが完璧ではないと思われても、より完璧な薬を求めることはできません。
わが国では、ベストインクラスの薬を期待する前に、ファーストインクラスの薬が必要なのです。そして、苦しんでいる人々には、アデュカヌマブの有用性に関する厳格で独立した規制当局の決定を、答えのない質問や検証されていない疑念に置き換えないプロセスが必要です。