
Contents
A scoping review of the pathophysiology of COVID-19
journals.sagepub.com/doi/full/10.1177/20587384211048026
Paul E Marik, Jose Iglesias, Joseph Varon, …
2021年9月26日初版発行 研究論文
概要
COVID-19は非常に異質で複雑な疾患である。実際、重度のCOVID-19は、医学的に知られている疾患の中で最も複雑なものの一つであろう。コロナウイルスに感染した患者に関わる分子経路の理解は飛躍的に進んでいるが,COVID-19の病態に関する包括的かつ総合的な理解は不足している。このような理解は、効果的な予防法や治療法の策定に不可欠である。臨床研究、プロテオミクス研究、ゲノム研究、および剖検データに基づいて、重症のCOVID-19感染症は、3つの基本的な病理学的プロセス、すなわち、制御不能な炎症を伴う肺マクロファージ活性化症候群、補体介在性内皮炎、および血栓性微小血管症を伴う凝固促進状態が関連していると考えられる。さらに、セロトニンの放出を伴う血小板の活性化、マスト細胞の活性化と脱顆粒が炎症亢進状態に寄与してる。また、多くの入院患者で自己抗体が検出されており、これが末端臓器の損傷と血栓促進状態を助長している。本稿では、重症化したCOVID-19感染症の主要な発症メカニズムを臨床的に紹介する。
キーワード
COVID-19,病因、剖検、マクロファージ活性化、微小血管炎、セロトニン、補体、NETosis
はじめに
COVID-19パンデミックは400万人以上の命を奪い、その勢いは衰える気配がない。SARS-CoV-2感染の大部分は自己限定的であるが、患者の約20%は症状を呈し、その多くは入院を必要とし、約3%の症状を呈した患者は致命的な転帰をたどっている。COVID-19に対する効果的な予防法や治療法を開発するためには、その発症メカニズムを正確に理解する必要がある。これまでに何万件もの論文が発表され、この病気の臨床的・基礎的な側面が研究されてきたが、この病気の発症メカニズムについては、統合的、包括的、かつ臨床的に焦点を当てたレビューはない。
COVID-19のフェーズ
COVID-19は、潜伏期、症候期、肺性フェーズの3つの段階を経て進行する(図1参照)5。SARS-CoV-2は感染力が強く、飛沫やエアロゾルによって感染する6 -9 SARS-CoVおよびSARS-CoV-2は、細胞表面タンパク質であるアンジオテンシン変換酵素2(ACE-2)にスパイクタンパク質の受容体結合ドメイン(RBD)を介して結合し、ヒト細胞に感染する10,11。ACE-2は、鼻咽頭や上気道の繊毛上皮、気管支上皮、II型肺細胞に加え、マクロファージ/単球、マスト細胞、血管内皮細胞などにも発現している10。ACE-2受容体の発現は、肺、脂肪、脳の微小血管系で最も多く、肝臓、腎臓、心臓では少ない10,13。ACE-2と結合したウイルスのスパイクタンパク質は、宿主の膜に結合したタンパク質である膜貫通型プロテアーゼセリン2(TMPRSS2)によるタンパク質分解分解を受ける14。TMPRSS2は、宿主とウイルスの膜融合に必要なスパイクタンパク質の構造変化を引き起こし、このスパイクを介した融合プロセスの後、体内に取り込まれたウイルス粒子はRNAゲノムを放出し、複製を開始する15。最近では、フーリンプロテアーゼや細胞内受容体であるニューロピリン-1(NRP1)が感染プロセスに関与していることが示されている16-18。
図1 COVIDの臨床段階

SARS-CoV-2は、潜伏期から症状が出るまでの間、鼻咽頭や上気道の繊毛上皮に感染する。12 ウイルスの拡散を制御するには、サイトカインのシグナル伝達や細胞間の接触を介した上皮細胞と免疫細胞の相互作用に依存している19。自然免疫は、ウイルス感染に対する免疫反応の第一段階である。ウイルスが侵入すると、感染細胞はいくつかのパターン認識受容体(PRR)を介して、異常なRNA構造の存在を検出する20。ウイルス特異的なRNA構造が関与すると、PRRがオリゴマー化し、下流の転写因子が活性化される。 21,22 IRFは、I型およびIII型のインターフェロン(IFN)を誘導し、IFNで刺激された遺伝子をアップレギュレートすることで、宿主の主要な抗ウイルス防御を指揮する様々なタンパク質の転写を引き起こす23。NF-κBが発現すると、炎症性サイトカインやケモカインが発現し、白血球の特定のサブセットが動員されるようになる。インフラマソームは、カスパーゼ-1を含む多タンパク質複合体であり、これが活性化されると、カスパーゼを介して前駆体サイトカイン分子が切断され、IL-1βやIL-18が放出される21,24。
SARS-CoV-2は、I型およびIII型インターフェロンの合成を阻害する。23,25,26 SARS-CoV-2の遺伝子産物には、非構造タンパク質1(NSP1)付属タンパク質ORF6,ORF3B、およびヌクレオカプシド(N)遺伝子産物が含まれており、これらの遺伝子産物は、シグナルトランスデューサーおよびアクチベーターオブトランスクリプション1(STAT1)の機能を低下させ、インターフェロンの合成を低下させる25,27。23,28,29 SARS-CoV-2の感染結果は、ウイルスの侵入量、ウイルスの複製速度、宿主のインターフェロンの産生、および炎症性メディエーターのバランスによって決まると考えられる。23,28,29 インターフェロン反応が活発で、自然免疫反応が有効な患者は、ウイルスを速やかに排除することができるが、大量のウイルスを接種した患者やインターフェロン反応が不十分または遅れている患者では、ウイルスが急速に複製され、上気道でのウイルス濃度が高くなってしまう。中程度の重症COVID-19患者では、分泌細胞がマクロファージ、T細胞、マスト細胞の動員を促進するケモカインの発現を有意に増加させている32。
中咽頭から肺へのウイルス接種物の吸引は、II型肺細胞や肺胞マクロファージに感染するウイルス量が多い患者で起こる可能性が高い12。IFN-IおよびIFN-IIIによる強力な反応が得られず、同時に高レベルのケモカインが誘導されるため、感染した肺組織に血液中の単球が集まることになる。マクロファージは、ACE-2受容体を発現している33。さらにマクロファージは、SARS-CoV-2の結合部位の露出と細胞膜との融合に必要な2つの酵素、furinとTMPRSS2を発現している34。COVID-19の肺性フェーズでは、「マクロファージがトロイの木馬の役割を果たし、肺実質内に特異的にウイルスを定着させることができる」と考えられている34。さらに、マクロファージにおけるACE-2の多様な発現は、SARS-CoV-2感染の重症度を左右する可能性がある34。下気道のマクロファージは、上気道のマクロファージに比べて、炎症性のケモカインやサイトカインをコードする遺伝子の発現が多いことが報告されている32。これらのマクロファージは、IL-8,IL-1β、TNF-αなどの炎症性サイトカインや、CCL2,CCL3,CCL5(RANTES)CXCL1,CXCL3,CXCL10などの様々なケモカインを発現しており、このマクロファージの亜集団は、単球のさらなる動員やマクロファージの分化を促進することで、過剰な肺の炎症に寄与していると考えられている32。
病因となる経路
36-39 インターフェロン反応の遅れと適応免疫の発達(中和抗体の出現)により、ウイルスの複製が停止し(ウイルスの死滅)40-42 ウイルスの複製が停止した後は、免疫系の過剰活性化や組織の継続的な損傷を防ぐために、活性化した免疫細胞を除去する必要がある。重症のCOVID-19患者で進行中の炎症反応は、ウイルスの除去が不十分であるというよりも、免疫系が過剰に活性化していることに起因する。23,43 これは、ナチュラルキラー(NK)細胞や細胞傷害性T細胞が活性化したマクロファージを除去できず、疲弊した細胞の表現型が形成されることと関係していると考えられる。Liら45は、SARS-CoVのssRNA GU(グアノシン、ウリジン)リッチフラグメントには強力な免疫賦活作用があり、TLR7およびTLR8経路を介してかなりのレベルの炎症性サイトカインTNF-α、IL-6,IL-12を誘導することを明らかにした45。ウイルスが除去されても炎症性メディエーターが産生され、マクロファージが継続的に活性化されることが、重度のCOVID-19感染患者に見られる進行性肺疾患の原因であると考えられる46。さらに、SARS-CoV-2感染マクロファージの代謝は、ミトコンドリアでの酸化的リン酸化から細胞質での解糖にリプログラムされる47。48,49 SARS-CoV-2感染者は、マクロファージ/単球の活性化が長期化している可能性がある。実際、Pattersonら50は、感染から15カ月後までの「Long-hauler患者」において、スパイクタンパクを含む活性化単球の存在を証明した。Orologas-Stavrouら51は、回復後2カ月の時点で、回復期の血漿提供者のCD4+ T細胞とB細胞のレベルが低下していることを示した。51 この研究では、以前に入院した回復期の血漿提供者は、CD8+制御細胞のレベルが非常に低く、Th17表現型とともに、炎症反応が長引いていることを示唆していた。追跡調査では、COVID-19感染後8カ月目にも同様の結果が得られている52。
図2 COVID COVIDのステージと免疫反応のタイム経過

COVID-19の肺性フェーズには複数の生物学的経路やプロセスが存在するが、我々は、重篤なCOVID-19を引き起こす2つの主要な病因プロセス、すなわち、i)肺における活性化マクロファージの蓄積(肺胞マクロファージ活性化症候群)と、その結果としての多臓器不全につながる高炎症状態、およびii)肺の微小血管系および脳や脂肪組織を巻き込む免疫血栓症を伴う内皮炎、があると考えている。この概念は、剖検調査、重症患者から採取した気管支肺胞洗浄液(BAL)のシングルセルプロファイリング、および重症COVID-19の臨床的特徴の評価に基づいている。重症COVID-19の病態は、肺への圧倒的な浸潤に加えて、脳、心臓、消化管、肝臓、腎臓、皮膚などを侵す多臓器疾患であることを強調しておく必要がある46。
重篤な COVID-19 感染症の病態について
COVID-19感染症の発症メカニズムを明らかにするには、剖検調査が有効である。一般的に、これらの研究では、主に活性化したマクロファージや単球、CD4+およびCD8+リンパ球からなる広範な免疫浸潤が認められ、びまん性肺胞障害(DAD)や組織性肺炎の特徴が見られた56-63 。一般的に、細胞浸潤は血管や血管周囲よりも肺実質領域で最も顕著である56。Wangら64は、剖検時に肺胞に見られた主な細胞タイプは、IL-6,IL-10,TNF-αを発現している活性化マクロファージであることを示した。Melmsら65は、COVID-19の死亡者の肺組織を対象に、一塩基RNA配列解析を行った。T細胞は異常な反応性を示した。肺胞2型細胞は、炎症に伴う一過性の前駆細胞状態をとり、肺胞1型細胞に完全に移行することができず、肺の再生が損なわれた。さらに、CTHRC1+の病的な線維芽細胞が拡大し、肺線維症の原因となっていることも確認された。56-59,61,62 マクロファージ/単球が主な免疫浸潤である一方で、好中球と好中球細胞外トラップ(NETs)が多くの剖検研究で報告されている66。 -NETsは、アポトーシスした好中球から放出されるdsDNA、ヒストン、抗菌ペプチド、プロテアーゼからなる細胞外の網目構造である。SARS-CoV-2で活性化した好中球から放出されたNETは、肺の上皮や内皮の細胞死を促進すると考えられている。 -68,70,71 さらに、NETsは免疫血栓症を促進し、COVID-19患者の血栓促進状態に寄与していると考えられる。COVID-19におけるマクロファージの機能不全は、ネトーシスをさらに促進する可能性がある。重度のCOVID-19感染は、典型的には微小血管血栓症を伴う内皮炎を伴い、主に肺、脳、皮膚、脂肪組織の血管系が侵される56。 -59,61,62 多くの著者が、肺組織におけるC3dおよびC5b-9複合体の強い染色を伴う補体介在性微小血管傷害を報告している。
剖検調査では、肺組織に豊富なウイルスRNAが検出され、肺胞マクロファージや隣接する中隔毛細血管の内皮に局在していた59。Nuovoら75は、皮下脂肪や脳の微小血管内のACE-2+内皮細胞にウイルスRNAを含まないウイルススパイクタンパクが局在することを示した75。これらの研究者は、肺毛細血管の内皮細胞が死滅すると、循環系に偽ウイルスが放出され、これらの偽ウイルスがACE-2+内皮細胞に付着することで、補体経路/凝固カスケードが活性化され、その結果、全身性の内皮炎と血液凝固促進状態が引き起こされると推測している59,75,76。
COVID-19の初期段階における肺の組織学的変化を記録した2つの報告があり、この疾患における単球とマクロファージの主な役割を示すさらなる証拠となっている77,78 Tian er al)。 Tianらは、SARS-CoV-2による感染が重なっていることが認識されていなかった時期に、肺の腫瘍に対して手術が行われた「偶発的な」肺採取の2例を報告している77。腫瘍のない肺の組織検査では、肺胞マクロファージが広範囲に浸潤し、好中球の浸潤は最小限であった。肺胞壁には、増殖した間質性線維芽細胞とII型肺細胞の過形成からなる、びまん性の肥厚が見られた。気腔内には局所的な線維芽細胞の栓と多核巨細胞が見られ、びまん性肺胞障害の増殖期の程度が異なることを示している。Zengら78は、良性の肺結節のために肺葉切除を受けたSARS-Cov-2感染「前症候性」患者の肺から採取した生検標本を評価し、マクロファージを主な細胞種とする肺浸潤を報告している78。
これらの組織学的所見は、COVID-19で挿管された重篤な患者から採取した気管支肺胞洗浄液(BAL)のシングルセルRNAシーケンシングによって強く裏付けられた79,80。BAL液を分析すると、マクロファージが豊富に存在することがわかった。さらに解析を進めると、マクロファージは主に炎症性単球由来で、肺胞に常駐するマクロファージは相対的に少ないことがわかった。末梢血のサイトカインパターンと同様に、マクロファージは、IL-1,IL-6,TNF-α、およびCCL2,CCL3,CCL4,CCL5(RANTES)CCL9などの複数のケモカインをコードする遺伝子の発現が増加しており、古典的なM1マクロファージに特徴的な遺伝子発現パターンを示していた。Xiongらは、BAL洗浄液や末梢血中の単核/マクロファージ細胞のトランスクリプトーム解析を行い、CXCL10やCCL2/MCP-1の産生が増加していることを明らかにした81。これらのケモカインやサイトカインは、組織由来や末梢血中の単球/マクロファージを感染部位に移動させ、衰えない炎症反応が続く中で、最終的に肺胞マクロファージに取って代わるように仕向けている可能性が高い。
これらのデータを総合すると、肺マクロファージが炎症性単球を肺に呼び込み、これがサイトカインやケモカインを産生し、炎症亢進の悪循環を助長していることが示唆される82,83。肺実質へのマクロファージの無秩序な呼び込みと活性化が、重篤なCOVID-19感染症の病態に中心的な役割を果たしていると考えられる。活性化されたマクロファージが過剰に蓄積されるのを阻止するのは、通常、細胞障害性のCD8+およびナチュラルキラー(NK)Tリンパ球である。重症のCOVID-19患者では、疲弊した表現型を持つCD8+およびNK T細胞の数が著しく減少している。43,84-86 炎症性サイトカイン(特にIL-6)の過剰産生は、T細胞の機能不全と関連している44。さらに、SARS-CoV-2のスパイクタンパクが肺上皮細胞に細胞内で発現すると、NK細胞の活性化と脱顆粒能力が低下する87。重症のCOVID-19患者で報告されている顕著なT細胞機能不全は、二次的な細菌・真菌感染症のリスクを高める。
重症COVID-19の臨床的特徴は、活性化されたマクロファージによる広範な肺浸潤がこの疾患の主要な病因であるという概念を裏付けるものである。まず、胸部CTスキャンで認められる進行性の多巣性ガラス混濁の特徴的なパターンは、組織性肺炎に典型的な単核細胞肺胞浸潤を強く支持している(図3参照)91。COVID-19肺炎の患者は、ほぼ例外なく血清フェリチン値が上昇している92。そして最後に、重症のCOVID-19の臨床的特徴と多臓器の病変は、マクロファージ活性化症候群/血球貪食性リンパ組織球症症候群と密接に重なっている。Bryceらの剖検シリーズでは、58’目立った血球貪食症と二次的な血球貪食性リンパ組織球症様症候群が多くの症例で見られた。
図3 COVID-19組織性肺炎のCT特徴の進行
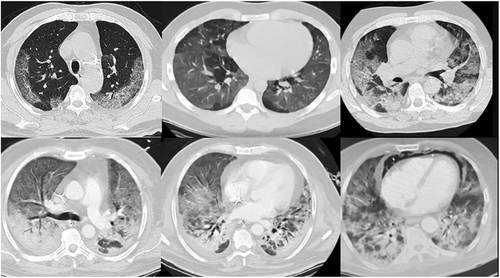
他の著者は、骨髄吸引で血球貪食症の特徴を報告している93。実際、重症のCOVIDは、マクロファージ活性化症候群のサブタイプと考えるべきである。
SARS-CoV-2微小血管症と補体活性化
重症COVID-19感染症は、上記の肺の特徴的な組織学的変化に加えて、典型的には内皮炎と微小血管血栓症を伴い、主に肺、脳、皮膚、脂肪組織の血管系が侵される56-59,61,94。中型動脈、細動脈、毛細血管の血栓は、多くの子孫の脳に広範囲の微小血栓と急性梗塞を伴って典型的に見られる。58 血栓には血小板が多く含まれていることが知られている。Magroらは、重症のCOVID-19患者において、肺の中隔毛細血管、皮膚や脳の毛細血管、静脈、動脈の微小血管系に補体を介した微小血管傷害が見られることを示した59,60。SARS-CoV-2における補体活性化の重要性は、実験的なSARS-CoVモデルにおいて、C3ノックアウトマウス(C3 -/-)では、野生型マウスに比べて肺の傷害や炎症性浸潤が有意に少ないことで示された95。
補体系は自然免疫系の一部であり、抗体依存性古典経路、マンノース結合レクチン(MBL)経路、代替経路の3つの経路を経て活性化される96。COVID-19では、SARS-CoV-2自身や、病気の後期に損傷を受けた組織や死にかけている細胞など、複数の経路を経て補体が活性化されていると考えられる60,96。MASP2は補体タンパク質C2およびC4を切断し、C3コンバーターゼを活性化してC5b-9複合体を形成する。MASP2は補体タンパク質C2とC4を切断し、C3コンベルターゼを活性化してC5b-9複合体を形成する。さらに、MASP2が補体経路と凝固経路の両方を活性化することにも注目したい98。さらに、補体が内皮を破壊すると、凝固促進因子であるvon Willebrand因子とFVIIIが放出される。したがって、補体の活性化は、SARS-Co-V-2感染症患者の凝固促進状態の発生と密接に関係している。
COVID-19で入院した患者は、通常、D-ダイマー濃度の上昇と血小板減少を特徴とする凝固性亢進状態に陥り、生命を脅かす播種性血管内凝固症候群(DIC)に進展する可能性がある99。したがって、低分子量ヘパリンを用いた早期かつ長期の薬理学的血栓予防が推奨される99。
COVID-19組織性肺炎とARDSではない
100,101 COVID-19 の肺性フェーズは典型的な ARDS というよりもむしろ組織性肺炎に特徴的な特徴を持っている。COVID-19の組織性肺炎は、ベルリン基準によるARDS症候群の非特異的診断基準を満たしているが、102 COVID-19の肺炎の臨床的、X線的、組織学的特徴は、古典的ARDS,103-105やAsbaughらによるARDSの原型とは大きく異なる106。COVID-19 の X 線画像は非常に独特であり、古典的な ARDS で見られる依存性の空 間圧密(スポンジ/ベビー・ラン)とは似ても似つかない107 。COVID-19の初期のX線像の特徴は、末梢のパッチ状の多葉性地溝浸潤であり、病状の進行に伴い、X線像の特徴はステレオタイプのパターンを示す(図3参照)。ARDSは肺コンプライアンスの低下を特徴とするが、COVID-19患者の肺は(少なくとも初期は)かなりコンプライアンスが高い108,109。最も注目すべきは、ARDSは高い血管外肺水(非心原性肺水腫)を特徴とすることである110,111。我々は、COVID-19組織性肺炎のICU患者のコホートにおいて、血管外肺水量指数(EVLWI)を測定したが、EVLWIが上昇した患者はいなかった(個人データはファイルにある)。最後に、COVID-19組織性肺炎と古典的なARDSの病態は全く異なるものである。上述したように、COVID-19肺炎は、好中球が少なくマクロファージが大量に浸潤しているのが特徴である。好中球減少はARDSの重症度を低下させ113,実験モデルではマクロファージの枯渇がコロナウイルス肺疾患の重症度を低下させる114。COVID-19肺炎とARDSの両方でびまん性肺胞損傷(DAD)が報告されているが、DADは進行した急性肺損傷の非特異的な所見である。COVID組織化肺炎とARDSの違いが治療に与える意味は大きい。標準的なARDSの治療(PEEPを段階的に増加させる)115はCOVID肺を傷つけ、予防しようとしている病気を引き起こす可能性があるからである。COVID-19患者(ARDSではない)に見られる不思議な所見は、呼吸反応が鈍くなる「サイレント低酸素症」である。69,116 この現象は、SARS-CoV-2が頸動脈小体の化学受容器や脳幹の機能障害に関与している可能性がある(図4)。
図4 重症COVID-19感染症の発症メカニズム
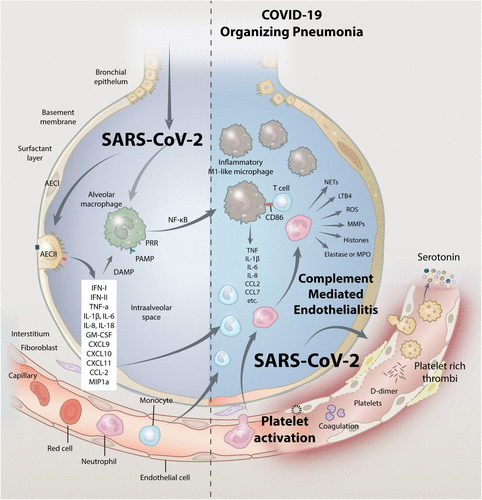
マクロファージの活性化と免疫介在性の内皮炎が重症COVID-19感染症の主要な発症メカニズムの根底にあるが、他の相互作用する経路も重要な役割を果たしていると考えられる。これらの経路には、血小板の活性化と血中セロトニン濃度の上昇、マスト細胞の活性化、自己抗体、レニン・アンジオテニン系の調節障害などが含まれる(ただし、これらに限定されるものではない)。
血小板の活性化と血中セロトニン濃度の上昇
SARS-CoV-2や偽ウイルスの内皮細胞への感染、および免疫システムの調節不全は、内皮を損傷し、血液凝固を活性化し、ミクロおよびマクロの血栓形成を伴う重篤な内皮炎を引き起こす。さらに、ACE-2受容体は血小板上に存在し、これが重症のCOVID-19感染症に特徴的な大量の血小板凝集に寄与している可能性がある。35,118,119 血小板の活性化は、血栓促進状態を助長し、炎症反応を増大させる。35 さらに、重症のCOVID-19患者の血小板は、健常者の単球においてex vivoで組織因子の発現を誘導することが示されている。35 COVID-19では血小板が過剰に活性化されるだけでなく、血小板の活性化の程度は疾患の重症度と相関しているようである。35,122 さらに、COVID-19 患者の血小板は、COVID-19 を発症していない ARDS 患者の血小板と比較して、トロンビンに反応して非常に効率的に活性化されることが示されている123。
COVID-19 患者では、血小板の活性化が進み、肺循環による除去が低下した結果、循環中のセロトニン(5-ヒドロキシトリプタミン、5HT)のレベルが上昇していると考えられている122-125 COVID-19 の活性化した血小板の顆粒から放出されるメディエーターの中でも、セロトニンは、全身のセロトニンプールの 95%が血小板顆粒内に貯蔵されているという点でユニークであり、放出されたセロトニンの除去には健康な肺内皮が必要である126。 -129-132 セロトニンは、肺血管の緊張および低酸素性肺血管収縮のメディエーターとして確立されている。129,132 さらに、セロトニン自体が血小板の凝集を促進し、免疫・血栓サイクルを伝播させる。133 セロトニンは肺線維化を促進し、重症COVID患者に発症する進行性の線維化に寄与している可能性がある。セロトニンの循環レベルの上昇は、COVID-19感染症に見られるいくつかのユニークな臨床観察を説明することができる。これらの観察には、説明のつかない重度の過呼吸(不適切な速い呼吸)の存在、135 足首のクローヌスと反射亢進の高い発生率、136 重度の下痢、冠状動脈の血管攣縮による心筋虚血などがある137。 -さらに、高解像度のCT分析によると、肺微小血管床のびまん性血管収縮は、COVID-19肺損傷における最も初期の異常の一つであり、顕著な肺浸潤の出現に先立つものであるようである。この所見にはセロトニンの増加が関与している可能性がある。血漿中のセロトニンの上昇は、血液脳関門の破壊に関与し、COVID-19で報告された神経学的所見の原因となる脳大血管の血管収縮に寄与している可能性がある。 140 セロトニン受容体(5HT-2)の効果的な拮抗は、セロトニンを介した肺血管収縮を逆転させ、肺血小板の捕捉を軽減し、血小板の活性化と凝集を抑制し、呼吸駆動の増加を正常化し、肺線維症のリスクを軽減し、重度のCOVID-19における腎、神経、心血管系の有害現象を抑制する可能性がある122。
抗うつ薬(SSRI)は、血小板のセロトニン量を減少させ、血小板凝集後のセロトニンの放出を減少させる141。 -146,147 フルボキサミンは、選択的セロトニン再取り込み阻害薬(SSRI)であり、シグマ-1受容体を活性化してサイトカインの産生を低下させる。これらの研究は、循環しているセロトニンの増加が COVID-19 疾患の病因に関与しているという概念を支持している。
マスト細胞の活性化
149 ウイルスは、ssRNAやdsRNAの複製中間体、補体、サイトカインなどのウイルス性または炎症性の生成物を介して、直接的または間接的にマスト細胞を活性化する。148 さらに、マスト細胞は、SARS-CoV-2の結合に必要なACE-2や、スパイクタンパクのプライミングに必要なTMPRSS2を発現している。活性化されたマスト細胞は、ヒスタミン、ロイコトリエンB4とLTC4,プロスタグランジンD2,血管内皮成長因子、トリプターゼやキマーゼなどのセリンプロテアーゼなどの血管作動性メディエーターを放出する148 。また、マスト細胞は、タイプ2サイトカインであるIL-4とIL-6を放出し、サイトカインのネットワーク化にも貢献している。さらに、COVID-19患者の血清や肺組織では、MC由来のプロテアーゼが上昇している153。ただし、重症のCOVID-19疾患におけるMCの病因的役割については、さらなる評価が必要である。
自己免疫疾患としてのCOVID-19
SARS-CoV-2 に感染すると、(おそらくスパイクタンパクによる)分子模倣の結果として、広範囲の自己抗体が産生され、これが COVID-19 感染症の病態生理に寄与する。Wang らは、入院中の COVID-19 患者 172 名を対象に、サイトカイン、ケモカイン、補体成分、細胞表面タンパク質などの免疫調整タンパク質に対する自己抗体が高頻度に認められたことを示した157。Zuoら158は、COVID-19で入院した患者の52%が抗リン脂質抗体を持っていたと報告している158。COVID-19感染症だけでなく、スパイクタンパクを産生するワクチンも、抗血小板抗体、血小板減少、および深遠な凝固促進状態と関連している159-161。
Pascoliniら162は、COVID-19肺炎患者33人のうち15人(45%)が少なくとも1つの自己抗体に陽性で、そのうち11人が抗核抗体(ANA)に陽性であったと報告している162。ANAの核小体パターンは、全身性硬化症の臨床経過を特徴づける間質性肺炎と関連することが多いことに留意すべきである163。抗好中球細胞質抗体(ANCA)を持っていた患者はいなかった。さらに、自己抗体を持っていた患者は予後が悪かった。164,165 I 型インターフェロンに対する自己抗体は、生命を脅かす重篤な感染症と関連している。166 交差反応性神経細胞抗体は、COVID-19 疾患に関連する多様な神経学的合併症と関連している167,168 SARS-CoV-2 に感染した後、I 型糖尿病と GAD65 A 抗体の存在が報告されている169。
ACE-2の発現変化、RAASのバランスの崩れ、ブラジキニンの増加
ACE-2は、アンジオテンシンIIのカルボキシル末端アミノ酸であるフェニルアラニンを切断して、血管拡張作用のあるアンジオテンシン1-7を生成する膜タンパク質である170。SARS-CoV-2によって誘導されたACE-2のダウンレギュレーションとそれに続く欠損は、アンジオテンシンIIからアンジオテンシン1-7への変換を阻害する。 171 SARS-CoVに感染したマウスを用いた研究では、ウイルスが上皮細胞に侵入した後、ACE-2が体内に取り込まれると、ACE-2の表面発現が低下し、肺の炎症が悪化することが示されている172。173 循環アンジオテンシンIとアンジオテンシンIIの増加は、炎症、酸化ストレス、線維化と関連している。また、アンジオテンシン1-7は、炎症性タンパク質のレベルを低下させることにより、血管系において抗炎症作用を示する。さらに、ACE-2はブラジキニンを分解し、ACE-2が欠損すると、循環中のブラジキニンが過剰になる174。動物モデルでは、組換えACE-2がマウスを重度の急性肺損傷から保護することが実証されている175。
本研究の限界
COVID-19 は非常に複雑でダイナミックな疾患である。本研究では、可能な限り最新の情報を提供するように努めたが、本レビューに含まれていない重要な新しい発症メカニズムが報告される可能性もある。さらに、SARS-CoV-2は急速に変異するウイルスであり、発表されたデータの多くは最初の「武漢変異株」に適用されている。したがって、この病気の病態生理は、新しい変異株ごとにダイナミックに変化する可能性がある。最後に、我々はマクロファージの活性化が重症のCOVID-19の病因の中心であると考えているが、マスト細胞、内皮細胞、好中球、リンパ球のサブポピュレーションの役割については、さらなる解明が必要である。
まとめと結論
重症COVID-19感染症は、制御不能な炎症を伴うマクロファージの活性化、補体を介する内皮炎、血小板の活性化と高循環セロトニンを伴う血栓性微小血管症が重なった結果として発症する。さらに、マスト細胞の活性化、自己抗体、RAASの不均衡が重篤なCOVID-19感染症の病因となる。パンデミックの最初の6ヶ月間、世界保健機関(WHO)とほとんどすべての国のガイドラインは、重症COVID-19の管理に「支持療法のみ」の戦略を推奨していた176。患者は、いくつかの異なるフェーズ(臨床段階)を経て移行するため、治療はそれぞれの特定のフェーズに合わせて行う必要がある。抗ウイルス療法が有効なのは、ウイルスが増殖している症状のある段階だけである。5,177,178 そして最後に、万能なプロトコルは存在せず、各患者の臨床表現型に応じて治療戦略を個別化しなければならないことが肝要である。
著者の貢献度
PM, 論文のコンセプト, 文献のレビュー, 論文の第一稿, 最終稿のレビューと承認.JI, 論文のコンセプト, 文献のレビュー, 修正原稿, 最終稿のレビューと承認.JV, 文献のレビュー, 修正原稿, 最終稿のレビューと承認.PK, 文献のレビュー, 修正原稿, 最終稿のレビューと承認.
利益相反の宣言
著者は,本論文の研究,執筆,出版に関して,潜在的な利益相反はないと宣言した。
資金調達
著者は,本論文の研究,執筆,および/または出版に関して,財政的支援を受けていない。
身元保証人について
Paul Marik, MDは、原稿の内容に責任を持つ。